

Abstract
Aims
The microbiome-derived metabolite trimethylamine-N-oxide (TMAO) has attracted major interest and controversy both as a diagnostic biomarker and therapeutic target in atherothrombosis.
Methods and results
Plasma TMAO increased in mice on ‘unhealthy’ high-choline diets and notably also on ‘healthy’ high-fibre diets. Interestingly, TMAO was found to be generated by direct oxidation in the gut in addition to oxidation by hepatic flavin-monooxygenases. Unexpectedly, two well-accepted mouse models of atherosclerosis, ApoE−/− and Ldlr−/− mice, which reflect the development of stable atherosclerosis, showed no association of TMAO with the extent of atherosclerosis. This finding was validated in the Framingham Heart Study showing no correlation between plasma TMAO and coronary artery calcium score or carotid intima-media thickness (IMT), as measures of atherosclerosis in human subjects. However, in the tandem-stenosis mouse model, which reflects plaque instability as typically seen in patients, TMAO levels correlated with several characteristics of plaque instability, such as markers of inflammation, platelet activation, and intraplaque haemorrhage.
Conclusions
Dietary-induced changes in the microbiome, of both ‘healthy’ and ‘unhealthy’ diets, can cause an increase in the plasma level of TMAO. The gut itself is a site of significant oxidative production of TMAO. Most importantly, our findings reconcile contradictory data on TMAO. There was no direct association of plasma TMAO and the extent of atherosclerosis, both in mice and humans. However, using a mouse model of plaque instability we demonstrated an association of TMAO plasma levels with atherosclerotic plaque instability. The latter confirms TMAO as being a marker of cardiovascular risk.
Keywords: TMAO, Bacterial microbiome, Atherosclerosis, Unstable plaque
Graphical Abstract
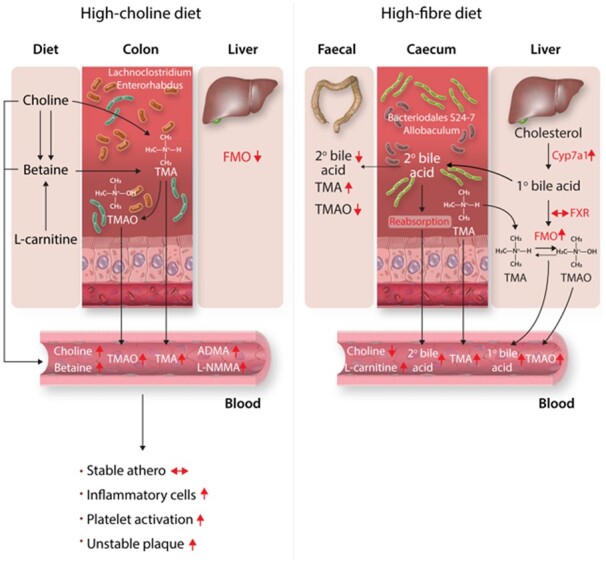
1. Introduction
Gut microbial-derived metabolites have attracted major interest as biomarkers and mediators of disease.1,2 The metabolite trimethylamine-N-oxide (TMAO), in particular, has received a lot of attention as a novel marker of cardiovascular disease (CVD) and mediator of atherosclerosis and increased platelet activation.3–6 It is proposed that increased dietary consumption of red meat or egg increases levels of nutrients such as choline, lecithin, and carnitine that are converted by gut bacteria to trimethylamine (TMA), which is oxidized by flavin-containing monooxygenases (FMOs), primarily in the liver, to TMAO,3 which then exerts pro-atherogenic and platelet activation effects in the vasculature.3–6
Choline-supplemented chow increased atherosclerosis and macrophage lipid content in mice.3 Furthermore, it was shown both in vitro and in vivo that TMAO increases platelet reactivity.6 Efforts to translate these findings are quite mature, with companies offering plasma TMAO measurement in individuals as a means of determining cardiovascular risk, and development of approaches to inhibit TMA production in the gut as a means of treating atherosclerosis.7 Yet, despite accumulating evidence of the association of TMAO with CVD events, its role in atherosclerosis remains controversial. Several reports did not support a correlation between TMAO and extent of atherosclerosis,8–12 one in fact showing an inverse correlation between plasma TMAO level and atherothrombosis.8 Likewise, in a population-based study in Norway, serum TMAO did not differ between patients with carotid atherosclerosis and healthy controls.13 Two recent publications addressing the effect of dietary choline in the two most commonly used mouse models of atherosclerosis—apolipoprotein E knockout (ApoE−/−) mice12 and both ApoE−/− and low-density lipoprotein receptor knockout (Ldlr−/−) mice11—concluded that atherosclerosis lesion size was not altered by a choline supplemented diet. Intriguingly, it was recently shown that TMAO administration in hypertensive rats had beneficial effects, significantly decreasing plasma NH2-terminal pro-B-type natriuretic peptide and vasopressin, left ventricular end-diastolic pressure, and cardiac fibrosis.14
Notably, plasma TMAO increases after apparently healthy dietary interventions.15,16 It has been shown that plasma TMAO is actually increased more by fish and vegetable diets, reported as cardioprotective, than by red meat and egg diets considered to increase cardiovascular risk.17 Other studies have shown that high-fibre diets in humans led to increased plasma TMAO,18 however, the underlying mechanism was unclear.
Therefore, in order to assess the relationship of TMAO to dietary context, and whether TMAO was related to atherosclerosis and cardiovascular risk, we examined divergent routes of TMAO generation in an ‘unhealthy’ (choline supplementation) and ‘healthy’ (high dietary fibre) dietary context, and explored its relationship to atherosclerosis in three different mouse models. We examined stable atherosclerosis using the most commonly used models of atherosclerosis: atherosclerosis-prone ApoE−/− and Ldlr−/− mice, and a tandem-stenosis model of plaque instability in ApoE−/− mice (see Supplementary material online, Figure S1). The aims of this study were: (i) to evaluate the effect of ‘unhealthy’ (high choline) and ‘healthy’ (high fibre) diets on plasma TMAO levels and describe the different generative pathways in each case; (ii) to examine the relationship between circulating TMAO levels and atherosclerosis in model systems and humans; and (iii) to characterize the relationship of TMAO and related metabolites to unstable plaque, the mechanism underlying cardiovascular events such as myocardial infarction. To address these specific aims, our sequence of experiments was: (i) administer high-choline and high-fibre diets to mice and examine the effects on microbiome, faecal/plasma metabolite changes, and TMA oxidation enzyme expression; (ii) in atherosclerosis-prone mice, examine the effect of choline supplementation on atherosclerosis, gut microbiome, and metabolites relevant to TMAO generation; (iii) in parallel, examine the relationship of plasma TMAO to atherosclerosis in a well-characterized human cohort; and (iv) in a tandem-stenosis mouse model, representing plaque instability, examine the effect of choline supplementation on plaque instability and its relationship to TMAO and related metabolites.
2. Methods
2.1 Mice and diets
Animal care and study protocols were approved by the institutional animal ethics committee at the University of Sydney (Protocol No. 2015/881) and the Alfred Research Alliance Animal Ethics committee (Protocol No. E/1721/2017/B), and all procedures conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes or the NIH Guide for the Care and Use of Laboratory Animals.
2.2 High-fibre diet mice
C57BL/6J male mice (4 weeks old) for the dietary fibre study were purchased from Animal Resources Centre (Western Australia) and were housed four per cage (five cages per diet, n = 20 mice/diet, six of which were used for this study). Commencing at 8 weeks of age, mice were fed ad libitum an isocaloric low protein-high carbohydrate diet (14 kJ/g) containing 10% energy from protein, 70% from carbohydrate, and 20% from fat (Specialty Feeds, Western Australia) for 18 weeks. This diet was a variation of AIN93G standard semi-pure rodent diet, which provides ∼19% of energy from protein, 17% from fat, and 64% from carbohydrates.19 Within the carbohydrate component, 35% energy was from sucrose and the remaining 65% from either native wheat starch or ‘gel-crisp’ high amylose resistant starch (RS) type-2 (CRISP FILM™ Starch from Ingredion, IL, USA).20 Detailed compositions of the diets are listed in Supplementary material online, Table S1. Mice were euthanized using overdose intraperitoneal injections of pentobarbital (Nembutal®, Abbott Laboratories, North Chicago, IL, USA; 25 mg/mL, 75 mg/kg) as approved by the Institutional Animal Ethics Committee at the University of Sydney.
2.3 Atherosclerosis in ApoE–/– and Ldlr–/– mice
Twenty-four male ApoE–/– and 24 Ldlr−/− mice obtained originally from the Animal Resource Centre in Western Australia, were bred in the Alfred Research Alliance animal facility. Six-week-old mice were fed a high-fat, high-cholesterol diet containing 22% fat and 0.15% cholesterol (SF00-219, Specialty Feeds, Western Australia) for 6 weeks. At 12 weeks of age, mice were provided high-choline, high-fat, high-cholesterol (SF17-200, 3% Choline, n = 12, Specialty Feeds) or low-choline, high-fat, high-cholesterol (SF00-219, 0.3% Choline, n = 12, Specialty Feeds) diets ad libitum until the endpoint. Mice were euthanized using overdose of 75 mg/kg intraperitoneal pentobarbital (Nembutal®, Abbott Laboratories, North Chicago, IL, USA). Detailed compositions of the diets are listed in Supplementary material online, Table S2.
2.4 Tandem stenosis (T/S) surgery in ApoE–/– mice
This surgery has been previously described.21 In short, male 6-week-old ApoE−/− mice were fed high-fat diet for 6 weeks. At 12 weeks of age, mice were anaesthetized by a ketamine (100 mg/kg) and xylazine (10 mg/kg) mixture through IP injection. An incision was made in the neck and the right common carotid artery was dissected from circumferential connective tissues. A T/S with 150 μm outer diameters was introduced with the distal point 1 mm from the carotid artery bifurcation and the proximal point 3 mm from the distal stenosis, stenosis diameter maintained with 150 μm sutured needle that was later removed. Animals were euthanized using overdose of 75 mg/kg intraperitoneal pentobarbital (Nembutal®, Abbott Laboratories, North Chicago, IL, USA) at 19 weeks of age.
2.5 Quantification of atherosclerosis
Frozen aortic sinuses were sectioned at 6 µm thickness for serial transverse sections, processed and stained as previously described.22 At least six samples per mouse at 100 µm intervals were used. In the T/S mode, serial cryosections were obtained at 100 µm intervals at 1500–2000 µm proximal to the proximal suture in the right common carotid artery and from the brachiocephalic trunk and stored at −80°C until staining was performed. All histological analyses were performed by an operator blinded to animal treatment.
2.6 Immunohistochemistry
The left and right common carotid arteries and the brachiocephalic trunk were sectioned as described above. Sections were incubated in primary antibody [rabbit anti-mouse vascular cell adhesion protein 1 (VCAM-1) (Santa Cruz sc-1504R, TX, USA) 1:200; rat anti-mouse TER-119 biotin (eBioscience 13-5921, CA, USA) 1:400; anti-smooth muscle actin antibody (ProteinTech 23081-1-AP, IL, USA) 1:200; and rat anti-mouse CD68 (Biorad MCA 1957, CA, USA) 1:200] at 4°C overnight, as described previously.
2.7 Blood lipid analysis
Total plasma cholesterol, LDL- and HDL cholesterol, and triglyceride concentrations were measured as described previously.21
2.8 Flow cytometry
Mouse blood was collected by cardiac puncture using the anticoagulant heparin (20 IU/mL). Blood was then centrifuged at low speed (250×g for 5 min using a soft break on a tabletop centrifuge) to remove red blood cells. Platelet-rich plasma (PRP) was obtained and diluted 1:50 in Phosphate-Buffered Saline (PBS) with 2 mM Ca2+ and Mg2+. The diluted PRP was stained for 15 min in the dark with anti-CD41 (APC, BD Biosciences, San Jose, CA, USA) and anti-CD62P (PerCp, BD Biosciences, San Jose, CA, USA). After incubation, Cell fix (BD Biosciences, USA) was added. All samples were analysed on a FACSCanto II flow cytometer (BD Bioscience, USA). CD41-positive events were identified as platelets and platelet activation was defined by P-selectin expression.
2.9 Sysmex haematology analysis
Mouse blood was diluted at 1:7 in Cellpack (Sysmex, Hamburg, Germany) for final total volume 100 μL. The diluted blood samples were then mixed and run on the Sysmex (XS-1000i/XS-800i, Wakinohama-Kaigandori Chuo-ku, Japan) in Capillary mode, complete blood count and differential measurements including white blood cell count; red blood cell count; haemoglobin; haematocrit; mean corpuscular volume; mean corpuscular haemoglobin; mean corpuscular haemoglobin concentration; neutrophil count; lymphocyte count; monocyte count; eosinophil count; basophil count; nucleated red blood cell count; red blood cell distribution width standard deviation; red blood cell distribution width coefficient of variation; mean platelet volume; reticulocyte count; and immature reticulocyte fraction data were collected.
2.10 Metabolomics
Blood was collected immediately by cardiac puncture using heparinized syringes and sample tubes, then centrifuged to isolate plasma. Aliquots of plasma were frozen on dry ice and stored at −80°C until analysis. Faecal samples upon collection were freeze-dried immediately and stored at −80°C until analysis.
Targeted metabolomic analysis measured hydrophilic metabolites in both positive and negative ionization mode using an LC–MS/MS system comprised of an Agilent 1260 Infinity liquid chromatography (LC) system coupled to a QTRAP 5500 mass spectrometer (MS) (AB SCIEX, Foster City, CA, USA), as previously described.20
2.11 Quantification of metabolites
Quantification using calibration curves based on the standard addition method was used to eliminate matrix effects and for quantitative analysis of choline, betaine, L-carnitine, TMA, TMAO, CA, and deoxycholic acid (DCA) in plasma and faecal samples, as previously described.23 In brief, pooled plasma and faeces extracts were divided into aliquots of equal volumes and were spiked with known amounts of the analytes (over the concentration range of 0.025 -10 μM) to build the calibration curve. Correlation coefficients of the calibration curve for all analytes weregreater than 0.995. The total endogeneous concentration of each metabolite in the pooled plasma and faeces is reported in Supplementary material online, Table S3.
2.12 qRT–PCR
Ten milligram of liver tissues was homogenized in Trizol (Life technologies, Carlsbad, CA, USA). Total RNA was isolated using 1-bromo 3-chloropropane for phase separation for total RNA precipitation according to published protocol.24 RNA was quantified spectrophotometrically using a NanoDrop (Thermo Scientific, Wilmington DE, USA). cDNA was synthesized ( iScript, BioRad, USA) and real-time PCR was performed using TaqMan® Gene Expression Assays (Thermo Scientific, MA, USA) to measure HMGCR (4331182; MM012824990_m1), CYP7A1 (4331182; MM00484150_m1), FMO1 (4331182; MM00515795_m1), FMO2 (4331182; MM00490159_m1), FMO3 (4331182; MM01306345_m1), and NR1H4/FXR (4331182; MM00436425_m1) according to manufacturer’s instructions. Reactions were performed in triplicate and target gene expression was normalized to the house keeping gene GAPDH (4331182; MM99999915_g1) and analysed using the ΔΔCt method.
2.13 Bacterial microbiome analysis
In the dietary fibre study, total DNA was extracted from caecal samples and the bacterial community was profiled using the 16S rRNA V4 region as previously described.20 In the choline study, total DNA was extracted from faecal samples using QIAamp ® Fast DNA Stool Mini Kit (QIAGEN, catalogue number: 51604), and the bacterial community was profiled using the 16S rRNA V1-V3 (27F-519 R) andV3-V4 region (341F–806R). Sequencing was performed Illumina MiSeq.
For each study, sequence analysis was performed in R v3.5.1.25 Sequence reads were aligned and partitioned into amplicon sequence variants (ASVs) using DADA2 v1.10.1.26 Taxonomy was assigned to ASVs against the SILVA database v128.27 Multiple sequence alignment was performed with the msa package v.1.14.0,28 phylogenetic tree construction was performed with the phangorn package v.2.5.3,27 and rooted using the ape package v.5.2.29 ASVs that contained less than 0.01% of total reads or <0.1% of reads in at least two samples were filtered out. After filtering, the minimum reads per sample was 14 799 for the choline study and 39 306 for the dietary fibre study. Principal co-ordinate analysis ordination of weighted UniFrac distances30 of gut microbial communities was performed using the phyloseq package v.1.26.131 and visualized with ggplot2 package v.3.1.0.32
The raw sequence reads for the RS vs. native starch (NS) study are available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive in BioProject PRJNA531040, under the accession numbers SRR8859087, SRR8859088, SRR8859091-SRR8859093, SRR8859099, SRR8859103, SRR8859106, and SRR8859109 and the raw sequence reads for the choline study are available under the accession numbers SRR10961733-10961756.
2.14 Framingham heart study offspring cohort
For participants from the Offspring cohort who attended Exam 5 in the Framingham Heart Study, LC–MS/MS metabolomic profiling was performed on plasma specimens collected from each individual. The concentration of plasma metabolites was normalized to pooled plasma samples, followed by a log2 transformation and centralization to the median. Carotid IMT measurements were obtained for subjects attending Exam 6, and coronary artery calcium scores (CAS) for those who attended Exam 7. To examine associations of TMAO with CAS and IMT, linear models stratified by age were fitted for log100 (CAS + 1) (with non-zero CAS only) and log-transformed IMT to account for right-skewed distributions. Analyses were performed with the lm function in R (3.5.2).
2.15 Statistical analyses
Data are expressed as the mean ± SEM and were compared using Student’s t-test, with P-values <0.05 considered statistically significant. Correlation of microbial abundance against metabolites was performed separately for the dietary fibre study and the choline study in R v3.5.1.25 Microbial abundance data was centre-log ratio transformed with the compositions package v.1.40.233 and partial least squares regression was performed with the plsdepot package v.0.1.17.34 Analysis of variance based on weighted UniFrac distances between microbial communities was performed with the vegan package v.2.5.4.35
3. Results
3.1 Choline-driven changes
To investigate atherosclerotic plaque progression, we used the two most common animal models of atherosclerosis. ApoE−/− and Ldlr−/− mice were studied at two time points: at 6 weeks of age, mice were given high-fat diet for 6 weeks. At 12 weeks of age, mice were randomly assigned to high-choline (3%) vs. low-choline (0.3%) high-fat, high-cholesterol diets for a further 7 weeks in ApoE−/− mice and for a further 14 weeks in Ldlr−/− mice. The durations of study diets were chosen according to the reported temporal development of atherosclerosis in each of these atherosclerosis-prone strains, with Ldlr−/− mice typically needing longer for atherosclerosis development as ApoE−/− mice.36,37 Several TMAO-related metabolites were significantly changed by choline supplementation. High-choline diets resulted in significant increases in plasma choline in both ApoE−/− [fold change (FC) = 1.8, P = 7.8 × 10−3] and Ldlr−/− mice (FC = 1.4, P = 2 × 10−2) (Figure 1A). Betaine, oxidative product of choline, was significantly elevated in ApoE−/− (FC = 1.3, P = 1.9 × 10−3) and Ldlr−/− mice (FC = 1.1, P = 6.0 × 10−3), consistent with previous reports revealing choline conversion to TMA via betaine (trimethylglycine).38 High-choline diets did not cause differences in carnitine levels in either strain (Figure 1A).
Figure 1.

Plasma metabolites and hepatic FMOs expression in low choline (LC) vs. high choline (HC)-fed mice. Normalized abundance of (A) choline, betaine, and carnitine; (B) TMA and TMAO; (C) CA and DCA in ApoE−/− and Ldlr−/− mice. Data are expressed as mean ± SEM and are representative of two independent experiments [n = 12 mice/diet for ApoE−/−; n = 9 mice/low choline, n = 12 mice/high choline diet (Ldlr−/−)]. (D) Relative liver mRNA of FMO1, FMO3, and FMO5 from Ldlr−/− mice (n = 12/diet). Data are from a single representative experiment. All data were analysed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. CA, cholic acid; DCA, deoxycholic acid; FMO, flavin-containing monooxygenase; TMA, trimethylamine; TMAO, trimethylamine-N-oxide.
TMA and TMAO were dramatically increased in the plasma. Plasma levels of TMA in ApoE−/− (FC = 18.4, P = 6 × 10−5) and Ldlr−/− (FC = 3.8, P = 9 × 10−4); and TMAO in ApoE−/− (FC = 8.9, P = 2 × 10−9) and Ldlr−/− (FC = 2.9, P = 3.5 × 10−5) mice (Figure 1B), significantly increased.
Bile acids can induce expression of FMOs, the enzyme that oxidizes TMA to TMAO, via their cognate receptor farnesoid X-receptor (FXR). Choline supplementation did not change bile acid concentrations. There were no significant changes in plasma primary bile acid (cholic acid) or secondary bile acid (DCA) in either strain (Figure 1C). The enzymes that oxidize TMA to TMAO, FMOs, decreased or did not change. In the Ldlr−/− mice fed high-choline diets for 14 weeks, hepatic expression of FMO3 (FC = 0.35, P = 4 × 10−3) significantly decreased, whereas FMO1 and FMO5 expression were non-significantly decreased (Figure 1D).
3.2 Fibre-driven changes
Plasma and faecal samples from both NS- and high-fibre RS-fed mice were collected, processed, and subjected to metabolomic analysis according to established protocols with minor modifications.20
The C57BL/6J mouse strain is an often used animal model for the study of dietary interventions and we found that C57BL/6J mice after 18 weeks of high-RS feeding had significantly higher circulating levels of TMA (FC = 3.6, P = 5 × 10−3) (Figure 2A) and TMAO (FC = 2.9, P = 4 × 10−4) (Figure 2A). Faecal TMA levels were significantly higher (FC = 3.2, P = 3 × 10−3) (Figure 2B) in RS-fed mice when compared with NS-fed mice. TMAO was readily detectable in the faeces, and levels were lower in the faeces of RS mice vs. NS mice (Figure 2B), although not statistically significant (FC = 0.34, P = 0.06). Mean absolute plasma and faecal concentrations of TMAO were determined using quantitative analysis by the standard addition method to compare NS-fed and RS-fed mice (both pooled across six samples) (Supplementary material online, Table S3). In NS mice, the plasma concentration of TMAO (0.5 µM) was similar to that in the faeces (0.2 µM); however, in RS mice, plasma TMAO was strikingly higher (6.2 µM) than faecal TMAO (0.07 µM). As faecal TMA was increased in RS mice, the dramatic reduction in faeces: plasma TMAO ratio suggests altered TMAO transport between the gut and the circulation.
Figure 2.

Plasma and faecal metabolites, and hepatic gene expression in resistant starch (RS) vs. native starch (NS) mice. Normalized abundance of plasma and faecal levels of (A, B) TMA and TMAO; (C, D) choline, betaine, and carnitine; (E, F) CA and DCA. Data are shown as mean ± SEM and are representative of two independent experiments with n = 6/diet. Relative liver mRNA of (G) HMGCR and CYP7A1, (H) FXR, FMO1, FMO2, and FMO3 in the liver of NS- and RS-fed mice (mean ± SEM, data are from a single representative experiment with n = 6/NS diet, n = 7/RS diet). All data were analysed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. CYP7A1, cholesterol 7α-hydroxylase; FXR, farnesoid X receptor; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase.
Despite an increase in plasma TMA and TMAO levels in RS mice, plasma choline levels were significantly decreased in RS-fed mice (FC = 0.72, P = 7 × 10−3) (Figure 2C). Faecal levels of choline were unchanged between the RS- and NS-fed mice (Figure 2D). There were no significant differences in plasma or faecal levels of betaine between RS- and NS-fed mice (Figure2C and D). Interestingly, L-carnitine, which is structurally similar to choline and can be synthesized from L-lysine and L-methionine, was significantly higher in the plasma of RS-fed mice (FC = 1.3, P = 1 × 10−2) (Figure 2C), but had a trend towards decreased concentration in the faeces (FC = 0.39, P = 0.1) (Figure 2D).
As mentioned above, bile acids are important inducers of expression of the FMO family of genes, which oxidize TMA to TMAO, and high-fibre diets caused increases in plasma bile acid concentrations. Plasma levels of the primary bile acid, cholic acid (CA), were significantly higher in RS-fed mice (FC = 6.6, P = 2 × 10−3) (Figure 2E) but not different in the faeces (Figure 2F). The microbial-derived secondary bile acid DCA significantly decreased (FC = 0.49, P = 2 × 10−2) (Figure 2F) in the faeces of RS-fed mice, but significantly increased (FC = 6.2, P = 2 × 10−5) (Figure 2E) in the plasma. Using plasma: faecal ratios based on absolute concentrations, on NS the plasma: faecal ratio of primary bile acid cholic acid was 0.28:1 and this increased on RS to 0.79:1. On NS, plasma: faecal ratio of secondary bile acid DCA was 1:1, and on RS this ratio increased dramatically to 5.1:1. This pattern of decreased faecal concentration in concert with increased plasma concentration suggests increased reabsorption in the ileum, cycling through the enterohepatic circulation.
Mice fed on RS diet also showed a significant increase in hepatic expression of cholesterol 7α-hydroxylase (CYP7A1) (FC = 3.7, P = 4 × 10−2) (Figure 2G), which catalyzes the formation of 7-α-hydroxycholesterol from cholesterol and is the rate-limiting step for the production of bile acids in the liver. This finding is consistent with our previous report showing significant reduction in plasma cholesterol in RS mice.20 We also observed a significant increase in the hepatic expression of the rate-limiting enzyme for cholesterol synthesis, 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) in RS mice (FC = 2.4, P = 1 × 10−2) (Figure 2G). On RS-fed mice, FXR (cognate hepatic bile acid receptor) and FMO1 expression did not change, but FMO2 (FC = 1.74, P = 0.046) expression was significantly increased whilst FMO3 expression was borderline significantly increased (FC = 2.94, P = 0.06) (Figure 2H).
3.3 Distinct microbiome-metabolite correlations
Principal co-ordinate analysis of the bacterial gut microbiota indicated that dietary fibre drove greater bacterial changes than dietary choline (Figure3A and C, Supplementary material online, Figure S2). The maximum variance explained in C57BL/6J mice fed RS vs. NS diets was 77.7% (P = 0.009) compared to 35.3% (P = 0.002) in ApoE−/− mice fed high-choline vs. low-choline diets.
Figure 3.

Correlations of the bacterial gut microbiome with metabolites that exhibit major changes upon dietary interventions. Principal co-ordinate analysis ordination of weighted UniFrac distances between (A) HC-fed or LC-fed ApoE−/− mice (n = 12 mice/diet), (C) RS-fed (n = 4) or NS-fed (n = 5) C57BL/6J mice. Ellipses represent 95% confidence intervals, and analysis of variance indicates significant difference between (A) HC vs. LC diets (P = 0.002) and (C) RS vs. NS diets (P = 0.009). Partial least squares correlations between metabolite and amplicon sequence variant (ASV) abundance in (B) HC-fed or LC-fed ApoE−/− mice (n = 12/diet), and (D) RS-fed (n = 4) or NS-fed (n = 5) C57BL/6J mice.
For further analyses, metabolites with major changes upon dietary interventions were chosen. Distinct bacterial taxa were associated with TMA and TMAO levels in the two studies. In high-choline vs. low-choline fed mice, partial least squares regression analysis indicated strong positive correlations between plasma choline, TMA, and TMAO levels with bacterial ASVs assigned to the genera Enterorhabdus (R = 0.76–0.88) and Lachnoclostridium (R = 0.70–0.80) (Figure 3B). In RS- vs. NS-fed mice, the strongest positive correlations to faecal TMA and plasma TMA levels was with ASVs assigned to the order Bacteroidales (R = 0.79–0.84) and the genus Allobaculum (R = 0.79–0.84) (Figure 3D). These ASVs showed a weaker positive correlation with plasma TMAO levels (R = 0.63–0.72) (Figure 3D). No ASVs showed strong positive correlated with faecal TMAO levels (R < 0.50).
3.4 High-choline diets did not alter atherosclerotic burden or plaque composition
High-choline (3%) vs. low-choline (0.3%) high-fat, high-cholesterol diets were administered in ApoE−/− and in Ldlr−/− mice as described in Section 2. There was no significant change in atherosclerotic plaque burden in either Ldlr−/− (Figure 4A) or ApoE−/− (Figure 4F) mice. Plaque composition determines stability,39 and we found no significant changes in plaque burden or histological features, including necrotic core (Figure4B and G), neutral lipids (Figure4C and H), macrophage content (CD68; Figure4D and I), or collagen (Figure4E and J) in 0.3% vs. 3% choline high-fat, high-cholesterol diets in both Ldlr−/− and ApoE−/− murine strains.
Figure 4.

Histological atherosclerotic plaque characteristics in LC vs. HC diet. Data plots showing (A, F) atherosclerotic plaque burden, (B, G) necrotic core size, (C, H) neutral lipid, (D, I) foam cell composition, and (E, J) collagen content in both strains of mice (n ≥ 8 per group). Shown are representative histological images of the different analysis for LC and HC diets in ApoE−/− mice. Data shown are representative of three independent experiments and all data are presented as median and interquartile range. Statistical analysis was performed using non-parametric Mann–Whitney test.
3.5 High-choline diets did not alter plasma lipids but increased circulating monocytes, neutrophils, and platelet activation
We next investigated circulating plasma lipids and immune cells in high-choline treated mice. High-choline supplementation did not alter plasma triglycerides (Figure 5A), cholesterol (Figure 5B), high-density lipoproteins (Figure 5C), low-density lipoproteins/very low-density lipoproteins (Figure 5D), lymphocytes (Figure 5E, Sysmex), or any other parameter determined in Sysmex (see Section 2), but did increase circulating levels of monocytes (Figure 5F, Sysmex), neutrophils (Figure 5G, Sysmex), and expression of P-selectin on the platelet surface (CD41+ P-selectin+) (Figure 5H, flow cytometry), the latter indicating increased platelet activation.
Figure 5.

Lipids, inflammatory cell count, platelet activation, and plasma levels of ADMA and L-NMMA in LC vs. HC. Data plots show (A) triglycerides, (B) total cholesterol, (C) HDL, (D) LDL, (E) circulating lymphocytes, (F) monocytes, (G) neutrophils (E–G: Sysmex data), (H) platelet activation (as indicated by surface expression of platelet P-selectin in flow cytometry) in ApoE−/−mice (n = 9–13 per group). Data are presented as median and interquartile range and statistical analysis was performed using non-parametric Mann–Whitney tests. (I) Plasma ADMA and L-NMMA. mean ± SEM of n = 11–12 per group. Metabolomic data were analysed using Student’s t-test and data shown are representative of a single experiment. *P < 0.05, **P < 0.01, ****P < 0.0001. ADMA, asymmetric dimethylarginine; HDL, high-density lipoprotein; LDL, low-density lipoprotein; L-NMMA, NG-monomethyl-l-arginine.
It has been previously shown that endothelial dysfunction characterized by reduced bioavailability of nitric oxide (NO) contributes to platelet activation.40,41 We found that NG,NG-dimethyl-l-arginine (ADMA) (FC = 1.3, P = 0.009) and NG-monomethyl-l-arginine (L-NMMA) (FC = 1.3, P = 0.01) (Figure 5I) (both inhibitors of NO synthase) were significantly higher in the plasma of high-choline-fed mice. Increased levels of ADMA and L-NMMA limit NO synthesis from L-arginine, which may contribute to platelet activation.
3.6 High-choline diets increased intra-plaque haemorrhage in a mouse model of plaque instability
We then investigated the effects of choline supplementation in a recently established mouse model of plaque instability, which is based on haemodynamic changes induced by a tandem-stenosis in the right carotid artery.21 This model uniquely reflects plaque instability/vulnerability as seen in patients and has been extensively validated histologically, in mRNA and miRNA expression profiling as well as by its use for imaging of unstable plaques and therapeutic plaque stabilization.12,21,22,42,43 In this mouse model, we found no changes in neutral lipids by oil red O (Figure 6A), CD68 (Figure 6B), VCAM-1 (Figure 6C), smooth muscle alpha actin (Figure 6D), platelet count by CD42c (Figure 6E), and collagen (Figure 6F) in 3% vs. 0.3% choline high-fat, high-cholesterol diets. Nevertheless, our investigation of one of the most prominent plaque instability markers—intraplaque haemorrhage22,44—indicated significantly increased plaque instability on high-choline supplementation as determined by increased ferritin (Perl’s iron) and erythrocyte marker TER-119 (Figure6G and H).
Figure 6.

Unstable plaque histology in tandem-stenosis model on LC vs. HC. Box-plots indicate (A) neutral lipids by oil red O staining, (B) CD68, (C) VCAM-1, (D) SMA, (E) platelet content by CD42c marker, (F) collagen, (G) iron deposition by Perl’s iron staining, and (H) erythrocyte marker TER-119 in ApoE−/−mice (n = 8–12 per group). *P < 0.05, **P < 0.01. CD68, cluster of differentiation 68; SMA, smooth muscle actin; VCAM, vascular cell adhesion protein 1. Representative images showing Perl’s and Ter-119 staining in LC and HC diets. Data shown are representative of three independent experiments and all data are presented as median and interquartile range. Statistical analysis was performed using non-parametric Mann–Whitney test.
3.7 Plasma TMAO did not associate with atherosclerosis in the Framingham Heart Study
Plasma TMAO was not associated with atherosclerosis, as defined by CAS (Figure 7A) and carotid IMT (Figure 7B) in the Framingham Heart Study Offspring cohort. Here, we performed logistic regression of CAS or carotid IMT with plasma TMAO levels in 628 and 587 individuals, respectively. We stratified analysis according to age and did not see a significant association across age groups. Grouping all individuals together, there remained no association (beta coefficient = 0.016, P = 0.45). In all age categories, there was no association between plasma TMAO levels and carotid IMT (Figure 7B), and grouping all age ranges together, there remained no association (beta coefficient = 0.003, P = 0.94).
Figure 7.

No association of plasma TMAO with coronary artery calcium score and carotid IMT in the Framingham Heart Study Offspring cohort, stratified to age tertiles. (A) Comparison of plasma TMAO levels and coronary artery calcium score (CAS) in 628 individuals, and (B) plasma TMAO levels and carotid intima-media thickness (IMT) in 587 individuals. CAS, coronary artery calcium score; IMT, intima-media thickness; TMAO, trimethylamine-N-oxide. Line represents linear regression and P-values were calculated from t statistics and are illustrated in each plot.
4. Discussion
Our data provide novel insights into the mechanisms underlying increased plasma TMAO levels in two divergent contexts. First, although we saw dramatic elevations of plasma TMA and TMAO, we didn’t observe any change in atherosclerotic plaque burden and composition induced by 3% choline-supplemented vs. 0.3% choline supplemented high-fat, high-cholesterol diets in two atherosclerosis-prone mouse strains at 7 or 14 weeks of feeding. ApoE−/− mice on a Western diet develop aortic fatty streaks by 5 weeks and plaques by 7–8 weeks37 and we saw no augmentation of this process using 3% compared to 0.3% choline supplementation. Similarly, Ldlr−/− mice on Western diets develop aortic plaques by 12 weeks,36 however, despite significantly-elevated TMA and TMAO in both strains, we did not see any change in atherosclerosis with 3% compared to 0.3% choline supplementation. TMAO levels were positively correlated with ASVs assigned to the bacteria genera Enterorhabdus and Lachnoclostridium. Lachnoclostridium is known to be involved in TMA synthesis from dietary choline by the action of choline TMA-lyase (CutC).45
TMA can be oxidized to TMAO via the action of gut bacterial enzymes or by hepatic enzyme FMO3.46,47 It is reported that almost all TMA is passively absorbed in the portal circulation and oxidized to TMAO in the liver,48 but our results suggest that this is dependent on dietary-microbiome context. In fact, we show that TMAO is readily detectable in the faeces at similar concentrations to the plasma. A significant decrease in hepatic expression of FMO3 accompanied by a significant increase in plasma levels of TMAO after 14 weeks of high-choline diet points towards the gut as the potential source of oxidation to TMAO. Notably, plasma TMAO levels of the mice fed on high-choline diet were highly positively correlated with the abundance of bacterial ASVs assigned to genus Enterorhabdus. Bacterial species within this genus are commonly observed in mouse models of inflammatory disease,49–51 and their active mucin-degrading capabilities can lead to inadvertent penetration of commensal bacteria that may come in direct contact with the gut epithelium,52 leading to elevated production of antimicrobial reactive oxygen species such as superoxide, hydrogen peroxide, and hydroxyl radical in the gut,53 which can oxidize TMA to TMAO.54,55 Consistent with these findings, the ratio of glutathione (GSH), an important scavenger of reactive oxygen species56 to the oxidized disulfide form of GSH (GSSG), a commonly used marker of oxidative stress, was decreased at borderline significance in high-choline fed mice (FC = 2.4, P = 0.06), suggesting an increase in oxidative stress (Supplementary material online, Figure S3).
There is a particular interest in the effects of high-fibre diets on the microbiome and consequent health benefits. RS, a type of dietary fibre found in a wide variety of food, such as whole grains, legumes, cooked and chilled pasta, potatoes and rice, and unripe bananas is highly representative of the most common dietary fibres ingested by humans.57 When mice were administered an RS diet, there was a strong increase in plasma bile acids and TMA compared to the NS-fed mice. Bile acids are known inducers of the FMO family of genes in the liver via FXR, which oxidize TMA to TMAO, and FMOs were increased on RS diet. We have previously shown that RS-fed mice had significantly lower plasma cholesterol,20 and lower plasma cholesterol accompanied by a marked increase in plasma bile acids suggests that dietary fibre may exert cholesterol-lowering effects through bile acid synthesis. Bile acids are predominantly synthesized from cholesterol via the neutral pathway involving CYP7A1,58 which catalyses the formation of 7-α-hydroxycholesterol and is the rate-limiting step of bile acid synthesis.58,59 The increased expression of hepatic CYP7A1 in RS-fed mice indicates increased conversion of cholesterol to bile acids, as reported previously using oat bran in the same murine model.60 Reduced liver cholesterol leads to the activation of HMGCR to increase cholesterol synthesis. High-fibre diets are known to increase metabolite absorption from the colon,20 likely explaining the increase in TMA and secondary bile acids in this context. Although we did not observe a change in FXR at the mRNA level, it is well established that elevated circulating bile acids activate the nuclear hormone receptor FXR in the liver, which induces hepatic FMO3 expression to increase conversion of TMA to TMAO,42 and we saw elevated hepatic FMO2 and FMO3 expression on RS diets. Furthermore, bacterial taxa showed stronger positive correlations to faecal and plasma TMA levels than TMAO levels. Together, these data suggest that high-choline diets lead to oxidation of TMA to TMAO in the gut, but high-fibre diets convert cholesterol to bile acids, which activate hepatic FMOs that oxidize the increased TMA to generate TMAO in the liver.
Intriguingly, we did not see any changes in atherosclerotic burden or plaque composition induced by high-choline diets with elevated plasma TMAO. This is in contrast to earlier studies describing an association of TMAO plasma levels and atherosclerosis in mice.3 However, more recent studies could not confirm this association and as such are consistent with our findings in mouse atherosclerosis.8–10,12 Furthermore, clinical association studies again initially described a positive correlation between cardiovascular risk and TMAO level, which was not confirmed in subsequent studies.9,43 Our investigation in the Framingham Heart Study did not reveal an association of TMAO with atherosclerosis.
There could be several reasons for these controversial results. In mice, these include differences in baseline bacterial gut microbiome between similar mouse strains from different vendors, diet (e.g. different choline levels), mouse age, animal housing conditions, and duration of experiments. In human studies, these include differences in ethnicity and patient age; and gender, diet, and medication usage that can introduce a change in the gut bacterial microbiome composition and alter the relationship between TMAO and atherosclerosis. However, arguably the most important reason is that TMAO is but one of many factors that can influence atherosclerosis and its complications. Depending on the magnitude of each factor, TMAO could be more or less dominant. Although plasma TMAO was increased in mice on a high-choline diet, so were other metabolites with known associations to platelet activation and vascular inflammation such as ADMA61,62 and L-NMMA.63 Overall, it is highly likely that, rather than a single mediator such as TMAO, high-choline diets exert their effects via a constellation of these pro-inflammatory and platelet-activating mediators.
Despite this, TMAO has been demonstrated to have clinical utility in predicting future cardiovascular events, independent of traditional risk factors and even in patients who are negative for troponin T64 and this is consistent with our findings in the tandem-stenosis model, which recreates the histological features of plaque instability seen in humans.22,65–67 Histopathological analysis of plaque rupture and the increased circulating TMAO in multiple human clinical studies suggest a relationship of TMAO with plaque instability,68–70 and these results were consistent with our mouse models of tandem stenosis, whereby high-choline diets led to greater intraplaque haemorrhage and platelet activation, two pathological features of unstable plaques. Although we found elevations in plasma levels of other relevant mediators such as ADMA and L-NMMA, these have not performed as well as TMAO as predictive biomarkers in terms of effect size and significance and have not been replicated in as large cohort sizes as has TMAO.71,72 Therefore, in the correct context, TMAO appears to be an excellent circulating biomarker of subclinical disease that manifests as atherothrombosis either in the short or long-term.6,64 As plaque instability cannot be studied in stable atherosclerosis models such as ApoE−/− and Ldlr−/−, our model provides insight into the context underlying the predictive capability of TMAO for atherothrombotic risk.
5. Conclusion
Our data reveal distinct dietary—bacterial microbiome interactions leading to elevated plasma TMAO, in both ‘healthy’ and ‘unhealthy’ dietary contexts and provide insight into the conflicting reports of TMAO’s relationship to atherothrombosis. Our data suggest that there is no direct association of plasma TMAO and the extent of atherosclerosis. However, we did demonstrate an association of TMAO plasma levels with atherosclerotic plaque instability. The latter is in accordance with TMAO being associated with an increased risk of cardiovascular events.
Data availability
The data underlying this article are available in the article and in its online supplementary material.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
Y.C.K. performed, acquired and analysed all metabolomic data, performed qRT–PCR in the high-fibre livers, and wrote the manuscript. Y.C.C. performed the high-choline mice experiments, including all atherosclerosis analysis and the tandem stenosis experiments. J.A.W. performed the high-fibre diet murine experiments. A.W.S.L. performed the high-fibre microbiome experiments. M.T.K.Z. conducted and analysed Sysmex and flow cytometry experiments. M.L. performed statistical analyses of the Framingham subjects. H.D., R.R., H.T.H., and A.E.F. performed and analysed microbiome experiments in the high-choline diet mice. G.F.A. performed qRT–PCR in the high-fibre livers. J.Y. supervised all statistical analyses. A.H. supervised microbiome experiments in the high-fibre mice. S.J.S. supervised high-fibre experiments and analysis, and wrote the manuscript. K.P. supervised the high-choline murine studies and wrote the manuscript. J.F.O.S. supervised the high-fibre metabolic experiments and wrote the manuscript. K.P. and J.F.O.S. conceived the study.
Conflict of interest: none declared.
Funding
This work was supported by the Heart Research Institute (J.F.O.S. and Y.C.K.), Sydney Medical School Foundation Chapman Fellowship (J.F.O.S.), NSW Health Early-Mid Career Fellowship (J.F.O.S., DOH1003) and by NSW Clinician-Scientist Award (J.F.O.S., DOH1006). K.P. was supported by a National Health and Medical Research Council Principle Research Fellowship. Y.C.C. was supported by a Future Leader Fellowship (Reference No. 102068) from the National Heart Foundation of Australia. Framingham data: The Framingham Heart Study (FHS) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01-HC-25195 and HHSN268201500001I). This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02-HL-64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding for Affymetrix genotyping of the FHS Omni cohorts was provided by Intramural NHLBI funds from Andrew D. Johnson and Christopher J. O’Donnell. Funding support for the Framingham Targeted and Untargeted Metabolomics—HILIC—Installment 1 dataset was provided by Massachusetts General Hospital Departmental funding. Funding support for the Framingham Metabolomics (HILIC—Installment 1, 2, and 3) datasets, Framingham Central Metabolomics—HILIC—Installment 1 and 2, and Lipid Platform—Installment 1 and 2, were provided by National Institutes of Health (R01 DK081572).
Supplementary Material
Translational perspective
There has been keen interest lately in the microbiome as a novel therapeutic target in the prevention and treatment of cardiovascular diseases. Trimethylamine-N-oxide (TMAO) is a microbiome-derived metabolite that has received a lot of interest as a novel biomarker and potential therapeutic target in atherothrombosis. However, there have been conflicting reports of its association with atherosclerosis and thrombosis, with some even showing an inverse association. In spite of this, in the correct context TMAO appears to be an excellent biomarker of subclinical atherosclerosis that can identify those at short-term and long-term risk of myocardial infarction. Herein, we reveal different dietary-microbiome interactions that help explain the apparent contradictions of TMAO’s association with atherothrombosis. This insight is critical for the appropriate use of TMAO as a potential diagnostic and therapeutic target. Furthermore, using a unique murine model, we reveal TMAO to be associated with several features of plaque instability, providing insight into its utility as a marker of cardiovascular risk.
References
- 1. Tang WHW, Li DY, Hazen SL.. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol 2019;16:137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schroeder BO, Backhed F.. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med 2016;22:1079–1089. [DOI] [PubMed] [Google Scholar]
- 3. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL.. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL.. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL.. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013;368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WHW, DiDonato JA, Brown JM, Lusis AJ, Hazen SL.. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016;165:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL.. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 2015;163:1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin J, Liao SX, He Y, Wang S, Xia GH, Liu FT, Zhu JJ, You C, Chen Q, Zhou L, Pan SY, Zhou HW.. Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J Am Heart Assoc 2015;4:e002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meyer KA, Benton TZ, Bennett BJ, Jacobs DR Jr, Lloyd-Jones DM, Gross MD, Carr JJ, Gordon-Larsen P, Zeisel SH.. Microbiota-dependent metabolite trimethylamine N-oxide and coronary artery calcium in the Coronary Artery Risk Development in Young Adults Study (CARDIA). J Am Heart Assoc 2016;5:e003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mueller DM, Allenspach M, Othman A, Saely CH, Muendlein A, Vonbank A, Drexel H, von Eckardstein A.. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015;243:638–644. [DOI] [PubMed] [Google Scholar]
- 11. Aldana-Hernández P, Leonard K-A, Zhao Y-Y, Curtis JM, Field CJ, Jacobs RL.. Dietary choline or trimethylamine N-oxide supplementation does not influence atherosclerosis development in Ldlr−/− and Apoe−/− male mice. J Nutr 2020;150:249–255. [DOI] [PubMed] [Google Scholar]
- 12. Lindskog Jonsson A, Caesar R, Akrami R, Reinhardt C, Fåk Hållenius F, Borén J, Bäckhed F.. Impact of gut microbiota and diet on the development of atherosclerosis in ApoE−/− mice. Arterioscler Thromb Vasc Biol 2018;38:2318–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Skagen K, Trøseid M, Ueland T, Holm S, Abbas A, Gregersen I, Kummen M, Bjerkeli V, Reier-Nilsen F, Russell D, Svardal A, Karlsen TH, Aukrust P, Berge RK, Hov JER, Halvorsen B, Skjelland M.. The Carnitine-butyrobetaine-trimethylamine-N-oxide pathway and its association with cardiovascular mortality in patients with carotid atherosclerosis. Atherosclerosis 2016;247:64–69. [DOI] [PubMed] [Google Scholar]
- 14. Huc T, Drapala A, Gawrys M, Konop M, Bielinska K, Zaorska E, Samborowska E, Wyczalkowska-Tomasik A, Pączek L, Dadlez M, Ufnal M.. Chronic, low-dose TMAO treatment reduces diastolic dysfunction and heart fibrosis in hypertensive rats. Am J Physiol Heart Circ Physiol 2018;315:H1805–H1820. [DOI] [PubMed] [Google Scholar]
- 15. Solanky KS, Bailey NJ, Beckwith-Hall BM, Bingham S, Davis A, Holmes E, Nicholson JK, Cassidy A.. Biofluid 1H NMR-based metabonomic techniques in nutrition research—metabolic effects of dietary isoflavones in humans. J Nutr Biochem 2005;16:236–244. [DOI] [PubMed] [Google Scholar]
- 16. Barton S, Navarro SL, Buas MF, Schwarz Y, Gu H, Djukovic D, Raftery D, Kratz M, Neuhouser ML, Lampe JW.. Targeted plasma metabolome response to variations in dietary glycemic load in a randomized, controlled, crossover feeding trial in healthy adults. Food Funct 2015;6:2949–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cheung W, Keski-Rahkonen P, Assi N, Ferrari P, Freisling H, Rinaldi S, Slimani N, Zamora-Ros R, Rundle M, Frost G, Gibbons H, Carr E, Brennan L, Cross AJ, Pala V, Panico S, Sacerdote C, Palli D, Tumino R, Kuhn T, Kaaks R, Boeing H, Floegel A, Mancini F, Boutron-Ruault MC, Baglietto L, Trichopoulou A, Naska A, Orfanos P, Scalbert A.. A metabolomic study of biomarkers of meat and fish intake. Am J Clin Nutr 2017;105:600–608. [DOI] [PubMed] [Google Scholar]
- 18. Bergeron N, Williams PT, Lamendella R, Faghihnia N, Grube A, Li X, Wang Z, Knight R, Jansson JK, Hazen SL, Krauss RM.. Diets high in resistant starch increase plasma levels of trimethylamine-N-oxide, a gut microbiome metabolite associated with CVD risk. Br J Nutr 2016;116:2020–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reeves PG, Nielsen FH, Fahey GC Jr.. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr 1993;123:1939–1951. [DOI] [PubMed] [Google Scholar]
- 20. Koay YC, Wali JA, Luk AWS, Macia L, Cogger VC, Pulpitel TJ, Wahl D, Solon-Biet SM, Holmes A, Simpson SJ, O'Sullivan FJ.. Ingestion of resistant starch by mice markedly increases microbiome-derived metabolites. FASEB J 2019;33:8033–8042. [DOI] [PubMed] [Google Scholar]
- 21. Chen YC, Bui AV, Diesch J, Manasseh R, Hausding C, Rivera J, Haviv I, Agrotis A, Htun NM, Jowett J, Hagemeyer CE, Hannan RD, Bobik A, Peter K.. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res 2013;113:252–265. [DOI] [PubMed] [Google Scholar]
- 22. Htun NM, Chen YC, Lim B, Schiller T, Maghzal GJ, Huang AL, Elgass KD, Rivera J, Schneider HG, Wood BR, Stocker R, Peter K.. Near-infrared autofluorescence induced by intraplaque hemorrhage and heme degradation as marker for high-risk atherosclerotic plaques. Nat Commun 2017;8:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang TJ, Ngo D, Psychogios N, Dejam A, Larson MG, Vasan RS, Ghorbani A, O’Sullivan J, Cheng S, Rhee EP, Sinha S, McCabe E, Fox CS, O’Donnell CJ, Ho JE, Florez JC, Magnusson M, Pierce KA, Souza AL, Yu Y, Carter C, Light PE, Melander O, Clish CB, Gerszten RE.. 2-Aminoadipic acid is a biomarker for diabetes risk. J Clin Invest 2013;123:4309–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kleinert M, Parker BL, Chaudhuri R, Fazakerley DJ, Serup A, Thomas KC, Krycer JR, Sylow L, Fritzen AM, Hoffman NJ, Jeppesen J, Schjerling P, Ruegg MA, Kiens B, James DE, Richter EA.. mTORC2 and AMPK differentially regulate muscle triglyceride content via Perilipin 3. Mol Metab 2016;5:646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. [Google Scholar]
- 26. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP.. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 2016;13:581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glöckner FO, Yilmaz P, Quast C, Gerken J, Beccati A, Ciuprina A, Bruns G, Yarza P, Peplies J, Westram R, Ludwig W.. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol 2017;261:169–176. [DOI] [PubMed] [Google Scholar]
- 28. Bodenhofer U, Bonatesta E, Horejš-Kainrath C, Hochreiter S.. msa: an R package for multiple sequence alignment. Bioinformatics 2015;31:3997–3999. [DOI] [PubMed] [Google Scholar]
- 29. Paradis E, Schliep K.. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019;35:526–528. [DOI] [PubMed] [Google Scholar]
- 30. Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R.. UniFrac: an effective distance metric for microbial community comparison. ISME J 2011;5:169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McMurdie PJ, Holmes S.. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013;8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016. [Google Scholar]
- 33. van den Boogaart KT-DT, Bren M.. Compositions: Compositional Data Analysis 2018. R Package v1. p40–42.
- 34. Sanchez G, Sanchez MG, Package ‘plsdepot’. Partial Least Squares (PLS) Data Analysis Methods, v 01. 2012;17.
- 35. Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, Simpson GL, Solymos P, Stevens MHH, Wagner H.. Package ‘vegan’. Community Ecology Package, Version. 2013;2:1–295. [Google Scholar]
- 36. Getz GS, Reardon CA.. Do the Apoe-/- and Ldlr-/- mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol 2016;36:1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cartland SP, Genner SW, Martínez GJ, Robertson S, Kockx M, Lin RC, O'Sullivan JF, Koay YC, Manuneedhi Cholan P, Kebede MA, Murphy AJ, Masters S, Bennett MR, Jessup W, Kritharides L, Geczy C, Patel S, Kavurma MM.. TRAIL-expressing monocyte/macrophages are critical for reducing inflammation and atherosclerosis. iScience 2019;12:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang A, Mitchell S, Smith R.. Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol 1999;37:515–520. [DOI] [PubMed] [Google Scholar]
- 39. Stefanadis C, Antoniou CK, Tsiachris D, Pietri P.. Coronary atherosclerotic vulnerable plaque: current perspectives. J Am Heart Assoc 2017;6:e005543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gkaliagkousi E, Passacquale G, Douma S, Zamboulis C, Ferro A.. Platelet activation in essential hypertension: implications for antiplatelet treatment. Am J Hypertens 2010;23:229–236. [DOI] [PubMed] [Google Scholar]
- 41. de Meirelles LR, Mendes-Ribeiro AC, Santoro MM, Mendes MA, da Silva MN, Mann GE, Brunini TM.. Inhibitory effects of endogenous L-arginine analogues on nitric oxide synthesis in platelets: role in platelet hyperaggregability in hypertension. Clin Exp Pharmacol Physiol 2007;34:1267–1271. [DOI] [PubMed] [Google Scholar]
- 42. Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ.. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab 2013;17:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kurilshikov A, van den Munckhof ICL, Chen L, Bonder MJ, Schraa K, Rutten JHW, Riksen NP, de Graaf J, Oosting M, Sanna S, Joosten LAB, van der Graaf M, Brand T, Koonen DPY, van Faassen MLifeLines Deep Cohort Study, BBMRI Metabolomics ConsortiumSlagboom PE, Xavier RJ, Kuipers F, Hofker MH, Wijmenga C, Netea MG, Zhernakova A, Fu J.. Gut microbial associations to plasma metabolites linked to cardiovascular phenotypes and risk. Circ Res 2019;124:1808–1820. [DOI] [PubMed] [Google Scholar]
- 44. Michel JB, Virmani R, Arbustini E, Pasterkamp G.. Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J 2011;32:1977–1985, 1985a, 1985b, 1985c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jameson E, Doxey AC, Airs R, Purdy KJ, Murrell JC, Chen Y.. Metagenomic data-mining reveals contrasting microbial populations responsible for trimethylamine formation in human gut and marine ecosystems. Microb Genom 2016;2:e000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Motika MS, Zhang J, Zheng X, Riedler K, Cashman JR.. Novel variants of the human flavin-containing monooxygenase 3 (FMO3) gene associated with trimethylaminuria. Mol Genet Metab Rep 2009;97:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen Y, Patel NA, Crombie A, Scrivens JH, Murrell JC.. Bacterial flavin-containing monooxygenase is trimethylamine monooxygenase. Proc Natl Acad Sci USA 2011;108:17791–17796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Janeiro MH, Ramirez MJ, Milagro FI, Martinez JA, Solas M.. Implication of trimethylamine N-oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients 2018;10:1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Clavel T, Charrier C, Braune A, Wenning M, Blaut M, Haller D.. Isolation of bacteria from the ileal mucosa of TNFdeltaARE mice and description of Enterorhabdus mucosicola gen. nov., sp. nov. Int J Syst Evol Microbiol 2009;59:1805–1812. [DOI] [PubMed] [Google Scholar]
- 50. Ye J, Lee JW, Presley LL, Bent E, Wei B, Braun J, Schiller NL, Straus DS, Borneman J.. Bacteria and bacterial rRNA genes associated with the development of colitis in IL-10−/− mice. Inflamm Bowel Dis 2008;14:1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D.. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA 2010;107:18933–18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Knights D, Lassen KG, Xavier RJ.. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut 2013;62:1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thaiss CA, Levy M, Korem T, Dohnalova L, Shapiro H, Jaitin DA, David E, Winter DR, Gury-BenAri M, Tatirovsky E, Tuganbaev T, Federici S, Zmora N, Zeevi D, Dori-Bachash M, Pevsner-Fischer M, Kartvelishvily E, Brandis A, Harmelin A, Shibolet O, Halpern Z, Honda K, Amit I, Segal E, Elinav E.. Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell 2016;167:1495–1510.e12. [DOI] [PubMed] [Google Scholar]
- 54. Balagam B, Richardson DE.. The mechanism of carbon dioxide catalysis in the hydrogen peroxide N-oxidation of amines. Inorg Chem 2008;47:1173–1178. [DOI] [PubMed] [Google Scholar]
- 55. Schöneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer’s disease. Biochim Biophys Acta 2005;1703:111–119. [DOI] [PubMed] [Google Scholar]
- 56. Ashfaq S, Abramson JL, Jones DP, Rhodes SD, Weintraub WS, Hooper WC, Vaccarino V, Harrison DG, Quyyumi AA.. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J Am Coll Cardiol 2006;47:1005–1011. [DOI] [PubMed] [Google Scholar]
- 57. Fuentes-Zaragoza E, Riquelme-Navarrete MJ, Sánchez-Zapata E, Pérez-Álvarez JA.. Resistant starch as functional ingredient: a review. Food Res Int 2010;43:931–942. [Google Scholar]
- 58. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 2009;50:1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Myant N, Mitropoulos K.. Cholesterol 7 alpha-hydroxylase. J Lipid Res 1977;18:135–153. [PubMed] [Google Scholar]
- 60. Andersson KE, Axling U, Xu J, Sward K, Ahrne S, Molin G, Holm C, Hellstrand P.. Diverse effects of oats on cholesterol metabolism in C57BL/6 mice correlate with expression of hepatic bile acid-producing enzymes. Eur J Nutr 2013;52:1755–1769. [DOI] [PubMed] [Google Scholar]
- 61. Gremmel T, Perkmann T, Kopp CW, Seidinger D, Eichelberger B, Koppensteiner R, Steiner S, Panzer S.. Interleukin-6 and asymmetric dimethylarginine are associated with platelet activation after percutaneous angioplasty with stent implantation. PLoS One 2015;10:e0122586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ferroni P, Guagnano MT, Falco A, Paoletti V, Manigrasso MR, Michetti N, Santilli F, Guadagni F, Basili S, Davi G.. Association of low-grade inflammation and platelet activation in patients with hypertension with microalbuminuria. Clin Sci (Lond) 2008;114:449–455. [DOI] [PubMed] [Google Scholar]
- 63. Gkaliagkousi E, Ritter J, Ferro A.. Platelet-derived nitric oxide signaling and regulation. Circ Res 2007;101:654–662. [DOI] [PubMed] [Google Scholar]
- 64. Li XS, Obeid S, Klingenberg R, Gencer B, Mach F, Raber L, Windecker S, Rodondi N, Nanchen D, Muller O, Miranda MX, Matter CM, Wu Y, Li L, Wang Z, Alamri HS, Gogonea V, Chung YM, Tang WH, Hazen SL, Luscher TF.. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: a prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur Heart J 2017;38:814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen YC, Rivera J, Peter K.. Tandem stenosis to induce atherosclerotic plaque instability in the mouse. Methods Mol Biol 2015;1339:333–338. [DOI] [PubMed] [Google Scholar]
- 66. Rashid I, Maghzal GJ, Chen YC, Cheng D, Talib J, Newington D, Ren M, Vajandar SK, Searle A, Maluenda A, Lindstedt EL, Jabbour A, Kettle AJ, Bongers A, Power C, Michaelsson E, Peter K, Stocker R.. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur Heart J 2018;39:3301–3310. [DOI] [PubMed] [Google Scholar]
- 67. Diehl P, Nienaber F, Zaldivia MTK, Stamm J, Siegel PM, Mellett NA, Wessinger M, Wang X, McFadyen JD, Bassler N, Puetz G, Htun NM, Braig D, Habersberger J, Helbing T, Eisenhardt SU, Fuller M, Bode C, Meikle PJ, Chen YC, Peter K.. Lysophosphatidylcholine is a major component of platelet microvesicles promoting platelet activation and reporting atherosclerotic plaque instability. Thromb Haemost 2019;119:1295–1310. [DOI] [PubMed] [Google Scholar]
- 68. Tan Y, Zhou J, Liu C, Zhou P, Sheng Z, Li J, Chen R, Song L, Zhao H, Xu B.. Association between plasma trimethylamine N-oxide and neoatherosclerosis in patients with very late stent thrombosis. Can J Cardiol 2019. 10.1016/j.cjca.2019.10.041. [DOI] [PubMed] [Google Scholar]
- 69. Liu X, Xie Z, Sun M, Wang X, Li J, Cui J, Zhang F, Yin L, Huang D, Hou J, Tian J, Yu B.. Plasma trimethylamine N-oxide is associated with vulnerable plaque characteristics in CAD patients as assessed by optical coherence tomography. Int J Cardiol 2018;265:18–23. [DOI] [PubMed] [Google Scholar]
- 70. Fu Q, Zhao M, Wang D, Hu H, Guo C, Chen W, Li Q, Zheng L, Chen B.. Coronary plaque characterization assessed by optical coherence tomography and plasma trimethylamine-N-oxide levels in patients with coronary artery disease. Am J Cardiol 2016;118:1311–1315. [DOI] [PubMed] [Google Scholar]
- 71. Zeller M, Korandji C, Guilland JC, Sicard P, Vergely C, Lorgis L, Beer JC, Duvillard L, Lagrost AC, Moreau D, Gambert P, Cottin Y, Rochette L.. Impact of asymmetric dimethylarginine on mortality after acute myocardial infarction. Arterioscler Thromb Vasc Biol 2008;28:954–960. [DOI] [PubMed] [Google Scholar]
- 72. Cavusoglu E, Ruwende C, Chopra V, Yanamadala S, Eng C, Pinsky DJ, Marmur JD.. Relationship of baseline plasma ADMA levels to cardiovascular outcomes at 2 years in men with acute coronary syndrome referred for coronary angiography. Coron Artery Dis 2009;20:112–117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.