


Abstract
Canonical non-homologous end-joining (cNHEJ) is the prominent mammalian DNA double-strand breaks (DSBs) repair pathway operative throughout the cell cycle. Phosphorylation of Ku70 at ser27-ser33 (pKu70) is induced by DNA DSBs and has been shown to regulate cNHEJ activity, but the underlying mechanism remained unknown. Here, we established that following DNA damage induction, Ku70 moves from nucleoli to the sites of damage, and once linked to DNA, it is phosphorylated. Notably, the novel emanating functions of pKu70 are evidenced through the recruitment of RNA Pol II and concomitant formation of phospho-53BP1 foci. Phosphorylation is also a prerequisite for the dynamic release of Ku70 from the repair complex through neddylation-dependent ubiquitylation. Although the non-phosphorylable ala-Ku70 form does not compromise the formation of the NHEJ core complex per se, cells expressing this form displayed constitutive and stress-inducible chromosomal instability. Consistently, upon targeted induction of DSBs by the I-SceI meganuclease into an intrachromosomal reporter substrate, cells expressing pKu70, rather than ala-Ku70, are protected against the joining of distal DNA ends. Collectively, our results underpin the essential role of pKu70 in the orchestration of DNA repair execution in living cells and substantiated the way it paves the maintenance of genome stability.
Graphical Abstract
Graphical Abstract.
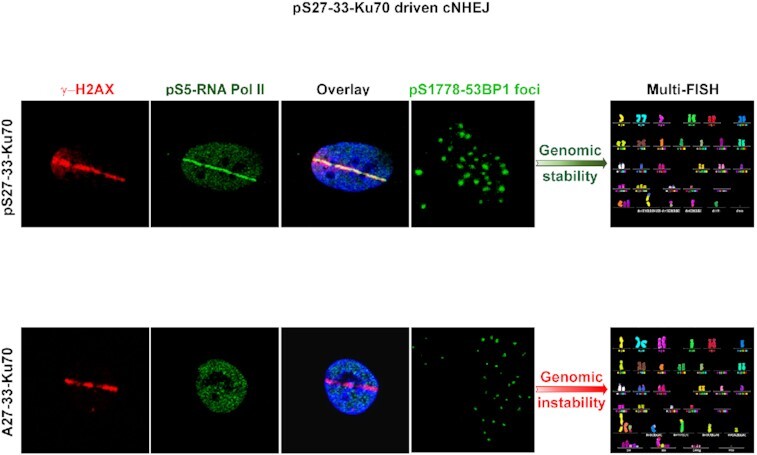
Following DNA damage, phospho-Ku70 promotes phospho-RNA Pol II and phospho-53BP 1 commitment to ensure genomic stability.
INTRODUCTION
DNA damage must be repaired rapidly and accurately to prevent genome instability that can lead to cancer development. Hence, it is not only the cellular DNA repair capacity but also simultaneous repair fidelity that needs to be orchestrated in a time-dependent manner at each DNA lesion. To avoid cell death and ensure legitimate survival, cells have developed multiple DNA repair systems that are specific for the type of DNA damage (1). DNA double strand breaks (DBs) are the most deleterious damages regarding possible cell outcomes (2,3). In response to DSBs, the cellular DNA damage response (DDR) is activated through three phosphatidylinositol-3-related family kinases, ATM and DNA-PKcs, which regulate the activities of proteins involved in non-homologous end joining (NHEJ), homology-directed recombination (HR) (3–10), and cell cycle checkpoints (11–16). In this context, BRCA1 and 53BP1, assisted by auxiliary factors, are two principal deciders for initiating HR and cNHEJ, respectively (17–20).
HR is considered accurate when it relies on homologous sister chromatid strands for finding identical DNA templates in late-S/G2 phases of the cell cycle. In contrast, NHEJ which is active throughout the cell cycle, has remained for a longtime as an error-prone pathway of DSBs repair. Nowadays, new data are precising that the accuracy of NHEJ repair is mainly depending on DNA end sequence features (21). Increasing evidences favour cNHEJ as a repair system that maintains the stability of the genome. Ku70 and Ku80 form the Ku heterodimer which displays high DNA end-binding affinity and forms a trimer with DNA-PKcs, referred to as the DNA-PK holoenzyme, which launches cNHEJ (22,23). The next step in NHEJ, which may require DNA end processing, involves the Artemis nuclease, polynucleotide kinase (PNK), DNA polymerases Pol λ and Pol μ, aprataxin, and polynucleotide kinase/phosphatase-like factor (APLF), MRN, and WRN helicase. The ligation step, which necessitates DNA ligase IV and XRCC4, as well as cofactors XLF (Cernunnos) and Paxx (9), follows the processing step. Thus, the recruitment and activation of all cNHEJ proteins are essentially ensured by the highly abundant (∼4 × 105 molecules/cell) Ku heterodimer through the Ku-binding motif (24) and DNA-PKcs kinase activity (25).
End-joining may result in inaccurate repair when non-cohesive DNA ends with 5’ or 3’ overhangs should be resolved by a search of sequence microhomology or by simple annealing before ligation, thus resulting in mutations and/or nucleotide loss (7,21,26). Subset of microhomology-directed DNA end-joining may be Ku independent [alternative NHEJ (aNHEJ)], and it represents powerful mechanisms of genome instability (rev. 27). The aNHEJ pathway is dependent on the MRN complex (27–30) and may be suppressed by the versatile protein WRN (regulating both, HR and cNHEJ) through the inhibition of Mre11/CtIP-mediated resection and subsequent large DNA deletions (31). Notably, the resection-dependent slow kinetics of end-joining during G1 may be of particular importance for genome stability, which involves the same initial components as HR. Effectively, this kind of end resection in DNA repair requires CtIP/Brca1 interaction and the exonuclease Exo1, as well as the endonuclease Artemis (32), which functions in both HR and NHEJ. This process is highly mutagenic, as it results in nucleotide loss and translocation events, thus representing a potential source of the illegitimate, disordered cancer genome. Therefore, depending on the DNA-end configuration at sites of damage, the availability of necessary factors, and the cell cycle phase, the end-processing step engages common proteins of different DNA repair pathways in tight dynamic interactions. This concept may also favour interplay and complementarity between the actors of different repair pathways in all phases of the cell cycle, rather than their competition (33). While irradiation induces multiple types of DNA DSB ends, an adaptability of DNA repair pathways, especially of the cNHEJ pathway, must be crucial for the final genome outcome. In this context, the engagement of 53BP1 at DNA DSBs appears to be a critical point of DDR fulfilment. 53BP1 is recruited by the methylated histone H4-K20 in the vicinity of DNA damage (34). It impedes BRCA1 activity, thus antagonising HR (35) and promoting cNHEJ through the downstream recruitment of RIF1 and shielding complex (36,37). In parallel to the inhibition of end resection and the protection of DNA ends’ degradation, including distant DNA ends (26), 53BP1 create[s] the environment nanodomains, facilitating access of repair factors to DNA damage (36). One new member of factors acceding DNA repair is RNA polymerase II (RNA Pol II). Indeed, recruitment of RNA Pol II at sites of DNA damage in actively transcribed genes (38), but also elsewhere in the genome (39,40), argued the accuracy of cNHEJ repair. Furthermore, these non-coding RNAs synthesised at DSBs by RNA Pol II were shown to be necessary for proper DNA damage response foci formation, as evidenced by 53BP1 recruitment (40).
Here, we have addressed the operative mode of action of phospho-ser27-ser33-Ku70 (named thereafter pKu70). Indeed, we have previously identified pKu70 in resistant primary leukemic cells (41), displaying upregulated cNHEJ (42) and short telomeres recognised as DSBs (43). Here we deepen the molecular analysis of the impact of pKu70 on DSB repair; we established that following DNA damage induced by irradiation, the final accuracy of cNHEJ is dependent on the Ku70 phosphorylation status. Effectively, Ku70 phosphorylation occurring at the sites of DNA DSBs appears necessary for both the interaction with RNA Pol II and the proper formation of phospho-53BP1 foci as well as its release from DNA. We showed that the lack of these last events compromises chromosomal stability.
MATERIALS AND METHODS
Cell lines and cell culture conditions
U2OS cancer cell lines (osteosarcoma) were purchased from the ATCC. U2OS-HR cells contain the truncated-GFP reporter gene and GFP gene harbouring the I-SceI cleavage site that allows the measurementof homology-directed repair (44). The human fibroblast cell line GC92 contains the CD4 reporter gene and two I-SceI restriction sites allowing distant end-joining assesment (30,45). These cells were cultured in Dulbecco's modified Eagle's medium (DMEM GlutaMAX, Life Technologies, Thermo Scientific, France) supplemented with 10% (v/v) foetal bovine serum and 1x non-essential amino acids (Life Technologies, Thermo Scientific, France). The non-cancerous human mammary epithelium derived HME cell line (46) was a kind gift from the R.A. Weinberg lab (Whitehead Institute for Biomedical Research, MIT). The HME cell line medium was additionally supplemented with insulin, epidermal growth factor (EGF), and hydrocortisone (purchased from Sigma-Aldrich). RNA Pol II inhibition by α-amanitin and 5,6-dichloro-1-b-d-ribofuranosylbenzimidazole (Sigma-Aldrich) was performed as previously described (40). To arrest GC92 cells in the G1 phase, cells were treated with 300 μM mimosine (Sigma-Aldrich, USA) overnight, followed by 2 h of mimosine-free cell culture before irradiation. For the inhibition of neddylation, cells were pre-treated with 3 μM MLN4924 (Interchim) for 1 h as previously described (47). For Rad6 inhibition, cells were treated with 10 μM TZ9 (Merck-Millipore) for 24 h.
Ku70 shRNA/cDNA vectors and cell transfection
We constructed EBV-based shRNA double-cassette vectors that enabled simultaneous inhibition of the expression of endogenous Ku70 (by shRNA) and episomal expression of different forms of exogenous proteins. Encoded mRNAs are resistant to shRNA due to codon changes in the cDNA (i.e. codons for aa54 (D) and aa57 (T) were converted from GAT to GAC and from ACA to ACG, respectively) (41). By this approach, the vector serine-Ku70 enables an expression of the wild-type form, alanine-Ku70 of a mutant form, and glutamic acid-Ku70 of a mutant phosphomimetic form. The same vectors were used to generate eGFP-Ku70 (pcDNA-eGFP, Addgene, UK) and mEos-Ku70 (mEos-2 pcDNA, Addgene, UK) fusion proteins. Cell transfection for all vectors was performed on the same day using the same JetPrime DNA transfection kit (Polyplus, Transfection SA, France) and antibiotic selection conditions (hygromycin or puromycin). As a control for antibiotic selection, non-transfected cells were treated with antibiotics, and cell death was monitored over time by light microscopy. Successful transfection was monitored in the different Ku70 mutants using SDS-PAGE and western blot analysis. Using the specific monoclonal anti-phospho-S27-Ku70 antibody, we showed that there was no expression of endogenous phospho-Ku70 in ala-Ku70 (non-phosphorylable) or glu-Ku70 (phosphomimetic)-expressing cells, while the exogenous expression of total Ku70 was the same in all cases. All stably transfected cell lines were frozen and stored in liquid nitrogen. eGFP-Ku70 and mEos-Ku70-expressing cells were freshly transfected before each live cell acquisition. All cell lines were kept under constant antibiotic selection pressure. Human POLR2D gene ORF cDNA clone C-GFPSpark tag and Human POLR2B gene ORF cDNA C-GFPSpark tag were purcahssed from Sino Biological.
Antibodies
Anti-pKu70 was generated in mouse hybridoma cells by BioGenes GmbH (Berlin, Germany); mouse anti-Ku70, clone N3H10 (ThermoFisher Scientific); rabbit anti-Ku70, ARG57851 (Arigo Biolaboratories); mouse anti-phospho-histone H2AX, clone JBW301 (Merck-Millipore), rabbit anti phospho-53BP1 (ser1778, #2675, Cell Signalling); rabbit anti-ubiquitin (ab137031, Abcam); mouse anti-ubiquitin, clone P4D1 (Cell Signalling); mouse anti-ubiquitinated proteins, clone FK2 and clone FK1 (Merck-Millipore); rat anti-RNA polymerase 2, CTD Ser5ph (Cosmo Bio Co. LTD); rabbit anti-phospho RNA Polymerase II (S5) (A304-408A-M-2, Bethyl); rabbit anti-NEDD8 (Cell Signalling); and rabbit anti-Rad6 (ab31917, Abcam).
γ-Irradiation, laser micro-irradiation, and real-time live cell imaging
Gamma irradiation was performed using the γ-irradiator IBL-637 (Cs137). The dose rate was 4.96 Gy/min for all experiments. For laser micro-irradiation and live cell imaging experiments, U2OS cells expressing eGFP-Ku70 and mEos2-Ku70 fusion proteins (ser-Ku70Ser, ala-Ku70) were transfected one week before each experiment. To stain nuclei, 1 μg/ml Hoescht 33342 (Sigma-Aldrich) was added 30 min before laser irradiation with a 405 nm laser beam (2 seconds of 10% laser intensity with NIS Photo-Activation software, Nikon) in a line of 5 μm in the nuclear region (avoiding nucleoli). The background eGFP-fluorescence intensity was measured before irradiation for each cell. The cells were followed at intervals of 10 min, for up to 4 h after irradiation. Data were acquired using a video-confocal inverted Nikon A1 microscope with an integrated chamber that maintained a 5% CO2 atmosphere and 37°C temperature. Data analysis was conducted using NIS Elements software (Nikon). Because of the observed interference of fluorescences, for DNA damage induction and subsequent colocalisation analysis of RNA Pol II and γ-H2AX, we have irradiated cells by Chameleon Vision II (Coherent) biphotonic laser at 800 nm and 30% of laser power. Images were acqisited and analysed by LSM 780 confocal microscopy (Zeiss). For cell localisation analysis of Ku70, PALM acquisitions were performed with an N-STORM (Nikon) system and an SR Apo TIRF 100x (1.49 numerical aperture) oil objective. Images were obtained using a TIRF inverted Nikon Eclipse Ti-E microscope equipped with a quad band emission filter (450/60–525/50–605/50–730/120, Chroma) coupled to an EMCCD camera (iXon DU897, Andor). Fixed cells expressing mEos2 constructs were used with sequential activation at 405 nm (Cube laser, 100 mW, Coherent), followed by excitation at 561 nm (Sapphir laser, 150 mW, Coherent). NIS-elements AR software (Nikon, v. 4.30.01) and the STORM analysis module (Nikon) were used to control the system and perform molecule detection, respectively. To prevent axial drift, we used a perfect focus system (Nikon); to correct for any lateral or axial drift during acquisition, we applied the auto-correlation algorithm of NIS-elements AR software (Nikon, v. 4.30.01). Local density analysis was performed using SR-Tesseler software. The local density of the mEos2 molecules was calculated using Voronoi tessellation and then displayed according to a colour scale that was normalised to the mean molecule density of each nucleus. To examine the kinetics of mEos2-Ku recruitment after laser irradiation, time-lapse experiments were performed on an inverted Nikon Ti Eclipse Eclipse-E microscope coupled with a spinning disk (Yokogawa, CSU-X1-A1), a 60× plan apo objective (Nikon, NA 1.49, oil immersion) and an sCMOS camera (Photometrics, Prime 95B). Cells expressing mEos2-Ku constructs grown in 35 mm Ibidi μ-dishes were imaged in a controlled atmosphere (37°C, 5% CO2). Before imaging, the cells were incubated for 30 min with 1 μg/ml Hoechst 33342. Hoechst fluorescence was detected with 405 nm excitation (Vortran, 100 mW laser) and a 450/50 bandpass filter (Chroma). mEos2 was excited at either 488 nm (Vortran, 150 mW laser) or 561 nm (Coherent, 100 mW laser), and a 525/45 bandpass filter (Semrock) or a 607/36 bandpass filter (Semrock) was used to detect its native or photoconverted form, respectively. Irradiation was performed by scanning a line of 10 μm with the 405 nm laser for 60 s. For each field of view, a time-lapse series was recorded with a pre-irradiation sequence of 60 s and a post-irradiation sequence of 600 seconds, acquiring an image every 20 s. A perfect focus system (Nikon) was used to keep the focus constant during the time-lapse series; the whole system was driven by Metamorph (version 7.7, Molecular Devices).
γ-H2AX and phospho-ser1778-53BP1 foci assessments
Cells were grown on 8-well covered slides (Millicell EZ Slides, Merck-Millipore) for 2 days until 70–80% confluency. The slides were irradiated at 2 or 4 Gy and left in the incubator for the indicated times (0.5–24 h). After anti-γ-H2AX and/or p53BP1 labelling, data acquisition was performed using a microscope (Zeiss, Imager Z2) as previously described (41). Each analysis was performed on at least 100 cells, and at least 10 images of each condition were analysed. Confocal microscopy optical slice sections of 8–20 μm were recorded from the apical to the basal pole of the cells, with each acquisition containing 26 stacks. Images were prepared and stacked with ImageJ software (Bethesda, MD) (48) by using the stacks tool. Then, TIFF images were converted to 8 bits before performing γ-H2AX and p53BP1 foci counts. Cell Profiler software (Cambridge, MA) (49) was used for the detection and scoring of foci in p53BP1 and γ-H2AX images. Representation of the data as boxplot was performed using GraphPad Prism 7. For statistical analysis, to compare the number of γ-H2AX and p53BP1 foci in each condition, a Mann-Whitney rank test based on at least 100 observations was performed. Western blot analysis was performed as previously described (41).
Chromatin-binding assay
Cells were seeded at approximately 70% confluency. For protein extraction, pre-extraction (PEB) buffer [10 mM PIPES (pH 7.0), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl, 0.7% (vol/vol) Triton X-100, H2O] was prepared on ice. Cells were washed three times with PBS without Ca2+/Mg2+ and then incubated in PEB-R buffer (PEB buffer, 7% RNAse A) for 3 min. The supernatant was collected as Fraction 1. The same procedure was repeated to obtain Fraction 2. After two PBS washes, Laemmli buffer (2x) [100 mM Tris–HCl (pH 6.8), 10% SDS, 20% glycerol, 100 mM DTT, 1% bromophenol blue, H2O] was added to obtain Fraction 3 (chromatin fraction). DNA was then digested with benzonase (Merck Millipore). To prepare the samples for SDS-PAGE, Laemmli 2× was added to Fractions 1 and 2, and all samples were heated for 5 min at 95°C.
Anti-pKu70/anti-Ku70 protein immunopurification
Proteins from at least 15 × 106 cells were extracted on ice for 1 h in hypertonic buffer containing 50 mM NaF, 450 mM NaCl, 20 mM HEPES, 0.2 mM EDTA, 0.5 mM DTT, 5 mM iodoacetamide, and a complete cocktail (Roche Diagnostic) inhibitor of phosphatases (100 μl extraction buffer per 107 cells). The cell lysates were centrifuged at 67 000 rpm for 1 h at 4°C. Next, 10% glycerol and β-mercaptoethanol (1:1250) were added to the supernatant, which was divided into three parts (two parts for immunoprecipitation and one for the input control). To equilibrate the salt content, equilibration buffer (EQ-buffer) [10 mM KCl, 10 mM MgCl, 20 mM HEPES, 2.5 mM EDTA, 0.5 mM DTT, 5 mM iodoacetamide, complete cocktail 1×, 10% (vol/vol) glycerol] was added at a ratio of 1:3, and the protein content was determined (Bradford method). Dynabeads (BE-M01/03, EMD Millipore) (1 μl per 10 μg total protein) were washed twice with PBS without Ca2+/Ca2+, and the cell lysate was added to the beads. To obtain a total volume of 500 μl, EQ buffer was added. Antibodies against Ku70 (NeoMarkers, Thermo-Fisher) and p-Ku70 (42) were mixed with the lysate/Dynabead mixture at a concentration of 1% of the total protein content and incubated overnight at 4°C with agitation. The supernatants were discarded, and the beads were washed three times in PBS before adding EQ buffer. Laemmli-buffer (5×) (0.255 M Tris–HCl (pH 6.8), 50% glycerol, 5% SDS, 0.05% bromophenol blue, 0.1 M DTT) was added, and the samples were heated at 95°C for 10 min. The immunoprecipitated proteins were resolved by SDS-PAGE for orbital-based shotgun mass spectrometry according to the standardised protocol (Proteomic Center, Rotterdam). Analysis was performed with Perseus software by varying FDR and S0 simultaneously. The S0 value is a statistical factor based on a paper referred to by the Max Quant developers and the t-test performed in SAM (from the microarray field). We chose values of S0 = 0.1 and FDR = 0.05 in the present study, which are the commonly used values for this type of analysis.
Anti-eGFP-Ku70 immunoprecipitation
eGFP-ser-Ku70 and eGFP-ala-Ku70 fusion proteins were immunoprecipitated using the ChromoTek GFP-Trap kit according to the supplier's protocol, except for the cell lysis step. Briefly, cells were lysed in the buffer specified above (see anti-Ku70 immunoprecipitation) supplemented with benzoase (15 U/assay). The final protein extracts at 1 mg per assay were adjusted to 0.5 M NaCl. Equilibrated GFPTrap beads (25 μl) were added to the lysates, followed by incubation for 1 h at 4°C. Magnetically separated beads were washed three times in lysis buffer adjusted to 1 M NaCl. The immunoprecipitated proteins were eluted from the beads in 100 μl 2× SDS–Laemmli buffer by boiling for 10 min at 95°C. The supernatants were used for SDS-PAGE followed by western blot analysis.
Multi-FISH analysis of chromosomal translocations
Human mammary epithelial cells, which were also used in the proteomic approach for the analysis of the Ku70 interactome, were used to assess chromosomal anomalies following irradiation. Cells were irradiated at 2 Gy, and metaphase cells were prepared after 24 h as described previously (50). Briefly, the slides were pre-treated with RNAse A (120 μl per slide; stock solution: 10 μg.ml–1) in 2× SSC buffer (AM9763, ThermoFisher Scientific) for 45 min at 37°C to eliminate RNA. To remove the remaining cytoplasm, pre-treatment with pepsin (P6887, Sigma Aldrich) was performed. The slides were incubated for 30 min at 37°C in 0.01 M HCl (pH 2) with 2 μg/ml pepsin. The slides were washed with PBS without Ca2+/Mg2+ (2×) and then with PBS with Ca2+/Mg2+ for 5 min. To fix pepsin-treated metaphases, the slides were incubated in 1% formaldehyde/PBS with Ca2+/Mg2+ for 10 min at RT. The slides were then washed with PBS without Ca2+/Mg2+ for 5 min. Dehydration was performed in three steps with 70, 90 and 100% ethanol at RT for 3 min, and the slides were left to dry for 2–3 min. mFISH probe denaturation and hybridisation were performed according to the manufacturer's protocol (24XCyte, D-0125-120-DI, MetaSystems). Metaphase cells were mounted with aqueous Flouromount-G (F4680, Sigma Aldrich). Data were acquired using a Zeiss Imager Z2 microscope. Data analysis was performed with ISIS software (Metasystem).
RESULTS
Phosphorylation of Ku70 occurs at the sites of DNA damage and fosters its dissociation
To establish how pKu70 may regulate the response to DNA damage, we decided to track it in living cells expressing three forms of Ku70 (ser-, ala- or glu-Ku70, Figure 1A), after DNA damage induction. At first instance, to explore the cell localisation of different forms of Ku70 at the molecular scale, we performed PALM (photoactivated localisation microscopy) experiments. We introduced cDNA in validated Ku70 vectors that enabled the expression of the photoconvertible molecule mEos2 fused to the N-terminal region of different forms of Ku70. These vectors were used to characterise the nuclear repartitioning of different forms of Ku70 in cells under non-stressed conditions. Figure 1B shows that ser-Ku70 (wild-type) filled the nucleolar structure and densely accumulated in a few nuclear subregions, while structural homologue glu-Ku70 was completely excluded from the nucleoli and showed homogenous nuclear localisation. Non-phosphorylable ala-Ku70 cells exhibited two different patterns: nucleolar with less dense regions in most cases or, less frequently, empty nucleoli with homogenous nuclear localisation (as observed for glu-Ku70). As a control, Supplemental Figure S1 shows enlarged microscopic fields of cells expressing eGFP-Ku70 fusion proteins, supporting the notion that the mEos2 fusion ligand does not affect Ku70 cellular localisation, which was also demonstrated using eGFP-Ku70 fusion proteins via a laser micro-irradiation approach and live cell imaging (see below).
Figure 1.

Ser-Ku70 preferentially localises in nucleoli and translocates to the sites of DNA damage where phosphorylation occurs; delayed kinetics of ala-Ku70 dissociation from the sites of damage. (A) Cells used in this study were transfected by the shRNA vectors double-cassette, and the extinction of endogenous Ku70 and re-expression of vectors-encoded Ku70 was verified by Western blots. (B, upper panel), mEos2 cDNA was fused into the above vectors to generate the mEos2-ser-Ku70, mEos2-ala-Ku70, or mEos2-glu-Ku70 vectors, and after 48 h, florescence was examined by photoactivated localisation microscopy (PALM). Scale bar = 5 μm. (B, Lower panel), Local density map analysis was also performed on PALM data to characterise the different patterns of the Ku70 nuclear distribution. (C, a) Localisation of mEos2-Ku70 (upper panels) and mEos2-Ku70-mut6E (lower panels) to DSBs after laser irradiation. Time-lapse observation of Hoechst (blue)-labelled cells expressing Ku70 or Ku70-mut6E fused to mEos2. Both native (green) and photoconverted (red) forms of mEos2 are recruited to the DSBs after 405 nm laser irradiation in the case of cells expressing mEos2-Ku70, but cells expressing mEos2-Ku70-mut6E exhibited no recruitment to the DSBs. Scale bar: 10 μm (b) Western blot analysis of protein extracts from cells expressing mEos2-Ku70 or mEos2-Ku70-mut6E. After SDS-PAGE, the membranes were probed with an anti-phospho-Ku70 antibody. An anti-total Ku70 antibody was used as a control. (D) Kinetics of the dissociation of Ku70 from the sites of DNA DSBs. Representative images of the dissociation kinetics in eGFP-ser-Ku70- and eGFP-ala-Ku70-expressing cells. Images depict cells before (NT), immediately after (t0), and at three time points following micro-irradiation (405 nm). Yellow arrows indicate the damage sites. Scale bar: 10 μM. (E) Relative intensity fold change based on the initial fluorescence intensity in NT cells. n = 20 (ser-Ku70); n = 27 (ala-Ku70). Unpaired t-test, *P < 0.01 .
Next, we sought to determine whether pKu70 may control clonogenic survival and cell growth following genotoxic stress. Effectively, both cell growth and clonogenic survival were affected in cells expressing ala-Ku70 or glu-Ku70 (Supplementarl Figure 2). Further, we addressed if the phosphorylation of Ku70 may occur when it was linked to DNA-ends at the sites of DNA damage. We modified above vectors, enabling the expression of mEos2-Ku70 fusion proteins, by introducing cDNA Ku70-mut6E (kind gift by S. Britton). In this vector, a positive charge in the Ku70 cavity domain was reversed by introducing six glutamic acids, giving rise to a defective Ku70 protein to bind DNA ends (51). Therefore, we generated a vector encoding the mEos2-Ku70-mut6E fusion protein. As shown in the lower panels of Figure 1C, a, this form was unable to co-localise with laser-induced DNA damage compared to mEos2-Ku70wt (upper panel). In parallel, western blots, shown in Figure 1C, b, validated that mEos2-Ku70-mut6E was not phosphorylated following irradiation (missing band at ∼95 kDa). Importantly, this form was highly toxic to cells (even in the absence of irradiation stress), impeding the complete hygromycin selection of transfected cells. This toxicity explains why there were two forms of Ku70, endogenous (wt) and exogenous, fused to mEos2 (corresponding to the ∼95 kDa band). Nevertheless, this construction allowed us to demonstrate that this form cannot undergo phosphorylation due to the absence of DNA binding, in contrast to the mEos2-Ku70 fusion protein.
Figure 2.

Phospho-Ku70 causes rapid dissociation from sites of DNA damage; inhibition of neddylation compromises both pKu70 release and DNA repair following laser micro-irradiation. (A) Ku70 release from DNA damage sites after the inhibition of neddylation by MLN4924 (3 μM, 1 h), followed by laser micro-irradiation as in Figure 1C. Relative fluorescence intensity fold change (established as in Figure 1D). Unpaired t-test, NS = non-significant. (B) Western blot of γ-H2AX after irradiation alone (a) or after combined cell treatment (b) with MLN4924 (3 μM, 1 h), followed by irradiation (4Gy). Anti-Ku70 (clone N3H10) was used as the loading control. (C, a) Western blot analyses of the neddylation patterns of untreated, irradiated (4 Gy), or MLN4924 (3 μM, 1 h) pre-treated and irradiated (4Gy) cells expressing eGFP-ser- or -ala-Ku70-fusion proteins. Cells were lysed 2 h post-irradiation. After SDS-PAGE, the membranes were probed with anti-NEDD8 or anti-Ku70 antibodies. The band below the 100 KDa-MW marker with decreased intensity upon MLN4924 treatment indicates decreased neddylation of cullin. (b) The ubiquitylation pattern analysed in protein extracts from cells expressing eGFP-Ku70-fusion proteins. Cells expressing eGFP-ser-Ku70 or eGFP-ala-Ku70 were irradiated with or without MLN4924 or TZ9 pre-treatment and lysed 2 h post-irradiation. Protein extracts were immunoprecitpitated by using a GFP-Trap_MA kit (Chromotek). After SDS-PAGE, the membranes were probed with an anti-ubiquitin, anti-Ku70, or anti-GFP antibody.
In parallel, demonstrating that pKu70 colocalized with DNA damage sites (Supplemental Figure S3, Supplemental Figure S4A), we have established an accelerated dynamic of disappearance of the γ-H2AX foci in both unsynchronised U2OS cells and in G1-arrested SV40-transformed human fibroblast GC92 (30), expressing ser-Ku70 as compared with cells expressing ala-Ku70 (Supplemental Figure S4B–F). We next performed real-time live-cell imaging to monitor the dynamics of Ku70 recruitment and release at DSBs. For this purpose, we transfected cells with vectors, enabling the expression of eGFP-Ku70 fusion proteins. DSBs were induced by micro-irradiation (405 nm UV in the presence of 1 μg ml–1 Hoechst 33342) under the same experimental conditions as applied in the experiments done for assessing phospho-Ku70 co-localisation with γ-H2AX. After micro-irradiation, fluorescence signals were followed for up to 4 h, and images were obtained every 10 min (Figure 1D and Supplemental ‘ser-Ku70’ and ‘ala-Ku70’ movies). The recruitment kinetics were rapid and quite similar, independent of introduced mutations in Ku70 cDNA vectors. Supplemental Figure S5 shows the micro-irradiation-induced laser strips at 2- and 12-s post-irradiation in cells expressing ser-Ku70 or glu-Ku70. eGFP-ser-Ku70- and ala-Ku70-expressing cells exhibited a maximal fluorescence signal between 30 and 50 min after irradiation. The quantified intensities indicated that ala-Ku70 provided a stronger signal at DSBs (i.e. 3.1-fold induction for ala-Ku70, whereas the maximal recruitment was 2.6-fold for ser-Ku70; Figure 1E; however, this difference is not statistically significant). The dissociation of eGFP-serKu70 from DNA damage sites occurred much faster and reached 50% at 150 min. The basal levels in the ser-Ku70-expressing cells occurred between 190 and 200 min. In contrast, the level in ala-Ku70 cells remained over 50% after 220 min and did not reach basal levels within 240 min. These data indicated that the phosphorylation of Ku70 favoured Ku70 release from sites of DNA damage and might implicate faster completion of the NHEJ process in these cells compared to ala-Ku70-expressing cells; these data are in accordance with data regarding γ-H2AX foci (Supplemental Figure S4).
Inhibition of neddylation-dependent ubiquitylation impairs pKu70 dissociation from DSBs
Neddylation has been reported to be an important prerequisite for Ku70 ubiquitylation and subsequent Ku70 release from DNA mediated by VCP/p97 3A + ATPase (47,52). Therefore, we asked whether the inhibition of neddylation by MLN4924 might also impair the release of phospho-Ku70 from the repair complex. Figure 2A shows the dissociation kinetics of eGFP-ser-Ku70 and eGFP-ala-Ku70 after treating cells with 3 μM MLN4924 for 1 h prior to micro-irradiation. The maximal recruitment of ser-Ku70 (6.4-fold) and ala-Ku70 (6.1-fold) after MLN 4924 treatment was ∼2-fold higher in comparison to the conditions without MLN 4924 (Figure 2A). The dissociation of ser-Ku70 was severely impaired, with only 20–25% of serKu70 having dissociated at 240 min, showing kinetics similar to that of ala-Ku70. These data indicated that ubiquitylation of Ku70 might act downstream of Ku70 phosphorylation to induce Ku70 release. Figure 2B, b shows the efficacy of MLN4924 inhibition on γ-H2AX protein levels at indicated times post-treatment. The level was sustained at 4 and 6 h after combined treatment with MLN4924 and irradiation (as compared to the irradiation treatment alone; Figure 2B, a), in ser-Ku70-expressing cells. Cells expressing ala-Ku70 exhibited similar protein levels to γ-H2AX, regardless of MLN4924 treatment, further supporting the necessity of neddylation-dependent ubiquitylation (Figure 2C, b) prior to pKu70 release from DSBs. Figure 2C,a shows the efficacy of MLN4924 inhibition of neddylation pattern.
In parallel, we performed interactome approach (see below) which revealed that several factors of the ubiquitin system interact with Ku70 depending on its phosphorylation status. Thus, the NEDD8-conjugating enzyme Ubc12 (UBE2M) and NEDD4-binding protein 1 (N4BP1), as well as the ubiquitin-conjugating enzyme UBE2G2 and the ubiquitin-ligase protein TRIM33, are new partners of Ku70, according to its phosphorylation status (see below Figure 3D). Notably, proteomic data also indicated an interaction between pKu70 and UBE2A, also known as the Rad6A ubiquitin-conjugating protein. Therefore, we tested possible neddylation-independent ubiquitylation of Ku70 through Rad6. We treated cells expressing eGFP-ser-Ku70 and eGFP-ala-Ku70 in parallel with the TZ9 Rad6-inhibitor or MLN4924 inhibitor. Immunopurification was performed by using the ChromoTek GFP-Trap kit. Figure 2C, b shows that MLN4924 inhibition affected the ubiquitination pattern of both the ser- and ala-Ku70 forms. In contrast, Rad6 inhibition seems to affect the ubiquitylation of the ala-Ku70 form only. This may raise the possibility that in the absence of any inhibition, Rad6 may exert an effect in cells expressing ala-Ku70, but not in cells expressing pKu70, since this should be dependent on completed repair complex formation.
Figure 3.

Phospho-Ku70 and mutant forms of Ku70 interact with core components of cNHEJ but only pKu70 recruits RNA Pol II in the repair complex. (A) Representative western blot of co-immunoprecipitation using anti-phospho-Ku70 shows that pKu70 interacts with the Ku80, ligase 4 and Paxx. (B) Similarly, using the anti-Ku70 antibody shows that all Ku70 forms (wild-type and mutants) interact with Ku80, ligase 4 and Paxx. The membranes were cut according to the corresponding molecular weights and probed with antibodies as previously described (see the Materials and Methods section). (C) Whole-cell protein extracts of transfected HME cells (46), expressing ser-Ku70 at 2 h post-irradiation (2 Gy), were co-immunoprecitpitated with anti-Ku70 or anti-pKu70 antibodies, resolved by SDS-PAGE and proteolyzed with trypsin. Peptides were analysed using a label-free-quantification (LFQ) approach with an orbitrap-based mass spectrometry analyser. The analysis was performed with Perseus software by varying FDR and S0 simultaneously. We chose values of S0 = 0.1 and FDR = 0.05, as the most commonly used values for this type of analysis. The corresponding genes, indicated by grey squares, are considered to exhibit non-significant differences in their affinity toward Ku70 and/or pKu70, whereas those in red squares are presumed to be more specific partners of total Ku70 or pKu70. (D) RNA Pol II localizes with γ-H2AX at laser microirradiation-induced DNA damage only in cells expressing phosphorylable ser-Ku70. Cells esxpressing ser-Ku70 or ala-Ku70 were irradiated by laser Chaameleon Vision II system and 1 min postirradiation cells were fixed and probed with anti-γ-H2AX (red) and anti-phospho-ser5-RNA Pol II (green). Hoechst 33342 was used to stain chromatin DNA. Scale bar = 5 μm. (E) RNA Pol II is recruited by pKu70 in the repair complex. Western blot analysis of immunopurified proteins from cells expressing ser-Ku70 or ala-Ku70. Cells were irradiated at 4Gy or left unirradiated, and following 30 min of post-irradiation culture, cells were lysed. The protein extracts were immunopurified by using a monoclonal anti-Ku70 antibody (clone N3H10) and magnetic beads coated with anti-mouse IgG (Estapor, Merck-Millipore). After SDS-PAGE, the membranes were probed with anti-RNA Pol II or anti-Ku70 antibodies.
pKu70 is essential for the recruitment of RNA Pol II
Following the above kinetic studies, we performed co-immunopurification assays to verify that pKu70 makes part of the cNHEJ complex. Using monoclonal anti-phospho-Ku70 and anti-Ku70 antibodies for immunoprecipitation, Figure 3A a shows the interactions of phospho-Ku70 with Ku80, Ligase 4, and PAXX, validating that phospho-Ku70 is one component of the cNHEJ complex required to repair DNA DSBs. However, ala-Ku70 and glu-Ku70 interacted equally with Ku80, Ligase 4, and PAXX (Figure 3B, where Ku70 antibodies recognaising ala- and glu-Ku70 forms, were used for protein immunoprecipitation). Furthermore, we applied a proteomics approach to analyse the proteins immunopurified with the same antibodies as in above co-immunoprecipitation assays, anti-phospho-ser27-Ku70 or anti-Ku70 (clone N3H10, NeoMarkers) antibodies. This approach allowed us to validate that there was no significant difference between the binding of Ku70 or phospho-Ku70 to the NHEJ core factors XRCC5 (Ku80), XRCC6 (Ku70), PRKDC (DNA- PKcs), LIG4 (Ligase 4) or XRCC4 (Figure 3B). These data indicated that phospho-Ku70 is not necessary for the formation and recruitment of cNHEJ factors to the break site but that it is a part of the cNHEJ complex and interacts with key partners. However, this interactome approach indicated the presence of other factors (most of them have been already reported in the literature to interact with Ku70). In addition the abowe results showing that Ku70 mutated in its DNA-binding domain was not phosphorylated, the observation that the core nucleolar factor nucleophosmin (NPM1) interacted less strongly with pKu70 further supported that phosphorylation occurs outside of nucleoli. Known auxiliary factors affecting cNHEJ were also found to be interactors with unphosphorylated (WRN and DNA-Polδ subunit 4) or phosphorylated Ku70 (CHAF1, RIF1; Figure 3B).
Of particular interest are the factors that may affect the fidelity of DNA repair (38–40), such as DNA-directed RNA polymerase I, II, and III subunit RPABC5, encoded by the POLR2L gene, which was indicated to be a specific partner of phospho-Ku70. While the other subunits specific of RNA Pol II have been found (in less significant manner) to be partners of pKu70 (data not shown), we have addressed whether RNA Pol II localizes at laser microirradiation-induced DNA DSBs. Thus, we have expressed POLR2B-eGFP and POLR2D-eGFP vectors in transient transfection assay. As shown in Supplemental Figure S7A both of these subunits localazed at microirradiation stretches in 10 seconds following irradiation. Next, we addressed whether transcriptionally active RNA Pol II localised at microirradiation-induced DNA damage. Because of the encountered interferences of fluorescences by laser at 405nm in presence of Hoechst 33342 as a photosensibilising agent and subsequent Alexaflour labellings, we have used the biphotonic laser Chameleon Vision II at 800nm to induce DNA DSBs without need of Hoechst treatment. At first, by using cells expressing eGFP-ser-Ku70 and eGFP-ala-Ku70, we established optimal laser power of 30% to recruit Ku70 at DNA damage (Supplemental Figure S7B). Following irradiation of cells expressing ser-Ku70 or ala-Ku70 in these conditions, cells were fixed at 1min or 10 min postirradiation and probed with anti-γ-H2AX and anti phospho-S5-RNA Pol II antibodies. The results shown in Figure 3D and Supplemental Figure S7C clearly evidenced that the transcriptionally active RNA Pol II was recruited to DNA damage site only in cells expressing phosphorylable ser-Ku70 but not in cells expressing ala-Ku70. This was further validatyed by co-immunoprecipitation assays followed by western blot analysis of protein fractions from cells expressing ser-Ku70 or ala-Ku70 before and after γ-irradiation stress. As shown in Figure 3E, an interaction between Ku70 and RNA Pol II was observed only after irradiation stress in extracts from U2OS cells expressing phosphorylable ser-Ku70. Indeed, unphosphorylated ser-Ku70 did not interact with RNA Pol II before irradiation. This interaction was also completely abolished in cells expressing the ala-Ku70 form, regardless of irradiation stress. These results indicated that even though the above experiments showed ala-Ku70 recruitment at the site of DNA damage and interaction with core elements of cNHEJ quasi-identical to those of ser-Ku70, the final repair complex was not the same for these two forms of Ku70.
Formation of phospho-ser1778-53BP1 foci depends on the phosphorylation status of Ku70
Small non-coding RNAs, generated at the DSBs (called DDRNAs) by RNA Pol II, have been reported as critical factors involved for proper activation of DDR. This involvement was evidenced by a defect in the formation of 53BP1 foci after ionising irradiation when RNA Pol II was inhibited (40). Proteomic data indicated no significant difference between 53BP1’s affinity towards Ku70- or pKu70-antibodies. However, RIF1, a downstream effector of 53BP1, was found in the group of proteins immunopurified by the anti-pKu70 antibody (Figure 3C). We sought to determine whether a defect in the formation of 53BP1 foci following irradiation stress could be discriminatory between cells expressing ser-Ku70 or ala-Ku70. We performed double immunofluorescence staining of γ-H2AX and p53BP1. The results shown in Figure 4A and B validated this hypothesis. Indeed, while ser-Ku70-expressing cells displayed no significant differences between γ-H2AX and p53BP1 number of foci following 1 h of 2 Gy post-irradiation culture (median values of 37 versus 35 foci, respectively; P = 0.997 ns), cells expressing ala-Ku70 displayed a significant discrepancy between the number of foci of γ-H2AX and p53BP1 (median values of 33.5 versus 26.5, respectively; **P = 0.009. In agreement with the involvement of DDRNAs in 53BP1 foci formation, we performed assays for the inhibition of RNA Pol II by α-amanitin (50 μM, 2 h) prior to irradiation treatment (2Gy) and performed the same immunostainings. In cells expressing ser-Ku70, the number of p53BP1 foci severely decreased compared to the number of γ-H2AX foci (Figure 4, median values 21 versus 30; ***P < 0.0001). Considering the defect in p53BP1 foci formation in the irradiation only treated cells expressing ala-Ku70, additional amanitin treatment, had a weak, if any, effect on the decrease in foci number (median values of 24.5 γ-H2AX vs 19,5 p53BP1 foci, *P = 0.024). Untreated cells displayed no significant differences in foci number, regardless of the Ku70 expression status. Differences in cell cycle phases should be excluded as a possible bias in above discrepancies, as all cell types exhibited similar cell cycle profiles post-irradiation (Supplemental Figure S6). In addition to their altered number, the size of p53BP1 foci also decreased in cells expressing ala-Ku70 in statistically significant maner; ***P = 0.0001, median values of 100 versus 52 for ser-Ku70- versus ala-Ku70-expressing cells, respectively. Of note, the p53BP1 foci size decreased in ser-Ku70-expressing cells following irradiation and amanitin treatments (median values of 100 for irradiation only versus 55 after double treatment; ****P = 0.0001). This was not observed in ala-Ku70-expressing cells (P = 0.3614) (Figure 4B).
Figure 4.

Phospho-53BP1 foci formation is impaired following irradiation in cells expressing unphosphorylable ala-Ku70; inhibition of RNA Poll II impairs foci formation in cells expressing the phosphorylable ser-Ku70. (A) Immunofluorescence labelling of γ-H2AX and p-53BP1 (S1778) foci in cells expressing ser-Ku70 or ala-Ku70. Foci were assessed in the untreated control (Control) or at 1 h post-irradiation (2Gy) without (IR) or with α-amanitin pre-treatment (2 h prior irradiation at 50 μg ml–1). Scale bar (white) corresponds to 10 μm. (B) Each analysis was performed on at least 100 cells, and at least 10 images of each condition were analysed. Confocal microscopy optical slice sections of 8–20 μm were recorded from the apical to the basal pole of the cells, with each acquisition containing 26 stacks. Images were prepared and stacked with ImageJ software (Bethesda, MD) (48) by using the stacks tool. Then TIFF images were converted into 8 bits before performing γ-H2AX and p53BP1 foci counts. Cell Profiler software (Cambridge, MA) (49) was used for the detection and scoring of foci in p53BP1 and γ-H2AX images. The representation of data as a box plot was performed using GraphPad Prism 7. For statistical analysis, to compare the number of γ-H2AX and p53BP1 foci as well as the size of p53BP1 foci, in each condition, a Mann–Whitney rank test based on at least 100 observations was performed. (C) The specific inhibition of RNA Pol II by α-amanitin delays the level of γ-H2AX in ser-Ku70-expressing cells. Cells were untreated (–) or pre-treated with α-amanitin (AM) at 50 μg ml–1 or with 5,6-dichloro-1b-d-ribofuransylbenzimidazole (DRB) at 50 μM for 2 h and then irradiated at 4 Gy. Total protein extracts were done at 2 or 4 h post-irradiation of the cell culture. After SDS-PAGE, membranes were probed with anti-γ-H2AX (ser-139) antibodies. Anti-Ku70 (clone N3H10), and anti-H2AX were probed as controls.
Following the above results evidencing a defect in p53BP1 foci formation in ala-Ku70-expressing cells, we further sought to validate the involvement of RNA Pol II as the specific partner of pKu70 in dynamics of the γ-H2AX level in two types of cells. We opted to perform western blots to validate the dynamic of γ-H2AX protein level because the kinetics were similar to the number of foci by the immunofluorescence approach. The timing was based on the results presented in Supplemental Figure S4, showing the significant differences in the remaining γ-H2AX foci between ser- and ala-Ku70-expressing cells at 4 h post-irradiation. Therefore, we again inhibited RNA Pol II by exposing cells to α-amanitin, a specific inhibitor, and 5,6-dichloro-1-b-d-ribofuranosylbenzimidazole (DRB), an inhibitor of RNA Pol II elongation (40). Cells pre-treated by α-amanitin (50 μg ml–1) for 2 h before irradiation slowed down the kinetics of DNA damage repair only in ser-Ku70-expressing cells. Effectively, Figure 4C shows a persistence of the γ-H2AX protein level at 4 h post-irradiation (4 Gy) in cells expressing ser-Ku70, whereas this did not occur in ala-Ku70-expressing cells. The lower concentrations of α-amanitin (25 μg ml–1, not shown), as well as those of DRB, had no effect on the level of the protein γ-H2AX and DNA repair kinetics in two types of cells. Therefore, these results further strengthened the requirement of the phosphorylation of Ku70 in the proper assembly of the repair complex.
Phospho-Ku70 protects against distal DNA DSB end junctions
Considering that the joining of distant ends inevitably results in genomic rearrangements and the fact that 53BP1 can affect the end joining of distant DNA DSBs (26), we used two specifically dedicated cell models, U2OS-HR and GC92 cells bearing intrachromosomal substrate monitoring HR or end-joining activities, respectively This approach allowed the targeting into the intrachromosomal substrates of DSBs by the I-SceI meganuclease. The DR-GFP substrate (44) contains a single I-SceI site inside of the GFP gene located downstream of the truncated GFP gene, while the NHEJ CD4 gene substrate (26,30,45) is located downstream of an insertion of 3.2 Kbp between the 5′ and 3′ I-SceI sites. The pCMV promoter was integrated upstream of this insertion (for detailed description see ref. 44 and 26,30). Expression of these substrates enabled a measure of efficacy of DNA break repair upon the expression of I-SceI. Seventy-two hours after I-SceI transfection, cells were probed for GFP, CD4-, and H2Kd expression by FACS analysis.
HR activity was based on the expression of GFP after a gene conversion event (Figure 5A, a). For U2OS-HR cells, equal expression of I-SceI was evaluated, and an empty transfection plasmid was used as a negative control (Figure 5A, c). To exclude any cell cycle-dependent events, the cell cycle was evaluated before and after transfection. The cell cycle was similar in all cell lines (Figure 5A, b). The percentage of cells expressing GFP increased in ser-Ku70-expressing cells compared with ala-Ku70-expressing cells, indicating that ser-Ku70-expressing cells had elevated HR activity under experimental conditions that was not due to an increased proportion of these cells in S/G2. Thus, these data indicated that phosphorylation of Ku70 can affect the HR repair pathway (commented in Discussion section). NHEJ activity was measured via the expression of CD4, which was induced upon I-SceI transfection and deletion of the internal fragment containing the H2Kd and CD8 genes. As a control, H2Kd expression before transfection was monitored to be ∼97–99% in all cell lines (Figure 5A and B). As for U2OS cells, equal expression of I-SceI was verified by western blotting (Figure 5B, b). As a negative control, an empty transfection plasmid was used. More elevated CD4 expression, which was observed in ala-Ku70-compared with ser-Ku70-expressing cells (Figure 5B, c), indicated that ala-Ku70 cells exhibited higher NHEJ activity that ligate two distals (3.2 kb) DNA ends. Thus, these data indicated that the phosphorylation of Ku70 can affect both the HR and end-joining DNA repair efficiency.
Figure 5.
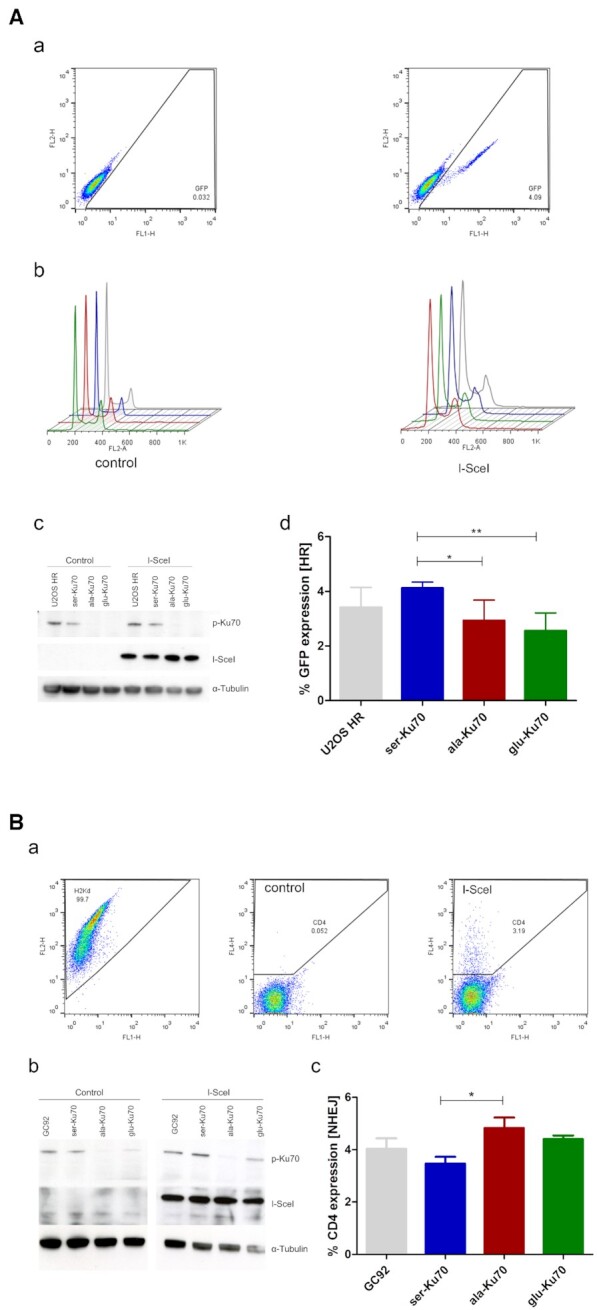
Ser-Ku70 stimulates HR activity while ala-Ku70 promotes DNA DSB distal end junctions. (A) HR activity and the cell cycle were assessed by flow cytometry of U2OS cell line containing the integrated reporter construct pDR-GFP (a and b). This cell line was transfected using ser-, ala-, or glu-Ku70 vectors as described in the methods section. (c) pKu70 expression was verified by western blot and is shown as cropped blots. The reporter is composed of two inactive eGFP genes. The upstream GFP gene is truncated; the gene downstream of the promoter has an integrated I-SceI cleavage site and is therefore inactive. Upon I-SceI expression, as verified by western blotting (c) (α-tubulin was used as the loading control), the cleaved GFP gene recombines with the truncated GFP gene on the sister chromatid, resulting in the expression of GFP, which was measured by flow cytometry 72h after I-SceI transfection (exemplified in a). The results (d) represent values that were calculated as follows: (I-SceI – transfection events) – (control – transfection events). n = 6. Unpaired t-test, *P < 0.01. (B) NHEJ activity was measured (a) using the human fibroblast cell line GC92 (SV40-transformed) expressing ser-, ala-, or glu-Ku70 [controlled by western cropped (b) blots] that contains the intrachromosomally integrated pCOH-CD4 (cohesive ends) reporter construct. Upon cleavage by I-SceI, a fragment containing the H2Kd and CD8 genes was excised. Re-joining of the flanking ends by NHEJ brought the pCMV promoter closer to the CD4 gene, which promoted its expression. (a) CD4 expression on the cell surface was quantified by flow cytometry 72 h after I-SceI transfection. (c) The results represent values that were calculated as follows: (I-SceI – transfection events) – (control – transfection events). n = 4. Unpaired t-test, *P < 0.01. Differences between values of cells expressing ser-Ku70 or glu-Ku70 are statisticaly non significant.
pKu70 promotes genome stability after genotoxic stress
Following the above results showing that pKu70 prevents distant DNA end junctions, we hypothesised that it should play a direct role in maintaining chromosomal stability. In addition to the established defect in RNA Pol II/53BP1 recruitment, starting from day 3 post-irradiation, U2OS cells expressing ala-Ku70 displayed significantly higher levels of hyperploid cells (data not shown). To address genome stability issues, we performed multi-FISH analysis. Because the U2OS and GC92 cell lines are inherently polyploid and harbour multiple chromosomal aberrations, we chose to use a human mammary epithelial diploid cell line (HME) (45), exhibiting a quasi normal karyotype. This approach allowed us to assess more precisely the type of chromosomal aberrations that may be indicative of both chromosomal translocations and loss (Figure 6A).
Figure 6.
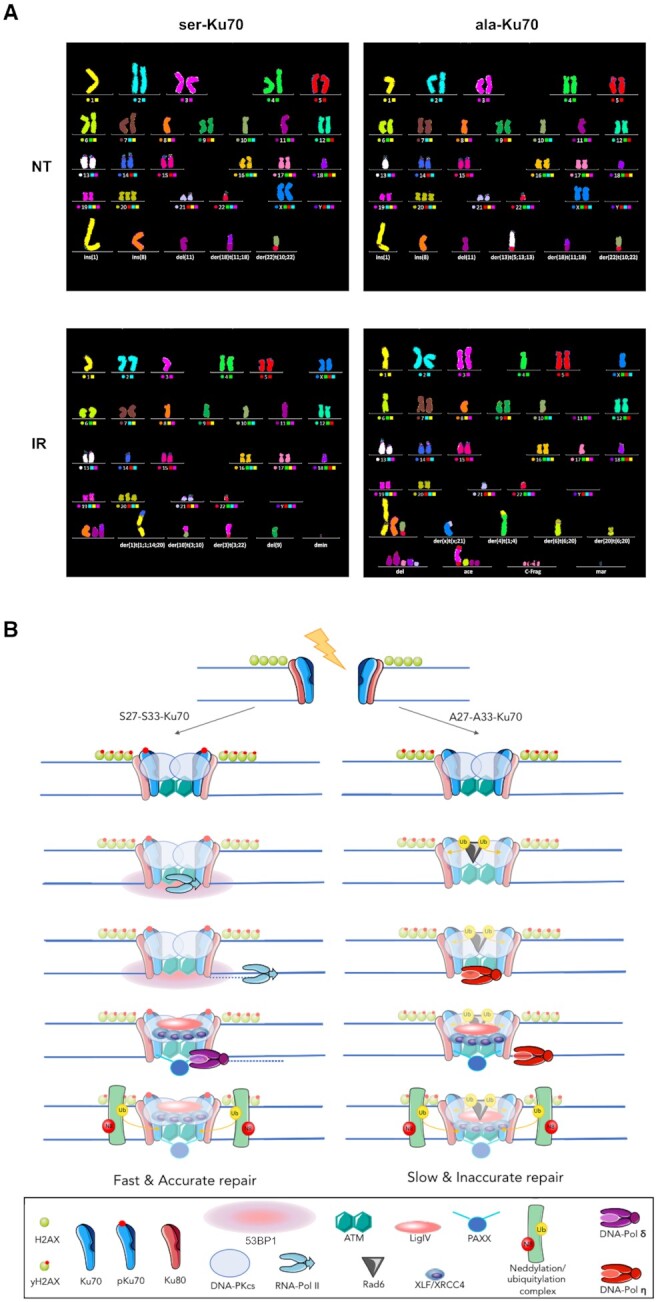
(A) pKu70 promotes genome stability after genotoxic stress. Human mammary epithelial (HME) cells expressing the ser-Ku70 or ala-Ku70 form were irradiated at 4 Gy. Following 24 h post-irradiation culture, metaphase cells were probed for chromosomal aberrations by the multi-FISH approach. Examples of 4 representative metaphase cells are shown. (Upper panel, left) Untreated (NT) ser-Ku70-expressing cells showing ins(1), ins(8–10), del(11), der(18)t(11:18), +20, der(22)t(10;22); (upper panel, right) ala-Ku70-expressing cells showing the same aberrations as in the left panel with an additional der(13)t((5;13) translocation. After irradiation (lower panels), only new translocation events (compared to untreated conditions) were considered. Other markers of genome instability are shown: dmin (double minute); del (deletion); ace (acentric fragment); C-Frag (chromosomal fragment); and mar (marker). (B). Schematic hypothetic pathway underlying the involvement of pKu70 in accurate cNHEJ. Ku70 interacts with ATM/DNA-PKcs kinases, resulting in its phosphorylation and interaction with RNA Pol II, which initiates bidirectional copying of complementary RNAs from the damage site with specific topological domains marked by 53BP1-RIF1. Newly synthesised DDRNAs are used in the next step by accurate DNA polymerases (encoded by POLD4 and POLD2; Figure 3B). Neddylation-dependent ubiquitylation precedes pKu70 dissociation from the repair complex. Unphosphorylable ala-Ku70 does not recruit RNAPII, exhibits a defect in phospho-53BP1 foci formation that allows distant end-junctions. Otherwise, its interaction with Rad6, a ubiquitin-conjugating enzyme that acts as a promotor of unfaithful DNA repair, hypothetically through Rad6/Rad18 association with translesion Y-family polymerases (i.e. Polη and Rev), may also compromise accurate repair and genome stability.
We performed two independent transfection assays with the previously described Ku70 vectors and analysed the resulting chromosomal aberrations via the multi-FISH technique. The transfections were performed using the same batch of HME cells with the same experimental protocols as previously described (50). The transfected cell lines exhibited the same proliferation rate as the parental (untransfected) HME cells (not shown). The mainline karyotype of parental HME (untransfected) cells was as follows: 46,XX,der(1)t(1;1),der(8)t(8;8), 10,del(11),der(18)t(11;18),+20,der(22)t(10;22). Multi-FISH analyses indicated that the karyotype of untransfected HME cells was found in all metaphases of ser- and ala-Ku70-expressing cells indicating that these two cell lines were isogenic. Interestingly, ala-Ku70 but not ser-Ku70-expressing cells, showed the presence of additional clonal aberrations, as represented by der(13)t(5;13;13) and der(13)t(13;13) in 26 and 24% of metaphase cells, respectively. After exposure to 2Gy irradiation and 24 h of post-irradiation cell culture, 34% of ser-Ku70 cells retained the initial non-treated cell karyotype, while only 20% of ala-Ku70 cells displayed the initial karyotype. These percentages decreased to 12% in ser-Ku70 cells after exposure to 4Gy irradiation, and no ala-Ku70 cells displayed this karyotype after exposure to 4Gy irradiation. Supplementary major chromosomal aberrations were represented by translocations and deletions in 23 and 62% of ala-Ku70 cells compared with 12 and 47% of ser-Ku70 cells, respectively, after exposure to 2Gy irradiation. These results clearly highlighted the genomic instability within cells expressing ala-Ku70 compared with those expressing ser-Ku70. This instability was observed both with and without exogenous irradiation stress.
DISCUSSION
Identified in primary leukemic cells disclosing several, somewhat conflicting, aspects reflecting an upregulated NHEJ and multiple chromosomal/telomeric aberrations (41–43), pKu70 remained for us a stumbling block. Previously, we showed accelerated repair kinetics due to pKu70 in a breast cancer cell line. Here, we sought to obtain mechanistic insights into how the regulation of cNHEJ by phospho-Ku70 may proceed. To perform this study, we again sought to exploit an advantageous experimental approach by constructing the EBV-based/shRNA vectors, enabling the simultaneous inhibition of endogenous Ku70 and expression of different forms of exogenous Ku70 in both cancer- and non-cancer-derived cell lines. Laser micro-irradiation and chromatin-binding approaches and live cell microscopy allowed us to demonstrate pKu70 co-localisation with DNA-damage sites/γ-H2AX (Figure 1C, D, Supplemental Figures S3, 4A). Moreover, we show that the phosphorylation of Ku70 occurred at the sites of DNA damage (Figure 1C, b). Most importantly, this phosphorylation appeared to be a crucial prerequisite for RNA polymerase II interaction which was shown to be recruited to DNA damage sites (Figure 3D, Supplemental Figure S7A, C). Effectively, the interaction Ku70-RNA Pol II was observed only after DNA damage induction in cells expressing ser-Ku70 but not in cells expressing ala-Ku70 (Figure 3E). This substantiates the evidence of Ku70 phosphorylation as an event occurring at DNA damage. Thus, concomitance of the above events should be regulated precisely and transiently. In consequence, inhibition of RNA Pol II by α-amanitin delayed DNA damage repair only in cells expressing ser-Ku70 (Figure 4C). These data agree with recent reports on the involvement of RNA Pol II in error-free DNA repair by cNHEJ (38,53,54). Coherently, the absence of RNA Pol II in the repair complex in cells expressing ala-Ku70 causes chromosomal rearrangements and the appearance of chromosomal instability in these cells (Figure 6). A central event in RNA Pol II activity at the sites of DNA damage is the bidirectional synthesis of damage-induced long non-coding RNA (dilncRNAs) as the precursors of small DDRNAs. These RNAs fuel DDR and 53BP1 foci formation (40,55). In this case, upon DSBs induced by I-SceI nuclease, RNA Pol II was recruited by binding to the MRE11/RAD50/NBS1 (MRN) complex prior to RNA synthesis (55). In our case, we showed that after irradiation-induced DSBs, pKu70, and not ala-Ku70, interacts with RNA Pol II. In addition to the lack of RNA Pol II interaction, ala-Ku70-expressing cells displayed a defect in the formation of 53BP1 foci that may be relevant to the appearance of chromosomal instability. Furthermore, we showed that inhibition of RNA Pol II in cells expressing the phosphorylable ser-Ku70 form impaired the DNA damage response in the assessed number of p53BP1 foci (Figure 4). 53BP1 and RIF1, as a downstream effector of 53BP1 activation, were recently shown by super-resolution microscopy (35) to form distinct nanodomains that are organised around DNA lesions. As was reported 53BP1-driven organisation of protein complex, underpins cNHEJ specific of DNA ends structure (56).
Here, we show that the phospho-ser1778-53BP1 foci are affected in number and in size in cells expressing ala-Ku70 (Figure 4A, B). This is in agreement with reports showing 53BP1 hyper-phosphorylation following DNA DSBs as well as that the dephosphorylation decreased NHEJ and favorised HR in BRCA1-dependent manner without affecting the level of 53BP1 protein (19,57). One could notice that POLR3C, the subunit of RNA Pol III interacts preferentially with Ku70 (Figure 3B) that appears quite interesting. Effectively, Liu et al., reported recently a role of RNA Pol III in DNA damage repair (58). According to the reported results, RNA Pol III was shown to participate in DNA damage repair through homologous recombination (HR) in S/G2 phase of cell cycle. RNA Pol III has been shwon to be recruited by MRN complex. While our work evidenced an involvement of pKu70 in RNA Pol II recruitment and cNHEJ fulfilment, and the fact that Ku70, through interaction with Mre11 (59), may be involved in initial steps of HR regulation, a speculation of a cell cycle-dependent engagement of RNA Pol II vs RNA Pol III in DNA damage repair desserves further investigations. In agreement with this hypothesis are the works repporting an inhibition of cNHEJ in G2 by CYREN or by phospho-RECQ4 (60,61).
The evidence of RNA Pol II recruitment by pKu70 and the formation of p53BP1 foci is certainly the central finding of this study, since γ-irradiation induces multiple types of DNA ends (‘dirty’ DSBs). The majority of these DSBs require end processing, thus leading to the possibility of nucleotide loss and error-prone repair, which is exacerbated in ala-Ku70-expressing cells but appears limited by the presence of pKu70. Furthermore, pKu70 enables the appropriate kinetics of DNA repair, while it is delayed in ala-Ku70-expressing cells (Figure 1 and Supplemental Figure S4). In addition to accelerated DNA repair kinetics, pKu70 contributed to its accelerated dissociation from the repair complex, as shown in Figure 1D, E. In accordance with recent reports (47), kinetics, as well as the dynamics of γ-H2AX protein levels, were impaired upon an the inhibition of neddylation by MLN4924 (Figure 2A, C, b).
Live cell imaging using eGFP- or mEos2-Ku70 fusion proteins allowed us to demonstrate preferential nucleolar ser-Ku70 cellular localisation that rapidly delocalised to laser induced DNA damage (Figure 1, Supplemental Figure S4, and Supplemental movies). Proteomic data (Figure 3B) highlighted that nucleophosmin and the proteins involved in ribosomal biogenesis are the main partners of Ku70. Similar nucleolar delocalisation has been reported for Rad52 (62). Other factors, such as WRN, p97/VCP, TRF2 and ABH2 (63–65), involved in DNA damage repair have already been shown to localise in nucleoli. Interestingly, Ku70 and WRN (demonstrated to interact with Ku70 in the interactome study, Figure 3B) share a common interaction with p97/VCP through which WRN may switch from cNHEJ to aNHEJ (52). Therefore, nucleolar membrane-less structures appear to be the essential storage organelles of the factors, including Ku70, which is involved in the maintenance of genome stability. Another factor, BRCA2 and CDKN1A interacting protein (BCCP1), involved in the maintenance of genome stability, was concurrent to 53BP1 loss in triple-negative breast cancers (34,66) and was one of new partners of Ku70 (Figure 3B). This interaction may be related to the promotion of homology-directed repair by cells expressing ser-Ku70 compared to cells expressing ala-Ku70 (Figure 5A). Accodingly, Ku protein was already reported to modulate HR (44) possibly though an inhibition of its phosphorylation by Wortmannin (67). More recently, the study of experimentally induced clusters of phosphorylation in Ku70 at the junction of its pillar and bridge regions, also evidenced the promotion of HR in the S phase without an effect on cNHEJ-dependent repair (68). All of above results prompted us to address genome (in)stability in two subsets of cells. For this purpose, we transfected and analysed the non-cancerous mammary epithelium derived HME cell line (46). This cell line was immortalised by hTert activation and displayed a quasi-normal diploid karyotype, thus enabling a relevant assessment of exogenously induced chromosomal rearrangements. Interestingly, untreated ala-Ku70-expressing cells displayed additional clonal aberrations, as represented by der(13)t(5;13;13) and der(13)t(13;13), suggesting the role of phospho-Ku70 in the replication process (69). After 2 or 4 Gy irradiation, the incidence of translocation/loss events was significantly more frequent in ala-Ku70-expressing cells. Thus, this instability was observed in ala-Ku70 expressing cells with and without exogenous stress from irradiation.
To maintain genome stability, cNHEJ should plays a critical role also in the late phase of the DNA repair process in G1 (30). Herein, we demonstrated that pKu70 contributes to NHEJ regulation in both unsynchronised and G1-arrested cells (Supplemental Figure S4). Together, the above findings instigate the question of how ala-Ku70 may promote resection-dependent end-joining. It promotes distant DNA end junctions (3200 bp of H2Kd/CD8 insert) (Figure 5B), favouring the role of 53BP1 that is defective in cells expressing this form of Ku70. Effectively defective Ku80 (70) and depleted 53BP1 (26) resulted in the same type of DNA end junctions. In parallel, the instability in cells expressing ala-Ku70 following G1-induced DSBs may also be a consequence of alternative NHEJ that would occur in a subset of cells undergoing the S phase in the absence of a fully functional Ku70 (71).
Another way by which DSB repair may result in genomic instability would imply Rad6 activity. Consequently, we tested the TZ9 inhibitor of the Rad6 ubiquitin-conjugating enzyme, which was indicated to be a partner of pKu70 through a proteomic approach. This type of inhibition affected only ala-Ku70 ubiquitylation but not ser-Ku70 ubiquitylation. While RNA Pol II recruitment occurs only in pKu70-expressing cells, we speculated that, in the absence of any inhibition of ubiquitylation, the multi-protein DNA repair complex is not the same in cells expressing ser-Ku70 or ala-Ku70. Therefore, this complex, including cullins/UBE2M/UBE2G2, allows Rad6 to interact with unphosphorylable ala-Ku70 and ubiquitylate it. Otherwise, the activity of Rad6 in the DNA repair complex in cells expressing ala-Ku70 may result in the recruitment of Rad18 and translesion polymerases (72), leading to inaccurate repair in a subset of cells in the S phase of the cell cycle. Considering that ‘[all] repair tools must be precisely regulated, because each in its own right can wreak havoc on the integrity of DNA if misused or allowed to access DNA at the inappropriate time or place’ (5), our hypothesis deserves further investigation to uncover the inappropriate auxiliary partners inactivated and/or excluded from pKu70 repair complex.
Supplementary Material
ACKNOWLEDGEMENTS
We thank P. Calsou and S. Britton for providing the Ku70-Mut6E vector and for their help. Thanks to C. Baldeyron for help and open access to laser Chameleon Vision II.
Author contributions: A.S. performed the main part of the experimental work; M.N.G. established the cell line models, performed the cytogenetic studies, and participated in manuscript writing; R.G., R.L.B. and V.P. conceived and performed the microscopy and flow cytometric analyses and participated in manuscript writing and figure design; C.M. participated in the experimental protocol evaluation and execution and in manuscript writing; D.B. designed and constructed all cDNA vectors; J.G.-B., M.H., V.B., E.R., P.B. and B.S.L. participated in project elaboration, cell model troubleshooting; B.S.L. and M.H. participated in manuscript redaction/edditing; V.P., Y.C. and B.T. developed the software for automatic scoring and data analysis of γ-H2AX and p53BP1 foci; R.L.B., T.K., H.S. and L.I. performed laser micro-irradiation kinetic studies; J.A.A.D. performed the proteomic studies and data analyses; S.C. and J.D. conceived the research and wrote the manuscript.
Contributor Information
Amelie Schellenbauer, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Marie-Noelle Guilly, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Romain Grall, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Romain Le Bars, Light Microscopy Facility, Imagerie-Gif, Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Univ. Paris-Sud, Université Paris-Saclay, 91198 Gif-sur-Yvette cedex, France.
Vincent Paget, IRS[N]/PSE-SANTE/SERAMED/LRMed, 31, Av. De la Division Leclerc, 92260 Fontenay aux Roses, France.
Thierry Kortulewski, Laboratoire de Radiopathologie, UMR Stabilité Génétique Cellules Souches et Radiations, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U1274, 18 Av. du Panorama, 92265 Fontenay aux Roses, France.
Haser Sutcu, IRS[N]/PSE-SANTE/SERAMED/LRAcc, 31, Av. De la Division Leclerc, 92260 Fontenay aux Roses, France.
Cécile Mathé, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Marie Hullo, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Denis Biard, Service d'étude des prions et maladies atypiques (SEPIA), DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, France.
François Leteurtre, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Vilma Barroca, Laboratoire Réparation et Transcription dans les cellules Souches, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U1274, 18, Av. du Panorama, 92265 Fontenay aux Roses, France.
Youenn Corre, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Lamya Irbah, Plateforme de Microscopie, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U12745, 18, Av. du Panorama, 92265 Fontenay aux Roses, France.
Emilie Rass, Laboratoire de Réparation et Vieillissement; Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U1274, 18, Av. du Panorama, 92265 Fontenay aux Roses, France.
Benoit Theze, Laboratoire de Réparation et Vieillissement; Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U1274, 18, Av. du Panorama, 92265 Fontenay aux Roses, France.
Pascale Bertrand, Laboratoire de Réparation et Vieillissement; Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U1274, 18, Av. du Panorama, 92265 Fontenay aux Roses, France.
Jeroen A A Demmers, Proteomics Center, Room Ee-679A | Faculty Building, Erasmus University Medical Center Wytemaweg 80, 3015 CN Rotterdam, The Netherlands.
Josée Guirouilh-Barbat, Université de Paris, INSERM U1016, UMR 8104 CNRS, Institut Cochin, Equipe Labellisée Ligue Contre le Cancer, 24 rue du Faubourg St Jacques, 75014 Paris, France.
Bernard S Lopez, Université de Paris, INSERM U1016, UMR 8104 CNRS, Institut Cochin, Equipe Labellisée Ligue Contre le Cancer, 24 rue du Faubourg St Jacques, 75014 Paris, France.
Sylvie Chevillard, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France.
Jozo Delic, Laboratoire de Cancérologie Expérimentale, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, 18, Av. du Panorama, 92265 Fontenay aux Roses, *Université Paris Descartes, 75006 Paris, France; Laboratoire de Radiopathologie, UMR Stabilité Génétique Cellules Souches et Radiations, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), Université Paris-Saclay, DRF, Institut de Biologie François Jacob (IBFJ), IRCM, UMRE008-U1274, 18 Av. du Panorama, 92265 Fontenay aux Roses, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A.S. benefited from a fellowship from the French Ministry of Research and Technology (MRT); CEA; Fondation de France, CEA ‘Segment n ° 4 Radiobiology’; EDF; light microscopy facility of Imagerie-Gif (http://www.i2bc.paris-saclay.fr), a member of IBiSA (http://www.ibisa.net), supported by ‘France-BioImaging’ [ANR-10-INBS-04-01]; Labex ‘Saclay Plant Science’ [ANR-11-IDEX-0003-02]. Funding for open access charge: Institut de Radiobiologie Cellulaire et Moleculaire.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chatterjee N., Walker G.C.. Mechanisms of DNA damage, repair and mutagenesis. Environ. Mol. Mutagen. 2017; 58:235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Le Guen T., Ragu S., Guirouilh-Barbat J., Lopez B.S.. Role of the double-strand break repair in the maintenance of genomic stability. Mol. Cell. Oncol. 2015; 2:e968020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jeggo P.A., Pearl L.H., Carr A.M.. DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer. 2017; 16:35–42. [DOI] [PubMed] [Google Scholar]
- 4. Riballo E., Kühne M., Rief N., Doherty A., Smith G.C., Recio M.J., Reis C., Dahm K., Fricke A., Krempler A.et al.. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol. Cell. 2004; 16:715–724. [DOI] [PubMed] [Google Scholar]
- 5. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Polo S.E., Jackson S.P.. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011; 25:409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mladenov E., Iliakis G.. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mut. Res. 2011; 711:61–72. [DOI] [PubMed] [Google Scholar]
- 8. Brandsma I., Gent D.C.. Pathway choice in DNA double strand break repair: observation of a balancing act. Genome Integrity. 2012; 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang H.H.Y., Pannunzio N., Adachi N., Lieber M.R.. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell. Biol. 2017; 11:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blackford A.N., Jackson S.P.. ATM, ATR and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017; 66:801–817. [DOI] [PubMed] [Google Scholar]
- 11. Hartwell L.H., Weinert T.A.. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989; 246:629–634. [DOI] [PubMed] [Google Scholar]
- 12. Warmerdam D.O., Kanaar R.. Dealing with DNA damage: relationship between checkpoint and repair pathways. Mut. Res. 2010; 704:2–11. [DOI] [PubMed] [Google Scholar]
- 13. Shaltiel I.A., Krenning L., Bruinsma W., Medema R.H.. The same, only different-DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015; 128:607–620. [DOI] [PubMed] [Google Scholar]
- 14. Jeggo P.A., Löbrich M.. How cancer cells hijack DNA double strand-break repair pathways to gain genomic instability. Biochem. J. 2015; 471:1–11. [DOI] [PubMed] [Google Scholar]
- 15. El-Deiry W.S. p21(WAF1) mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 2016; 76:5189–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Visconti R., Della Monica R., Grieco D.. Cell cycle checkpoint in cancer: a therapeutic targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016; 27:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chapman J.R., Sosick A.J., Boulton S.J., Jackson S.P.. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012; 125:3529–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Panier S., Boulton S.J.. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014; 15:7–18. [DOI] [PubMed] [Google Scholar]
- 19. Isono M., Niimi A., Oike T., Hagiwara Y., Sato H., Sekine R., Yoshida Y., Isobe S.Y., Obuse C., Nishi R.et al.. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017; 18:520–532. [DOI] [PubMed] [Google Scholar]
- 20. Mackay D.R., Howa A.C., Werner T.L., Ullman K.S.. Nup153 and Nup50 promotes recruitment of 53BP1 to DNA repair foci by antagonizing BRCA1-dependent events. J. Cell Sci. 2017; 130:3347–3359. [DOI] [PubMed] [Google Scholar]
- 21. Bétermier M., Bertrand P., Lopez B.S.. Is non-homologous end-joining really an inherently error-prone process. PLos Genet. 2014; 10:e1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gottlieb T.M., Jackson S.P.. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993; 72:131–142. [DOI] [PubMed] [Google Scholar]
- 23. Walker J.R., Corpina R.A., Goldberg J.. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001; 412:607–614. [DOI] [PubMed] [Google Scholar]
- 24. Grundy G.J., Rulten S.L., Arribas-Bosacoma R., Davidson K., Kozik Z., Oliver A.W., Pearl L.H., Caldecott K.W.. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat. Commun. 2016; 7:11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis A.J., Chen B.P.C., Chen D.J.. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst.). 2014; 17:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guirouilh-Barbat J., Gelot C., Xie A., Dardillac E., Scully R., Lopez B.S.. 53BP1 protects against CtIP-dependent capture of ectopic chromosomal sequences at the junctions of distant double-strand breaks. PLos Genet. 2016; 12:e1006230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deriano L., Roth D.B.. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013; 47:451–473. [DOI] [PubMed] [Google Scholar]
- 28. Dinkelmann M., Spehalski E., Stoneham T., Buis J., Wu Y., Sekiguchi J.M., Ferguson D.O.. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009; 16:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie A., Kwok A., Scully R.. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009; 16:814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rass E., Grabarz A., Plo I., Gautier J., Bertrand P., Lopez B.S.. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009; 16:819–824. [DOI] [PubMed] [Google Scholar]
- 31. Shamanna R.A., Lu H., de Freitas J.K., Tian J., Croteau D.L., Bohr V.A.. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016; 7:13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Biehs R., Steinlage M., Barton O., Juhász S., Künzel J., Spies J., Shibata A., Jeggo P.A., Löbrich M.. Double–strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell. 2017; 65:671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foster S.S., Balestrini A., Petrini J.H.. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol. Cell. Biol. 2011; 31:4379–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Butoyan V.M., Lee J., Ward I.M., Kim J.-E., Thompson J.R, Chen J., Mer G.. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and crb2 in DNA repair. Cell. 2006; 127:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q.et al.. 53BP1 loss rescues BRCA deficiency and is associated with triple negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010; 17:688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ochs F., Karemore G., Miron E., Brown J., Sedlackova H., Rask M.B., Lampe M., Buckle V., Schermelleh L., Lukas J.et al.. Stabilization of chromatin topology safeguards genome integrity. Nature. 2019; 574:571–574. [DOI] [PubMed] [Google Scholar]
- 37. Setiaputra D., Durocher D.. Shieldin-the protector of DNA ends. EMBO Rep. 2019; 20:e47560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chakraborty A., Tapryal N., Venkova T., Horikoshi N., Pandita R.K., Sarker A.H., Sarkar P.S., Pandita T.K., Hazra T.K.. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016; 7:13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T.. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016; 167:1001–1013. [DOI] [PubMed] [Google Scholar]
- 40. Michelini F., Pitchiaya S., Vitelli V., Sharma S., Gioia U., Pessina F., Cabrini M., Wang Y., Capozzo I., Iannelli F.et al.. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017; 19:1400–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bouley J., Saad L., Grall R., Schellenbauer A., Biard D., Paget V., Morel-Altmeyer S., Guipaud O., Chambon C., Salles B.et al.. A new phosphorylated form of Ku70 identified in resistant leukemic cells confers fast but unfaithful DNA repair in cancer cell lines. Oncotarget. 2015; 29:27980–28000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deriano L., Guipaud O., Merle-Béral H., Binet J.L., Ricoul M., Potocki-Veronese G., Favaudon V., Maciorowski Z., Muller C., Salles B.et al.. Human chronic lymphocytic leukemia B cells can escape DNA damage-induced apoptosis through the nonhomologous end-joining DNA repair pathway. Blood. 2005; 105:4776–4783. [DOI] [PubMed] [Google Scholar]
- 43. Brugat T., Nguyen-Khac F., Grelier A., Merle-Béral H., Delic J.. Telomere dysfunction-induced foci arise with the onset of telomeric deletions and complex chromosomal aberrations in resistant chronic lymphocytic leukemia cells. Blood. 2010; 116:239–249. [DOI] [PubMed] [Google Scholar]
- 44. Pierce A.J., Johnson R.D., Thomson L.H., Jasin M.. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999; 13:2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gelot C., Guirouilh-Barbat J., Le Guen T., Dardillac E., Chailleux C., Canitrot Y., Lopez B.S.. The cohesin complex prevents the end joining of distant DNA double-strand ends. Mol. Cell. 2016; 61:15–26. [DOI] [PubMed] [Google Scholar]
- 46. Elenbaas B., Spirio L., Koerner F., Fleming M.D., Zimonjic D.B., Donaher J.L., Popescu N.C., Hahn W.C., Weinberg R.A.. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001; 15:50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brown J.S., Lukashchuk N., Sczaniecka-Clift M., Britton S., le Sage C., Calsou P., Beli P., Galanty Y., Jackson S.P.. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015; 11:704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abramoff M.D., Magalhaes P.J., Ram S.J.. Image processing with ImageJ. Biophotonics Int. 2004; 11:36–42. [Google Scholar]
- 49. Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J.et al.. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Bio. 2006; 7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Padilla-Nash H.M., Barenboim-Stapleton L., Difilippantonio M.J., Ried T.. Spectral karyotyping analysis of human and mouse chromosomes. Nat. Protoc. 2006; 1:3129–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Britton S., Coates J., Jackson S.P.. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2014; 202:579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Van den Boom J., Wolf M., Weimann L., Schulze N., Li F., Kaschani F., Riemer A., Zierhut C., Kaiser M., Iliakis G.et al.. VCP/p97 extracts sterically trapped Ku70/80 rings from DNA in double-strand break repair. Mol. Cell. 2016; 64:189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jang Y., Elsayed Z., Eki R., He S., Du K-P., Abbas T., Kai M.. Intrinsically disordered protein RBM14 plays a role in generation of RNA:DNA hybrids at double-strand break sites. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thapar R., Wang J.L., Hammel M., Ye R., Liang K., Sun C., Hnizda A., Liang S, Maw S.S., Lee L.et al.. Mechanism of efficient double-strand break repair by a long non-coding RNA. Nucleic Acids Res. 2020; 48:10953–10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pessina F., Giavazzi F., Yin Y., Gioia U., Vitelli V., Galbiati A., Barozzi S., Garre M., Oldani A., Flaus A.et al.. Functional transcription poromoters at DNA double-strand breaks mediate RNA-driven phase separation of damage response factors. Nat. Cell Biol. 2019; 21:1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ghezraoui H., Oliveira C., Becker J.R., Bilham K., Moralli D., Anzilotti C., Fisher R., Deobagkar-Lele M., Sanchiz-Calvo M., Fueyo-Marcos E.et al.. 53BP1 cooperation with the REV7-Shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018; 560:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kang Y., Lee J-H., Hoan NN., Sohn H-M., Chang I-Y., You J.. Protein phosphatase 5 regulates the function of 53BP1 after neocarzinostatin-induced DNA damage. J. Biol. Chem. 2009; 284:9845–9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu S., Wang J., Li L., Yuan J., Zhang B., Wang Z., Ji J., Kong D.. RNA Polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell. 2020; 184:1314–1329. [DOI] [PubMed] [Google Scholar]
- 59. Goedecke W., Eijpe M., Offenberg HH., van Aalderen M., Heyting C.. Mre11 and Ku70 interact in somatic cells, but are differentially expressed in early meiosis. Nat. Genet. 1999; 23:194–198. [DOI] [PubMed] [Google Scholar]
- 60. Arnoult N., Correia A., Ma J., Merlo A., Garcia-Gomez S., Maric M., Tognetti M., Benner CW., Boulton SJ., Saghatelian A., Karlseder J.. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature. 2017; 549:548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu H., Shamanna R., de Freitas JK., Okur M., Khadka P., Kulikowic T., Holland PP., Tian J., Croteau DL., Davis A.J., Bohr V.A.. Cell cycle-dependent phosphorylation regulates RECQL4 pathway choiced and ubiquitination in DNA double-strand break repair. Nat. Commun. 2017; 8:2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Koike M., Yutoku Y., Koike A.. The C-terminal region of Rad52 is essential for Rad52 nuclear and nucleolar localization, and accumulation at DNA damage sites immediately after irradiation. Biochem. Biophys. Res. Commun. 2013; 435:260–266. [DOI] [PubMed] [Google Scholar]
- 63. Partridge J.J., Lopreiato J.O. Jr, Latterich M., Indig F.E.. DNA damage modulates nucleolar interaction of the Werner protein with the AAA ATPase p97/VCP. Mol. Biol. Cell. 2003; 14:4221–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang S., Hemmerich P., Grosse F.. Nucleolar localization of the human telomeric repeat binding factor 2 (TRF2). J. Cell Sci. 2004; 117:3935–3945. [DOI] [PubMed] [Google Scholar]
- 65. Li P., Gao S., Wang L., Yu F., Li J., Wang C., Li J., Wong J.. ABH2 couples regulation of ribosomal DNA transcription with DNA alkylation repair. Cell Rep. 2013; 4:817–829. [DOI] [PubMed] [Google Scholar]
- 66. Droz-Rosario R., Lu H., Liu J., Liu N.-A., Ganesan S., Xia B., Haffty B.G., Shen Z.. Roles of BCCIP deficiency in mammary tumorigenesis. Breast Cancer Res. 2017; 19:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Delacote F., Han M., Stamato T.D., Jasin M., Lopez B.S.. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acid Res. 2002; 30:3454–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee K.-J., Saha J., Sun J., Fattah K.R., Wang S.C., Jakob B., Chi L., Wang S.Y., Taucher-Scholz G., Davis A.J.et al.. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res. 2015; 44:1732–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mukherjee S., Chakraborty P., Saha P.. Phosphorylation of Ku70 subunit by cell cycle kinases modulates the replication related function of Ku heterodimer. Nucleic Acids Res. 2016; 44:7755–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guirouilh-Barbat J., Huck S., Bertrand P., Prizio L., Desmaze C., Sabatier L., Lopez B.S.. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell. 2004; 14:611–623. [DOI] [PubMed] [Google Scholar]
- 71. Yu W., Lescale C., Babin L., Bedora-Faure M., Lenden-Hasse H., Baron L., Demangel C., Yelamos J., Brunrt E., Deriano L.. Repair of G1 induced DNA double-strand breaks in S-G2/M by alternative NHEJ. Nat. Commun. 2020; 11:5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hedglin M., Benkovic S.J.. Regulation of Rad6/Rad18 activity during DNA damage tolerance. Ann. Rev. Biophys. 2015; 44:207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.