

Abstract
The Oklahoma Shock Nathan Shock Center is designed to deliver unique, innovative services that are not currently available at most institutions. The focus of the Center is on geroscience and the development of careers of young investigators. Pilot grants are provided through the Research Development Core to junior investigators studying aging/geroscience throughout the USA. However, the services of our Center are available to the entire research community studying aging and geroscience. The Oklahoma Nathan Shock Center provides researchers with unique services through four research cores. The Multiplexing Protein Analysis Core uses the latest mass spectrometry technology to simultaneously measure the levels, synthesis, and turnover of hundreds of proteins associated with pathways of importance to aging, e.g., metabolism, antioxidant defense system, proteostasis, and mitochondria function. The Genomic Sciences Core uses novel next-generation sequencing that allows investigators to study the effect of age, or anti-aging manipulations, on DNA methylation, mitochondrial genome heteroplasmy, and the transcriptome of single cells. The Geroscience Redox Biology Core provides investigators with a comprehensive state-of-the-art assessment of the oxidative stress status of a cell, e.g., measures of oxidative damage and redox couples, which are important in aging as well as many major age-related diseases as well as assays of mitochondrial function. The GeroInformatics Core provides investigators assistance with data analysis, which includes both statistical support as well as analysis of large datasets. The Core also has developed number of unique software packages to help with interpretation of results and discovery of new leads relevant to aging. In addition, the Geropathology Research Resource in the Program Enhancement Core provides investigators with pathological assessments of mice using the recently developed Geropathology Grading Platform.
Keywords: Geropathology Grading Platform, Genomic Sciences Core, Geroscience
Introduction
The Oklahoma Nathan Shock Aging Center, which was initially funded in 2015, is a collaboration between the three major research institutions in Oklahoma City: The University of Oklahoma Health Sciences Center, the Oklahoma Medical Research Foundation, and the Oklahoma City VA Medical Center. The focus of our Shock Center is on geroscience because this is a growth area in aging and fits well with the emphasis on geroscience on our three campuses. The goal of our Center is to maximize our outreach to investigators throughout the USA in two ways. First, the Research Development Core directs a Pilot Grant Program that focuses on providing funds to junior investigators outside Oklahoma City. Second, the research cores in the Center provide unique research services that use frozen tissue/cells, which can be collected at the investigator’s institution and shipped to Oklahoma. The Oklahoma Shock Center has four research cores and a Geropathology Research Resource which are described below and can be viewed at https://oklahomanathanshockcenteronaging.org/.
Multiplexing Protein Analysis Core
Measuring changes in protein abundance is a central part of aging and geroscience research because proteins are key drivers of all processes that occur in a cell. Changes in protein abundance are usually measured with immunochemical methods such as Western blotting. While immunochemical methods can function well and have a rich history of excellent use, all have modest to severe limitations. Indeed, problems of unreliable and/or unproven specificity and lot-to-lot variability of antibodies have produced a backlash in the protein quantification field and a renewed emphasis on the need for greater rigor in protein quantification methods [2]. The targeted methods used by the Multiplexing Protein Analysis Core (selected reaction monitoring (SRM), parallel reaction monitoring (PRM), and high-resolution accurate mass (HRAM)) are mass spectrometry (MS) methods that are specific, precise, and have a wide dynamic range and high throughput. Further, we have perfected the ability to develop, optimize, and validate new assays for any protein from any animal. Studies that are otherwise limited by the availability of antibodies can be conducted more quickly and at lower costs using MS methods. In addition, the specificity of analysis and documented validation steps inherent to the method development directly address the question of rigor in quantitative analysis of protein expression.
Figure 1 shows how targeted quantitative proteomics works, using catalase from Drosophila as an example. The assay selectively targets peptides generated by trypsin digestion from the protein being measured. Because trypsin is highly purified with high specific activity, the digestion produces a quantitative conversion of all proteins in the sample to peptides. A subset of the peptides from any given protein is utilized as surrogates for the abundance of that protein. The Core uses two or three peptides per protein. Our example of a Drosophila protein demonstrates that these assays can be developed for any protein from any animal, including invertebrates, which are used widely in aging research. While the majority of our assays have been developed for mice, they are equally useful for rats due to the high homology between mouse and rat proteins. Similarly, analysis of the homology of the mouse peptides used in our assays shows they also function well for other animal models, e.g., naked mole rats. We have made a continuous effort to develop and validate new targeted assays for mouse proteins, giving us the current capability to measure approximately 500 proteins based on the detection of over 1200 peptides. We have also adapted these assays to rat and human samples. These assays are organized to interrogate specific biochemical pathways, e.g., TCA cycle, β-oxidation, glycolysis, gluconeogenesis, and antioxidant and mitochondrial proteins. For a typical experiment, investigators prepare and submit tissue homogenates made using the methods familiar to their laboratory along with carefully measured protein concentrations. By measuring multiple proteins involved in a pathway, which is possible with the panels developed by the Core, one has a better indication of how aging or an experimental manipulation affects that pathway. An example is shown in Fig. 2 where the expression of proteins in glycolysis and the Krebs cycle are measured in wild type and the -/- too high mice. The data generated by the Core clearly show that the knocking out Sod1 (Cu/Zn-superoxide dismutase) had no impact on glycolysis but increased the expression of almost all proteins involved in the TCA cycle.
Fig. 1.

LC-tandem MS analysis of a whole homogenate from Drosophila. A The raw LC-tandem MS data showing the detection of the 3 peptides in scheduled time windows at characteristic retention times. B and C The raw data are analyzed with the program Pinpoint, which finds and integrates the chromatographic peaks via the overlap of the selected reactions. Only 2 peptides are shown because of space. D The amount of each protein is determined by normalizing to a known amount of bovine serum albumin added to the sample as a non-endogenous internal standard. It is notable that the 50% decrease in catalase abundance expected in Drosophila heterozygous for catalase is observed
Fig. 2.

The ability of targeted quantitative proteomics to interrogate pathways. The levels of proteins involved in glycolysis, fatty acid metabolism, the Krebs cycle, and mitochondrial function are shown for skeletal muscle from 10-month-old wild type and Sod1−/− mice. Red bars represent increased expression, blue bars represent decreased expression, and black bars represent non-significant changes in proteins in Sod1−/− mice compared to WT mice. Data were taken from Sataranatarajan et al. [58]
The Multiplexing Protein Analysis Core also uses mass spectrometry to measure the turnover of individual proteins using the stable isotope, D2O. The measurement of the synthesis rates of individual protein uses the same proteomic approaches employed in measuring protein abundance with additional determination of isotope incorporation for calculation of synthesis rates. By simultaneously measuring both protein content and protein synthesis rates, the rates of protein breakdown and turnover (half-life) can be calculated. The measurement of the synthesis and breakdown of specific proteins allows investigators to determine the mechanism responsible for changes in the abundance of a protein. In addition, the measurement of these dynamic processes is important because the synthesis and breakdown of proteins are two of the primary regulatory mechanisms of proteostatic maintenance and even in the absence of changes in protein abundance, changes in turnover can occur that are indicative of a pro- or slowed-aging state.
Isotopic labeling is the favored approach for measuring protein turnover and has been in use since the early twentieth century [76]. The concept of stable isotope labeling is relatively simple in that a precursor pool (amino acids) is enriched with isotope that is incorporated into proteins so that the proteins become enriched as a product of time (giving a rate). In practice, isotope experiments are much more complicated and involve careful study design that requires knowledge of assumptions and limitations inherent in the method used.
Because of limitations of classic analysis methods, traditional protein turnover measurements have assumed that a protein pool is homogeneous and constant over the experimental period [57]. However, within a tissue homogenate or subcellular fraction, there are hundreds to thousands of individual proteins that are independently regulated. Dr. Miller’s group, along with other notable geroscience researchers, have taken advantage of long-term labeling procedures, such as those using D2O in water or labeled amino acids in food. Using long-term labeling, it is possible to measure the synthesis rates of individual proteins when combined with proteomic analysis. Figure 3 shows the value of this approach to explore mechanisms of proteostatic changes. These data are comparisons of rapamycin versus rapamycin plus metformin treatment in male and female mice. The skeletal muscle proteins are grouped by GO terms to demonstrate changes by subcellular compartment (A and B), within the mitochondrial proteome (C and D), and within mitochondrial complexes (E and F). It is important to note that D2O labeling has been used in many models important for the study of the basic biology of aging including cells, yeast, C. elegans, mice, rats, and humans.
Fig. 3.

Feasibility of measuring protein synthesis rates of individual proteins in skeletal muscle. Normalized (Log2) fold change data are presented from control male and female HET3 mice treated with rapamycin (Rap) or rapamycin plus metformin (Rap + Met). These data were obtained using a time course approach with three mice for time point and five time points. Using GO terms, we separate by cellular compartment (A and B), mitochondrial compartment (C and D) and mitochondrial complex (E and F). Note, there are differences between treatments and relative changes between sexes
The use of D2O for labeling experiments has advantages over other methods (e.g., labeled amino acids) making D2O the preferred tracer for proteomic studies [24, 28, 38, 39, 44, 50, 56]. First, at the enrichments used for a typical labeling experiment, D2O is biologically safe facilitating its use in various models, e.g., rodents and humans, without constraints. Second, compared to other labeling procedures, D2O is relatively inexpensive. Depending on complexity of study designs, experiments using yeast and C. elegans, are typically $100–$400, while labeling in rodents is < $1000. Third, perhaps more than any other tracer, D2O lends itself to long-term labeling. Therefore, experiments can be designed for up to weeks to months to capture proteins that turn over slowly. Fourth, D2O is easy to administer by adding to liquid media or regular drinking water thus expanding the number of model organisms amendable to labeling. Fifth, D2O labeling is sensitive, and there are several software programs that facilitate its use proteomic analyses [24, 28, 44, 56]. Sixth, D2O is a “universal tracer” and with the appropriate planning can be used to capture additional dynamic events, such as synthesis of DNA and RNA without conducting additional experiments to measure the synthesis of these molecules.
A typical experiment using the Multiplexing Protein Analysis Core to measure protein synthesis begins with the investigator contacting the Core about the goals of his/her study. The Core advises the investigator on the appropriate study design and directs the investigator to where to purchase and how to administer the D2O. The investigator will collect samples in a manner consistent with the study goals and the model organism, which can range from hours to weeks. The researcher will collect water samples (media, plasma, etc.) for determination of precursor enrichment. When an experiment is completed in its entirety, the researcher will mail frozen samples to the Core for analysis. The Core will finish any necessary sample processing and run both the precursor and product for calculation of synthetic rates.
Genomic Sciences Core
The goal of the Genomic Sciences Core is to provide researchers in aging and geroscience with access to state-of-the-art genomic, epigenomic, and transcriptomic analyses that are not typically provided by institutional cores but are central to biology of aging research. Specifically, the Core offers services in the analysis of, DNA modifications (methylation and hydroxymethylation), mitochondrial genomes, and single-cell transcriptomics. In coordination with the GeroInformatics Core, these services will take investigator submitted samples from data generation through analysis and interpretation. Epigenetic and mitochondrial variant analyses have been a part of molecular studies in aging and geroscience for some time. However, a significant evolution of the methods to perform these studies has been occurring with new levels of accuracy, throughput, and comprehensiveness now possible. These advancements offer the opportunity to resolve outstanding questions in the field such as the epigenomic contribution to age-related gene expression dysregulation, how oxidative stress affects mitochondrial genomic stability, and the molecular profile of cellular senescence.
DNA methylation/hydroxymethylation
Epigenetics is regulation of gene expression at the genomic level without changes in DNA sequence. Histone and DNA modifications and genomic accessibility are key epigenetic regulators of genomic structure and gene expression, and the dysregulation of epigenomic patterns and genomic organization is a hallmark of aging [4, 74]. However, epigenetic analyses are challenging for individual investigators as they require specialized instrumentation and advanced bioinformatic analyses of sequencing data. Epigenomic studies have therefore generally been limited to a few high throughput sequencing laboratories.
The Genomic Sciences Core services are designed to allow investigators with a wide variety of model species, experimental interventions, and organ systems to address epigenetic hypotheses in their studies. We have developed a suite of techniques to not only allow analysis of methylation but also hydroxymethylation at the whole genome (WG-ox/BS-Seq), genome-wide (ox/BOCS), and gene specific (ox/BSAS) levels [36]. We developed the BSAS approach [35] and have the only BOCS capture probeset for the rat genome (Masser et al., 2016b). This combination of approaches allows investigators to tailor their DNA modification analyses to analyzing the portions of the genome needed to meet experimental goals without sacrificing accuracy and precision. Even with the rapidly decreasing cost of sequencing, focusing on the specific parts of the genome needed for a study keeps costs down. The combination of hydroxymethylation and non-CpG methylation analyses is a focus not often found in the field. Most studies only measure methylation of CpG regions [74]. Cytosines in CpH sites are also methylated, and because the number of CpH sites in the genome is much greater than CpG sites, methylation at CpH sites could potentially play an important role in gene expression. The oxBS chemistry allows us to measure the distribution of both mC and hmC on across regions or on a base-by-base basis, as shown in Fig. 4. The ability to differentiate between mC and hmC is important because DNA hydroxymethylation is relatively high in the CNS [37], and is often differentially associated with gene expression, as compared to mC when found in enhancers and gene bodies.
Fig. 4.
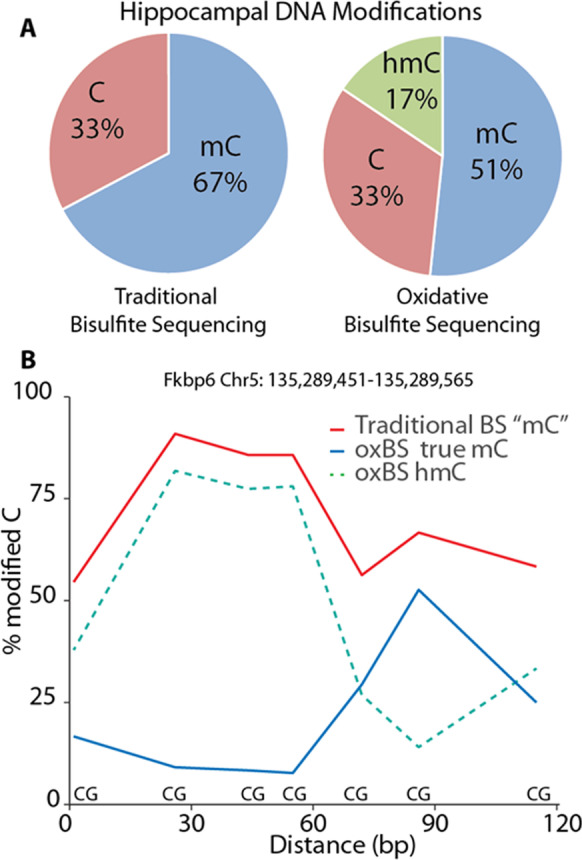
Levels of methylation and hydroxymethylation in the genome of the hippocampus. DNA isolated from the hippocampus from mice was analyzed for methylation of the traditional bisulfite sequencing and oxBOCS. A Data showing the 5mC and 5hmC contend of DNA isolated from brain. Approximately 25% of the methylation that is normally identified as 5mC is 5hmC. B Data showing a region of the genome where bisulfite sequencing gives an incorrect analysis of DNA modifications on a base-by-base basis
Mitochondrial heteroplasmy
A single mitochondrion contains multiple circular mitochondrial genomes (mtDNA), which encode critical electron transport chain subunits, as well as transfer and ribosomal RNAs. The mtDNA copy number varies by cell type, and normally all mtDNA copies are identical within a single mitochondrion, termed homoplasmy. Heteroplasmy occurs when a portion of the mtDNA within a mitochondrion/cell accumulates mutations or deletions. Accumulation of mitochondrial heteroplasmy and a reduction in mtDNA copy number with age are proposed to play a role in mitochondrial dysfunction [20, 63],therefore, quantitative measurements of mtDNA copy number and mutations/deletions are important to many aging studies.
The Genomics Sciences Core is using new next-generation-sequencing methods [59] that allow investigators to determine the location and frequency of mitochondria mutations in any tissue and any animal model from invertebrates to rodents to humans. Our approach of targeting sequencing to mtDNA is through an amplification enrichment approach that allows for higher throughput and lower costs. Our digital PCR technology is unique in that it gives the absolute number of mtDNA genome copies per cell, as measured in relation to the number of nuclear genomes (Masser et al., 2016a). An example of the power of this analysis in measuring mtDNAheteroplasmy is shown in Fig. 5. The heteroplasmy mtDNA was measured in skeletal muscle from young and old mice and from young Sod1−/− mice, which show accelerated aging and increased oxidative stress/damage. The data in Fig. 5 show that not only the location and frequency of mutated sites change with age but that loss of Sod1 accelerates the accumulation of heteroplasmy.
Fig. 5.

Example of mtDNA heteroplasmy. Muscle tissue from young (8 months) wild type (WT, blue), old (27 months) WT (red), and young (8 months) Sod1−/− (KO, green) mice was analyzed for mtDNA mutations using next-generation-sequencing. Mutation locations and frequencies are shown in the circos plot for each group. More heteroplasmic sites were found in old WT and KO mice than young WT (inset, ANOVA, SNK *p < 0.05, n = 5–8 group)
Single-cell transcriptomics
While molecular studies of aging have explored gene expression with qPCR, microarrays, and RNAseq, these data have generally been at the tissue level and have not examined cell-to-cell heterogeneity. Recent advances in single cell RNAseq technology [65] allow researchers to measure the molecular phenotypes of hundreds to thousands of individual cells from a sample. This technology opens the door to understanding cellular heterogeneity/subpopulations with aging [62]. A prime example of this need is cellular senescence studies where only a small subpopulation of individual cells are believed to senesce.
The Genomic Sciences Core’s approach to scRNAseq uses a combination of services that better suits the wide variety of needs of investigators. Many investigators are in a rush to use the new 10X Genomics technology [78] resulting in technical issues and at times expensive and poor quality data [22, 82]. To provide a route for investigators new to single cell work, we offer a range of scRNAseq services in addition to 10X Genomics including droplet-based ddSeq [23] and SMART-Seq. These allow investigators to begin with smaller more tractable studies and also for more focused studies of lower number of cells or physiologically characterized cells that need transcriptomic characterization at a greater depth. Combining the experience of the Genomic Sciences Core in the intricacies of cell preparation and library generation with capabilities of the GeroInformatics Core to analyze the resultant datasets allows investigators to perform rigorous scRNAseq experiments.
Geroscience Redox Biology Core
The oxidative stress theory advanced by Denham Harman proposed that increased oxidative damage during aging is causally related to aging and lifespan and has been a long-standing and intellectually appealing concept that has been tested in organisms ranging from yeast to humans. While many studies in a variety of animal models show that an increase in lifespan is correlated to an increased resistance to oxidative stress and reduced oxidative damage, several studies show that oxidative stress plays a limited role in the lifespan of mice, suggesting that the role of oxidative stress in aging is not as simple as once believed [41, 47]. What is clear however is that redox status and oxidative stress/damage may contribute to the pathogenesis of a variety of age-related diseases including diabetes, neurodegenerative and cardiovascular disease, and cancer, suggesting that the age-related increase in oxidative stress/oxidative damage is an important contributing factor in the mechanism for why age is major risk factor in these diseases and a critical factor in the field of geroscience. Therefore, there is a continued need and demand to measure oxidative stress and damage in tissues/cells. The Geroscience Redox Biology Core provides investigators with assays needed to measure to oxidative stress, including live imaging of reactive oxygen species and imaging of structural and functional effects of changes in oxidative insults.
A key strength of the Geroscience Redox Biology Core is that we provide investigators with a comprehensive profile of the interrelated components of metabolism and oxidative homeostasis. The contribution of free radical mediated processes cannot be accurately evaluated simply by measuring oxidative damage. In fact, redox status reflects the balance between pro-oxidant production and antioxidant capacity. Thus, the Geroscience Redox Biology Core takes into account the highly integrative nature of redox and energy dependent processes to gain molecular insight into underlying factors and functional consequences. Measures of redox status can be coordinated with results from the Multiplex Protein Analysis Core, which can measure changes in levels of proteins involved in antioxidant systems, mitochondria function, and various metabolic pathways. Thus, this integrated approach allows an investigator to assess global networks that influence redox status, oxidative stress, and cellular homeostasis.
Redox status
The Geroscience Redox Biology Core offers the direct analyses of a panel of redox couples (GSH/GSSG, NADP/NADPH, NAD/NADH) and oxidative damage to lipid and protein with a degree of specificity and sensitivity not readily available within a single laboratory or available through institutional cores. Coordinated measurements of critical redox couples provide insight into the source of oxidative stress and the cellular compartment(s) affected.
Redox couples
We have developed methods for analysis of redox couples as summarized below. A major advantage is that numerous interrelated metabolites can be analyzed with relatively small amounts of tissue. The types of samples amenable to analyses include flash frozen tissue, tissue homogenates, cells, and isolated organelles from invertebrates and vertebrates. Samples are extracted with 5% meta-phosphoric acid (GSH, GSSG) or 150 mM KOH (NADP+, NAD+, NADPH, NADH). Compounds are resolved by ion pairing reverse phase HPLC and quantified using electrochemical, fluorescence, or UV/VIS detection. Routine spiking of experimental samples with known quantities of standards is utilized to ensure accurate peak assignment. The limits of detection for each metabolite are in the nanomole range.
GSH/GSSG ratio. Glutathione is the most abundant thiol in animal cells making it a major antioxidant in cells. The ratio of GSH to GSSG, therefore, provides a measure of redox status and antioxidant capacity [51, 52]. GSH and GSSG are separated using a HPLC system is equipped with a boron-doped diamond electrode electrochemical cell and a reverse-phase column as previously described by Park et al. [46] with modifications.
NADPH/NADP+ and NADH/NAD+ ratio. Regeneration of GSH and reduced thioredoxin requires oxidation of NADPH to NADP+. Thus, the relative levels of NADPH and NADP+ provide an additional index of antioxidant capacity of a cell. NADH is the primary carrier of electrons derived from the oxidation of glucose and fatty acids, and the relative ratio of NADH to NAD+ is intimately linked to the redox couples GSH/GSSG and NADPH/NADP+. In addition, NADH and NADPH can be interconverted by the nicotinamide nucleotide transhydrogenase or NAD+- and NADP+-dependent isoforms of isocitrate dehydrogenase. NAD+, NADH, NADP+, and NADPH are measured using an HPLC system as previously by Rindler et al. [53].
Oxidative damage
One consequence of changes in redox couples or oxidative stress is the occurrence of oxidative damage to macromolecules, which is believed to play an important role in the physiological consequences of increased oxidative stress [45, 60]. Although there are various kits available to measure oxidative damage in tissues, these assays are neither specific for a specific type of damage nor sensitive enough to detect damage occurring normally in vivo. The assays the Geroscience Redox Biology Core provides are sensitive and reproducible in part because the data is collected for specific types of damage using methodologies and equipment not readily available in many labs. Figure 6 shows the ability of the assays described below to detect lipid peroxidation and protein oxidation in liver tissue from young and old rats.
Lipid peroxidation (F2-isoprostanes). F2-isoprostanes are formed in membranes as a result of free radical attack on arachidonic acid in membrane phospholipids. F2-isoprostanes are chemically stable end-products of lipid peroxidation and reliable and sensitive markers of lipid peroxidation [40, 54, 55]. Because isoprostanes are produced in every tissue, plasma levels of free F2-isoprostanes provide a measure of endogenous production of F2-isoprostanes from all sites in the body, thus providing an excellent marker of whole-body oxidative stress levels. Traditional measures of lipid peroxidation, e.g., TBARS or MDA, are unstable and, therefore, difficult to accurately measure. The discovery of F2-isoprostanes and neuroprostanes as a stable and sensitive marker of lipid peroxidation has been a major improvement over other assays of lipid peroxidation. The sample preparation for the isoprostane assay is critical and in the case of isoprostanes is also very labor intensive. The isoprostane level can be measured in frozen samples of 100 mg of tissue, 1 ml of plasma, or 300 μl of urine. Studies from the Gerosciences Redox Biology Core have previously shown that that F2-isoprostanes levels increase with age in the plasma, and also in liver and kidney in F344 rats, and the changes in F2-isoprostanes levels in liver and kidney tissue can be directly correlated with oxidative damage to DNA [75]. Thus, F2-isoprostanes levels are an excellent biomarker of lipid peroxidation for aging studies. Details of the assay can be found in Ward et al. [75].
Protein oxidation (carbonyls). Oxidative modifications to proteins can lead to mis-folded proteins that are prone to forming deleterious oligomers or aggregates that can alter cellular homeostasis and contribute to age-related pathologies and to the aging process itself. A predominant form of protein oxidative modification is the formation of carbonyl groups on specific amino acid residues, e.g., lysine, arginine, proline, and threonine. Carbonyl groups can also be formed by the reaction of amino acid residues with aldehydes (malondialdehyde, 4-hydroxy-2-nonenal) and reactive carbonyl derivatives generated through the reaction of reducing sugars or their oxidation products with lysine residues of proteins. The majority of assays available to measure protein oxidation involve derivatization of the carbonyl group with dinitrophenylhydrazine (DNPH) leading to the formation of a stable dinitrophenyl hydrazine product. This product can be detected spectrophotometrically, by HPLC or using antibodies to DNP. However, these assays often suffer from high background (contamination from unreacted DNP) and low reproducibility. To circumvent the deficiencies in these assays, the Core uses a modified assay in which oxidized proteins are measured using a fluorescent-based assay to detect protein carbonyls [6]. This method is very sensitive and allows quantitative detection of proteins with even very low levels of protein carbonyls. Global changes in oxidized proteins are measured in tissue homogenates treated with fluorescein-5-thiosemicarbazide (FTC) and subjected to electrophoresis on a 12% gel to resolve fluorescence-labeled proteins from FTC. Importantly, samples can also be analyzed by two-dimensional gel electrophoresis to identify carbonyl levels of individual proteins. The detailed methods for this analysis can be found in Chaudhuri et al. [6] and Pierce et al. [49].
Fig. 6.
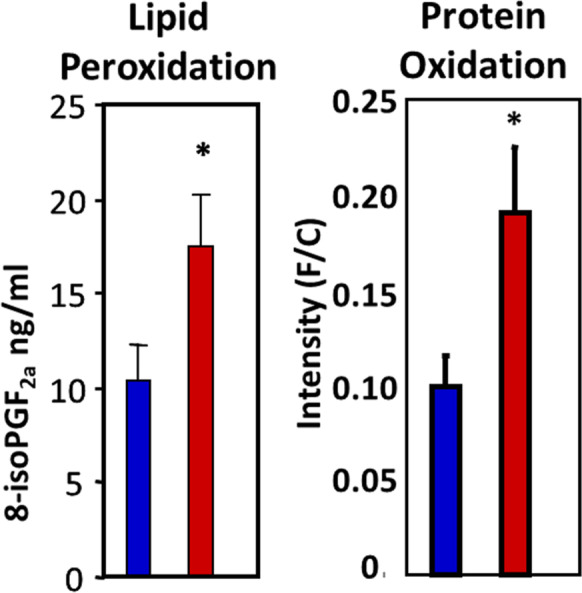
Effect of age on the levels of oxidative damage in liver tissue. The levels of lipid peroxidation (isoprostanes) and protein oxidation (carbonyls) were measured in liver tissue from 4- to 6-month-old (blue bars) and 22- to 24-month-old (red bars) rats. Data taken from Ward et al. [75] and Chaudhuri et al. [6]
In vivo analysis of free radical production
The Geroscience Redox Biology Core also provides investigators with in vivo approaches to assess oxidative stress, either directly or indirectly. Direct measurements include in vivo free radical assessment, as well as determining oxygen use. Indirect measures include in vivo vascular and tissue structural alterations (free radical–initiated or via inflammation) and metabolic evaluations (as a result of mitochondrial function or tissue response to free radicals and/or inflammation). These techniques allow investigators to directly detect cellular/tissue changes in vivo and in situ, so that regional sites of free radical production can be pin-pointed to specific regions/cells in tissue samples.
The Geroscience Redox Biology Core uses the novel method developed by Dr. Towner for detecting in vivo free radicals distributed within various tissues/organs as well as heterogeneously within specific tissues/organs [9, 66–71, 73]. The approach involves the use of immuno-spin trapping (IST) in conjunction with molecular-targeted magnetic resonance imaging (mt-MRI). This combined IST-mt-MRI approach initially utilizes a spin-trapping agent, DMPO (5,5-dimethyl-1-pyrroline N-oxide), which traps free radicals in an oxidative-stress-related disease model, and then administration of a mt-MRI probe, called an anti-DMPO probe, that uses an antibody against DMPO-radical adducts (anti-DMPO antibody) and a MRI contrast agent, resulting in targeted free radical adduct mt-MRI. This combined IST-mt-MRI approach has been used in several rodent disease models, including diabetes, ALS, gliomas, and septic encephalopathy [9,66–71 , 73]. The contrast agent used in this approach includes an albumin-Gd-DTPA-biotin construct, where the anti-DMPO antibody is covalently linked to cysteine residues of albumin, forming an anti-DMPO-adduct antibody-albumin-Gd-DTPA-biotin entity. The Gd-DTPA moiety is the MRI signaling component, which increases MRI signal intensity (SI), or decreases T1 relaxation (MRI contrast parameter). Both of these parameters, MRI SI or T1 relaxation, can be used to assess the presence of the anti-DMPO probe. The biotin moiety can be used for ex vivo validation of the presence of the anti-DMPO probe in tissues, by using streptavidin-fluorescence (e.g., Cy3) or streptavidin-HRP (horse radish peroxidase) to tag the biotin in the anti-DMPO probe. Figure 7 shows the measurement of in vivo free radical levels in the skeletal muscle Sod1−/− mice, which develop skeletal muscle atrophy [42]. Free radical levels were > twofold higher in Sod1−/− mice compared to wild-type, control mice.
Fig. 7.
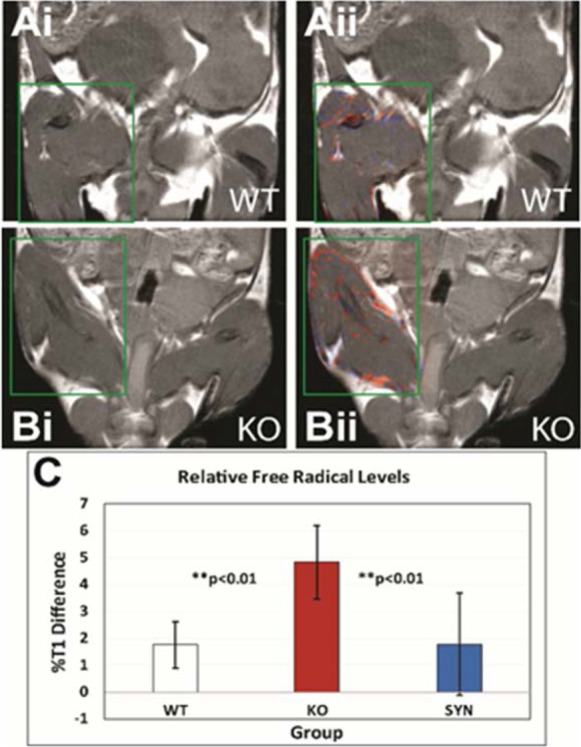
Measurement of free radical levels in vivo. Levels of free radicals were measured is skeletal muscle from wild type (WT), Sod−/− (KO), and SynTgSod−/− (SYN) mice. Free radical maps (red-false-coloring) (ii) overlaid on top of MR images for WT (A) and KO (B) mice are shown. The quantitative levels of free radicals (C), as measured by a percent change in T1 relaxation differences. Free radical levels were found to significantly increase in KO mice compared to WT or SynTgSod−/− (SYN) mice, which have Sod1 expressed in the neurons of Sod1−/− mice. Data taken from Ahn et al. [1]
Mitochondrial function
The Geroscience Redox Biology Core also provides investigators with services to measure mitochondrial function (oxidant generation, ATP production, respiration) in fresh tissue samples or isolated mitochondria. This includes in vitro analyses of mitochondrial function in isolated mitochondria, respirometry analysis in fresh tissue using the Oroborus respirometer, as well as measurement of mitochondrial function in cells using the Seahorse XF24 Extracellular Flux Analyzer. While energy status can be measured on flash frozen tissues/cells, assays of mitochondria function must be performed with fresh tissue/cells or freshly isolated mitochondria. As a result, outside investigators can send cohorts of mice to the Core for all available analyses on live animals and tissues.
Alterations in mitochondrial function and increased mitochondrial generation of reactive oxygen species have long been implicated in the reduced capacity in cellular function that occurs with aging. In fact, two of the most popular theories in aging research, the oxidative stress theory of aging and the mitochondrial theory of aging, are based on this concept. The Core has optimized an array of assays to measure various aspects of mitochondrial function, including ROS generation, mitochondrial respiration, and ATP production. ATP production and H2O2 release are measured in isolated mitochondria using a luciferase/luciferin-based system and the fluorogenic probe, Amplex Red respectively, as previously described [3, 34]. Mitochondrial respiratory function is assessed in isolated mitochondria and permeabilized tissue using the Oroborus O2K high resolution respirometer which allows simultaneous analysis of mitochondrial respiration and ROS generation in tissues, cells suspension, or isolated mitochondria [1]. The Core has the capability to measure oxygen consumption rate (OCR) in cultured cells using the Seahorse Extracellular Flux Analyzer. Many laboratories have used this methodology to measure mitochondrial function in cells [7, 15]. The Seahorse Analyzer also contains a probe to measure acidification of the media and to measure the proton production rate or rate of extracellular acidification (ECAR) due to lactic acid production during glycolytic energy metabolism. By measuring OCR and ECAR simultaneously, one can get a more detailed picture of cellular energetic flux through these two pathways. This technique is a high throughput analysis that allows the Geroscience Redox Biology Core to measure the bioenergetic state and physiology of the cell without disruption of the cells.
GeroInformatics Core
Over the past decade, the number of data points per experiment that scientists are capable of gathering has grown exponentially. The importance of identifying and understanding patterns within this data has become more and more common for all areas of science, including aging. Analysis and interpretation of experimental data are the cornerstone of scientific progress; however, large-scale data provides unique challenges. NIH has called for increased rigor and reproducibility, which requires unbiased experimental design, methodology, analysis, interpretation, and reporting of results. Although investigators often master specific statistical and analytical techniques required for the most commonly performed experiments, they frequently have limited or no expertise in bioinformatics or statistics, which can be rate-limiting for their research. The GeroInformatics Core fills this gap by providing an established expertise in molecular biology and bioinformatics methods development. Especially important are the efforts of the Core to provide these services specifically for researchers in aging/geroscience and developing novel methods for interrogating and analyzing aging data bases.
Standard bioinformatic analyses
The GeroInformatics Core assists researchers with “standard” types of bioinformatics analyses for high-throughput sequencing data, such as alignment and QC of RNA-seq data, identification of differentially expressed (DE) genes, enriched GO categories and KEGG pathways, and ingenuity pathway analysis for analyses such as identifying potential upstream regulators. Although these are capabilities present in bioinformatics cores at other institutions, they are nonetheless services that remain in high demand and are often a good starting point for further data exploration. Besides receiving data from the other Cores in our Center for further analysis, we help outside investigators who have already gathered their own data. In addition, the Core is well-staffed to assist with statistical issues, such as pre-experimental statistical design of experiments to help investigators ensure that their experiment is sufficiently powered to detect the effects they are looking for, post-experimental statistical analysis, and the generation of figures and tables for publications. An example of how the GeroInformatics Core can assist investigators in analyzing their data is shown by our interaction with Dr. Kirkland who was searching for drugs (called “senolytics”) that can selectively kill senescent cells, in part by overcoming their resistance to apoptosis. Dr. Kirkland approached our Core to help him analyze the transcriptional data he generated after knocking down genes with siRNA. We conducted a multi-omics analysis of the subnetwork of genes that increase cellular susceptibility to apoptosis as shown in Fig. 8. HIF1A was identified as a gene highly interconnected with known senolytic targets [80]. HIF1A was subsequently validated in a later study to have senolytic activity [79].
Fig. 8.

Protein–protein interactions shared by known members of the BCL-2 family that can overcome cellular senescence. The size of the node correlates with the number of connections in the network. Yellow denotes known apoptotic proteins and the white nodes are highly connected on multiple omics levels. The arrow points to the node for HIF1A. Data from Zhu et al. [80]
Database searches
The GeroInformatics Core keeps up to date with widely used databases and software packages published to help with analysis of the most common data types that researchers in aging work with, e.g., mRNA, miRNA, genomic, proteomic, and metabolomics. In addition, the Core has developed several software packages, designed to condense, summarize, and identify patterns within large datasets. Briefly, the programs we have developed and how they can be helpful to investigators in the aging field are described below.
IRIDESCENT
The amount of scientific literature is vast (> 29 million papers in MEDLINE) and growing exponentially (~ 6%/year). With almost all journals offering online access to publications and supplementary data, the size of the average publication is also growing rapidly. The concept behind implicit relationship identification by software compilation of an entity-based network from text (IRIDESCENT), which Dr. Wren developed, is to condense this growing knowledge into conceptual association networks. It searches for “entities” (e.g., genes, diseases, chemicals, phenotypes, cell types, diseases) co-mentioned within publication abstracts and sentences, and weights them probabilistically on the basis of frequency and proximity. Once this network is constructed, Fig. 9 illustrates examples of how IRIDESCENT can be used to assist researchers in the analysis of the literature. Commonalities (B) are identified and ranked given two entities of interest (A and C). Figure 9b shows how novel associations can be discovered by starting with a concept of interest (A), for which all known literature relationships to A are compiled (A–B), and then all known relationships to B are then compiled (B–C) and, importantly, each implied relationship (C) is ranked for how specific the bridging concepts are based upon their connectivity in the network. Commercial recommendation systems (e.g., Netflix, Amazon) use similar mathematics to infer customer interest. Figure 9c shows an example of how upregulated genes in an RNAseq experiment are connected to two concepts of interest, cell adhesion and neutrophil adhesion (yellow), and to each other. Line width correlates with literature abundance, and node size correlates with the number of connections in this network. Blue nodes are genes associated with neutrophil adhesion, and white nodes are those only associated with a higher-level concept, cell adhesion. Therefore, IRIDESCENT allows experimental results to be both analyzed and visualized. These types of analyses help interpret observations, summarize how new experimental data fits into known (published) relationships, and prioritize long lists of potential findings.
Fig. 9.

Algorithmic analysis of the scientific literature. a Identifying which concepts/entities (B) connect two other concepts (A and C) in MEDLINE. b Identifying implied relationships by finding and assessing the strength of all A–B and B–C connections. c Visualizing how entities/concepts are connected in the literature
GAMMA
In 2019, ~ 53% of human transcripts (coding and non-coding) had not been mentioned in PubMed abstracts, indicating that they are either uncharacterized or poorly characterized (i.e., these genes have not been a significant focus or finding of scientific studies to date). Furthermore, ~ 10% of known genes account for 75% of gene name mentions in the literature, showing a strong scientific preference towards studying well-characterized genes, such that the rate of characterizing unannotated genes proceeds extremely slowly [11]. In fairness, when an investigator observes a well-characterized gene respond in their study, prior literature on it provides a wealth of potential hypotheses for how it affects their experimental system. Conversely, uncharacterized or poorly characterized genes make hypothesis formation challenging.
In 2009, global microarray meta-analysis (GAMMA), a program developed by the GeroInformatics Core, was developed to identify gene–gene correlations within NCBI’s publicly available GEO database [77]. Using a “guilt by association” approach for each transcript, it first identifies a set of the most highly correlated genes across all conditions and experiments analyzed. Then, statistically enriched commonalities for the correlated transcripts are identified using IRIDESCENT (and/or gene ontology), which then serves as the predicted functions, phenotypes, and genetic networks for every transcript. For known genes, computational analysis shows highly significant overlap between functions of the correlated genes and those of the known gene. The GeroInformatics Core initiated a 4- to 5-year project to systematically test GAMMA’s predictions experimentally, mostly via siRNA knockdowns in cell lines. We found that 48 out of 56 (86%) of the predicted phenotypes were validated experimentally, and this has resulted in 11 co-authored publications thus far, reporting the characterization of a formerly unknown genes [8, 10, 16–18, 32, 48, 70, 71]. Thus, GAMMA is now being used by investigators who have found an unknown or poorly known gene to be of interest in their research. Predicting phenotype, function, and pathway relevance, enables investigators to test specific hypotheses.
GenomeRunner.
Many studies focus on changes in the genome, regions that are usually not amenable to literature searches or to transcriptional analysis, for example, GWAS, ChIP-seq, and studies on methylation changes. To this end, the GeroInformatics Core developed the GenomeRunner program [12] and later deployed it as a freely available online web server [13]. GenomeRunner iteratively searches regions of interest for their overlap with genome track annotations and experimental results available from UCSC’s Genome Browser. In a Nature Genetics paper reporting the GWAS SNP’s associated with Sjogren’s syndrome, GenomeRunner was instrumental in identifying RFX5, an important transcription factor binding site enriched near the SNPs. We were able to show experimentally that several of the risk alleles significantly altered RFX5-dependent transcription [30]. The GeroInformatics Core also uses GenomeRunner to identify common genomic features, such as histone modification marks, and surrounding regions that displayed sexual dimorphism during aging in their methylation patterns [37].
Transcriptional Module Detection
To enable greater reproducibility, we can analyze differentially expressed (DE) transcripts from an investigator’s experiment by calculating how correlated the DE transcripts are in other, publicly available datasets. This system was used in a study on patients with an autoimmune syndrome, thrombotic thrombocytic purpura (TTP), which have flares versus those that do not [14]. Because the initial GO enrichment analysis only returned ribosome-related hits, it was puzzling as to how ribosome production could cause flare-ups, and therefore, the Core analyzed their correlations. The top 90 upregulated genes were all ribosome-related and highly correlated across other GEO microarray experiments. The bottom set of eight genes, however, are normally anti-correlated with the ribosomal genes in other experiments. Here, however, they are all upregulated in TTP and all significantly related to immune system activation. This result suggested that these eight genes are the most immune-relevant within the study and that their expression, plus increased ribosomal expression, is what characterizes the pathogenesis of TTP flares [14].
ALE
Automated label extraction (ALE) is a program the GeroInformatics Core recently developed to identify reported ages, sexes, and tissue types within the meta-data (author-written text regarding the experimental conditions and samples) of experiments in GEO [19] with reasonable precision and recall when run on over 861,000 samples. This enables one to conduct a meta-analysis on how (or if) transcription might change with age and, presuming enough samples exist, the analysis can be broken down by tissue type and sex. For example, only one gene (CD248) was common between two studies on the effect of age on the transcriptome for blood from human subjects [21, 43]. The GeroInformatics Core conducted our own meta-analysis of GEO data (not including the samples in either study) and found most of the genes had high variance across studies, suggesting cohort-dependency, except for CD248. We have used this meta-analytic approach to help investigators prioritize genes for further study on the transcriptional level and to identify potentially sex-dependent transcriptional changes.
Single cell transcriptomics
The ability to quantify transcripts that are present within a single cell (scRNAseq), like most new technologies, simultaneously creates exciting opportunities to answer new scientific questions but also presents challenges in data processing and analysis. This technology is especially important in aging research because changes in the relative proportion of cells in tissues or even appearance/disappearance of new cell populations with age (e.g., senescent cells, stem cells) could play an important role in tissue dysfunction, which would not be observed when using bulk RNAseq data. Currently, scRNAseq data is subjected to dimensionality reduction techniques, like principle component analysis (PCA), but more commonly t-stochastic neighbor embedding (t-SNE) to visualize clusters, and t-SNE has seen increased use since its introduction [33] because of its superior ability (relative to other algorithms) to find structure within high-dimensional data. In addition, t-SNE is very helpful when one knows beforehand how many clusters to expect, but unfortunately less helpful when one does not. In any “discovery” type of experiment where little or nothing is known, at least two very important questions remain unresolved: (1) for every cluster identified, what (if anything) could be said about its biological significance? (2) How could the biological differences (if any) between clusters be identified and evaluated statistically?
The GeroInformatics Core is currently working on two approaches to help with interpretive analysis of scRNAseq data. First, we are developing a transcriptional similarity analysis for clusters — similar to existing approaches such as CMAP [29], LINCS [31], and ExpressionBLAST [81], which compare transcriptional profiles. Our analysis will do something similar, except using scRNAseq data. This analysis would enable a researcher to approximate what cell types a cluster might contain if the answer is not already known and similar expression patterns were found in another experiment. It would also help approximate if any potentially novel cell types are present, on the basis of having transcriptional profiles unlike those reported in prior data. Second, we are developing a cluster-specific gene ontology (GO) differential enrichment analysis, which will use weighted centroids from each cluster as “representative” of the group and search for enriched GO categories by using the genes that best define the difference between the two clusters. If no significant enrichments are found between any clusters, it would suggest that the t-SNE parameters may not be optimal. If two nearby clusters have no difference, it suggests there is probably only one cluster instead of two. When cluster differences are reflected by significant ontology differences, it provides researchers with a means to both categorize the clusters (e.g., “inflamed” vs “non-inflamed” epithelial cells) and engage in further hypothesis testing.
Geropathology Research Resource
It is widely acknowledged that pathological information is an important component of animal studies focused on aging. Lifespan data without pathological data limits insight into the aging process [5], which becomes critical when aging interventions are studied. It is difficult to conclude from survival data alone if changes in lifespan of an animal arise because aging has been altered by the experimental manipulation, e.g., are a broad spectrum of disease processes or pathological lesions modified, which is predicted if underlying mechanisms of aging have been altered. Currently, pathological analyses of aging rodents have focused on identifying the number of mice with specific pathological lesions and in some cases scoring the severity of the lesions. However, the types of lesions that occur and the tissues in which they occur vary greatly from animal to animal making it difficult to statistically determine if a significant change occurs in each lesion without a large number of animals per group. This problem is largely eliminated by the Geropathology Grading Platform (GGP), which was recently developed by the Geropathology Grading Committee. This Committee was chaired by Dr. Ladiges and composed of a group of board-certified veterinary pathologists, including Dr. Snider, the Leader of the Geropathology Research Resource. The Geropathology Grading Committee was part of the NIA-funded Geropathology Research Network, which had the goal of developing a grading system that would allow investigators to assess the pathological status of a wide range of tissues in old mice [25–27], and validating the grading system [64]. The Geropathology Grading Committee evaluated a large number of mice at various ages to identify the types of pathological lesions that changed with age and could be reliably used in the GGP. The GGP is based on a standardized set of guidelines to (1) detect the presence or absence of low-impact histopathological lesions that occur with age and (2) determine the level of severity of high-impact lesions in tissues of aged mice. The GGP allows one to generate a numerical score for each lesion in a tissue, which is then summed to give a score for the total lesions in the tissue that are averaged for the mice in the cohort such that a composite lesion score for that tissue as well as a composite lesion score for the whole animal can be obtained. In other words, the composite lesion score gives one a numerical value for the burden of lesions, which can be compared to similar values from mice from other cohorts and be used to measure changes in pathological status with age or with an aging intervention.
Ladiges et al. [25] demonstrated that the composite lesion score for the heart, lung, kidney, and liver increased dramatically with age in two strains of mice. They also reported that rapamycin treatment, which has been shown to increase the lifespan of mice, reduced the composite lesion score in multiple tissues. In a project with faculty at the University of Oklahoma Health Sciences Center, Dr. Snider evaluated the pathological status of mice lacking Cu/Zn-superoxide dismutase (Sod1−/− mice) that show accelerated aging (Deepa et al., 2017). As shown in Fig. 10, the whole body composite lesion score was 2- to 3.5-fold higher for Sod1−/− mice compared wild type mice, demonstrating that the GGP predicted the accelerated aging phenotype observed in the Sod1−/− mice . These data also suggest that that GGP can be used to assess the health span of mice because the increase in the composite lesion score for the whole animal mirrors the decline in physiological functions that were observed in Sod1−/− mice. Thus, the GGP is potentially a new tool for evaluating the effect of an intervention on the pathological status of an animal, which can also give insight into the health span of the mice.
Fig. 10.
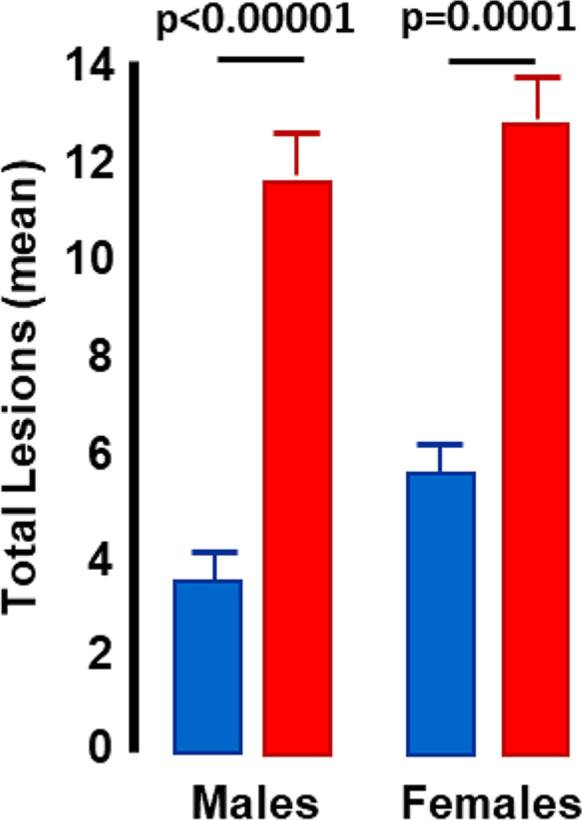
Geropathology analysis of wild type and Sod1−/− mice. Whole body geropathology composite scores are shown for male and female wild type (blue bars) and Sod1−/− (red bars) mice at 9 to 10 months of age. Data taken from Snider et al. [61]
The Geropathology Research Resource provides investigators studying aging across the country with the geropathological analysis of mice using the newly developed GGP or standard pathology analyses. Dr. Snider is an active member of the Geropathology Grading Committee and, therefore, is in a position to add new tissues to the pathological analysis when the Geropathology Grading Committee develops scoring system for these tissues. The pathological analysis of mice will be conducted as described by Snider et al. [61]. For example, the carcasses of the mice will be collected immediately after death by the investigators at their home institutions and the whole body immersion-fixed in 10% buffered neutral formalin. The carcasses in formalin will be shipped to the geropathology resource where the tissues will be collected and processed on a fee for service basis, e.g., paraffin embedded, sectioned via microtomy at 4 microns, stained with hematoxylin and eosin, and cover-slipped. The slides will be labeled by mouse number and group number; however, identifying details of groups will be blinded to the scoring pathologists until the analysis is completed. It is also possible for investigators to provide the Geropathology Research Resource directly with slides from processed tissues.
Funding
The services described above are supported by the Oklahoma Nathan Shock Center P30 AG050911 grand and Senior Career Research Awards 1IK6BX005238 (AR) and IK6BX005234 (HVR) from the Department of Veterans Affairs.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ahn B, Smith N, Saunders D, Ranjit R, Kneis P, Towner RA, Van Remmen H. Using MRI to measure in vivo free radical production and perfusion dynamics in a mouse model of elevated oxidative stress and neurogenic atrophy. Redox Biol. 2019;26:101308. [DOI] [PMC free article] [PubMed]
- 2.Baker M. Reproducibility crisis: blame it on the antibodies. Nature. 2015;521:274–276. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya A, Muller FL, Liu Y, Sabia M, Liang H, Song W, Jang YC, Ran Q, Van Remmen H. Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J Biol Chem. 2009;284:46–55. doi: 10.1074/jbc.M806311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Booth LN, Brunet A. The aging epigenome. Mol Cell. 2016;62:728–744. doi: 10.1016/j.molcel.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bronson T, Lipman RD. The role of pathology in rodent experimental gerontology. Aging (Milano) 1993;5:253–257. doi: 10.1007/BF03324169. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri AR, de Waal EM, Pierce A, Van Remmen H, Ward WF, Richardson A. Detection of protein carbonyls in aging liver tissue: a fluorescence-based proteomic approach. Mech Ageing Dev. 2006;127:849–861. doi: 10.1016/j.mad.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Chacko BK, Kramer PA, Ravi S, Johnson MS, Hardy RW, Ballinger SW, Darley-Usmar VM. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013;93:690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clemmensen SN, Bohr CT, Rorvig S, Glenthoj A, Mora-Jensen H, Cramer EP, Jacobsen LC, Larsen MT, Cowland JB, Tanassi JT, et al. Olfactomedin 4 defines a subset of human neutrophils. J Leukocyte Biol. 2012;91:495–500. doi: 10.1189/jlb.0811417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coutinho de Souza P, Smith N, Atolagbe O, Ziegler J, Nijoku C, Lerner M, Ehrenshaft M, Mason RP, Meek B, Plafker SM, Saunders D, Mamedova N, Towner RA. OKN-007 decreases free radicals levels in a preclinical F98 rat glioma model. Free Radical Biol Med. 2015;87:157–168. doi: 10.1016/j.freeradbiomed.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, Gorbsky GJ. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Current Biol. 2009;19:1467–1472. doi: 10.1016/j.cub.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolgin E. The most popular genes in the human genome. Nature. 2017;551:427–431. doi: 10.1038/d41586-017-07291-9. [DOI] [PubMed] [Google Scholar]
- 12.Dozmorov MG, Cara LR, Giles CB, Wren JD. GenomeRunner: automating genome exploration. Bioinformatics. 2012;28:419–420. doi: 10.1093/bioinformatics/btr666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dozmorov MG, Cara LR, Giles CB, Wren JD. GenomeRunner web server: regulatory similarity and differences define the functional impact of SNP sets. Bioinformatics. 2016;32:2256–2263. doi: 10.1093/bioinformatics/btw169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar CE, Terrell DR, Vesely SK, Wren JD, Dozmorov IM, Niewold TB, Brown M, Zhou F, Frank MB, Merrill JT, et al. Ribosomal and immune transcripts associate with relapse in acquired ADAMTS13-deficient thrombotic thrombocytopenic purpura. PloS One. 2015;10:e0117614. [DOI] [PMC free article] [PubMed]
- 15.Espinosa JA, Pohan G, Arkin MR Markossian S. Real-time assessment of mitochondrial toxicity in HepG2 cells using the Seahorse Extracellular Flux Analyzer. Curr Protoc. 2021;1:e75. [DOI] [PubMed]
- 16.Fields E, Wren JD, Georgescu C, Daum JR, Gorbsky GJ. Predictive bioinformatics identifies novel regulators of proliferation in a cancer stem cell model. Stem Cell Res. 2018;26:1–7. doi: 10.1016/j.scr.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisch AS, Yerges-Armstrong LM, Backman JD, Wang H, Donnelly P, Ryan KA, Parihar A, Pavlovich MA, Mitchell BD, O’Connell JR, et al. Genetic variation in the platelet endothelial aggregation receptor 1 gene results in endothelial dysfunction. PloS One. 2015;10:e0138795. [DOI] [PMC free article] [PubMed]
- 18.Gandhapudi SK, Tan C, Marino JH, Taylor AA, Pack CC, Gaikwad J, Van De Wiele CJ, Wren JD, Teague TK. IL-18 acts in synergy with IL-7 to promote ex vivo expansion of T lymphoid progenitor cells. J immunol. 2015;194:3820–3828. doi: 10.4049/jimmunol.1301542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giles CB, Brown CA, Ripperger M, Dennis Z, Roopnarinesingh X, Porter H, Perz A, Wren JD. ALE: automated label extraction from GEO metadata. BMC Bioinformatics. 2017;18:509. [DOI] [PMC free article] [PubMed]
- 20.Gonzalez-Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P, Ferrucci L. Reconsidering the role of mitochondria in aging. J Gerontol A Biol Sci Med Sci. 2015;70:1334–1342. doi: 10.1093/gerona/glv070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harries LW, Hernandez D, Henley W, Wood AR, Holly AC, Bradley-Smith RM, Yaghootkar H, Dutta A, Murray A, Frayling TM, et al. Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell. 2011;10:868–878. doi: 10.1111/j.1474-9726.2011.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haque A, Engel J, Teichmann SA, Lonnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9:75–87. doi: 10.1186/s13073-017-0467-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasumov T, Dabkowski ER, Shekar KC, Li L, Ribeiro RF, Walsh K, Previs SF, Sadygov RG, Willard B, Stanley WC. Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2013;304:H1201–H1214. doi: 10.1152/ajpheart.00933.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ladiges W, Ikeno Y, Niedernhofer L, McIndoe RA, Ciol MA, Ritchey J, Liggitt D. The Geropathology Research Network: an interdisciplinary approach for integrating pathology into research on aging. J Gerontol A Biol Sci Med. Sci. 2016;71:431–434. doi: 10.1093/gerona/glv079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ladiges W, Snyder JM, Wilkinson E, Imai DM, Snider T, Ge X, Ciol M, Pettan-Brewer C, Pillai SPS, Morton J, Quarles E, Rabinovitch P, Niedernhofer L, Liggitt D. A new preclinical paradigm for testing anti-aging therapeutics. J Gerontol A Biol Sci Med Sci. 2017;72:760–762. doi: 10.1093/gerona/glx019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ladiges W. The emerging role of geropathology in preclinical aging studies. Pathobiol Aging Age Relat Dis. 2017b;7:1304005. [DOI] [PMC free article] [PubMed]
- 28.Lam MPY, Wang D, Lau E, Liem DA, Kim AK, Ng DCM, Liang X, Bleakley BJ, Liu C, Tabaraki JD, et al. Protein kinetic signatures of the remodeling heart following isoproterenol stimulation. J Clin Invest. 2014;124:1734–1744. doi: 10.1172/JCI73787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 30.Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, Kelly JA, Dozmorov MG, Miceli-Richard C, Bowman S, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren’s syndrome. Nat Genet. 2013;45:1284–1292. doi: 10.1038/ng.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu C, Su J, Yang F, Wei K, Ma J, Zhou X. Compound signature detection on LINCS L1000 big data. Mol Biosyst. 2015;11:714–722. doi: 10.1039/c4mb00677a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lupu C, Zhu H, Popescu NI, Wren JD, Lupu F. Novel protein ADTRP regulates TFPI expression and function in human endothelial cells in normal conditions and in response to androgen. Blood. 2011;118:4463–4471. doi: 10.1182/blood-2011-05-355370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maaten L, Hinton G. Visualizing data using t-SNE. J Machine Learning Res. 2008;9:2579–2605. [Google Scholar]
- 34.Mansouri A, Muller FL, Liu Y Ng, R, Faulkner J, Hamilton M, Richardson A, Huang TT, Epstein CJ, Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev. 2006;127:298–306. [DOI] [PubMed]
- 35.Masser DR, Berg AS, Freeman WM. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenetics Chromatin. 2013;6:33. [DOI] [PMC free article] [PubMed]
- 36.Masser DR, Hadad N, Porter H, Stout MB, Unnikrishnan A, Stanford DR, Freeman WM. Analysis of DNA modifications in aging research. Geroscience. 2018;40:11–29. doi: 10.1007/s11357-018-0005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masser DR, Hadad N, Porter HL, Mangold CA, Unnikrishnan A, Ford MM, Giles CB, Georgescu C, Dozmorov MG, Wren JD, et al. Sexually divergent DNA methylation patterns with hippocampal aging. Aging Cell. 2017;16:1342–1352. doi: 10.1111/acel.12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathis AD, Naylor BC, Carson RH, Evans E, Harwell J, Knecht J, Hexem E, Peelor FF, Miller BF, Hamilton KL, et al. Mechanisms of in vivo ribosome maintenance change in response to nutrient signals. Mol Cell Proteomics. 2017;16:243–254. doi: 10.1074/mcp.M116.063255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller BF, Pharaoh GA, Hamilton KL, Peelor FF, Kirkland JL, Freeman WM, Mann SN, Kinter M, Price JC, Stout MB. Short-term calorie restriction and 17α-estradiol administration elicit divergent effects on proteostatic processes and protein content in metabolically active tissues. J Gerontol a Biol Sci Med Sci. 2020;75:849–857. doi: 10.1093/gerona/glz113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morrow JD, Roberts LJ. Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- 41.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;15:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 42.Muller FL, Song W, Jang Y, Liu Y, Sabia M, Richardson A, Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura S, Kawai K, Takeshita Y, Honda M, Takamura T, Kaneko S, Matoba R, Matsubara K. Identification of blood biomarkers of aging by transcript profiling of whole blood. Biochem Biophys Res Commun. 2012;418:313–318. doi: 10.1016/j.bbrc.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 44.Naylor BC, Porter MT, Wilson E, Herring A, Lofthouse S, Hannemann A, Piccolo SR, Rockwood AL, Price JC. DeuteRater: a tool for quantifying peptide isotope precision and kinetic proteomics. Bioinformatics. 2017;33:1514–1520. doi: 10.1093/bioinformatics/btx009. [DOI] [PubMed] [Google Scholar]
- 45.Olga B, Virolainen E, Fagerstedt KV. Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot. 2003;91:179–194. doi: 10.1093/aob/mcf118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park HJ, Mah E, Bruno RS. Validation of high-performance liquid chromatography-boron-doped diamond detection for assessing hepatic glutathione redox status. Anal Biochem. 2010;407:151–159. doi: 10.1016/j.ab.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piccini A, Castroflorio E, Valente P, Guarnieri FC, Aprile D, Michetti C, Bramini M, Giansante G, Pinto B, Savardi A, et al. APache is an AP2-interacting protein involved in synaptic vesicle trafficking and neuronal development. Cell Rep. 2017;21:3596–3611. doi: 10.1016/j.celrep.2017.11.073. [DOI] [PubMed] [Google Scholar]
- 49.Pierce A, deWaal E, Van Remmen H, Richardson A, Chaudhuri A. A novel approach for screening the proteome for changes in protein conformation. Biochemistry. 2006;45:3077–3085. doi: 10.1021/bi052031i. [DOI] [PubMed] [Google Scholar]
- 50.Price JC, Khambatta CF, Li KW, Bruss MD, Shankaran M, Dalidd M, Floreani NA, Roberts LS, Turner SM, Holmes WE, et al. The effect of long term calorie restriction on in vivo hepatic proteostatis: a novel combination of dynamic and quantitative proteomics. Mol Cel Proteomics. 2012;11:1801–1814. doi: 10.1074/mcp.M112.021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;57:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rebrin I, Bregere C, Gallaher TK, Sohal RS. Detection and characterization of peroxynitrite-induced modifications of tyrosine, tryptophan, and methionine residues by tandem mass spectrometry. Methods Enzymol. 2008;441:283–294. doi: 10.1016/S0076-6879(08)01215-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rindler PM, Cacciola A, Kinter M, Szweda LI. Catalase-dependent H2O2 consumption by cardiac mitochondria and redox-mediated loss in insulin signaling. Am J Physiol Heart Circ Physiol. 2016;311:H1091–H1096. doi: 10.1152/ajpheart.00066.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts LJ, Morrow JD. The isoprostanes: novel markers of lipid peroxidation and potential mediators of oxidant injury. Adv Prostaglandin Thromboxane Leukot Res. 1995;23:219–224. [PubMed] [Google Scholar]
- 55.Roberts LJ, Morrow JD. Measurement of F(2)-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med. 2000;28:505–513. doi: 10.1016/s0891-5849(99)00264-6. [DOI] [PubMed] [Google Scholar]
- 56.Sadygov RG, Avva J, Rahman M, Lee K, Ilchenko S, Kasumov T, Borzou A. d2ome, software for in vivo protein turnover analysis using heavy water labeling and LC-MS, reveals alterations of hepatic proteome dynamics in a mouse model of NAFLD. J Proteome Res. 2018;17:3740–3748. doi: 10.1021/acs.jproteome.8b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samarel AM. In vivo measurements of protein turnover during muscle growth and atrophy. FASEB J. 1991;5:2020–2028. doi: 10.1096/fasebj.5.7.2010055. [DOI] [PubMed] [Google Scholar]
- 58.Sataranatarajan K, Pharaoh G, Brown JL, Ranjit R, Piekarz KM, Street K, Wren JD, Gorgescu C, Kinter C, Kinter M, Freeman WM, Richardson A, Van Remmen H. Molecular changes in transcription and metabolic pathways underlying muscle atrophy in the CuZnSOD null mouse model of sarcopenia. GeroScience. 2020;42:1101–1118. doi: 10.1007/s11357-020-00189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sevini F, Giuliani C, Vianello D, Giampieri E, Santoro A, Biondi F, Garagnani P, Passarino G, Luiselli D, Capri M, et al. mtDNA mutations in human aging and longevity: controversies and new perspectives opened by high-throughput technologies. Exp Gerontol. 2014;56:234–244. doi: 10.1016/j.exger.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 60.Sies H, Berndt C, Jones DP. Oxidative stress. Ann Rev. Biochem. 2017;86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 61.Snider TA, Richardson A, Stoner JA, Deepa SS. The Geropathology Grading Platform demonstrates that mice null for Cu/Zn-superoxide dismutase show accelerated biological aging. Geroscience. 2018;40:97–103. doi: 10.1007/s11357-018-0008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song R, Sarnoski EA, Acar M. The systems biology of single-cell aging. iScience. 2018;7:154–169. doi: 10.1016/j.isci.2018.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 64.Snyder JM, Snider TA, Ciol MA, Wilkinson JE, Imai DM, Casey KM, Vilches-Moure JG, Pettan-Brewer C, Pillai SPS, Carrasco SE, Salimi S, Ladiges W. Validation of a geropathology grading system for aging mouse studies. Geroscience. 2019;42:455–465. doi: 10.1007/s11357-019-00088-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanay A, Regev A. Scaling single-cell genomics from phenomenology to mechanism. Nature. 2017;541:331. [DOI] [PMC free article] [PubMed]
- 66.Towner RA, Smith N, Saunders D, Henderson M, Downum K, Lupu F, Silasi-Mansat R, Ramirez DC, Gomez-Mejiba SE, Bonini MG, Ehrenshaft M, Mason RP. In vivo imaging of immune-spin trapped radicals with molecular MRI in a mouse diabetes model. Diabetes. 2012;61:2405–2413. doi: 10.2337/db11-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Towner RA, Smith N, Saunders D, Lupu F, Silasi-Mansat R, West M, Ramirez DC, Gomez-Mejiba SE, Bonini MG, Mason RP, Ehrenshaft M, Hensley K. In vivo detection of free radicals using molecular MRI and immuno-spin-trapping in a mouse model for amyotrophic lateral sclerosis (ALS) Free Radic Biol Med. 2013;63:351–360. doi: 10.1016/j.freeradbiomed.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 68.Towner RA, Smith N, Saunders D, De Souza PC, Henry L, Lupu F, Silasi-Mansat R, Ehrenshaft M, Mason RP, Gomez-Mejiba SE, Ramirez DC. Combined molecular MRI and immuno-spin-trapping for in vivo detection of free radicals in orthotopic mouse GL261 gliomas. Biochim Biophys Acta. 2013;1832:2153–2161. doi: 10.1016/j.bbadis.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 69.Towner RA, Garteiser P, Bozza F, Smith N, Saunders D, d’Avila JCP, Magno F, Oliveira MF, Ehrenshaft M, Lupu F, Silasi-Mansat R, Ramirez DC, Gomez-Mejiba SE, Mason RP, Faria-Neto HCC. In vivo detection of free radicals in mouse septic encephalopathy using molecular MRI and immuno-spin-trapping. Free Radica Biol Med. 2013;65:828–837. doi: 10.1016/j.freeradbiomed.2013.08.172. [DOI] [PubMed] [Google Scholar]
- 70.Towner RA, Jensen RL, Colman H, Vaillant B, Smith N, Casteel R, Saunders D, Gillespie DL, Silasi-Mansat R, Lupu F, et al. ELTD1, a potential new biomarker for gliomas. Neurosurgery. 2013;72:77–90. doi: 10.1227/NEU.0b013e318276b29d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Towner RA, Jensen RL, Vaillant B, Colman H, Saunders D, Giles CB, Wren JD. Experimental validation of 5 in-silico predicted glioma biomarkers. Neuro Oncol. 2013;15:1625–1634. doi: 10.1093/neuonc/not124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Towner RA, Smith N, Saunders D, Carrizales J, Lupu F, Silasi-Mansat R, Ehrenshaft M, Mason RP. In vivo targeted molecular magnetic resonance imaging of free radicals in diabetic cardiomyopathy within mice. Free Radic Res. 2015;49:1140–1146. doi: 10.3109/10715762.2015.1050587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Towner RA, Smith N. In vivo and in situ detection of macromolecular free radicals using immuno-spin trapping and molecular MRI. Antioxid Redox Signal. 2018;28:1404–1415. doi: 10.1089/ars.2017.7390. [DOI] [PubMed] [Google Scholar]
- 74.Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, Richardson A. The role of DNA methylation in epigenetics of aging. Pharmacol Ther. 2019;195:172–185. doi: 10.1016/j.pharmthera.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ward WF, Qi W, Van Remmen H, Zackert WE, Roberts LJ, Richardson A. Effects of age and caloric restriction on lipid peroxidation: measurement of oxidative stress by F2-isoprostane levels. J Gerontol A Biol Sci Med Sci. 2005;60:847–851. doi: 10.1093/gerona/60.7.847. [DOI] [PubMed] [Google Scholar]
- 76.Wilkinson DJ, Brook MS, Smith K, Atherton PJ. Stable isotope tracers and exercise physiology: past, present and future. J Physiol. 2017;595:2873–2882. doi: 10.1113/JP272277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wren JD. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics. 2009;25:1694–1701. doi: 10.1093/bioinformatics/btp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed]
- 79.Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, Kirkland JL. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 2017;9:955–963. doi: 10.18632/aging.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zinman GE, Naiman S, Kanfi Y, Cohen H, Bar-Joseph Z. ExpressionBlast: mining large, unstructured expression databases. Nat Methods. 2013;10:925–926. doi: 10.1038/nmeth.2630. [DOI] [PubMed] [Google Scholar]
- 82.Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, Leonhardt H, Heyn H, Hellmann I, Enard W. Comparative analysis of single-cell RNA sequencing methods. Mol Cell. 2017;65:631–643. doi: 10.1016/j.molcel.2017.01.023. [DOI] [PubMed] [Google Scholar]