

Abstract
Neural tube defects are among the most common birth defects, with a prevalence of close to 19 per 10,000 births worldwide. The etiology of neural tube defects (NTD) is complex involving the interplay of genetic and environmental factors. Since nutrient deficiency is a risk factor and dietary changes are the major preventative measure to reduce the risk of NTDs, a more detailed understanding of how common micronutrient imbalances contribute to NTDs is crucial. While folic acid has been the most discussed environmental factor due to the success that population-wide fortification has had on prevention of NTDs, folic acid supplementation does not prevent all NTDs. The imbalance of several other micronutrients have been implicated as risks for NTDs by epidemiological studies and in vivo studies in animal models. In this review, we highlight recent literature deciphering the multifactorial mechanisms underlying NTDs with an emphasis on mouse and human data. Specifically, we focus on advances in our understanding of how too much or too little retinoic acid, zinc, and iron alter gene expression and cellular processes contributing to the pathobiology of NTDs. Synthesis of the discussed literature reveals common cellular phenotypes found in embryos with NTDs resulting from several micronutrient imbalances. The goal is to combine knowledge of these common cellular phenotypes with mechanisms underlying micronutrient imbalances to provide insights into possible new targets for preventative measures against NTDs.
Keywords: Neural tube closure, neural tube defects, micronutrients, environmental factors, multifactorial threshold, mouse models, human genetics
Introduction
According to the World Health Organization, neural tube defects (NTD) are among the three most prevalent congenital anomalies. Simply, NTDs are a failure to close the neural tube, the precursor of the brain and spinal cord, during embryonic development. In humans, neural tube closure takes place during the third and fourth gestational week, thus the onset of neural tube closure usually occurs before pregnancy is realized. Neural tube defects can be categorized based on the location of the neural tube affected. Craniorachischisis is the most severe form of NTD and is characterized by a fully open neural tube, exencephaly is a NTD in the cranial region of the neural tube and leads to anencephaly, whereas spina bifida is a NTD in the caudal region and is the most common form. Craniorachischisis and exencephaly result in infant mortality whereas infants with spina bifida can survive, but with higher risk of mortality and life-long complications. Given its relatively common occurrence, NTDs are a significant worry for families and a significant public health issue. Thus, it is critical to communicate about risks and preventative measures. While NTDs are common and there are recommendations for prevention, the etiologies of NTDs are complex and are still poorly understood.
Neural tube closure (NTC) entails taking a flat sheet of columnar cells and rolling it into a tube with regional specificity. This tube of neuroectodermal cells will continue to develop into the brain and the spinal cord. While this process seems simple, NTC is a complicated process and in humans NTDs are considered complex multifactorial disorders. NTC must utilize signaling gradients, transcriptional cascades, and coordinated cellular movements to accomplish this goal (Araya García, 2017; Nikolopoulou et al., 2017). First, neural ectoderm cells must be specified. This highly proliferative population of cells undergoes convergent extension to facilitate anterior-posterior elongation and medial-lateral narrowing, while apical constriction leads to the bending of the neural ectoderm folds at hinge points toward the midline. When the neural folds meet in the middle, the neural ectoderm and non-neural ectoderm or surface ectoderm separate, and each tissue layer then seals together by a mix of cellular rearrangements including the extension and intercalation of regional-specific cellular protrusions and cell adhesion. Each one of these steps is under discrete transcriptional control brought on, in part, by dorsoventral and anteroposterior morphogen gradients. The choreography of these processes is necessary for NTC, making it highly susceptible to failure when any one of these steps is dysregulated.
NTDs are a multifactorial disorder
Genetic factors
Neural tube closure is sensitive to both genetic and environmental insults and it is widely agreed that NTDs have multifactorial inheritance (Finnell et al., 2021; Lee & Gleeson, 2020; Zohn, 2012). As recently reviewed in Lee and Gleeson 2020, several lines of evidence suggest NTDs are a genetic disease, with the most impactful evidence being that siblings of NTD affected infants are more likely to develop a NTD than the general population (Lee & Gleeson, 2020). These authors also point out that infants with cranial neural tube defects do not survive and those that survive with spinal neural NTDs are not likely to reproduce suggesting that NTDs do not follow a Mendelian inheritance pattern, but instead there is a more complex genetic component (Lee & Gleeson, 2020). Studies in mice have been informative in uncovering genetic pathways that span different protein functions including cell adhesion, cell cycle, cell mitosis, cell migration, cell transport, cell polarity, cilia, cytoskeleton, epigenetic regulators, metabolism, signaling, transcription, translation, as well as yet unrecognized proteins (Harris & Juriloff, 2007, 2010). However, mouse studies have largely focused on single and largely homozygous recessive gene mutations, which is not representative of the complex genetic etiology of human NTDs. Instead, it will likely be more informative to consider related processes, rather than individual mutations, in directing future investigations to particular cellular processes in the context of NTC. A list of published mouse NTD models, their cellular functions, NTD type, penetrance, and study reference is being updated and can be found on the Niswander Lab website as a resource https://www.niswanderlab.org/ntd-mouse-models.
Genetic sequencing of human NTD cases suggests that the genetic contributions to NTD etiology follow a threshold model where an embryo can suffer a discrete number of genetic insults before NTC fails (Lee & Gleeson, 2020). Thus, NTDs are regarded as polygenic where many gene variants with small effects can act additively to reach the threshold that changes the trajectory of NTC from successful to a NTD (Copp & Greene, 2010; Lee & Gleeson, 2020; Zohn, 2012). Evaluation of whole genome sequencing from three different regional NTD cohorts against controls revealed that risk of NTDs could be predicted by nine singleton loss of function variants, regardless of background (Z. Chen et al., 2018). Intriguingly, they also found that comparing the number of variants in anencephaly versus spina bifida revealed a potential difference where the more severe phenotype requires a higher threshold of variants. Whether this variant threshold model holds true for other cohorts has not been evaluated. Since gene variants are generally a product of unrepaired DNA damage, special attention should be paid to the rate of DNA damage and efficiency of DNA damage repair in NTDs. While an increased rate of DNA damage may be manageable with a fully functioning DNA damage repair system, a compromised DNA damage repair system may lead to damage that exceeds the genetic threshold acceptable for proper NTC.
In addition to genomic insults, changes in gene expression by aberrant transcription factor regulation, epigenetic regulation, and miRNA expression can have severe effects on the outcome of NTC. Opposing morphogen gradients are responsible for setting up transcription factor networks that give regional specificity to the neural tube (Cohen et al., 2013; Delile et al., 2019). Disruption to these networks can have severe consequences on the outcome of NTC. Furthermore, DNA methylation and other epigenetic modifications are dynamic during embryonic development (Breton-Larrivée et al., 2019). As such, the effect of aberrant methylation has also been considered in NTDs, especially given the connection of methylation to folic acid metabolism (Li & Niswander, 2018). Moreover, transcriptional repression by miRNA have also been considered in NTD etiology due to the role of some miRNAs in neural development (Cao et al., 2006; Cho et al., 2019, 2019; Kosik & Krichevsky, 2005). MicroRNAs can have multiple targets, meaning disruption of one miRNA can have a broad effect on gene expression (B. Liu et al., 2014). Therefore, how miRNAs are expressed and the effect on their downstream targets in NTDs warrants more investigation. Hence, the source of transcriptional variability should be considered. Overall, evaluation of the degree of gene expression changes that can be tolerated during NTC may give us insight into a threshold effect specific to transcriptional regulation.
Environmental factors
Environmental factors have long been considered in NTD etiology and generally refer to exogenous factors that can act as teratogens. These may include dietary imbalances of micronutrients, treatment with anticonvulsant drugs, and indirect exposure to toxins (Finnell et al., 2021). In this review we will focus our attention on micronutrient imbalance. Micronutrients are vitamins and minerals needed in small amounts for cells in the body to carry out cellular processes. Thus, imbalance of micronutrients can have adverse effects (Tulchinsky, 2010). Folic acid has been the prominent micronutrient in NTD research due to the success of fortification on reducing the rates of NTDs (Blom et al., 2006). Following national grain fortification with folic acid in the United States in 1998, a 26% reduction in NTDs was reported by the CDC (Centers for Disease Control and Prevention (CDC), 2004; Williams et al., 2015). Due to the success of the United States’ national fortification, 86 countries have followed national grain fortification programs (Blom et al., 2006; Global Progress, n.d.; Wallingford et al., 2013; Wilde et al., 2014). Although folic acid fortification resulted in a rapid reduction in NTDs, we have not seen a further reduction in prevalence after the initial decline (Williams et al., 2015). In general, it can be considered that there are folic-acid responsive NTDs, but it is clear that folic acid by itself is not sufficient to prevent all causes of NTDs. Moreover, despite the efficacy of folic acid in reducing the risk for NTDs, we do not know the molecular basis for its role in NTD prevention. This public policy impetus has led to a folic-acid centric view of NTDs with a large fraction of work focusing on the effects of folic acid deficient or supplemented diets and treating the one-carbon metabolism pathway as the main suspect. One way researchers have studied gene-environment interactions in mouse models has been to challenge the pregnant dam with folic acid-supplemented diet and identify if the embryo is folic acid responsive (reduced NTDs), non-responsive (same NTD penetrance), or is detrimental (increased penetrance of NTDs) (summarized in Wilde et al., 2014). The list of tested models is still small, and the community could benefit from more widespread testing to attempt to identify common processes that are folic acid sensitive. Given that many authors have reviewed the deep literature describing the potential roles of folic acid and folic acid metabolism in NTDs, (Au et al., 2017; Blom et al., 2006; Copp et al., 2013; Copp & Greene, 2010; Greene et al., 2011; Greene & Copp, 2014; Wallingford et al., 2013; Wilde et al., 2014; Zhu et al., 2009) we will place our focus on other environmental factors that have not been as thoroughly discussed. Case-control epidemiological studies of human NTD cohorts and previous functional studies in animal models have suggested retinoic acid, zinc, and iron as potential risk factors for NTDs. More thorough investigation of how wildtype and mutant animals respond to micronutrient imbalance will provide insight into the mechanisms that are involved in NTC and indicate whether other micronutrients should be pursued by public health professionals as a preventative measure.
In the multifactorial model of NTDs, it is clear that trying to fix any one genetic or environmental deficiency at a time will not prevent all cases of NTD. Unlike the lab mouse, humans do not have homogenous genetic backgrounds or standardized environmental exposure. Instead of focusing on individual components, more success may come from embracing the idea of multistep mechanisms for the different NTD etiologies and paying close attention to how they converge or diverge downstream. By identifying common cellular, molecular and/or biochemical phenotypes that may act as a committing step in the process of developing a NTD, we can suggest new targets for preventative measures. The goal of this review is to highlight recent literature studying how retinoic acid, zinc, and iron imbalances may play a role in NTDs. We then speculate on the common downstream phenotypes and mechanisms they share and finish with a discussion of how we can better identify these convergent phenotypes.
The case for retinoic acid, zinc, and iron imbalance in NTDs
Micronutrient deficiencies or excess in pregnant people are a public health concern world-wide (Gernand, 2019; Gernand et al., 2016; Keats et al., 2019; Pike & Zlotkin, 2019; Tulchinsky, 2010). Cells have several compensatory pathways, including absorption, excretion, and storage, to control the level of micronutrients available; thus an exact concentration of a micronutrient is not needed in order for the cell to obtain an appropriate level necessary for function (Gernand, 2019). However, there are upper and lower limits for each micronutrient, where crossing the threshold can have toxic effects. Here we will refer to micronutrients crossing the threshold to being teratogenic as a micronutrient imbalance. Because deficiency or excess of the same micronutrient can have teratogenic effects, it is important to assess how either direction of micronutrient imbalance impacts NTC. Due to the higher micronutrient demands of a developing embryo, pregnant people are at risk for deficiency and they are suggested to increase their consumption of essential micronutrients (Keats et al., 2019). However, the possibility of over-supplementation in resource-rich countries should also be considered. Vitamin A, zinc, and iron are among the noted micronutrients to have teratogenic effects and specifically connected to an increased risk of NTDs when they are deficient or taken in excess in pregnancy (Gernand, 2019; Gernand et al., 2016; Pike & Zlotkin, 2019) Thus, this review focuses on the current understanding of how vitamin A (retinoic acid), zinc, and iron factor into the etiology of NTDs. With more research and understanding, we hope that informed supplementation of these micronutrients might have the potential to further reduce the risk of NTDs in a folic acid fortified population.
Retinoic Acid
All-trans-retinoic acid (ATRA) belongs to a group of vitamin A derivatives called retinoids (Kam et al., 2012). Vitamin A is derived from diet and when it is ingested, it is converted to the active form, ATRA, which regulates gene expression through interacting with retinoic acid receptors (Kam et al., 2012). While ATRA is a product of vitamin A in the body, it can also be directly applied, such as in topical creams used for skin care or orally by supplements. In neural development, proper spatial concentrations of ATRA along the anteroposterior axis are critical to development of the hindbrain and spinal cord (Araya García, 2017; Maden, 2006; Wilson et al., 2003). Both retinoic acid (RA) deficiency and overexposure are known to result in embryonic defects (Ross et al., 2000). The teratogenic effects of excess ATRA on the central nervous system and other fetal tissues have been acknowledged in humans when the acne treatment, Accutane (isotretinoin), was classified by the FDA as category X (positive evidence showing human fetal risk) due to its correlation with higher rates of birth defects (W. H. Chen et al., 1994; FDA Pregnancy Categories - CHEMM, n.d.; Tantibanchachai, 2014). In mouse and rat, overexposure of RA has been shown to result in NTDs (Figure 1-green) (Alles & Sulik, 1990; Wei et al., 2012). NTD penetrance upon overexposure of ATRA can differ depending on the genetic background, suggesting there is an interplay between RA and genetics. For example, treating pregnant dams with RA at E8 resulted in increased rate of exencephaly in SELH/Bc and curly tail mice, but a decrease in spina bifida in splotch mice (Tom et al., 1991). While genetics may play a factor in the differential responsiveness to RA, it is worth noting that the differences in the rate of regional NTDs connected to RA exposure may be an effect of the anteroposterior gradient of RA necessary to properly pattern the hindbrain and spinal cord (Araya García, 2017; Maden, 2006; Wilson et al., 2003). It may also be the case that some genetic perturbations lead to changes in genes involved in RA metabolism. The curly tail NTD model, due to a hypomorphic allele of Grhl3 (De Castro et al., 2018; Gustavsson et al., 2007), is a widely studied NTD model and the incidence of NTD can be influenced by genetic and environmental factors suggesting a multifactorial or threshold model, much like human NTDs (Harris, 2009; van Straaten & Copp, 2001). Curly tail mice show reduced expression of RA receptor (RAR) gamma in the tail bud and posterior neuropore (W. H. Chen et al., 1994, 1995). Supplementation of the curly tail model with RA at Embryonic Day 9 (E9, but not E8 when neurulation begins) reduced the incidence of NTD (W. H. Chen et al., 1994). Given this evidence, studying how RA imbalance and dysregulation of the RA pathway contributes to NTDs may reveal an important factor of NTD etiology.
Figure 1.
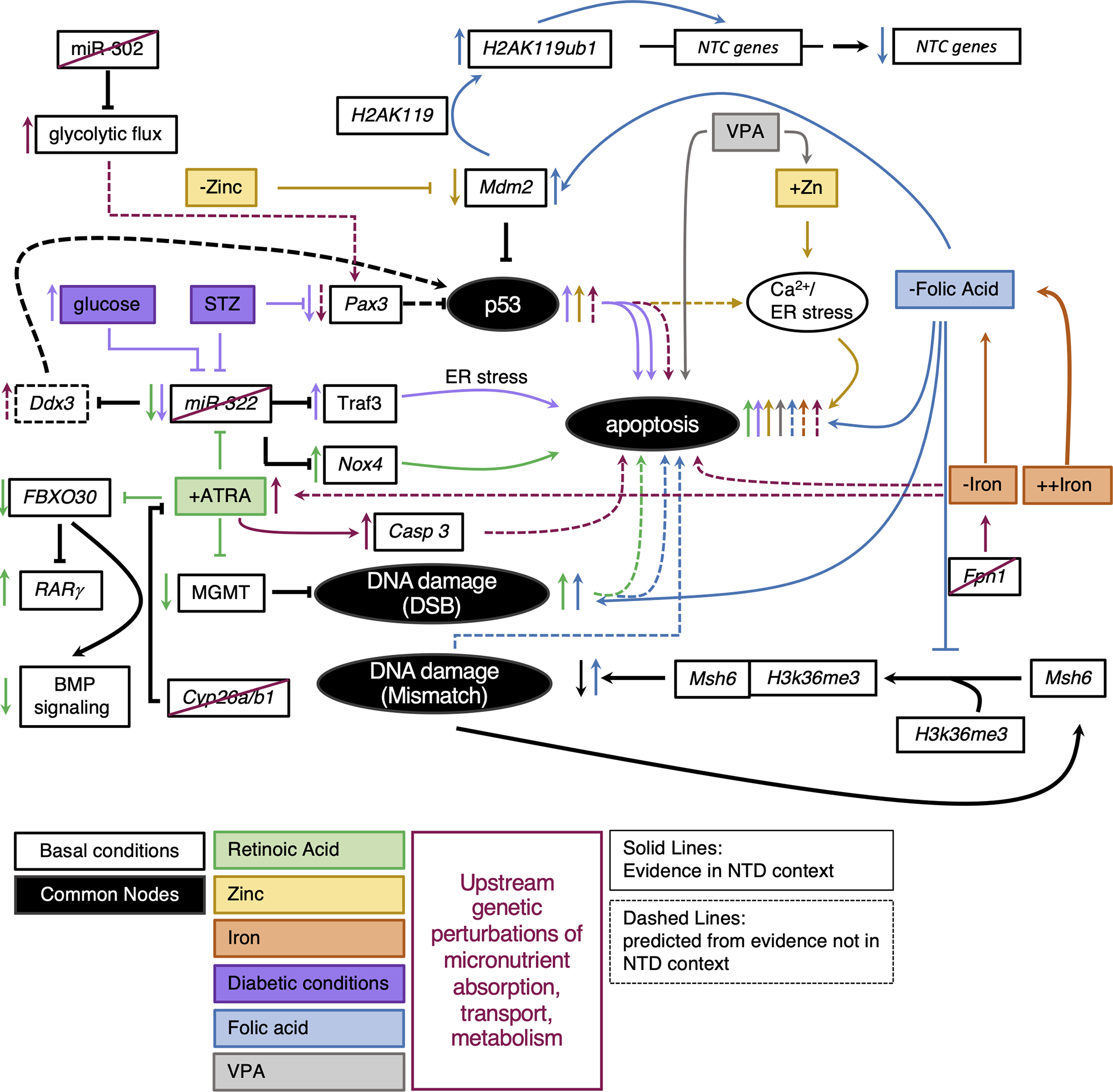
Integrated model of convergent phenotypes across micronutrient imbalances. Black circles represent common nodes discussed. Basic pathways diagrams were drawn for mechanisms reviewed. The black lines indicate regulation under normal conditions. Colored lines correspond to action in micronutrient imbalance. Solid lines represent mechanism described in an NTD context. Dashed lines described mechanism described elsewhere that we predict could be involved in mechanism leading to NTD.
It is necessary to maintain the spatial concentrations of ATRA for proper NTC. RA metabolism entails localized synthesis and destruction, leading to transcriptional regulation and target gene expression. Cyp26a1 is one of several enzymes responsible for maintaining precise concentrations of RA by degrading it. When Cyp26a1 is knocked out in mouse embryos, NTC fails in the caudal neural tube (Figure 1-red/green) (Abu-Abed et al., 2001). Sequencing of human NTD cases also revealed an enrichment for polymorphisms in Cyp26a1 in spina bifida cases (Rat et al., 2006). More recently, a case-control study of human NTD cohorts revealed several variants in ATRA related genes enriched in NTD cases. The majority of missense mutations enriched in the NTD cohort were in the gene Cyp26b1, which is also involved in RA degradation (Li, Zhang, Chen, et al., 2018). Although maternal diet and fetal levels were not evaluated, disruption of Cyp26b1 would likely result in the accumulation of RA in affected cases which would be predicted to be detrimental to NTC (Figure 1-red/green). The authors found that neuroepithelial progenitor cells carrying human variants of Cyp26b1 and treated with ATRA show increased expression of RA target genes when normalized to wild-type, ATRA-treated cells. This indicates the Cyp26b1 variants have reduced function in degrading RA. Interestingly, cells carrying the Cyp26b1 variants and treated with RA show increased expression of the apoptotic marker, Casp3, which is consistent with the finding of increased apoptosis in ATRA-induced NTD mouse models described in the next paragraph (Figure 1-red/green). Furthermore, neuroepithelial progenitor cells transfected with the human Cyp26b1 variants and treated with RA had dysregulated neural differentiation raising the possibility of another process, the balance of proliferation versus differentiation, as connected in the etiology of RA-involved NTDs. Another strategy for the regulation of RA transcriptional activity is through regulation of RARγ. RARγ is one of several RA receptors responsible for binding to RA to initiate RA-related transcription. Knocking out RARγ in the Cyp26a1 null mutant is able to reduce the prevalence of NTDs and restore proper tailbud patterning by balancing the increase in RA levels by loss of Cyp26a1 with decreased transcriptional activity by loss of RARγ (Abu-Abed et al., 2003). RARγ levels are regulated via targeting for ubiquitination and proteasomal degradation by the E3 ligase, FBXO30 (Figure 1-green). (Cheng et al., 2019). Intriguingly, in both ATRA-induced mouse NTDs and human anencephalic fetal tissues with high levels of RA, FBXO30 expression is reduced, and RARγ expression increased (Figure 1-green). Additionally, (Cheng et al., 2019) suggested that FBXO30 promotes BMP signaling by targeting RARγ for degradation, thus aberrant BMP signaling may also be involved in the etiology of RA-induced NTDs (Figure 1-green). Together, these data indicated that in both humans and mouse, excess RA as a result of dysregulation of ATRA inhibitory mechanisms can act as a teratogen.
Maternal diabetes is a known risk factor for NTDs and has been modeled in mice by treating with high glucose or chemical induction by streptozotocin (STZ) (Gao & Gao, 2007; Zabihi & Loeken, 2010). One hallmark of the diabetic NTD model is an elevated level of cell apoptosis, which is also seen in ATRA-induced NTDs. Intriguingly, some mechanisms that lead to elevated apoptotic levels in excess ATRA conditions and diabetic conditions involve common molecular players. One mechanism leading to the increased incidence of apoptosis in excess ATRA conditions is by way of repression of miR-322 and enrichment of the NADPH oxidase NOX4 (Figure 1-green) (Y.-S. Liu et al., 2020). How NOX4 induces apoptosis is unknown. Impressively, injection of a miR-322 mimic into an ATRA treated mouse prevented both the increase in apoptosis and NTD phenotype (Y.-S. Liu et al., 2020). Alternatively, miR-322 is also a known repressor of TRAF3 and in the diabetic mouse model or in high glucose cultured cells, miR-322 expression is reduced and TRAF3 expression is enriched resulting in an increase in apoptotic cells (Figure 1-purple)(Gu et al., 2015). Treating high glucose cultured cells with the antioxidant enzyme, Superoxide Dismutase (SOD1), can mitigate the expression changes of miR-322 and TRAF3, suggesting that the miR-322/TRAF3/Apoptosis circuit is responsive to oxidative stress (Gu et al., 2015). While the STZ-induced diabetic mouse model is known to induce oxidative stress, ATRA has not been shown to induce oxidative stress in NTDs, although ATRA can induce reactive oxygen species in cultured ES cells (Castro-Obregón & Covarrubias, 1996). TRAF4/NOX form a complex to alter the microenvironment by producing endosomal reactive oxygen species (ROS) in a tumor microenvironment in lung fibroblasts (Kim et al., 2017). Perhaps the increase in apoptosis observed in the ATRA and diabetic mouse model is due to the increase in expression in TRAF and/or NOX that give rise to an increase in oxidative stress and ultimately apoptosis. This would suggest the etiology of NTDs in two different environmental backgrounds could be explained by the same hierarchy: Repression of miR-322 leads to increased expression of multiple genes involved in oxidative stress resulting in apoptosis that limits the ability of the neural tube to close.
A broader speculation given the previously described miR-322 circuits is that aberrant expression of a miRNA caused by an environmental factor, such as RA, may change the expression of enough targets to surpass a threshold of transcriptional insults and result in disruption of several processes required for NTDs. Another example of how ATRA might induce a substantial genetic burden in NTDs is through reduction of mRNA expression of the DNA damage repair gene, MGMT (Zhang et al., 2019). In this work, the authors showed that treating rats with ATRA resulted in spina bifida, reduced MGMT expression, and an increase in DNA double-strand breaks (Figure 1-green) (Zhang et al., 2019). These results indicate that the reduction in MGMT may give rise to an increased DNA damage burden. While it is known in other contexts that both inhibition of MGMT and accumulation of double strand breaks can result in cell apoptosis (Norbury & Zhivotovsky, 2004; Ochs & Kaina, 2000), it is unknown if they are part of the mechanism for an increase in cell apoptosis in ATRA-induced NTDs. If MGMT inhibition and double strand breaks were factors in the increase in apoptosis in ATRA-induced NTDs, would they be responsible for all apoptosis or be a contributing factor along with the dysregulated miR-322 circuit?
Taken together, excess RA can act as a teratogen resulting in NTDs. How each independent mechanism described fits into the complex etiology of NTDs induced by RA remains unknown. A speculative synthesis of this data highlighted in Figure 1 might suggest that when ATRA is in excess, variants in Cyp26b1 fail to degrade ATRA resulting in in excess ATRA above an acceptable threshold which has been demonstrated to result in repression of miR-322, MGMT, and FBXO30 which would increase oxidative stress, DNA damage, and RA-mediated transcription as well as reduce BMP signaling; ultimately this confluence of aberrant processes would increase apoptosis leading to the failure of NTC. Transcriptional profiling of ATRA-induced NTDs may be the most efficient way to identify if all of the genes discussed here are dysregulated concurrently in individual embryos. Furthermore, using cell culture or possibly neural tube organoids to test the epistasis of the Cyp26b1 variants and the mechanisms discussed here could provide additional evidence as to whether these mechanisms are independent of one another or work together in an apoptotic constellation.
Zinc
Zinc is a critical micronutrient necessary for human function. Close to 2800 human proteins interact with zinc mediating a wide range of processes (Velie et al., 1999). Once zinc is ingested, it is absorbed into cells via zinc transporters which regulate cytoplasmic concentrations of zinc. ZnT transporters reduce the concentration of zinc in the cytoplasm whereas Zip transporters increase concentrations (Bafaro et al., 2017). In mouse, a maternal diet deficient in zinc causes an NTD in about 4% of genetically normal embryos, however a zinc-deficient diet in mice with null alleles for zinc transporters Zip1 or Zip1/Zip3 increases the risk for NTDs (Dufner-Beattie et al., 2006). This suggests that not only is dietary zinc availability important, but the proteins necessary to transport zinc should also be considered. As further evidence of the importance of zinc, depleting cultured mouse embryos of zinc with the zinc chelator TPEN resulted in 100% NTD incidence (Figure 1-yellow) (Li, Zhang, & Niswander, 2018). The neural folds had a wider inflection angle and did not close. Human case-control studies also revealed that higher maternal zinc intake resulted in lower risk for NTDs and lower levels of zinc in neonatal serum were associated with increased risk of NTDs (Dey et al., 2010). On the other hand, higher levels of zinc measured in the second trimester toenails of mothers with NTD offspring suggest that high zinc concentrations can also be a risk factor for NTDs (Milunsky et al., 1992). In the SELH/BC mouse line, supplementing the diet with zinc-enriched water with concentrations up to 500ppm had no effect on NTD penetrance, however 1500ppm showed a significant increase in NTDs indicating that too much zinc can be detrimental (Harris & Juriloff, 2005). Valproic acid (VPA) treatment, which increases the risk for NTDs in humans and mice, has been correlated in mouse with increased serum zinc levels (Figure 1-grey/yellow) (Wegner et al., 1990). Perhaps VPA’s interplay with zinc may act alongside its other cellular effects, such as inhibition of histone deacetylases, in the development of NTDs (Finnell et al., 2021). Moreover, treatment with zinc-oxide (ZnO) nanoparticles can cause NTDs in mouse and chick embryos (Figure 1-yellow) (Yan et al., 2021). Zinc-oxide nanoparticles are a manufactured form of zinc used in food, cosmetics and medicines that are more bioavailable than conventional sources of zinc and have antibacterial properties (Singh, 2019). While the main goal of using ZnO in food production is to deliver zinc that is more bioavailable, it is unclear whether the nanoparticle form has other toxic effects and at what concentrations (Singh, 2019). Mouse and chick embryos exposed to ZnO experienced a dose-dependent failure of NTC, with the worst outcomes at the highest dose (Yan et al., 2021). Human neuroblastoma cells treated with ZnCl2 at concentrations similar to ZnO were also shown to have a significant reduction in cell viability. This is consistent with the hypothesis that varying concentrations of a micronutrient (zinc) is tolerable until it reaches a threshold where it becomes toxic.
Intriguingly, in both cases of zinc deficiency and overexposure to ZnO nanoparticles, the authors determined that the phenotype likely at fault for NTC failure was an increase in apoptosis. Using a neuroepithelial cell model of zinc deficiency, a reduction in zinc led to reduced binding of the ubiquitin E3 ligase, Mdm2, to p53, which stabilized p53 expression and increased apoptosis (Figure 1-yellow) (Li, Zhang, & Niswander, 2018). Interestingly, it is known that Zn2+ ions are involved in proper folding of p53, however zinc-free p53 can still fold but with significantly reduced binding affinity to DNA (Loh, 2010). Thus, having more p53 with partial function can potentially still induce apoptosis. In the case of ZnO treated chick embryos, expression profiling suggested that endoplasmic reticulum (ER) stress may be leading to an increase in apoptosis (Figure 1-yellow) (Yan et al., 2021). They found that exposing human neuroblastoma cells to ZnO resulted in bursts of calcium that could be inhibited by an ER stress inhibiting drug, 4-PBA. The addition of 4-PBA to chick embryos treated with ZnO resulted in fewer apoptotic cells in both the cranial and trunk regions suggesting the ZnO induced apoptosis occurs by way of calcium-mediated ER stress. Given the involvement of p53 in zinc-deficient NTDs, could p53 modulation also be involved in the etiology of NTDs resulting from ZnO exposure? In mouse embryonic fibroblasts, stress stimuli have been shown to induce p53 accumulation at the ER and stabilize Ca2+ pumps that give rise to the calcium dependent apoptosis, thus it may be possible that p53 and ER stress are common to both excess and deficient zinc NTD environments (Giorgi et al., 2015).
While these studies make a case that zinc imbalance factors into the etiology of NTDs on its own, it is important to consider how zinc imbalance may influence molecular processes in different mouse mutant lines. Could supplementation with zinc prevent NTDs in models that are folic acid non-responsive or detrimental? Assessing maternal or fetal zinc status when sequencing for variants in NTD cohorts will enable new hypotheses about the potential connections between zinc and NTD etiology. Working out commonalities between processes affected by zinc imbalance and considering the responsiveness of those same processes to folic acid may uncover potential ways for zinc supplementation to be complementary to folic acid in preventing NTDs.
Iron
Iron is an essential micronutrient involved in oxygen transport, DNA synthesis, and electron transport (Puig et al., 2017). Dietary iron comes in a heme and a non-heme form depending on the source (meats vs vegetables and grains). Iron deficiency is extremely common in pregnancy with the WHO estimating that 40% of pregnant people are iron-deficient-anemic worldwide (https://www.who.int/health-topics/anaemia#tab=tab_1). Although iron deficiency is common, especially in pregnancy, how it affects NTC has scarcely been studied. Retrospective epidemiological studies have demonstrated the correlation between iron status and NTDs are not straight-forward in humans. NTD cases were found to have higher odds of low iron stores in data collected from a cohort along the Texas-Mexico border from 1995–2000 and low iron intake was found to be associated with higher risk of spina bifida in data from a Dutch cohort in 1998 (Felkner et al., 2005; Groenen et al., 2004). In contrast, analysis of a cohort from 1986–1990 in Dublin did not reveal iron status as a risk factor for NTDs. More evaluation of NTD cases with respect to iron status will need to be performed to better calculate iron-associated risks of NTDs (Molloy et al., 2014).
Similar to RA and zinc, efficient transport of iron and iron availability is critical. The curly tail NTD mouse model exhibits reduced uptake and/or binding of transferrin, the protein which meditates the transfer of iron through blood, on the hindgut epithelial surface (Hoyle et al., 1996). However, knockout of the transferrin receptor (TFRC) which moves transferrin from the bloodstream to the cell only results in a kinked neural tube phenotype (Levy et al., 1999). Although the authors saw an increase in apoptosis in neural tissue in the TFRC mutant, they found no neural tube closure defects suggesting that reduced Transferrin transport by the Transferrin receptor is not likely a significant contributor to the curly tail mouse NTD. However, this evidence does not exclude that reduced Transferrin transport is a contributing factor to the multigenic curly tail NTD etiology. Ferroportin 1 (Fpn1) is responsible for iron transport from the visceral endoderm to embryo. When Fpn1 is reduced in the Fpn1 flatiron mouse model, the embryos have less iron and develop exencephaly at a rate around 70% (Figure 1-red/orange) (Mao et al., 2010; Stokes et al., 2017; Zohn et al., 2007). In this model, the Fpn1ffe/+ dams did not have significantly different stores of iron than wildtype, suggesting that transport of iron to the embryo may be the problem (Stokes et al., 2017). In agreement with this evidence, wild-type embryos cultured in an iron chelator also result in NTDs (Mao et al., 2010). To test if increasing the maternal iron stores can influence the embryonic NTD outcome, Fpn1ffe/+ dams were fed an iron-fortified diet resulting in a reduced rate of exencephaly in mutant embryos. This result suggests that supplementation of iron can prevent NTDs when iron transport is compromised.
Lrp2 is also a known transporter of ferritin bound iron and knockout of Lrp2 results in 60% of embryos with exencephaly. However, in these mutant embryos, there was not a deficiency in serum iron levels and maternal iron fortification of Lrp2 heterozygous dams did not reduce the rate of exencephaly (Sabatino et al., 2017). In addition to iron transport, Lrp2 is also known to be involved in folic acid homeostasis. Supplementation of Lrp2 maternal diet with folic acid has only a mild effect on NTD rates whereas intraperitoneal injection with folic acid significantly reduces the rate of exencephaly in Lrp2 mutants from 60 to 20% incidence. Interestingly, supplementing with iron in addition to folic acid negated the positive effects of folic acid supplementation (diet or IP) (Sabatino et al., 2017), In fact, high iron supplementation reduced the folate levels in pregnant Lrp2+/− mice. While the connection between iron and folic acid has only been minimally studied in NTD models and human cases, it is known that iron can influence both folate status and metabolism in other contexts (Herbig & Stover, 2002). The complexity of the iron-folate balance relative to genetic background is highlighted by studies of the Fpn1 mutants which have reduced folate levels, suggesting that iron deficiency may influence folate deficiency (Stokes et al., 2017). Yet fortifying the maternal diet of Fpn1ffe/+ dams with a high concentration of iron resulted in a further reduction of folate in both the pregnant dam and her mutant embryos (Figure 1-orange/blue) (Stokes et al., 2017). Unlike the Lrp2 mutants, supplementation of Fpn1 dams with high folic acid did reduce the prevalence of NTDs and there was an even greater reduction when folic acid was given in combination with iron. Thus, while both Lrp2 and Fpn1 are known transporters of iron, high iron supplementation and high folic acid supplementation differentially affect NTD rate in the different backgrounds, highlighting a delicate interplay between genetic and environmental factors. This raises an interesting concern about the effect that one micronutrient may have on another and how the complexity of this interaction, in conjunction with genetic causes, may influence the desired effect.
While iron deficiency is generally linked to abnormal neural development after the neural tube has closed, its influence over folate levels suggests that it could be a stronger contributing factor to failed NTC (Georgieff, 2008; Lozoff & Georgieff, 2006). Although speculative, in cases where a folic acid fortified diet may prevent NTDs, additional supplementation with high iron with consequent effects in reducing serum folate levels may negate the positive effect of folic acid supplementation. On the other hand, in those cases seen in a few mouse mutants where folic acid supplementation increases the NTD risk, perhaps high iron supplementation may prevent NTDs. Similar to how iron deficient-anemic patients may present with lower folate levels (Herbig & Stover, 2002), serum levels of zinc have been indicated as lower in iron deficient-anemic patients, though these correlations have not been tested in NTDs (Abdelhaleim et al., 2019). Analysis of a NTD cohort did suggest that both iron and magnesium levels were significantly reduced in NTD cases, thus the interplay between iron and magnesium should also be considered in NTDs (Groenen et al., 2004). Moreover, iron deficiency in mouse embryos can result in congenital heart defects and this was associated with increased RA signaling (Kalisch-Smith et al., 2020). Perhaps iron deficiency in the Fpn1 hypomorphs may lead to an increase in RA. Alternatively, it may be the case that an iron deficient diet in a wildtype mouse could lead to higher RA levels, which we described earlier can lead to failure of NTC. Together these data and the overwhelming prevalence of iron deficiency in pregnancy suggest that iron deficiency needs more consideration in studying the etiology of NTDs.
Identifying common nodes in connecting micronutrient imbalance to NTD etiology
In synthesis of the papers reviewed, we noticed patterns in the types of transcriptional regulators and NTC processes that were dysregulated in NTDs across different conditions. By conceptualizing common mechanisms or nodes by which several micronutrient imbalances lead to NTD pathobiology, this may help to identify key genes and processes as possible targets for preventative measures across multiple micronutrient imbalance conditions.
Apoptosis as a central phenotype in NTDs
The number of cells is critical for successful closure of the neural tube. Too little or too much proliferation or too much or too little apoptosis each can lead to a NTD (Juriloff & Harris, 2018). Focusing on aberrant apoptosis as a common theme, mouse mutants with null alleles for apoptosis-related genes are biased toward cranial neural tube defects suggesting a regional specificity (Juriloff & Harris, 2018). Indeed, programmed cell death occurs at the time of neural tube fusion and contributes to this process (Yamaguchi et al., 2011). While disrupting apoptotic mechanisms directly is the most obvious way to evaluate the impact of too little cell death in NTDs, we have reviewed different instances where environmental factors can indirectly increase the degree of apoptosis leading to NTDs (Figure 1). In these cases, an increase in apoptosis is found in varying regions of the neural tube based on the type of micronutrient imbalance. ATRA-induced NTDs in mice and rats often result in caudal NTDs and the incidence of apoptosis is increased in that area (Figure 1-green) (Y.-S. Liu et al., 2020; Wei et al., 2012). In contrast, zinc deficiency by TPEN results in cranial NTDs accompanied by an increase in apoptosis in the cranial neural tube (Figure 1-yellow) (Li, Zhang, & Niswander, 2018). Excess ZnO induced an increase in apoptosis in mouse and chick embryos and in chick the increase in apoptosis occurred in both the cranial and trunk regions (Figure 1-yellow) (Yan et al., 2021). In addition to the environmental cases, we highlighted in this review that an increase in apoptosis is also a known factor into the etiology of NTDs in mice resulting from 1) induction of diabetes by STZ (Figure 1-purple), 2) folate dysregulation by MTX (Figure 1-blue), and 3) treatment with the antiepileptic drug VPA (Figure 1-grey) (Gu et al., 2015; Mallela & Hrubec, 2012; Tung & Winn, 2011; X. Wang et al., 2014). The increase in apoptotic cells may represent a common phenotypic node that each of these different etiologies manifests, resulting in failure of the neural tube to close. While the individual genes involved in the mechanisms responsible for inducing an increase in apoptosis might be divergent, there may be similarities in the types of genes or hierarchy of signaling cascades brought on by the different environmental factors.
Single protein node: dysregulation of p53
One potential node upstream of the apoptosis phenotype is activation of p53 (Figure 1). P53 is best known for its role in protection against cancer, however there is growing interest in its role in embryonic development (Jain & Barton, 2018; Kastenhuber & Lowe, 2017). Studying p53 has provided ample insight into the relationship of environmental factors and cancer, thus there is a plausible connection in the multifactorial etiology of NTDs. In mice, p53 null mutant embryos and p53S mutants (modeling the human variant without DNA binding ability), develop exencephaly (Armstrong et al., 1995; Lozano, 2010; Sah et al., 1995; Zhao et al., 2019, p. 23). The p53S mutant has less apoptotic cells in NTD cases suggesting in this context p53 promotes apoptosis (Zhao et al., 2019). In zinc-deficient embryos there is also a p53-dependent increase in apoptosis (Figure 1-yellow) (Li, Zhang, & Niswander, 2018; Yan et al., 2021). While p53 expression was not queried in the ZnO treated embryos, the authors found that the increase in apoptosis was dependent on a Ca/ER stress circuit, which involves p53 in a different context (Figure 2-yellow) (Giorgi et al., 2015; Yan et al., 2021). Furthermore, RA is known to increase p53 expression in keratinocytes and glioma cells (Lu et al., 2013; Mrass et al., 2004). Perhaps p53 expression is enriched in ATRA-induced NTDs and works in a parallel or same pathway as the miR-322/NOX4 circuitry to contribute to a multifactorial threshold of increased apoptosis, causing NTC to fail (Figure 1-green). As noted previously, miR-322 expression was also repressed in the STZ-diabetic model upstream of an increase in apoptosis (Figure 2-purple) (Gu et al., 2015, p. 322). In addition to the miR-322/TRAF3/apoptosis circuit, the STZ model is also known to result in Pax3 repression (Figure 1-purple) (Phelan et al., 1997). Pax3 deficient splotch mouse embryos with NTDs exhibit an increase in apoptosis which can be mitigated by crossing the splotch mice with p53 null mice (Pani et al., 2002), indicating the excess apoptosis experienced in the Pax3 deficient mouse is a result of an increase in p53. This suggests that in the STZ model the increase in apoptosis is likely a result of Pax3 deficiency leading to p53 enrichment (Figure 1-purple). This is consistent with a known role of Pax3 in targeting p53 for ubiquitination and subsequent degradation (X. D. Wang et al., 2011). It is important to note that the hypothesis that elevated apoptosis is downstream of reduced expression of Pax3 is still unresolved. Different reports have shown in splotch mutant mice that apoptosis is only increased in the dermomyotome (Mansouri et al., 2001), and there is no change in apoptosis levels in Sp2H mice (Conway et al., 2000; Greene et al., 2009; Sudiwala et al., 2019). Given this contrary evidence, it will be of interest to definitively define how apoptosis changes in splotch mice under discrete conditions. If apoptosis is elevated when Pax3 expression is reduced, it will be of interest to determine if the Pax3/p53 circuitry works in the same pathway with the miR-322/TRAF3 circuitry or in parallel in STZ induced NTD. Intriguingly, studies in spermatocytes illustrate a mechanism in which repression of miR-322 leads to the enrichment of Ddx3, which when highly expressed has a known role in promoting apoptosis by positive regulation of p53 (Figure 2-maroon) (Che et al., 2019, p. 3). Repression of miR-322 leading to an increase in p53 may be a common mechanism in NTDs resulting from maternal diabetes and excess RA, or in a Pax3 depleted background. Together, these studies suggest a common node in NTD etiology is the dysregulation of p53 downstream of micronutrient imbalance (Figure 1).
Hierarchical nodes: dysregulation of DNA damage repair and miRNAs as root causes for NTD
Another convergent mechanism to consider for the increase in apoptosis does not revolve around the dysregulation of a single protein, but rather the dysregulation of a process. DNA damage repair (DDR) is necessary to reduce the accumulation of DNA damage in cells and preserve the correct DNA sequences to encode functionally intact proteins. A compromised DDR system could have broad repercussions in the complex choreography necessary for proper NTC. The rapid cellular proliferation taking place during embryogenesis requires engagement of DDR genes to limit the number of mutations and genomic instability. Disrupted DDR can act at the top of a cascade leading to NTDs by introducing deleterious mutations to genes necessary for processes that underlie NTC or resulting in the accumulation of unrepaired DNA damage that can lead to apoptosis. A number of mouse mutants for genes across different DDR pathways result in embryonic lethality or a variety of embryonic malformations including NTDs (Friedberg & Meira, 2006; Tran et al., 2012). For example, knock-out of DDR genes Brca1, Xrcc2, p53, and Rad9b result in embryos with neural tube defects, however the incidence for each of these examples is less than 100% (Armstrong et al., 1995; Cao et al., 2020; Deans et al., 2000; Gowen et al., 1996; Leloup et al., 2010; Sah et al., 1995). Knockout of Rad9b, a DDR repair gene involved in double strand break detection, in mouse ES cells results in an increase in genomic instability and decrease in cell viability when cells were challenged with a damaging agent (Leloup et al., 2010). Would the incidence of NTDs increase if these examples of mice with reduced DDR capacity are challenged with an environmental stressor which could contribute to the DNA damage burden? Indeed, zinc deficiency, iron deficiency, folate deficiency, and excess RA have the capability to induce DNA damage (Duthie et al., 2002; Ho, 2004; Puig et al., 2017; Zhang et al., 2019), but is the increase in DNA damage sufficient or must the DNA damage be in the presence of a compromised DDR system to contribute to NTD etiology? As mentioned in our discussion of RA, excess ATRA results in an increase in double strand breaks by way of reducing expression of MGMT (Figure 1-green) (Zhang et al., 2019). Furthermore, human sequencing revealed that NTD cases were enriched for deleterious rare variants in occupancy regions of H3K36me3, which recruits DNA mismatch repair machinery (mmr) to the DNA (Li et al., 2020). In the assessed NTD cohort, fetal brain concentrations of folate were lower and levels of microsatellite instability (MSI, a read out of compromised mismatch repair) were higher suggesting a connection between folate deficiency and aberrant mismatch repair. This connection was tested in folate deficient cells, where the authors observed reduced binding of the mmr protein Msh6 to genes critical to NTC even though there were higher levels of MSI (Figure 1-blue). While excess ATRA resulted in dysregulation of a specific DDR gene (Zhang et al., 2019), folate deficiency disrupted the binding of DDR machinery to DNA mismatches (Li et al., 2020). Interestingly, folate deficiency induced by treatment with MTX in mouse ES cells was also shown to lead to an increase in double-strand breaks and a reduction in expression of neural progenitor-related genes by way of mono-ubiquitination of histone-H2A by Mdm2 (Figure 1-blue) (Pei et al., 2019). Although this work did not directly assess apoptosis, MTX-induced NTDs show increased apoptosis (X. Wang et al., 2014), thus the accumulation of double-strand breaks may be one factor in triggering apoptosis and eventual failure to close the neural tube in folate deficient mice. While in this paragraph we speculated on how micronutrient imbalance can lead to DNA damage-induced apoptosis, evidence from Rad9b mutants and MTX-induced NTDs suggest reduced expression of neural progenitor-related genes may be a common phenotype when DDR is dysregulated (Cao et al., 2020; Pei et al., 2019). The reduced expression of neural progenitor genes is consistent with the enrichment of neural differentiation genes found in Cyp26b1-deficient cells treated with ATRA (Li, Zhang, Chen, et al., 2018). Together these data suggest the dysregulation of neural progenitor cell genes is common to the pathobiology of NTDs induced by different micronutrient imbalances—however, we will not further discuss this point in this review. In short, we suspect a hierarchical framework where compromised DDR is not able to resolve additional DNA damage induced by micronutrient imbalance and therefore results in dysregulation of NTC processes leading to NTDs. This model fits well with the multifactorial threshold where micronutrient imbalance may increase the accumulation of genetic insults necessary to surpass the threshold and confer a NTD (Lee & Gleeson, 2020).
Similar to the hierarchical etiology of aberrant DDR discussed above, the dysregulation of microRNAs by micronutrient imbalance may also be at the top of a cascade leading to NTDs. MicroRNAs are responsible for post-transcriptional repression of mRNAs, and the web of miRNA-mediated repression is predicted to be highly connected (B. Liu et al., 2014). An example of miRNA dysregulation being at the top of a misexpression cascade was discussed earlier where dysregulation of miR-322 expression is upstream of several genes which cause apoptosis when dysregulated (Figure 1-purple/green) (Gu et al., 2015; Y.-S. Liu et al., 2020, p. 3). MiRNAs often have multiple targets across different cellular processes, thus the disruption of one miRNA may lead to many compromised processes. In the nervous system, miRNAs regulate neural progenitor cell proliferation, differentiation, and axonal growth (Cho 2019). Analysis of E8.5, E9, and E9.5 mouse embryos detected around 70 miRNAs, some of which target genes implicated as critical for neural tube closure (Mukhopadhyay et al., 2011). MiRNAs have also been implicated in several multifactorial neurodevelopmental disorders such as Fragile X syndrome, Rett syndrome, and Autism Spectrum Disorder (Ardekani & Naeini, 2010). In mouse and rat, it has been reported that miRNAs are dysregulated in NTDs induced by ATRA, p53 null mutation, and STZ (diabetic model) suggesting that miRNA dysregulation is common to NTDs arising from different environmental factors (Gu et al., 2015, p. 322; Hosako et al., 2009; Y.-S. Liu et al., 2020). Furthermore, sequencing in human NTD and control cases revealed 6 miRNAs that were enriched during pregnancy in maternal serum from mothers carrying a fetus with a NTD (Gu et al., 2012). Of the six enriched miRNAs, two were significantly enriched in both spina bifida and anencephaly compared to controls, 3 were significantly enriched in only spina bifida, and 1 was significantly reduced in anencephaly. These data suggest that miRNAs help regulate NTC and perhaps in a regionally-specific manner. Continued genome-wide sequencing efforts of NTD cohorts with closer examination of possible contributions by miRNA variants will be necessary to substantiate this idea. Several miRNAs have been indicated as dysregulated and reported in at least two NTD-related studies: miR142–3p, miR124a, miR9–5p, miR331–3p. Intriguingly, miR-142–3p was found to be dysregulated in both human NTDs and E8.5 p53 null embryos (11% NTD penetrance in the mouse study), though the direction of dysregulation differed between the two studies (Gu et al., 2012; Hosako et al., 2009). Continued genome-wide sequencing efforts of larger NTD cohorts with closer examination of possible contributions by miRNA variants will be necessary to substantiate this idea. Finally, studying NTD mouse models with null mutations of miRNAs will help to identify the processes that they regulate. For example, transcriptional profiling of the cranial neural tube of miR-302 null NTD mice revealed that miR-302 regulates glucose metabolism and when knocked out, there is higher flux through glycolysis. This suggests an NTD etiology similar to embryos from diabetic dams (Figure 1-maroon) (Keuls et al., 2020). Identifying common dysregulated miRNAs across different mouse NTD models and their range of effect will illustrate a broader picture of the processes involved in specific NTD etiologies while potentially narrowing the number of biomarkers needed to be sequenced in humans.
Looking forward in studying gene-environment interactions in NTDs
Folic acid deficiency, pre-existing diabetes, and hyperthermia are all examples of known common factors in NTD cases. Commonality of these risk factors across populations and NTD types make them good targets for preventative measures ranging from diet supplementation to avoidance of certain practices while pregnant. Currently, the CDC has a page named “5 Ways to Lower the Risk of Having a Pregnancy Affected by a Neural Tube Defect”. This page has a list of suggestions for reducing the risk of NTDs summarized as such:
To be aware of folate in your diet and to take measures to supplement diet with folic acid.
Manage pre-existing health conditions such as diabetes and obesity.
Take into consideration family history of NTDs and be tested for MTHFR C677T (a genetic variant in the one-carbon metabolism pathway).
Avoid certain medications known to be correlated with NTDs including anticonvulsants.
Avoid overheating.
Although these factors are widely recognized as risk factors for NTDs and communicated as such, much of what we understand about the etiology of NTDs has not been well communicated. The lack of communication about the nuances and more detailed understanding of NTDs, and that NTDs are not due to any single genetic or environmental factor does a disservice to families learning that their fetus has a NTD, despite following the clearly communicated recommendations. The CDC aptly states at the end of this page: “Understanding the characteristics that are more common among babies with a neural tube defect will help us learn more about the causes and the ways to prevent these types of birth defects from occurring.” Indeed, identifying convergent phenotypes and mechanisms by systematic interrogation of NTD models or thorough synthesis of the literature will continue to provide insight into how NTDs occur and potential population-level preventions to consider.
In order to determine if a micronutrient imbalance is a considerable risk factor or supplementation is preventative, more investigation needs to be done. While challenging several mouse mutants with folic acid fortified diets has revealed differential responsiveness, without more models tested it is hard to predict the types of processes that are most benefitted or adversely affected by folic acid fortification. Since folic acid does not prevent 100% of NTD cases, it is imperative to test the supplementation of other nutrients with more rigor. It would be interesting to first revisit mouse NTD models where folic acid fortification was non-responsive or detrimental and test other nutrients to query if they have beneficial effects. Furthermore, considering the wealth of mouse NTD models, it is surprising how limited our knowledge is of the potential of other micronutrients to prevent NTDs. Testing many models with a set of nutrients and categorizing effects into beneficial, non-responsive, and detrimental will give us better insight into how a particular model responds to supplementation and broadly the types of processes likely affected by micronutrient supplementation. Although this review has focused on work in mice and rats, Xenopus and zebrafish present animal models with embryos that are easily accessible for testing complex conditions of micronutrient imbalance and genetic perturbations by direct embryonic manipulation (Borodinsky, 2017; Guo, 2009). Both models are relatively high throughput for in vivo work and would make an excellent platform for testing the effect of multiple micronutrients independently or together. For example, it was recently shown that zebrafish embryos exposed to VPA had smaller midbrains and hindbrain midline gap, however when the VPA treated embryos were also given folic acid the brain size increased, and midline gap decreased (Muhsen et al., 2021). Though NTC in these models does not mimic human NTC as well as mouse, the relative quickness and accessibility make them an excellent model to test many conditions (Nikolopoulou et al., 2017). Chick embryos are also classically studied in NTC research due to their relative similarity in NTC to mouse (Nikolopoulou et al., 2017). Chick embryos can be cultured ex vivo allowing them to be accessible to manipulation with different nutrients (Bjørnstad et al., 2015). Last, the sophistication of neural tube organoids has been progressing over the years presenting a model to test variants from sequenced cohorts in a human background (Wu et al., 2021). Although organoids represent specific neural tube regions and do not completely recapitulate the mechanical aspects of human neural tube closure, they are accessible for treatment with exogenous nutrients allowing an evaluation of potential effects of nutrients on NTC processes in a human genome.
In addition to testing how micronutrient imbalances and prevention strategies affect NTC, thorough interrogation of the downstream effects must be parsed, and information integrated between different NTD models. Next-generation sequencing has come of age and is advancing rapidly allowing for profiling of the genome, methylome, and transcriptome in bulk or single cell tissue. Observing differences in data from micronutrient deficient and fortified samples will provide insight into overall transcriptional dysregulation, specific changes in gene regulation, and the processes which are affected downstream. Comparison of these analyses across different micronutrients may provide insight into commonly dysregulated processes or nodes, and this has the potential to pinpoint possible supplementation schemes to directly test for the ability to decrease NTD risk. Furthermore, spatial single cell technologies are maturing allowing for the measure of gene expression and chromatin accessibility in a given NTD region (Marx, 2021). Given that different regions of the neural tube have different mechanisms of closure (Juriloff & Harris, 2018), it may be beneficial to profile NTD models by taking into account the affected region and comparing it to the unaffected region as a pseudo-internal control to identify genes and processes only dysregulated in the NTD-affected region. Furthermore, region-specific mutations in DDR genes may result in localized increases in DNA damage that surpass a mutation threshold that result in regionally restricted NTDs. Region-specific analysis may provide better resolution to what is happening to gene expression at the site of NTC failure. Together, testing more micronutrient conditions, profiling their downstream effects and comparing this data across conditions and models will help identify how different micronutrient imbalances contribute to the etiology of NTDs.
Conclusions
In this review, we re-visited the multifactorial etiology of neural tube defects. We highlighted how imbalances in retinoic acid, zinc, and iron from either diet or genetic causes contribute to NTD risk. We touched on examples of known common nodes in NTD etiology by integrating the mechanistic interpretations of multiple micronutrient imbalances. We emphasized shared similarities in the downstream processes affected, including the enrichment of p53 and increase in apoptotic cells, as well as dysregulation of DNA damage repair and miRNA activity in conjunction with or downstream of micronutrient imbalances to lead to failed neural tube closure. Finally, we appeal to the research community to test more micronutrient conditions and to examine the effect that micronutrient imbalances have on downstream genes and processes. With these considerations, this motivated field should be able to better understand the etiology of neural tube defects and inform how we might prevent more cases worldwide.
Acknowledgements
We would like to acknowledge Irene Zohn for inviting us to contribute to this special issue. We would also like to acknowledge members of the Niswander lab for thoughtful conversations about the literature. Last, ADK thanks the greater neural tube closure and defect field for writing excellent reviews which have enabled me (a new postdoc) to gather a quick and thoughtful grasp of where the field stands.
This work was funded by NIH NICHD P01-HD104436 & NIH NICHD R01HD081562
References
- Abdelhaleim AF, Abdo Soliman JS, Amer AY, & Abdo Soliman JS (2019). Association of Zinc Deficiency with Iron Deficiency Anemia and its Symptoms: Results from a Case-control Study. Cureus, 11(1), e3811. 10.7759/cureus.3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Abed S, Dollé P, Metzger D, Beckett B, Chambon P, & Petkovich M (2001). The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes & Development, 15(2), 226–240. 10.1101/gad.855001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Abed S, Dollé P, Metzger D, Wood C, MacLean G, Chambon P, & Petkovich M (2003). Developing with lethal RA levels: Genetic ablation of Rarg can restore the viability of mice lacking Cyp26a1. Development (Cambridge, England), 130(7), 1449–1459. 10.1242/dev.00357 [DOI] [PubMed] [Google Scholar]
- Alles AJ, & Sulik KK (1990). Retinoic acid-induced spina bifida: Evidence for a pathogenetic mechanism. Development (Cambridge, England), 108(1), 73–81. [DOI] [PubMed] [Google Scholar]
- Araya García CA (2017). Formation of Neural Tube. In Reference Module in Biomedical Sciences. Elsevier. 10.1016/B978-0-12-801238-3.11055-4 [DOI] [Google Scholar]
- Ardekani AM, & Naeini MM (2010). The Role of MicroRNAs in Human Diseases. Avicenna Journal of Medical Biotechnology, 2(4), 161–179. [PMC free article] [PubMed] [Google Scholar]
- Armstrong JF, Kaufman MH, Harrison DJ, & Clarke AR (1995). High-frequency developmental abnormalities in p53-deficient mice. Current Biology: CB, 5(8), 931–936. 10.1016/s0960-9822(95)00183-7 [DOI] [PubMed] [Google Scholar]
- Au KS, Findley TO, & Northrup H (2017). Finding the genetic mechanisms of folate deficiency and neural tube defects-Leaving no stone unturned. American Journal of Medical Genetics. Part A, 173(11), 3042–3057. 10.1002/ajmg.a.38478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafaro E, Liu Y, Xu Y, & Dempski RE (2017). The emerging role of zinc transporters in cellular homeostasis and cancer. Signal Transduction and Targeted Therapy, 2. 10.1038/sigtrans.2017.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørnstad S, Austdal LPE, Roald B, Glover JC, & Paulsen RE (2015). Cracking the Egg: Potential of the Developing Chicken as a Model System for Nonclinical Safety Studies of Pharmaceuticals. The Journal of Pharmacology and Experimental Therapeutics, 355(3), 386–396. 10.1124/jpet.115.227025 [DOI] [PubMed] [Google Scholar]
- Blom HJ, Shaw GM, den Heijer M, & Finnell RH (2006). Neural tube defects and folate: Case far from closed. Nature Reviews. Neuroscience, 7(9), 724–731. 10.1038/nrn1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodinsky LN (2017). Xenopus laevis as a Model Organism for the Study of Spinal Cord Formation, Development, Function and Regeneration. Frontiers in Neural Circuits, 11. 10.3389/fncir.2017.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton-Larrivée M, Elder E, & McGraw S (2019). DNA methylation, environmental exposures and early embryo development. Animal Reproduction, 16(3), 465–474. 10.21451/1984-3143-AR2019-0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Tian T, Steele JW, Cabrera RM, Aguiar-Pulido V, Wadhwa S, Bhavani N, Bi P, Gargurevich NH, Hoffman EN, Cai C-Q, Marini NJ, Yang W, Shaw GM, Ross ME, Finnell RH, & Lei Y (2020). Loss of RAD9B impairs early neural development and contributes to the risk for human spina bifida. Human Mutation, 41(4), 786–799. 10.1002/humu.23969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Yeo G, Muotri AR, Kuwabara T, & Gage FH (2006). Noncoding RNAs in the mammalian central nervous system. Annual Review of Neuroscience, 29, 77–103. 10.1146/annurev.neuro.29.051605.112839 [DOI] [PubMed] [Google Scholar]
- Castro-Obregón S, & Covarrubias L (1996). Role of retinoic acid and oxidative stress in embryonic stem cell death and neuronal differentiation. FEBS Letters, 381(1–2), 93–97. 10.1016/0014-5793(96)00088-9 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC). (2004). Spina bifida and anencephaly before and after folic acid mandate—United States, 1995–1996 and 1999–2000. MMWR. Morbidity and Mortality Weekly Report, 53(17), 362–365. [PubMed] [Google Scholar]
- Che Q, Wang W, Duan P, Fang F, Liu C, Zhou T, Li H, Xiong C, & Zhao K (2019). Downregulation of miR-322 promotes apoptosis of GC-2 cell by targeting Ddx3x. Reproductive Biology and Endocrinology: RB&E, 17(1), 63. 10.1186/s12958-019-0506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WH, Morriss-Kay GM, & Copp AJ (1994). Prevention of spinal neural tube defects in the curly tail mouse mutant by a specific effect of retinoic acid. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 199(2), 93–102. 10.1002/aja.1001990203 [DOI] [PubMed] [Google Scholar]
- Chen WH, Morriss-Kay GM, & Copp AJ (1995). Genesis and prevention of spinal neural tube defects in the curly tail mutant mouse: Involvement of retinoic acid and its nuclear receptors RAR-beta and RAR-gamma. Development (Cambridge, England), 121(3), 681–691. [DOI] [PubMed] [Google Scholar]
- Chen Z, Lei Y, Zheng Y, Aguiar-Pulido V, Ross ME, Peng R, Jin L, Zhang T, Finnell RH, & Wang H (2018). Threshold for neural tube defect risk by accumulated singleton loss-of-function variants. Cell Research, 28(10), 1039–1041. 10.1038/s41422-018-0061-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Pei P, Yu J, Zhang Q, Li D, Xie X, Wu J, Wang S, & Zhang T (2019). F-box protein FBXO30 mediates retinoic acid receptor γ ubiquitination and regulates BMP signaling in neural tube defects. Cell Death & Disease, 10(8). 10.1038/s41419-019-1783-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KHT, Xu B, Blenkiron C, & Fraser M (2019). Emerging Roles of miRNAs in Brain Development and Perinatal Brain Injury. Frontiers in Physiology, 10, 227. 10.3389/fphys.2019.00227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Briscoe J, & Blassberg R (2013). Morphogen interpretation: The transcriptional logic of neural tube patterning. Current Opinion in Genetics & Development, 23(4), 423–428. 10.1016/j.gde.2013.04.003 [DOI] [PubMed] [Google Scholar]
- Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, & Will BM (2000). Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovascular Research, 47(2), 314–328. 10.1016/s0008-6363(00)00098-5 [DOI] [PubMed] [Google Scholar]
- Copp AJ, & Greene NDE (2010). Genetics and development of neural tube defects. The Journal of Pathology, 220(2), 217–230. 10.1002/path.2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Stanier P, & Greene NDE (2013). Neural tube defects: Recent advances, unsolved questions, and controversies. The Lancet. Neurology, 12(8), 799–810. 10.1016/S1474-4422(13)70110-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Castro SCP, Hirst CS, Savery D, Rolo A, Lickert H, Andersen B, Copp AJ, & Greene NDE (2018). Neural tube closure depends on expression of Grainyhead-like 3 in multiple tissues. Developmental Biology, 435(2), 130–137. 10.1016/j.ydbio.2018.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans B, Griffin CS, Maconochie M, & Thacker J (2000). Xrcc2 is required for genetic stability, embryonic neurogenesis and viability in mice. The EMBO Journal, 19(24), 6675–6685. 10.1093/emboj/19.24.6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delile J, Rayon T, Melchionda M, Edwards A, Briscoe J, & Sagner A (2019). Single cell transcriptomics reveals spatial and temporal dynamics of gene expression in the developing mouse spinal cord. Development (Cambridge, England), 146(12). 10.1242/dev.173807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey AC, Shahidullah M, Mannan MA, Noor MK, Saha L, & Rahman SA (2010). Maternal and neonatal serum zinc level and its relationship with neural tube defects. Journal of Health, Population, and Nutrition, 28(4), 343–350. 10.3329/jhpn.v28i4.6040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufner-Beattie J, Huang ZL, Geiser J, Xu W, & Andrews GK (2006). Mouse ZIP1 and ZIP3 genes together are essential for adaptation to dietary zinc deficiency during pregnancy. Genesis (New York, N.Y.: 2000), 44(5), 239–251. 10.1002/dvg.20211 [DOI] [PubMed] [Google Scholar]
- Duthie SJ, Narayanan S, Brand GM, Pirie L, & Grant G (2002). Impact of folate deficiency on DNA stability. The Journal of Nutrition, 132(8 Suppl), 2444S–2449S. 10.1093/jn/132.8.2444S [DOI] [PubMed] [Google Scholar]
- FDA Pregnancy Categories—CHEMM. (n.d.). Retrieved June 29, 2021, from https://chemm.nlm.nih.gov/pregnancycategories.htm
- Felkner MM, Suarez L, Brender J, Scaife B, & Hendricks K (2005). Iron status indicators in women with prior neural tube defect-affected pregnancies. Maternal and Child Health Journal, 9(4), 421–428. 10.1007/s10995-005-0017-3 [DOI] [PubMed] [Google Scholar]
- Finnell RH, Caiaffa CD, Kim S-E, Lei Y, Steele J, Cao X, Tukeman G, Lin YL, Cabrera RM, & Wlodarczyk BJ (2021). Gene Environment Interactions in the Etiology of Neural Tube Defects. Frontiers in Genetics, 12, 659612. 10.3389/fgene.2021.659612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, & Meira LB (2006). Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair, 5(2), 189–209. 10.1016/j.dnarep.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Gao Q, & Gao Y-M (2007). Hyperglycemic condition disturbs the proliferation and cell death of neural progenitors in mouse embryonic spinal cord. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience, 25(6), 349–357. 10.1016/j.ijdevneu.2007.08.002 [DOI] [PubMed] [Google Scholar]
- Georgieff MK (2008). The Role of Iron in Neurodevelopment: Fetal Iron Deficiency and the Developing Hippocampus. Biochemical Society Transactions, 36(Pt 6), 1267–1271. 10.1042/BST0361267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernand AD (2019). The upper level: Examining the risk of excess micronutrient intake in pregnancy from antenatal supplements. Annals of the New York Academy of Sciences, 1444(1), 22–34. 10.1111/nyas.14103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernand AD, Schulze KJ, Stewart CP, West KP, & Christian P (2016). Micronutrient deficiencies in pregnancy worldwide: Health effects and prevention. Nature Reviews. Endocrinology, 12(5), 274–289. 10.1038/nrendo.2016.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di Virgilio F, Zito E, Pandolfi PP, Wieckowski MR, Mammano F, Del Sal G, & Pinton P (2015). P53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proceedings of the National Academy of Sciences of the United States of America, 112(6), 1779–1784. 10.1073/pnas.1410723112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Progress. (n.d.). Food Fortification Initiative. Retrieved June 29, 2021, from https://www.ffinetwork.org/globalprogress
- Gowen LC, Johnson BL, Latour AM, Sulik KK, & Koller BH (1996). Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nature Genetics, 12(2), 191–194. 10.1038/ng0296-191 [DOI] [PubMed] [Google Scholar]
- Greene NDE, & Copp AJ (2014). Neural tube defects. Annual Review of Neuroscience, 37, 221–242. 10.1146/annurev-neuro-062012-170354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene NDE, Massa V, & Copp AJ (2009). Understanding the causes and prevention of neural tube defects: Insights from the splotch mouse model. Birth Defects Research. Part A, Clinical and Molecular Teratology, 85(4), 322–330. 10.1002/bdra.20539 [DOI] [PubMed] [Google Scholar]
- Greene NDE, Stanier P, & Moore GE (2011). The emerging role of epigenetic mechanisms in the etiology of neural tube defects. Epigenetics, 6(7), 875–883. 10.4161/epi.6.7.16400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen PMW, van Rooij IALM, Peer PGM, Ocké MC, Zielhuis GA, & Steegers-Theunissen RPM (2004). Low maternal dietary intakes of iron, magnesium, and niacin are associated with spina bifida in the offspring. The Journal of Nutrition, 134(6), 1516–1522. 10.1093/jn/134.6.1516 [DOI] [PubMed] [Google Scholar]
- Gu H, Li H, Zhang L, Luan H, Huang T, Wang L, Fan Y, Zhang Y, Liu X, Wang W, & Yuan Z (2012). Diagnostic role of microRNA expression profile in the serum of pregnant women with fetuses with neural tube defects. Journal of Neurochemistry, 122(3), 641–649. 10.1111/j.1471-4159.2012.07812.x [DOI] [PubMed] [Google Scholar]
- Gu H, Yu J, Dong D, Zhou Q, Wang J-Y, & Yang P (2015). The miR-322-TRAF3 circuit mediates the pro-apoptotic effect of high glucose on neural stem cells. Toxicological Sciences: An Official Journal of the Society of Toxicology, 144(1), 186–196. 10.1093/toxsci/kfu271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S (2009). Using zebrafish to assess the impact of drugs on neural development and function. Expert Opinion on Drug Discovery, 4(7), 715–726. 10.1517/17460440902988464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson P, Greene NDE, Lad D, Pauws E, de Castro SCP, Stanier P, & Copp AJ (2007). Increased expression of Grainyhead-like-3 rescues spina bifida in a folate-resistant mouse model. Human Molecular Genetics, 16(21), 2640–2646. 10.1093/hmg/ddm221 [DOI] [PubMed] [Google Scholar]
- Harris MJ (2009). Insights into prevention of human neural tube defects by folic acid arising from consideration of mouse mutants. Birth Defects Research. Part A, Clinical and Molecular Teratology, 85(4), 331–339. 10.1002/bdra.20552 [DOI] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2005). Maternal diet alters exencephaly frequency in SELH/Bc strain mouse embryos. Birth Defects Research. Part A, Clinical and Molecular Teratology, 73(8), 532–540. 10.1002/bdra.20170 [DOI] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2007). Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Research. Part A, Clinical and Molecular Teratology, 79(3), 187–210. 10.1002/bdra.20333 [DOI] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2010). An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(8), 653–669. 10.1002/bdra.20676 [DOI] [PubMed] [Google Scholar]
- Herbig AK, & Stover PJ (2002). Regulation of Folate Metabolism by Iron. In Massaro EJ & Rogers JM (Eds.), Folate and Human Development (pp. 241–262). Humana Press. 10.1007/978-1-59259-164-0_12 [DOI] [Google Scholar]
- Ho E (2004). Zinc deficiency, DNA damage and cancer risk. The Journal of Nutritional Biochemistry, 15(10), 572–578. 10.1016/j.jnutbio.2004.07.005 [DOI] [PubMed] [Google Scholar]
- Hosako H, Martin GS, Barrier M, Chen YA, Ivanov IV, & Mirkes PE (2009). Gene and microRNA expression in p53-deficient day 8.5 mouse embryos. Birth Defects Research. Part A, Clinical and Molecular Teratology, 85(6), 546–555. 10.1002/bdra.20565 [DOI] [PubMed] [Google Scholar]
- Hoyle C, Henderson DJ, Matthews DJ, & Copp AJ (1996). Transferrin and its receptor in the development of genetically determined neural tube defects in the mouse embryo. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 207(1), 35–46. [DOI] [PubMed] [Google Scholar]
- Jain AK, & Barton MC (2018). p53: Emerging roles in stem cells, development and beyond. Development (Cambridge, England), 145(8). 10.1242/dev.158360 [DOI] [PubMed] [Google Scholar]
- Juriloff DM, & Harris MJ (2018). Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. Journal of Developmental Biology, 6(3). 10.3390/jdb6030022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisch-Smith JI, Ved N, Szumska D, Munro J, Troup M, Harris SE, Jacquemot A, Miller JJ, Stuart EM, Wolna M, Hardman E, Prin F, Lana-Elola E, Aoidi R, Fisher EMC, Tybulewicz VLJ, Mohun TJ, Lakhal-Littleton S, Giannoulatou E, & Sparrow DB (2020). Maternal iron deficiency perturbs embryonic cardiovascular development. BioRxiv, 2020.08.03.230615. 10.1101/2020.08.03.230615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam RKT, Deng Y, Chen Y, & Zhao H (2012). Retinoic acid synthesis and functions in early embryonic development. Cell & Bioscience, 2(1), 11. 10.1186/2045-3701-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, & Lowe SW (2017). Putting p53 in Context. Cell, 170(6), 1062–1078. 10.1016/j.cell.2017.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keats EC, Haider BA, Tam E, & Bhutta ZA (2019). Multiple‐micronutrient supplementation for women during pregnancy. Cochrane Database of Systematic Reviews, 3. 10.1002/14651858.CD004905.pub6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuls RA, Kojima K, Lozzi B, Steele JW, Chen Q, Gross SS, Finnell RH, & Parchem RJ (2020). MiR-302 Regulates Glycolysis to Control Cell-Cycle during Neural Tube Closure. International Journal of Molecular Sciences, 21(20). 10.3390/ijms21207534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Kim W, Lee S, Chun J, Kang J, Park G, Han I, Yang HJ, Youn H, & Youn B (2017). TRAF4 promotes lung cancer aggressiveness by modulating tumor microenvironment in normal fibroblasts. Scientific Reports, 7(1), 8923. 10.1038/s41598-017-09447-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, & Krichevsky AM (2005). The Elegance of the MicroRNAs: A Neuronal Perspective. Neuron, 47(6), 779–782. 10.1016/j.neuron.2005.08.019 [DOI] [PubMed] [Google Scholar]
- Lee S, & Gleeson JG (2020). Closing in on Mechanisms of Open Neural Tube Defects. Trends in Neurosciences, 43(7), 519–532. 10.1016/j.tins.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup C, Hopkins KM, Wang X, Zhu A, Wolgemuth DJ, & Lieberman HB (2010). Mouse Rad9b is essential for embryonic development and promotes resistance to DNA damage. Developmental Dynamics, 239(11), 2837–2850. 10.1002/dvdy.22415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JE, Jin O, Fujiwara Y, Kuo F, & Andrews NC (1999). Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nature Genetics, 21(4), 396–399. 10.1038/7727 [DOI] [PubMed] [Google Scholar]
- Li H, & Niswander L (2018). Does DNA methylation provide a link between folate and neural tube closure? Epigenomics, 10(10), 1263–1265. 10.2217/epi-2018-0116 [DOI] [PubMed] [Google Scholar]
- Li H, Wang X, Zhao H, Wang F, Bao Y, Guo J, Chang S, Wu L, Cheng H, Chen S, Zou J, Cui X, Niswander L, Finnell RH, Wang H, & Zhang T (2020). Low folate concentration impacts mismatch repair deficiency in neural tube defects. Epigenomics, 12(1), 5–18. 10.2217/epi-2019-0279 [DOI] [PubMed] [Google Scholar]
- Li H, Zhang J, Chen S, Wang F, Zhang T, & Niswander L (2018). Genetic contribution of retinoid-related genes to neural tube defects. Human Mutation, 39(4), 550–562. 10.1002/humu.23397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang J, & Niswander L (2018). Zinc deficiency causes neural tube defects through attenuation of p53 ubiquitylation. Development (Cambridge, England), 145(24). 10.1242/dev.169797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li J, & Cairns MJ (2014). Identifying miRNAs, targets and functions. Briefings in Bioinformatics, 15(1), 1–19. 10.1093/bib/bbs075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-S, Gu H, Huang T-C, Wei X-W, Ma W, Liu D, He Y-W, Luo W-T, Huang J-T, Zhao D, Jia S-S, Wang F, Zhang T, Bai Y-Z, Wang W-L, & Yuan Z-W (2020). MiR-322 treatment rescues cell apoptosis and neural tube defect formation through silencing NADPH oxidase 4. CNS Neuroscience & Therapeutics, 26(9), 902–912. 10.1111/cns.13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh SN (2010). The missing zinc: P53 misfolding and cancer. Metallomics: Integrated Biometal Science, 2(7), 442–449. 10.1039/c003915b [DOI] [PubMed] [Google Scholar]
- Lozano G (2010). Mouse Models of p53 Functions. Cold Spring Harbor Perspectives in Biology, 2(4). 10.1101/cshperspect.a001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozoff B, & Georgieff MK (2006). Iron deficiency and brain development. Seminars in Pediatric Neurology, 13(3), 158–165. 10.1016/j.spen.2006.08.004 [DOI] [PubMed] [Google Scholar]
- Lu J, Zhang F, Yuan Y, Ding C, Zhang L, & Li Q (2013). All-trans retinoic acid upregulates the expression of p53 via Axin and inhibits the proliferation of glioma cells. Oncology Reports, 29(6), 2269–2274. 10.3892/or.2013.2391 [DOI] [PubMed] [Google Scholar]
- Maden M (2006). Retinoids and spinal cord development. Journal of Neurobiology, 66(7), 726–738. 10.1002/neu.20248 [DOI] [PubMed] [Google Scholar]
- Mallela M, & Hrubec T (2012). Reduction in valproic acid-induced neural tube defects by maternal immune stimulation: Role of apoptosis. Birth Defects Research. Part B, Developmental and Reproductive Toxicology, 95(4), 296–303. 10.1002/bdrb.21018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Pla P, Larue L, & Gruss P (2001). Pax3 acts cell autonomously in the neural tube and somites by controlling cell surface properties. Development, 128(11), 1995–2005. 10.1242/dev.128.11.1995 [DOI] [PubMed] [Google Scholar]
- Mao J, McKean DM, Warrier S, Corbin JG, Niswander L, & Zohn IE (2010). The iron exporter ferroportin 1 is essential for development of the mouse embryo, forebrain patterning and neural tube closure. Development (Cambridge, England), 137(18), 3079–3088. 10.1242/dev.048744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx V (2021). Method of the Year: Spatially resolved transcriptomics. Nature Methods, 18(1), 9–14. 10.1038/s41592-020-01033-y [DOI] [PubMed] [Google Scholar]
- Milunsky A, Morris JS, Jick H, Rothman KJ, Ulcickas M, Jick SS, Shoukimas P, & Willett W (1992). Maternal zinc and fetal neural tube defects. Teratology, 46(4), 341–348. 10.1002/tera.1420460405 [DOI] [PubMed] [Google Scholar]
- Molloy AM, Einri CN, Jain D, Laird E, Fan R, Wang Y, Scott JM, Shane B, Brody LC, Kirke PN, & Mills JL (2014). Is low iron status a risk factor for neural tube defects? Birth Defects Research. Part A, Clinical and Molecular Teratology, 100(2), 100–106. 10.1002/bdra.23223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrass P, Rendl M, Mildner M, Gruber F, Lengauer B, Ballaun C, Eckhart L, & Tschachler E (2004). Retinoic acid increases the expression of p53 and proapoptotic caspases and sensitizes keratinocytes to apoptosis: A possible explanation for tumor preventive action of retinoids. Cancer Research, 64(18), 6542–6548. 10.1158/0008-5472.CAN-04-1129 [DOI] [PubMed] [Google Scholar]
- Muhsen M, Youngs J, Riu A, Gustafsson J-Å, Kondamadugu VS, Garyfalidis E, & Bondesson M (2021). Folic acid supplementation rescues valproic acid-induced developmental neurotoxicity and behavioral alterations in zebrafish embryos. Epilepsia. 10.1111/epi.16915 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Brock G, Appana S, Webb C, Greene RM, & Pisano MM (2011). MicroRNA gene expression signatures in the developing neural tube. Birth Defects Research. Part A, Clinical and Molecular Teratology, 91(8), 744–762. 10.1002/bdra.20819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulou E, Galea GL, Rolo A, Greene NDE, & Copp AJ (2017). Neural tube closure: Cellular, molecular and biomechanical mechanisms. Development, 144(4), 552–566. 10.1242/dev.145904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ, & Zhivotovsky B (2004). DNA damage-induced apoptosis. Oncogene, 23(16), 2797–2808. 10.1038/sj.onc.1207532 [DOI] [PubMed] [Google Scholar]
- Ochs K, & Kaina B (2000). Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Research, 60(20), 5815–5824. [PubMed] [Google Scholar]
- Pani L, Horal M, & Loeken MR (2002). Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: Implications for Pax-3- dependent development and tumorigenesis. Genes & Development, 16(6), 676–680. 10.1101/gad.969302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei P, cheng X, Yu J, Shen J, Li X, Wu J, Wang S, & Zhang T (2019). Folate deficiency induced H2A ubiquitination to lead to downregulated expression of genes involved in neural tube defects. Epigenetics & Chromatin, 12(1), 69. 10.1186/s13072-019-0312-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan SA, Ito M, & Loeken MR (1997). Neural tube defects in embryos of diabetic mice: Role of the Pax-3 gene and apoptosis. Diabetes, 46(7), 1189–1197. 10.2337/diab.46.7.1189 [DOI] [PubMed] [Google Scholar]
- Pike V, & Zlotkin S (2019). Excess micronutrient intake: Defining toxic effects and upper limits in vulnerable populations. Annals of the New York Academy of Sciences, 1446(1), 21–43. 10.1111/nyas.13993 [DOI] [PubMed] [Google Scholar]
- Puig S, Ramos-Alonso L, Romero AM, & Martínez-Pastor MT (2017). The elemental role of iron in DNA synthesis and repair. Metallomics: Integrated Biometal Science, 9(11), 1483–1500. 10.1039/c7mt00116a [DOI] [PubMed] [Google Scholar]
- Rat E, Billaut‐Laden I, Allorge D, Lo‐Guidice J-M, Tellier M, Cauffiez C, Jonckheere N, Seuningen I. van, Lhermitte M, Romano A, Guéant J-L, & Broly F (2006). Evidence for a functional genetic polymorphism of the human retinoic acid–metabolizing enzyme CYP26A1, an enzyme that may be involved in spina bifida. Birth Defects Research Part A: Clinical and Molecular Teratology, 76(6), 491–498. 10.1002/bdra.20275 [DOI] [PubMed] [Google Scholar]
- Ross SA, McCaffery PJ, Drager UC, & De Luca LM (2000). Retinoids in embryonal development. Physiological Reviews, 80(3), 1021–1054. 10.1152/physrev.2000.80.3.1021 [DOI] [PubMed] [Google Scholar]
- Sabatino JA, Stokes BA, & Zohn IE (2017). Prevention of neural tube defects in Lrp2 mutant mouse embryos by folic acid supplementation. Birth Defects Research, 109(1), 16–26. 10.1002/bdra.23589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, & Jacks T (1995). A subset of p53-deficient embryos exhibit exencephaly. Nature Genetics, 10(2), 175–180. 10.1038/ng0695-175 [DOI] [PubMed] [Google Scholar]
- Singh S (2019). Zinc oxide nanoparticles impacts: Cytotoxicity, genotoxicity, developmental toxicity, and neurotoxicity. Toxicology Mechanisms and Methods, 29(4), 300–311. 10.1080/15376516.2018.1553221 [DOI] [PubMed] [Google Scholar]
- Stokes BA, Sabatino JA, & Zohn IE (2017). High levels of iron supplementation prevents neural tube defects in the Fpn1ffe mouse model. Birth Defects Research, 109(2), 81–91. 10.1002/bdra.23542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudiwala S, Palmer A, Massa V, Burns AJ, Dunlevy LPE, de Castro SCP, Savery D, Leung K-Y, Copp AJ, & Greene NDE (2019). Cellular mechanisms underlying Pax3-related neural tube defects and their prevention by folic acid. Disease Models & Mechanisms, 12(11), dmm042234. 10.1242/dmm.042234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantibanchachai C (2014). Isotretinoin (Accutane) as a Teratogen. In Embryo Project Encyclopedia. [Google Scholar]
- Tom C, Juriloff DM, & Harris MJ (1991). Studies of the effect of retinoic acid on anterior neural tube closure in mice genetically liable to exencephaly. Teratology, 43(1), 27–40. 10.1002/tera.1420430105 [DOI] [PubMed] [Google Scholar]
- Tran S, Wang L, Le J, Guan J, Wu L, Zou J, Wang Z, Wang J, Wang F, Chen X, Cai L, Lu X, Zhao H, Guo J, Bao Y, Zheng X, & Zhang T (2012). Altered methylation of the DNA repair gene MGMT is associated with neural tube defects. Journal of Molecular Neuroscience: MN, 47(1), 42–51. 10.1007/s12031-011-9676-2 [DOI] [PubMed] [Google Scholar]
- Tulchinsky TH (2010). Micronutrient Deficiency Conditions: Global Health Issues. Public Health Reviews, 32(1), 243–255. 10.1007/BF03391600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung EWY, & Winn LM (2011). Valproic acid increases formation of reactive oxygen species and induces apoptosis in postimplantation embryos: A role for oxidative stress in valproic acid-induced neural tube defects. Molecular Pharmacology, 80(6), 979–987. 10.1124/mol.111.072314 [DOI] [PubMed] [Google Scholar]
- van Straaten HWM, & Copp AJ (2001). Curly tail: A 50 year history of the mouse spina bifida model. Anatomy and Embryology, 203(4), 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velie EM, Block G, Shaw GM, Samuels SJ, Schaffer DM, & Kulldorff M (1999). Maternal supplemental and dietary zinc intake and the occurrence of neural tube defects in California. American Journal of Epidemiology, 150(6), 605–616. 10.1093/oxfordjournals.aje.a010059 [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Niswander LA, Shaw GM, & Finnell RH (2013). The continuing challenge of understanding, preventing, and treating neural tube defects. Science (New York, N.Y.), 339(6123), 1222002. 10.1126/science.1222002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Morgan SC, & Loeken MR (2011). Pax3 Stimulates p53 Ubiquitination and Degradation Independent of Transcription. PLoS ONE, 6(12). 10.1371/journal.pone.0029379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang J, Guan T, Xiang Q, Wang M, Guan Z, Li G, Zhu Z, Xie Q, Zhang T, & Niu B (2014). Role of methotrexate exposure in apoptosis and proliferation during early neurulation. Journal of Applied Toxicology: JAT, 34(8), 862–869. 10.1002/jat.2901 [DOI] [PubMed] [Google Scholar]
- Wegner C, Drews E, & Nau H (1990). Zinc concentrations in mouse embryo and maternal plasma. Effect of valproic acid and nonteratogenic metabolite. Biological Trace Element Research, 25(3), 211–217. 10.1007/BF02990416 [DOI] [PubMed] [Google Scholar]
- Wei X, Li H, Miao J, Zhou F, Liu B, Wu D, Li S, Wang L, Fan Y, Wang W, & Yuan Z (2012). Disturbed apoptosis and cell proliferation in developing neuroepithelium of lumbo-sacral neural tubes in retinoic acid-induced spina bifida aperta in rat. International Journal of Developmental Neuroscience: The Official Journal of the International Society for Developmental Neuroscience, 30(5), 375–381. 10.1016/j.ijdevneu.2012.03.340 [DOI] [PubMed] [Google Scholar]
- Wilde JJ, Petersen JR, & Niswander L (2014). Genetic, epigenetic, and environmental contributions to neural tube closure. Annual Review of Genetics, 48, 583–611. 10.1146/annurev-genet-120213-092208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J, Mai CT, Mulinare J, Isenburg J, Flood TJ, Ethen M, Frohnert B, Kirby RS, & Centers for Disease Control and Prevention. (2015). Updated estimates of neural tube defects prevented by mandatory folic Acid fortification—United States, 1995–2011. MMWR. Morbidity and Mortality Weekly Report, 64(1), 1–5. [PMC free article] [PubMed] [Google Scholar]
- Wilson L, Gale E, & Maden M (2003). The role of retinoic acid in the morphogenesis of the neural tube. Journal of Anatomy, 203(4), 357–368. 10.1046/j.1469-7580.2003.00230.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Peng S, Finnell RH, & Zheng Y (2021). Organoids as a new model system to study neural tube defects. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 35(4), e21545. 10.1096/fj.202002348R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Shinotsuka N, Nonomura K, Takemoto K, Kuida K, Yosida H, & Miura M (2011). Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. The Journal of Cell Biology, 195(6), 1047–1060. 10.1083/jcb.201104057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Wang G, Luo X, Zhang P, Peng S, Cheng X, Wang M, & Yang X (2021). Endoplasmic reticulum stress-related calcium imbalance plays an important role on Zinc oxide nanoparticles-induced failure of neural tube closure during embryogenesis. Environment International, 152, 106495. 10.1016/j.envint.2021.106495 [DOI] [PubMed] [Google Scholar]
- Zabihi S, & Loeken MR (2010). Understanding Diabetic Teratogenesis: Where Are We Now and Where Are We Going? Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(10), 779–790. 10.1002/bdra.20704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H-N, Guo Y, Ma W, Xue J, Wang W-L, & Yuan Z-W (2019). MGMT is down-regulated independently of promoter DNA methylation in rats with all-trans retinoic acid-induced spina bifida aperta. Neural Regeneration Research, 14(2), 361–368. 10.4103/1673-5374.244799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Tian Y, Zhang H, Qu L, Chen Y, Liu Q, Luo Y, & Wu X (2019). P53 Mutant p53N236S Induces Neural Tube Defects in Female Embryos. International Journal of Biological Sciences, 15(9), 2006–2015. 10.7150/ijbs.31451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Kartiko S, & Finnell RH (2009). Importance of gene-environment interactions in the etiology of selected birth defects. Clinical Genetics, 75(5), 409–423. 10.1111/j.1399-0004.2009.01174.x [DOI] [PubMed] [Google Scholar]
- Zohn IE (2012). Mouse as a model for multifactorial inheritance of neural tube defects. Birth Defects Research. Part C, Embryo Today: Reviews, 96(2), 193–205. 10.1002/bdrc.21011 [DOI] [PubMed] [Google Scholar]
- Zohn IE, De Domenico I, Pollock A, Ward DM, Goodman JF, Liang X, Sanchez AJ, Niswander L, & Kaplan J (2007). The flatiron mutation in mouse ferroportin acts as a dominant negative to cause ferroportin disease. Blood, 109(10), 4174–4180. 10.1182/blood-2007-01-066068 [DOI] [PMC free article] [PubMed] [Google Scholar]