

Abstract
Understanding the mRNA life cycle requires information about the dynamics and macromolecular composition and stoichiometry of mRNPs. Fluorescence correlation and cross-correlation spectroscopy (FCS and FCCS) are appealing technologies to study these macromolecular structures because they have single molecule sensitivity and readily provide information about their molecular composition and dynamics. Here, we demonstrate how FCS can be exploited to study cytoplasmic mRNPs with high accuracy and reproducibility in cell lysates. Cellular lysates not only recapitulate data from live cells but provide improved readings and allow investigation of single mRNP analysis under particular conditions or following enzymatic treatments. Moreover, FCCS employing minute amounts of cells closely corroborated previously reported RNA dependent interactions and provided estimates of the relative overlap between factors in the mRNPs, thus depicting their heterogeneity. The described lysate-based FCS and FCCS analysis may not only complement current biochemical approaches but also provide novel opportunities for the quantitative analysis of the molecular composition and dynamics of single mRNPs.
Graphical Abstract
Graphical Abstract.

The use of cell lysates in combination with Fluorescence Correlation and Cross-correlation Spectroscopy allows a precise analysis and quantification of RNA-binding protein diffusion, stoichiometry and heterogeneity in mRNPs.
INTRODUCTION
Understanding post-transcriptional control of gene expression and its role in pathological processes requires an understanding of the molecular composition of particular mRNPs. mRNP analysis has to a large extent relied on biochemical analysis of bulk mRNPs - employing antibodies or tagged baits in pull-down analysis or fluorescence sorting in combination with western analysis and/or mass spectrometry (1–5). Although these methods have provided an impressive insight into the components of particles, they have a number of limitations with respect to stoichiometry, heterogeneity and single mRNP resolution.
Fluorescence Correlation and Cross-Correlation Spectroscopy (FCS and FCCS, respectively) are appealing alternatives for the analysis of complex protein assemblies and protein interactions and the composition of complex protein assemblies. By recording the fluorescence fluctuations produced by labelled molecules or proteins entering and leaving a small focal volume over time and their subsequent analysis by autocorrelation, FCS instantly provides information about the diffusion time, concentration and aggregation of fluorescent molecules (6) (Figure 1A). FCCS further determines the cross-correlation between factors labelled with two spectrally distinct fluorophores allowing you to establish if two factors are part of the same macromolecular complex (7) (Figure 1A). Compared to immunoprecipitation (IP), the duration of FCS and FCCS experiments is significantly shorter since measurements only take minutes to execute and the amount of starting material is one or two orders of magnitude lower (∼104–106 versus 107–108 cells). Moreover, the sensitivity is high, facilitating detection of non-abundant protein-protein interactions. Finally, in contrast to assays involving recombinant proteins, the use of lysates overcome common problems connected to bioactivity, purity and production yield. Previously, FCS and FCCS have been successfully employed in RNA research to compare diffusion of cytoplasmic and nuclear EGFP-Ago2 and to quantify loading of siRNA guide strand into the RISC complex (8) and test the effectiveness of Mango-based (fluorogenic aptamer family) single-molecule analysis (9).
Figure 1.

Schematic representation of fluorescence correlation spectroscopy (FCS) and fluorescence cross-correlation spectroscopy (FCCS) methods used in the study. (A) FCS measurements are performed either in cells expressing a protein of interest fused to a GFP tag or in cell lysates from cells expressing the GFP fusion protein. Focal volume is positioned in a specific cellular location (live cell FCS) or in a fluorescent protein solution (cell lysate FCS). Fluorescence fluctuations are recorded and are subsequently analyzed by the autocorrelation function, which results in the autocorrelation curve, used to determine the diffusion time of the examined GFP-tagged protein. FCCS is based on the combined FCS measurements of two spectrally-distinct fluorescent proteins, in this case GFP and mCherry. Autocorrelation function is used for analyzing diffusion of GFP- and mCherry-tagged proteins and also applied between channels, generating the cross-correlation curve. Interacting GFP- and mCherry-tagged proteins diffuse synchronously through the focal volume, therefore the cross-correlation curve (grey) is positive. In contrast, non-interacting GFP- and mCherry-tagged proteins diffuse independently from each other yielding a flat cross-correlation curve. (B) Representative scheme of the steps performed in cell lysate FCS in order to determine RNA-binding protein stoichiometry in single mRNPs. (C) Representative scheme of the steps followed in FCCS to determine protein-protein associations and heterogeneity of mRNPs. *Figure partially created with BioRender.com
Employing a number of well-established cytoplasmic mRNA binding proteins, including the mRNP core proteins YBX1, IMP1 and PABPC1 as well as IMP2, IMP3, ELAVL2 (HuB), STAU1 and FMRP (10–16) (Figure 2 and Supplementary Figure S1), we established and validated a novel lysate-based FCS and FCCS assay to accurately resolve RNA-binding protein stoichiometry and protein-protein interactions in single mRNPs (Figure 1B and C). Due to the nature of cell lysates, which would fall in a category between in vivo and in vitro, protein composition analysis can be simplified and at the same time complemented with stoichiometric and heterogeneity information of single mRNPs. Cellular lysates not only recapitulate data from live cells but provide improved readings and allow single mRNP analysis under particular conditions or following enzymatic treatments. We consider that cell lysate-based FCS and FCCS provide a precise and reproducible way of analysing protein-RNA complexes with many advantages compared to current analysis techniques.
Figure 2.

Overview of RNA-binding proteins used in the study. (A) Structured illumination microscopy (SIM) image of HeLa cell cytoplasmic mRNPs obtained by indirect immunofluorescence staining of their major protein components: YBX1 (Alexa Fluor 647, red), IMP1 (Alexa Fluor 568, green) and PABPC1 (Alexa Fluor 488, cyan). Scale bar = 0.2 μm. (B) Schematic representation of the different proteins used in this study with their RNA-binding domains and predicted low complexity sequences (CSD: cold-shock domain; RRM: RNA recognition motif, KH: hnRNP K-homology, DSRBD: double-stranded RNA-binding domain).
MATERIALS AND METHODS
Cell lines
HeLa cells (ATCC® CCL-2™) were grown in phenol red-free Dulbecco's Modified Eagle Medium (DMEM), high glucose, supplemented with GlutaMAX™, 1 mM sodium pyruvate, 10% FBS (Biowest) and penicillin/streptomycin. A Flp-In™ T-REx™ 293 cell line (Invitrogen) stably carrying a 3xFLAG-IMP1 under a tetracycline inducible CMV promoter was generated and is referred in the text as TREX 293 3xFLAG-IMP1. TREX 293 3xFLAG-IMP1 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), high glucose, supplemented with GlutaMAX™, 10% FBS (Biowest), 5 μg/ml Blasticidin, 100 μg/ml hygromycin and penicillin/streptomycin. 3xFLAG-IMP1 expression was induced by the addition of 1 μg/ml tetracycline for 24 h. Cell lines were cultured in a humidified incubator at 37ºC and 5% CO2.
Vectors
Coding sequences of IMP1, IMP2, IMP3, ELAVL2, STAU1 and FMRP were PCR-amplified and cloned into pEGFP-C1 (Clontech), and PABPC1 coding sequence was cloned into pEGFP-N1 (Clontech) by restriction enzyme digestion. GFP-IMP1KH1-4mut construct was obtained by site-directed mutagenesis of the GXXG loops in the four KH domains (17) from GK(E/K/G)G to GELG. YBX1 was cloned into pcDNA3.1 + N-eGFP (Genscript) inserting a 25 aminoacid flexible linker (5× GGGGS) (18) between the fluorescent tag and YBX1. IMP1 and YBX1 (including flexible linker) coding sequences were also cloned into pmCherry-C1 (Clontech). EGFP was PCR-amplified from pEGFP-C1 and cloned into mCherry-C1 by restriction enzyme digestion in order to obtain mCherry-GFP fusion protein. 2xGFP oligomer was obtained by cloning an EGFP sequence with a flexible linker (3xGGGGS) upfront into a pEGFP-C1 vector. 5xGFP oligomer was obtained by cloning a 4xGFP sequence with 3xGGGGS flexible linkers in between each EGFP unit into a pEGFP-C1 vector by restriction enzyme digestion.
Cell lysate FCS/FCCS protocol
Plasmid transfections
400.000 HeLa cells were seeded in Nunc™ cell-culture treated 6-well dishes. After 4–5 h, cells were transfected with the plasmids described above in ‘Vectors’ section using FuGene 6 and following the manufacturer's instructions. Per dish, 1.3 μg of plasmid DNA and 3 μl of FuGene 6 (Promega) were mixed in 100 μl Opti-MEMTM medium before the addition to the dish. For co-transfections, pEGFP and pmCherry vectors were mixed at 2:1 ratio. 18–22 h post-transfection, cells were lysed at room temperature in 500 μl for FCS or 300 μl for FCCS of lysis buffer containing 20 mM Tris–HCl pH 7.5, 140 mM KCl, 1,5 mM MgCl2, 1 mM DTT and 0,5% NP-40 supplemented with 1:300 mammalian protease inhibitor cocktail (Sigma). Cell lysates were briefly centrifuged at 800 × g for 1 min and supernatant was transferred to a 35 mm glass bottom dish (No. 1.5 Coverslip, uncoated, MatTek) before being subjected to FCS or FCCS. When indicated, cell lysates were treated with 100 μg/ml of RNase A (DNase and protease-free, EN0531, Thermo Scientific) and FCS/FCCS measurements were recorded 1–2 min after RNase A treatment. For IMP stoichiometric measurements (FCS), cells were also lysed in 500 μl of hyperosmolar lysis buffer containing 20 mM Tris–HCl pH 7.5, 500 mM KCl, 1.5 mM MgCl2, 1 mM DTT and 0,5% NP-40 supplemented with 1:300 mammalian protease inhibitor cocktail (Sigma).
Fluorescence correlation spectroscopy (FCS)
FCS measurements were performed using a Zeiss LSM780 confocal microscope using a C-Apochromat 40×/1.2 W Corr M27 objective and using a water-phase immersion oil Immersol W 2010 (Zeiss). GFP measurements were performed with an argon laser with a 488 nm excitation wavelength and with a detection window between 500 and 633 nm (Supplementary Figure S2). Before each measurement, average molecular count rate (in kHz per molecule) was checked at different laser powers to ensure that fluorescence count signal was linear with laser power and not in saturation. Cell lysate and live cell fluorescence measurements were recorded during 60 s and experimental autocorrelation curves, average count rate (kHz) and counts per particle (kHz) were obtained and analysed in ZEN 2011 software (Zeiss). FCS in cell lysates was performed at room temperature (21–23ºC) whereas for live cell measurements, dishes were taken out from the incubator (37ºC) and sealed, and measurements were taken 5–20 min after. Counts per particle or CPP were calculated as follows: CPP (kHz) = amplitude × average intensity. Experimental autocorrelation curve fitting to free 3D diffusion 1-component and 2-component models was performed in ZEN 2011 software (Zeiss) starting from 1E-05 s (10 μs). Diffusion coefficients were calculated assuming a range between 150 nm and 190 nm as the confocal radius and the model equations for 1- and 2-component models are the following:
1-component fit
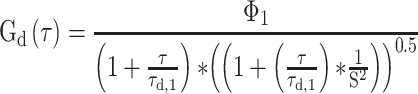 |
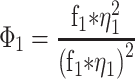 |
2-component fit
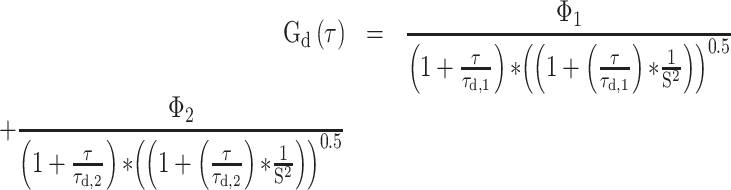 |
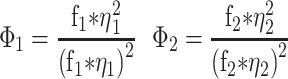 |
i - Index of component (1, 2)
Φi - Fractional intensity
fi - Fraction of molecules
ηi - Molecular brightness
τd,i - Diffusional correlation time
S - Structural parameter (ratio between radial and axial waist)
Fluorescence cross-correlation spectroscopy (FCCS)
All FCCS measurements (live cell and cell lysate) were performed in a Zeiss LSM780 confocal microscope using a C-Apochromat 40×/1.2 W Corr M27 objective and Immersion oil Immersol W 2010 (Zeiss). GFP or mCherry measurements were performed with an argon laser with a 488 nm excitation wavelength and a DPSS laser with a 561 nm excitation wavelength, respectively. GFP fluorescence was captured with a detection window of 482–553 nm and mCherry fluorescence was captured with a detection window of 590–695 nm (Supplementary Figure S2). Before each measurement, average molecular count rate (kHz per molecule) was checked at different laser powers to ensure that fluorescence count signal was linear with laser power and not in saturation. Cell lysate and live cell fluorescence measurements were recorded for 60 s in the case of cell lysate FCCS. In live cells, measurements were performed at randomly picked volumes in the cytoplasm for 20 s. FCCS in cell lysates was performed at room temperature (21–23ºC) whereas for live cell measurements, dishes were removed from the incubator (37ºC) and sealed, and measurements were taken 5–20 min later. Experimental autocorrelation and cross-correlation curves were obtained and analysed in ZEN 2011 software (Zeiss). Cross-correlation and autocorrelation amplitude values, needed to calculate the cross-correlation/autocorrelation ratios, were extracted from the average of the amplitude values G(τ) from either curve in the area of maximum amplitude (5E–06 to 1E–05 s). Cross-correlation (CC)/autocorrelation (AC) ratio was calculated with the following formula:
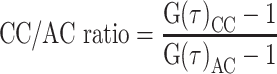 |
Confocal microscopy imaging
HeLa cells were seeded in 35 mm glass bottom dishes (No. 1.5 Coverslip, uncoated, MatTek), transfected with GFP or GFP/mCherry vectors and subsequently imaged ∼24 h after transfection. Cells were fixed with 3,7% formaldehyde in PBS, washed and mounted in non-hardening VECTASHIELD antifade mounting medium (H-1000) with a refractive index n = 1.45. Confocal images were obtained using a Zeiss LSM780 confocal microscope with a Plan-Apochromat 63×/1.4 NA oil objective.
Immunofluorescence staining and structured illumination microscopy (SIM) imaging
HeLa cells were seeded in glass-bottom coverslips (P35G-0.170-14-C, MatTek) and fixed 24 h after with 3.7% formaldehyde solution in PBS, followed by a permeabilization step with 0.5% Triton X-100 in PBS. Indirect immunofluorescence staining of YBX1, IMP1 and PABPC1 was performed using the following primary antibodies: goat anti-IMP1 (E-20, Santa Cruz), rabbit anti-YBX1 (ab12148, Abcam) and mouse anti-PABPC1 (ab6125, Abcam). Coverslips were washed 3x with PBS prior to incubation with Alexa Fluor 488, 568 and 647 conjugated secondary antibodies (Thermo Fisher Scientific) for an hour at RT. After that, coverslips were washed 3× with PBS and mounted in VECTASHIELD mounting media (RI = 1.45). Structured Illumination Microscopy (SIM) was performed using a Zeiss ELYRA PS.1 microscope with a Plan-apochromat 63×/1.4 NA oil objective. Raw SIM images were taken using three rotations and reconstructed using ZEN 2011 software (Zeiss). After reconstruction, channel correction was applied using a channel alignment file created by imaging 0.1 μm TetraSpeck Microspheres (Thermo Fisher Scientific). Images were thresholded to remove diffuse background (honeycombs) caused by stray pollutants or some residual autofluorescence on the SIM reconstructed pictures before contrasting.
3xFLAG-IMP1 immunoprecipitation
TREX 293 3xFLAG-IMP1 cell line was used for immunoprecipitation. 5 × 106 cells were seeded in 140 mm diam. Nunc® petri dishes and expression of 3xFLAG-IMP1 was induced by the addition of 1 μg/ml tetracycline for 24 h. Cells were transfected 4–5 h after seeding using FuGene 6 following the manufacturer's instructions with pEGFP plasmids described in ‘Vectors’ section. Per dish, 16.75 μg of plasmid DNA and 37.5 μl of FuGene 6 were mixed in 1200 μl Opti-MEM™ medium before addition to the cells. 48 h after seeding, cells were lysed in 1 ml of lysis buffer containing 20 mM Tris–HCl pH 7.5, 140 mM KCl, 1.5 mM MgCl2, 1 mM DTT and 0.5% NP-40 supplemented with 1:300 mammalian protease inhibitor cocktail (Sigma) on ice. Cell lysates were cleared at 8200 × g for 5 min at 4ºC. 50 μl of Anti-FLAG® M2 Magnetic beads (Sigma) were added to each cleared cell lysate and samples were incubated for 3 h with rotation at 4ºC. Beads were washed 3× with lysis buffer and finally immunoprecipitated material was collected by addition of 2× SDS buffer directly to the beads.
Western blot
Total lysate and FLAG immunoprecipitation samples were loaded into a 10% RunBlue™ SDS protein gel (Expedeon) and separated by electrophoresis. Proteins were transferred to a PVDF membrane using the iBlot™ 2 Gel Transfer system (Invitrogen). After transfer, membrane was blocked in wash buffer with 5% skim milk powder for 1 h at room temperature. After blocking, primary antibodies against GFP (rabbit mAb, Cell Signaling) and FLAG (M2 mouse mAb, Sigma) were incubated overnight at 4ºC. The day after, membranes were washed and incubated with either anti-rabbit or anti-mouse HRP-conjugated secondary antibodies (Cell Signaling). After incubation, membranes were washed, incubated 2–3 min with SuperSignal™ West Pico Chemiluminiscent Substrate (Thermo Scientific) and imaged using C-DiGit® Blot Scanner (LI-COR).
RNA-seq dataset alignment and analysis
Two public datasets corresponding to an IGF2BP1/IMP1 eCLIP experiment in HepG2 cells (sample GSM2423912 under series GSE92021 and SRA accession nr. SRR4112015) (19,20) and a YBX1 RIP-seq experiment in HEK293 cells (sample GSM3753525 under series GSE130781 and SRA accession nr. SRR9019707) (21) were downloaded from SRA and aligned using CLC Genomics Workbench to the hg19 genome with the program's default parameters. Genes were sorted by ‘transcript_biotype’ and ‘total gene reads’ corresponding to each gene were used to calculate the % of reads mapping to each ‘transcript_biotype’ category.
RESULTS
FCS of mRNPs in live cells and cell lysates
In order to compare the dynamics of single mRNPs in live cells with cell lysates, fluorescence intensity fluctuations in HeLa cells expressing GFP-tagged YBX1, IMP1 and IMP1KH1-4mut, an IMP1 mutant with impaired RNA-binding (19) (Figure 3A), were recorded in the cytoplasm of live cells and in cell lysates (Figure 3C–E). GFP was included as a reference in both live cells and cell lysates (Figure 3F). Live cells expressing GFP-tagged proteins were depicted by confocal imaging and the focal volume was positioned at randomly picked volumes in the cytoplasm as represented in Figure 3A. As described above, corresponding cell lysates were prepared by the addition of an iso-osmolar lysis buffer containing non-ionic detergent, to release the cytoplasmic content. In the case of cell lysates, localization of the focal volume was not needed since the fluorescent molecules or complexes were not confined in a specific place but were diffusing homogeneously in the lysate solution. Live cell FCS measurements were carried out in different cells from a single transfection and also from different transfections, and cell lysate FCS measurements were recorded from different cell transfections. Measurements in both live cells and cell lysates were highly reproducible and in lysates mRNPs were stable for at least 2 h at room temperature (Supplementary Figure S3). Representative measurements of each GFP-tagged factor are shown in Figure 3.
Figure 3.

Comparison of live cell and cell lysate Fluorescence Correlation Spectroscopy (FCS). (A) Confocal microscopy images of HeLa cells expressing GFP-YBX1, GFP-IMP1, GFP-IMP1KH1-4mut (RNA-binding impaired mutant) and GFP. Crosses illustrate how arbitrary points in the cytoplasm were chosen to perform live cell FCS. Scale bar = 5 μm. (B) Autocorrelation curves (normalized from 1 to 2) from GFP-IMP1 (mRNP), GFP-IMP1KH1-4mut and GFP measurements in live cells (top) and cell lysates (bottom). (C–F) Representative autocorrelation curves from live cells (top) and cell lysates (bottom) of GFP-YBX1, GFP-IMP1, GFP-IMP1KH1-4mut and GFP, respectively. Fittings to 1-component (blue) and 2-component (red) models are displayed in the graphs together with the experimental autocorrelation curves. Blue and red arrows on x axis (lag time, sec) represent the diffusion coefficients corresponding to the fit to 1-component (blue) and 2-component (red) diffusion models.
In comparison to live cell diffusion, the relative distribution of the autocorrelation curves of GFP, GFP-IMP1KH1-4mut and GFP-IMP1 was reproduced in cell lysates, but in agreement with the lower viscosity and unrestrained motions in lysates, diffusion was in general faster (Figure 3B). The diffusion constant of the mRNP component GFP-IMP1 was about an order of magnitude slower than GFP, in accordance with the embedment in the mRNP complex. Disruption of IMP1 RNA-binding ability increased as expected diffusion almost to the level of free GFP (Figure 3B). The average difference between live cells and lysates was first quantified and compared by fitting the experimental autocorrelation curves to a single-component diffusion model (Figure 3C–F, blue line and arrows), and diffusion time was about one order of magnitude smaller in cell lysates than in live cells. The GFP-YBX1 and GFP-IMP1 autocorrelation curves clearly did not fit with a 1-component diffusion model, and in both live cells and cell lysates addition of a second component improved the experimental fitting (Figure 3C and D, red line and arrows). Consequently, the analysis reveals that mRNP motion may be explained by at least two diffusing subpopulations, diffusing at ∼4E–03 and ∼3E–01 s in live cells and ∼4E–04 and ∼8E–03 s in cell lysates. These correspond to 0.056–0.16 μm2/s and 4.12–7.35 μm2/s in live cells and 1.16–1.71 μm2/s and 7.09–14.76 μm2/s in cell lysates. GFP-IMP1KH1-4mut fitted better to a 1-component diffusion model but the fit was still significantly better with a 2-component model (Figure 3E), probably reflecting a residual RNA-binding activity of the protein. In the case of GFP (Figure 3F), the single component model in both conditions was sufficient, reconciling the unrestrained diffusion of this protein. Residuals of the fittings to 1-component and 2-component diffusion models are shown in Supplementary Figure S4. Taken together, we find that FCS is able to depict the motion of macromolecular mRNP assemblies, and lysates largely recapitulate the in vivo conditions but at the same time improve accuracy, reproducibility and simplicity.
Determination of RNA-binding protein stoichiometry of mRNPs
Since mRNAs frequently exhibit several binding sites for a particular RBP, we exploited the cell lysates to derive average and single mRNP stoichiometric data. IMP1 binds to widespread single-stranded CA-rich stretches (22), so a particular mRNA may in principle harbour one or more handfuls of IMPs depending on the size of the transcript.
Cells expressing GFP-IMP1 or GFP-IMP1KH1-4mut were either lysed in iso-osmolar lysis buffer and treated with RNase A or simply lysed in hypertonic lysis buffer, after which lysates were subjected to FCS. Plotting of the count rate (kHz) as a function of time shows the fluorescent peaks of different intensities representing mRNPs containing varying numbers of GFP-tagged IMP1. mRNP peaks are not observed in the recorded fluctuations for GFP-IMP1KH1-4mut (Figure 4A, top row). The same is observed for GFP-IMP1 after treatment with RNase A (Figure 4A, middle row) or lysis in a hypertonic buffer, which disrupt RNA-protein interactions (Figure 4A, bottom row), while fluorescence intensities remain unchanged for GFP-IMP1KH1-4mut. The two other members of IMP RNA-binding protein family, GFP-IMP2 and GFP-IMP3, exhibited the same pattern as GFP-IMP1 (Supplementary Figure S5A).
Figure 4.

IMP1 protein stoichiometry in mRNPs. (A) Fluorescence intensity measurements (kHz) in time (5 s) of cell lysates from HeLa cells transfected with GFP-IMP1 or GFP-IMP1KH1-4mut. Cells were lysed using iso-osmolar lysis buffer (blue, top row), subsequently treated with 100 μg/ml RNase A (red, medium row) or lysed with hyperosmolar lysis buffer in order to break protein-RNA interactions (green, bottom row). Counts per particle (CPP) averages ± standard deviations of n = 6 biological replicates in cell lysates are shown in the graphs. (B) Fluorescence autocorrelation curves of HeLa cell lysates transfected with GFP-IMP1 or GFP-IMP1KH1-4mut under different buffer conditions: iso-osmolar lysis buffer (140 mM KCl), iso-osmolar lysis buffer (140 mM KCl) + RNAse A treatment and hyperosmolar lysis buffer (500 mM KCl). (C) Distribution of the fluorescent intensities of cell lysates from cells expressing GFP-IMP1 obtained with iso-osmolar lysis buffer (blue) or treated with 100 μg/ml RNase A (red). Histograms of GFP-IMP1 cell lysate (mRNP) and GFP-IMP1 cell lysate treated with RNase A (monomer) showing the distribution of fluorescence intensity (kHz) per millisecond and the number of events (∼frequency).
The amplitude of the autocorrelation curve is inversely proportional to the concentration of the fluorescent molecules or particles in solution, so a higher concentration of fluorescent particles yields lower amplitudes and vice versa. As shown in Figure 4A, when IMP1 is part of mRNPs, multiple IMPs are bound to the same mRNA. Therefore, the concentration of fluorescent particles in the lysate is low but the brightness of each particle is high because it contains several IMP1 molecules, yielding a high G(0) amplitude (Figure 4B, top plot). After treatment with RNase A or lysis in hypertonic lysis buffer, IMPs dissociate from mRNPs. Consequently, the concentration of the fluorescent species rises—the G(0) amplitude decreases—whereas the molecular brightness also decreases (as each individual particle now becomes monomeric). In contrast, the amplitude and brightness of GFP-IMP1KH1-4mut remained unchanged in all conditions before and after treatment with RNase A because it exists as a monomer to begin with in all conditions (Figure 4B, bottom plot). A scheme illustrating the changes in brightness and amplitude in oligomers and monomers upon treatment with RNase A can be found in Supplementary Figure S6.
The average brightness of a given complex decorated by fluorescent labels (counts per particle or CPP, or sometimes referred to as counts per molecule or CPM) can be derived from the FCS analysis. The number of particles is obtained from the inverse of the amplitude and CPP is proportional to the average fluorescence intensity divided by the inverse of the amplitude. In other words, CPP = amplitude × average fluorescence intensity. In order to determine the absolute molecular brightness of the mRNPs, a gamma factor can be introduced to the formula to correct for the detection efficiency given by the illumination and detection profile (the point spread function, PSF), but in this case we were merely interested in the brightness increase per particle (mRNP/monomer), therefore this correction cancels out. By comparing the mRNP brightness (CPP avg.) of GFP-IMP1 with the brightness of the monomer (GFP-IMP1KH1-4mut) or the GFP-IMP1 lysate treated with RNase A (Figure 4A), we can show that mRNPs on average contain 10 GFP-IMP1 molecules.
Fluorescent GFP oligomers have been reported to self-quench (23), resulting in a non-linear relation between the number of GFPs in the oligomer and the brightness of GFP oligomers. We observed a similar behaviour in our own experimental settings when the oligomer size was 5xGFP (Supplementary Figure S5C), indicating that quenching could potentially lead to erroneous conclusions when trying to deduce the number of GFP-tagged IMPs from mRNP brightness.
We therefore checked if there were indications of quenching in the mRNPs by comparing the brightness per particle (counts per particle) of GFP-IMP1 and GFP-IMP2, that quantified by the average amount of fluorescence in the lysate (average count rate) is expressed at 2-fold higher levels than GFP-IMP1. The brightness per particle was also doubled, so we infer there is a linear relation between expression level of the protein and the number of GFP-IMP molecules in the complexes (Supplementary Figure S5B). Quenching is highly dependent on the distance between fluorescent molecules and the small GFP oligomers may not reconcile the binding of RBPs to independent binding sites in a much larger extended mRNA.
An approximation can be made in order to get a deeper insight about the brightness and distribution of number of GFP-IMP1 molecules per mRNP. While the focal volume in live cells contains about 30 mRNPs, the focal volume in lysates only contains single mRNPs due to the dilution factor. As shown in Figure 4C, measurements with counts below the average count rate are frequently observed. Fluorescence intensities per millisecond can be obtained directly from the raw fluorescence intensity plot (before applying autocorrelation function), and the frequency of events of a certain brightness can be plotted so the range of fluorescence intensities becomes apparent (Figure 4C). This can be useful and complementary to the average counts per particle in a situation where the number of molecules per complex is not homogeneous.
As shown in Figure 4A, GFP-IMP1 measurements in cell lysates exhibit peaks of different heights, which can be attributed to different number of GFP-IMP1 molecules per mRNP. The distribution of GFP-IMP1 fluorescence intensities exhibited a wider range than the same sample treated with RNase A (Figure 4C). The brightness or CPP of monomeric GFP-IMP1 is about 5.5 kHz (Figure 4A). Therefore, a fluorescence measurement of 100 kHz would correspond to approximately 18 GFP-IMP1 molecules. In this way, we can determine that the bulk range of IMP1 molecules in mRNPs range from 1 to 36 molecules—corresponding to a brightness of 5.5–200 kHz.
Determination of mRNP core protein interactions by FCCS in cell lysates
Once we established the behaviour of the mRNP components by FCS, we subsequently explored the feasibility of fluorescence cross-correlation spectroscopy (FCCS) to depict associations between different RNA-binding proteins in the cell lysates. We initially tested the interactions between YBX1 and IMP1 or PABPC1, that previously have been demonstrated to constitute the major elements of cytoplasmic mRNPs (10). Cytoplasmic YBX1 coats mRNAs along their entire length by attachment to the sugar-phosphate backbone (13,15), whereas IMPs have a preponderance for loops and 3’UTRs (24).
Cells were co-transfected with mCherry-YBX1 and GFP-IMP1 and the reverse pair mCherry-IMP1 and GFP-YBX1. PABPC1-GFP was co-transfected with mCherry-YBX1. By analysing the total cellular content when performing cell lysis, measurements are more homogeneous since the complexity is reduced compared to the situation in a live cell. Moreover, a lower concentration of the tagged complexes or proteins yields a higher amplitude. As a positive control for cross-correlation, a mCherry-GFP fusion protein emitting in both green and red channels was employed, whereas the negative control consisted of a co-transfection of mCherry and GFP encoded in separate plasmids. As shown in Figure 5A, cross-correlation was observed with the mCherry-GFP fusion protein while cross-correlation was not observed when fluorescent proteins are expressed as separate proteins. Due to photobleaching, misfolding or fluorescent proteins being ‘off’ or in dark states (25), the cross-correlation is not expected to be 100%, as illustrated by the mCherry-GFP fusion protein.
Figure 5.

Protein-protein interaction quantification in cell lysates between three major mRNP components: YBX1, IMP1 and PABPC1. (A) Cell lysate fluorescence cross-correlation spectroscopy (FCCS) measurements of cells expressing a mCherry-GFP fusion protein (positive control for cross-correlation, left) and mCherry/GFP transfected in separate plasmids (negative control for cross-correlation, right). (B) Cell lysate FCCS measurements of cells expressing GFP-IMP1 and mCherry-YBX1 (top), GFP-YBX1 and mCherry-IMP1 (middle) and PABPC1-GFP and mCherry-YBX1 (bottom). Autocorrelation and cross-correlation curves shown on the right side represent a re-scaled data from the curves on the left side, as indicated with the discontinued line. Green and red lines represent GFP and mCherry autocorrelation curves, respectively, and grey line represents the cross-correlation curve.
As expected, mCherry-YBX1 exhibited a positive cross-correlation with both GFP-IMP1 and PABPC1-GFP. Cross-correlation was high with GFP-IMP1, meaning that GFP-IMP1 containing mRNPs encompass mCherry-YBX1 to a great extent (Figure 5B, top). At the same time, we could see that not all GFP-YBX1 containing mRNPs encompassed mCherry-IMP1, as observed in a positive but slightly lower cross-correlation with GFP-YBX1 (Figure 5B, middle). Positive cross-correlation between PABPC1-GFP and mCherry-YBX1 indicates interaction between the factors, but in agreement with the widespread and general distribution of PABPC1, to a lesser extent.
FCCS analysis unveils mRNP heterogeneity
Different mRNA-binding proteins have been described to interact by immunoprecipitation analysis, but whether these mRNP complexes are homogeneous or heterogeneous, as well as the likelihood of the interaction between the RNA-binding proteins, are issues that are not fully understood and not possible to address by bulk pull-downs. Consequently, we examined the feasibility of analysing the binding between the core component IMP1 or YBX1 and other RNA-binding proteins as well as the heterogeneity of the mRNPs by FCCS in cell lysates.
Cells were transfected with mCherry-IMP1 in combination with GFP-tagged IMPs, ELAVL2, STAU1 or FMRP and fluorescence measurements were recorded after lysis (Figure 6A). From the analysis, we observed that mCherry-IMP1, as expected, cross-correlated to a high degree with GFP-IMP1, as well as with GFP-IMP2 and GFP-IMP3, in agreement with the similar binding patterns of these proteins. The cross-correlation ratio for IMP1 tagged with two different fluorescent proteins was close to the maximum cross-correlation, and it was even higher than the cross-correlation of the mCherry-GFP fusion protein (Figures 6A and 7A, left plot). This was probably due to the presence of multiple IMP molecules in the mRNP, which makes the occurrence of completely ‘off’ states of the complexes less likely. GFP-ELAVL2 also cross-correlated with mCherry-IMP1 but to a lower extent than IMP1, IMP2 and IMP3 (Figure 6A and 7A, left plot). On the other side, cross-correlation with GFP-STAU1 or GFP-FMRP and mCherry-IMP1 was very low, indicating that the proteins associate significantly less with mCherry-IMP1 containing mRNPs (Figures 6A and 7A, left plot). Moreover, we could determine that the interaction of all the tested GFP-tagged factors was RNA dependent since cross-correlation was lost after treatment with RNase A (Figure 6A, right column). The results obtained by FCCS were validated by immunoprecipitation of 3xFLAG-IMP1 from TREX 293 3xFLAG-IMP1 cells. As shown in Figure 6B, GFP-IMP1, GFP-IMP2, GFP-IMP3 and GFP-ELAVL2 were immunoprecipitated with 3xFLAG-IMP1 while GFP-STAU1, GFP-FMRP and GFP alone (negative control) were not detected in the immunoprecipitate. Taken together, we found that FCCS measurements are in fine agreement with immunoprecipitation data although FCCS appears to be more sensitive.
Figure 6.

Protein-protein interaction quantification in cell lysates. (A) Representative Fluorescence Cross-correlation Spectroscopy measurements of HeLa cell lysates co-transfected with mCherry-IMP1 and GFP-IMP2, GFP-IMP3, GFP-ELAVL2, GFP-STAU1 or GFP-FMRP (left). Relative cross-correlation in respect to mCherry-IMP1 autocorrelation before (middle) or after treatment with 100 μg/mL RNase A (right). Green and red lines represent GFP and mCherry autocorrelation curves, respectively, and grey line represents the cross-correlation curve. (B) Western Blot of total lysate (left) and FLAG immunoprecipitated (right) fractions of TREX 293 3xFLAG-IMP1 cells transiently transfected with GFP-IMP1, GFP-IMP2, GFP-IMP3, GFP-ELAVL2, GFP-STAU1, GFP-FMRP and GFP (negative control).
Figure 7.

Cross-correlation quantification of cell lysates from co-transfections with mCherry-IMP1 and mCherry-YBX1. (A) Average cross-correlation / mCherry-IMP1 (left) or mCherry-YBX1 (right) autocorrelation ratios ± SD showing different degrees of interaction depending on the GFP-tagged protein across n = 6 biological replicates. (B) Summary of all the factors tested and the corresponding % overlap with YBX1 and IMP1 according to FCCS results in cell lysates. Average % are obtained from average cross-correlation ratios shown in panel A. The maximum cross-correlation of 0.7, obtained in the GFP-IMP1 and mCherry-IMP1 FCCS measurement, is considered to be 100% and all the percentages shown are adjusted accordingly.
We also performed FCCS with the same GFP-tagged proteins but this time employing mCherry-YBX1 (Supplementary Figure S7). We calculated the average ratio between the cross-correlation with the different GFP-tagged factors and either mCherry-IMP1 or mCherry-YBX1 in cell lysates. Ratios were obtained from six biological replicates and the results are shown in Figure 7A. The percentage overlaps with IMP1 and YBX1 are summarized in Figure 7B, taking the GFP-IMP1/mCherry-IMP1 pair as the maximum (100%) and adjusting the rest accordingly. Similar to mCherry-IMP1, GFP-tagged IMPs presented a high cross-correlation with mCherry-YBX1 mRNPs, while GFP-ELAVL2 cross-correlation with mCherry-IMP1 was lower than with mCherry-YBX1. GFP-tagged STAU1 and FMRP cross-correlation were the lowest both with mCherry-IMP1 and mCherry-YBX1.
Finally, FCCS with mCherry-YBX1 was performed in live cells to compare the above-described associations (Figure 8). In agreement with the data shown in Figure 3, the autocorrelation curves shifted approximately an order of magnitude. With the exception of GFP-FMRP, live cell cross-correlation experiments corroborated the results obtained from lysates (Figure 8B and C). In the case of FMRP, we observed a prominent cross-correlation between GFP-FMRP and mCherry-YBX1 in live cells, but the interaction was significantly lower in cell lysates. Finally, no interaction (no cross-correlation) was found between GFP-IMP1KH1-4mut and mCherry-YBX1, confirming the RNA-dependent association between the factors (Figure 8D).
Figure 8.

Protein-protein interaction quantification in live cells. (A) Confocal microscopy images of HeLa cells co-transfected with mCherry-YBX1 and GFP-IMP1, GFP-IMP2, GFP-IMP3, GFP-ELAVL2, GFP-STAU1 or GFP-FMRP. Scale bar = 5 μm. (B) Cytoplasmic live cell Fluorescence Cross-correlation Spectroscopy measurements of HeLa cells co-transfected with mCherry-YBX1 and GFP-IMP1, GFP-IMP2, GFP-IMP3, GFP-ELAVL2, GFP-STAU1 or GFP-FMRP. (C) Average cross-correlation / mCherry-YBX1 autocorrelation ratios ± SD showing different degrees of interaction depending on the GFP-tagged protein across n = 3 different cells. (D) Cytoplasmic live cell FCCS measurement of a HeLa cell co-transfected with mCherry-YBX1 and GFP-IMP1KH1-4mut as a negative control for cross-correlation (no interaction).
DISCUSSION
FCS and FCCS are broadly employable and versatile technologies, that have been applied to the molecular characterisation of a number of fundamental biological processes such as plasma membrane organization, cellular transport, morphogenic gradients, chromatin organization in intact cells and organisms (8,25). Although FCS and FCCS per se do not provide direct structural information like other image-based fluorescence microscopy modalities, they have single-molecule sensitivity and make it possible to analyse molecular interactions under physiological conditions. Moreover, the procedures are quantitative and provide information about concentration and diffusion and interactions of proteins. From a practical perspective, FCS and FCCS are also appreciated as fast and highly reproducible methods that can be applied to study a wide range of fluorescent molecules. Consequently, FCS and FCCS are appealing approaches to be used in the context of post-transcriptional processes, governed by a series of combinatorial assemblies of proteins on RNA from transcription to mRNA transport, translation and decay.
Here, we demonstrate how FCS and FCCS can be exploited to analyse single mRNPs in cell lysates that offer a number of assets compared to live cell recordings. Whereas live cells portray subcellular trafficking, docking or signalling events, we find that lysates have less spatial constraints and provide optimal readings of diffusion, stoichiometry and protein associations (Figure 3). Due to the dilution factor inherent to cell lysis, the concentration of fluorescent particles can be adjusted and optimised for single mRNP analysis. Moreover, different buffer conditions or enzymatic treatments can easily be employed to investigate biochemical characteristics of the macromolecular complexes (Figures 4-6). The inter- and intra-assay reproducibility is very high (Supplementary Figure S3). We noticed minor day to day variations that we consider related to small differences in temperature and dilution artefacts. Consequently, we propose that true biological replicates should be used.
A limitation of FCS is obviously the need for the attachment of a fluorescent molecule that can affect the function of the protein under scrutiny. Some RNA-binding proteins are sensitive to the position of the tag. In general, we experienced that separation of the fluorescent tag from the protein of interest by the addition of a flexible linker composed of Gly and Ser residues, i.e. (GGGGS)n (24) was helpful in order to preserve the function of the RNA-binding protein. Photobleaching and quenching or fluorescent proteins being ‘off’ or in dark states are other inherent issues of FCS that should be considered because they may lead to underestimation of the number of factors in a particular complex. This is particularly important in the case of mRNP analysis because mRNAs frequently exhibit multiple overlapping and mutually exclusive binding elements, making it difficult to predict the composition of the individual mRNP (16,22,26). Recent analyses have demonstrated that GFP oligomers do not emit a linear range of photons corresponding to the number of GFPs (23) and we observed a similar pattern when five GFP molecules were combined in an oligomer (Supplementary Figure S5C). However, in the case of mRNPs, the amount of photons emitted appears to be reasonably linear with the number of molecules in the complex, perhaps because the individual GFP-tagged RBPs are separated by a larger distance. Therefore, quenching is less likely compared to GFP oligomers, where the distance between GFPs is shorter.
Another factor that may influence the stoichiometric analysis is the concentration of the tagged RNA-binding protein. In our experiments, we had the chance to observe the changes in the number of IMP molecules per mRNP due to the expression pattern of two different IMP protein family members - IMP1 and IMP2. GFP-IMP2 was expressed or present in the cells at a 2-fold higher level compared to GFP-IMP1 under the same transfection conditions. We found that when the level of GFP-IMP was doubled, the brightness per mRNP (counts per particle or CPP) also doubled, reinforcing the linearity between the number of GFP-IMP molecules and the brightness of the individual mRNP complexes (Supplementary Figure S5B). Finally, as described above, we observed a wide distribution of IMP molecules per mRNP, which underscores the ability to measure different oligomer sizes through brightness (Figure 4C).
The heterogeneity of the individual mRNPs is observed directly in the fluorescence intensity fluctuations where the individual mRNPs are visible as bright peaks resulting from the binding of several molecules of the tagged RBP to mRNA. The monomeric brightness is obtained by the usage of an RNA binding mutant, by cleavage of mRNA by RNase A or the use of hypertonic buffer and this allows quantification of the number of RBPs per mRNP (Figure 4). This approach may basically be applied to any RNA-binding protein. In live cells, the focal volume contains around 30 mRNPs while in lysates, the concentration can be reduced to less than 1 mRNP per focal volume as mentioned in the results section. The average number of bound IMPs in lysates was similar to the one in live cells (16) reinforcing that they are representative of the in vivo situation. By plotting the frequency of events having a particular count rate, we could show that the bulk of mRNPs contained 1–36 molecules of GFP-IMP1, but it was evident that the range of bound IMPs is wide and a particular mRNP can contain up to 50 molecules (Figure 4C). The FCS data are roughly in agreement with cross-linking immunoprecipitation (CLIP) data, showing that an mRNA contains on average about 7 IMP1 binding sites (16).
Although changes in the diffusion coefficient of a protein may serve as an indication of its interaction with another protein- or molecular complex, changes in diffusivity can be small or misleading, for example if the transported species is the oligomeric form it will appear to be faster than a bound monomeric species – this is certainly an important factor to be considered in vivo. Moreover, FCS does not identify the binding partners of a protein. Consequently, the preferred method for detection of molecular interaction is FCCS. Compared to immunoprecipitation assays that typically require 107–108 million cells (Figure 6B), FCCS requires only a single live cell, or about 1000 cells/μl from a transient transfection if performed in lysates. A very important addition that FCCS has to offer, and that other protein-protein interaction methods may not uncover, is the heterogeneity of molecular complexes since FCCS provides a direct measure of the proportion of a particular protein in a given complex (Figure 7). We could show that the vast majority of IMP1 positive particles contain YBX1, while ELAVL2 showed a strong cross-correlation with YBX1 (similar to IMPs) but less overlap with IMP1. In this way, IMP1 and ELAVL2 could share the binding to some mRNAs, but also bind to different mRNA subsets or potentially compete for binding to the same binding sites in mRNAs. Moreover, only a small proportion of STAU1 and FMRP was bound to YBX1 and IMP1 mRNPs in an RNA-dependent manner. In the case of FMRP, we noted a difference between live cells and lysates since the cross-correlation between FMRP and YBX1 was significantly lower in lysates (Figure 8 and Supplementary Figure S7). Cell lysis could disrupt a putative RNA-independent association between the two factors such as membrane vesicles. RNA granules have in fact been described to be tethered onto vesicles in a mechanism that promotes RNA transport to distal cell sites (27). An alternative explanation could be that there is an immobile non-mRNP bound fraction of FMRP in live cells, that will not be detected by FCCS.
Taken together, we devise a method for single mRNP analysis based on FCS and FCCS in cell lysates. Due to their stability cytoplasmic mRNPs (Supplementary Figure S3B-C) are in many ways suitable for FCS and FCCS analysis, but in principle, we see no hindrance for the analysis of mRNPs in other defined subcellular domains. FCS is moreover applicable for the study of assemblies that otherwise cannot be examined by methods relying on physical separation. Finally, we consider it an important feature of our method that it generates highly accurate and reproducible quantitative data on the dynamics and protein-protein interactions of the mRNPs.
DATA AVAILABILITY
Vectors and TREX 293 3xFLAG-IMP1 cell line used in this study are available upon request. All data is available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Stine Østergaard for her technical assistance.
Author contributions: À.M.-R. and F.C.N. designed the study. À.M.-R. performed the experiments. À.M.-R. and F.C.N analysed the data. À.M.-R., J.C., F.O.B., C.H. and F.C.N. wrote the manuscript.
Contributor Information
Àngels Mateu-Regué, Center for Genomic Medicine, Rigshospitalet, Blegdamsvej 9, 2100 Copenhagen, Denmark.
Jan Christiansen, Department of Biology, University of Copenhagen, Ole Maaloes Vej 5, 2200 Copenhagen, Denmark.
Frederik Otzen Bagger, Center for Genomic Medicine, Rigshospitalet, Blegdamsvej 9, 2100 Copenhagen, Denmark.
Christian Hellriegel, Carl Zeiss RMS / Harvard Center for Biological Imaging, 16 Divinity Avenue, Cambridge, MA 02138, USA.
Finn Cilius Nielsen, Center for Genomic Medicine, Rigshospitalet, Blegdamsvej 9, 2100 Copenhagen, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Infrastructure Program from the Danish Medical Research Council. Funding for open access charge: Copenhagen University Hospital.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hubstenberger A., Courel M., Benard M., Souquere S., Ernoult-Lange M., Chouaib R., Yi Z., Morlot J.B., Munier A., Fradet M.et al.. P-body purification reveals the condensation of repressed mRNA regulons. Mol. Cell. 2017; 68:144–157. [DOI] [PubMed] [Google Scholar]
- 2. Jain S., Wheeler J.R., Walters R.W., Agrawal A., Barsic A., Parker R.. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016; 164:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jonson L., Vikesaa J., Krogh A., Nielsen L.K., Hansen T., Borup R., Johnsen A.H., Christiansen J., Nielsen F.C.. Molecular composition of IMP1 ribonucleoprotein granules. Mol. Cell. Proteomics. 2007; 6:798–811. [DOI] [PubMed] [Google Scholar]
- 4. Markmiller S., Soltanieh S., Server K.L., Mak R., Jin W., Fang M.Y., Luo E.C., Krach F., Yang D., Sen A.et al.. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018; 172:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Villace P., Marion R.M., Ortin J.. The composition of Staufen-containing RNA granules from human cells indicates their role in the regulated transport and translation of messenger RNAs. Nucleic Acids Res. 2004; 32:2411–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S.A., Heinze K.G., Schwille P.. Fluorescence correlation spectroscopy in living cells. Nat. Methods. 2007; 4:963–973. [DOI] [PubMed] [Google Scholar]
- 7. Bacia K., Kim S.A., Schwille P.. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods. 2006; 3:83–89. [DOI] [PubMed] [Google Scholar]
- 8. Ohrt T., Mutze J., Staroske W., Weinmann L., Hock J., Crell K., Meister G., Schwille P.. Fluorescence correlation spectroscopy and fluorescence cross-correlation spectroscopy reveal the cytoplasmic origination of loaded nuclear RISC in vivo in human cells. Nucleic Acids Res. 2008; 36:6439–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Panchapakesan S.S.S., Ferguson M.L., Hayden E.J., Chen X., Hoskins A.A., Unrau P.J.. Ribonucleoprotein purification and characterization using RNA Mango. RNA. 2017; 23:1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh G., Pratt G., Yeo G.W., Moore M.J.. The clothes make the mRNA: past and present trends in mRNP fashion. Annu. Rev. Biochem. 2015; 84:325–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dictenberg J.B., Swanger S.A., Antar L.N., Singer R.H., Bassell G.J.. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell. 2008; 14:926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nielsen J., Christiansen J., Lykke-Andersen J., Johnsen A.H., Wewer U.M., Nielsen F.C.. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell. Biol. 1999; 19:1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Skabkin M.A., Kiselyova O.I., Chernov K.G., Sorokin A.V., Dubrovin E.V., Yaminsky I.V., Vasiliev V.D., Ovchinnikov L.P.. Structural organization of mRNA complexes with major core mRNP protein YB-1. Nucleic Acids Res. 2004; 32:5621–5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weidensdorfer D., Stohr N., Baude A., Lederer M., Kohn M., Schierhorn A., Buchmeier S., Wahle E., Huttelmaier S.. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA. 2009; 15:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kretov D.A., Clement M.J., Lambert G., Durand D., Lyabin D.N., Bollot G., Bauvais C., Samsonova A., Budkina K., Maroun R.C.et al.. YB-1, an abundant core mRNA-binding protein, has the capacity to form an RNA nucleoprotein filament: a structural analysis. Nucleic Acids Res. 2019; 47:3127–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mateu-Regue A., Christiansen J., Bagger F.O., Winther O., Hellriegel C., Nielsen F.C.. Single mRNP analysis reveals that small cytoplasmic mRNP granules represent mRNA singletons. Cell Rep. 2019; 29:736–748. [DOI] [PubMed] [Google Scholar]
- 17. Hollingworth D., Candel A.M., Nicastro G., Martin S.R., Briata P., Gherzi R., Ramos A.. KH domains with impaired nucleic acid binding as a tool for functional analysis. Nucleic Acids Res. 2012; 40:6873–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen X., Zaro J.L., Shen W.C.. Fusion protein linkers: property, design and functionality. Adv. Drug. Deliv. Rev. 2013; 65:1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Nostrand E.L., Freese P., Pratt G.A., Wang X., Wei X., Xiao R., Blue S.M., Chen J.Y., Cody N.A.L., Dominguez D.et al.. A large-scale binding and functional map of human RNA-binding proteins. Nature. 2020; 583:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lyabin D.N., Eliseeva I.A., Smolin E.A., Doronin A.N., Budkina K.S., Kulakovskiy I.V., Ovchinnikov L.P.. YB-3 substitutes YB-1 in global mRNA binding. RNA Biol. 2020; 17:487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Conway A.E., Van Nostrand E.L., Pratt G.A., Aigner S., Wilbert M.L., Sundararaman B., Freese P., Lambert N.J., Sathe S., Liang T.Y.et al.. Enhanced CLIP uncovers IMP protein-RNA targets in human pluripotent stem cells important for cell adhesion and survival. Cell Rep. 2016; 15:666–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dunsing V., Luckner M., Zuhlke B., Petazzi R.A., Herrmann A., Chiantia S.. Optimal fluorescent protein tags for quantifying protein oligomerization in living cells. Sci. Rep. 2018; 8:10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eliscovich C., Shenoy S.M., Singer R.H.. Imaging mRNA and protein interactions within neurons. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E1875–E1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Machan R., Wohland T.. Recent applications of fluorescence correlation spectroscopy in live systems. FEBS Lett. 2014; 588:3571–3584. [DOI] [PubMed] [Google Scholar]
- 26. Goodarzi H., Liu X., Nguyen H.C., Zhang S., Fish L., Tavazoie S.F.. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015; 161:790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liao Y.C., Fernandopulle M.S., Wang G., Choi H., Hao L., Drerup C.M., Patel R., Qamar S., Nixon-Abell J., Shen Y.et al.. RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell. 2019; 179:147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Vectors and TREX 293 3xFLAG-IMP1 cell line used in this study are available upon request. All data is available upon request.