

ABSTRACT
Heat-shock protein 90 (Hsp90) is a central regulator of cellular proteostasis. It stabilizes numerous proteins that are involved in fundamental processes of life, including cell growth, cell-cycle progression and the environmental response. In addition to stabilizing proteins, Hsp90 governs gene expression and controls the release of cryptic genetic variation. Given its central role in evolution and development, it is important to identify proteins and genes that interact with Hsp90. This requires sophisticated genetic and biochemical tools, including extensive mutant collections, suitable epitope tags, proteomics approaches and Hsp90-specific pharmacological inhibitors for chemogenomic screens. These usually only exist in model organisms, such as the yeast Saccharomyces cerevisiae. Yet, the importance of other fungal species, such as Candida albicans and Cryptococcus neoformans, as serious human pathogens accelerated the development of genetic tools to study their virulence and stress response pathways. These tools can also be exploited to map Hsp90 interaction networks. Here, we review tools and techniques for Hsp90 network mapping available in different fungi and provide a summary of existing mapping efforts. Mapping Hsp90 networks in fungal species spanning >500 million years of evolution provides a unique vantage point, allowing tracking of the evolutionary history of eukaryotic Hsp90 networks.
Keywords: protein–protein interactions, proteomics, chemogenomics, Hsp90, synthetic lethality, mutant libraries, Candida, Cryptococcus neoformans, Aspergillus fumigatus
Hsp90 networks in fungi provide a unique opportunity for the study of network evolution.
INTRODUCTION
Proteins facilitate diverse cellular processes, such as metabolism, the electron transport chain and movement of organelles. It is, thus, critical that proteins remain folded and stable, yet they are inherently unstable. Their folding is maintained by the equivalent energy of approximately three hydrogen bonds, meaning they exist on a thermodynamic knife-edge. As such they are vulnerable to denaturation or aggregation, especially in the crowded cellular environment (Ellis 2001) or when cells are experiencing environmental stress. In response to these challenges, chaperone proteins evolved to assist with protein folding and stabilization, thereby maintaining proteostasis and ultimately ensuring survival of the organism.
Hsp90 is an essential chaperone and evolutionary capacitor
The highly conserved and ubiquitous molecular chaperone Heat-shock protein 90 (Hsp90) was first described in the 1970s as a protein of approximately 90 kDa that was significantly upregulated during heat stress in Drosophila cell culture and salivary gland tissue (Moran et al. 1978). Subsequently, orthologs have been identified in eubacteria and all eukaryotes but not archaebacteria (Stechmann and Cavalier-Smith 2004). Hsp90 protein sequences are 60% identical between human and the model eukaryote Saccharomyces cerevisiae. Mammalian Hsp90 can complement the otherwise lethal phenotype of Hsp90 depletion in S. cerevisiae (Minami et al. 1994; Nathan, Harju Vos and Lindquist 1999). Hsp90 is amongst the 20 most abundant proteins in the eukaryotic cell (Ghaemmaghami et al. 2003) and in response to thermal stress and other environmental stimuli, Hsp90 levels increase further (Taipale, Jarosz and Lindquist 2010). Lack of Hsp90 results in severe susceptibility to elevated temperatures (Borkovich et al. 1989). Under both non-stress and stress conditions, the chaperone functions by stabilizing a specific set of proteins, called target proteins or clients. Hsp90 forms a complex with its client until the client reaches its cellular destination or is required in its active state. At this point, the chaperone cycle is completed, and the mature client released (Zuehlke and Johnson 2010). Hsp90 is involved in numerous fundamental processes such signal transduction, cell growth and cellular differentiation in the eukaryotic cell (Zhao et al. 2005; Taipale et al. 2012).
Beyond functioning as a traditional chaperone, Hsp90 acts as an evolutionary capacitor. As such, it facilitates the storage and release of genetic variation. Under non-stress conditions, Hsp90 correctly folds its clients, masking any mutations their DNA sequences may accumulate because Hsp90 recognizes the 3D-structure of partially folded proteins rather than their sequence. Depleting Hsp90 either genetically or pharmacologically, however, removes the evolutionary buffer and causes clients to fold based solely on their amino acid sequence, causing the mutations to be expressed. Consequently, aberrations in plant (Queitsch, Sangster and Lindquist 2002) and fly (Rutherford and Lindquist 1998) morphology, as well as eye size in cave fish (Rohner et al. 2013) can be detected. In fungi, reducing Hsp90 function abolishes antifungal drug resistance (Cowen and Lindquist 2005). Hsp90’s ability to buffer a multitude of traits is due to its capacity to control expression of ∼20% of the pre-existing genetic variation (Jarosz and Lindquist 2010).
In addition to chaperoning client proteins, Hsp90 regulates gene expression in evolutionarily diverse organisms. For example, mammalian circadian clock genes (Schneider, Linka and Reinke 2014), plant phytohormone genes (Shigeta et al. 2015) and fungal stress-responsive kinase genes (Diezmann et al. 2012) require Hsp90 for expression. Due to Hsp90’s central role in fundamental cellular processes and its ability to shape evolutionary trajectories, it is critical to identify Hsp90 interactors. Elucidating Hsp90 interaction networks, of either direct or indirect nature, would allow for a comprehensive understanding of the protein complexes and cellular pathways Hsp90 is involved in. Yet, mapping Hsp90 networks in eukaryotes is technically challenging and requires extensive genetic and biochemical tools and techniques, such as genome-scale mutant libraries. Until recently these were only available in S. cerevisiae. Yet, with the development of molecular biology tools in other fungal species, Hsp90 interaction networks can now be mapped in a diverse range of fungi.
Fungi represent an opportunity to study Hsp90 function and interaction networks in an evolutionary context
Fungi are ancient. Fungal microfossils from the Canadian Northwest Territories date back to ∼1 billion years (Loron et al. 2019). Thus, fungi evolved before land plants, which emerged ∼470 million years ago on the fossil record (Edwards et al. 2014). Fungal species for which tools required for Hsp90 network mapping exist, span >500 million years of evolution (Fig. 1). Being able to map Hsp90 interaction networks in multiple fungal species across evolutionary time scales allows for powerful comparisons of how Hsp90 networks evolved, their evolutionary trajectories and the impact of ecology on network evolution. Within the subphylum Saccharomycotina, S. cerevisiae and Candida glabrata diverged ∼50 million years ago. They split from the ancestor of Candidaalbicans and Candida parapsilosis ∼250 million years ago. The Saccharomycotina diverged from the Pezizomycotina, the sub-phylum containing moulds such as Aspergillus fumigatus, ∼400 million years ago and the Dikarya, the group formed by the Ascomycota and the Basidiomycota, emerged ∼560 million years ago (Beimforde et al. 2014; Shen et al. 2020). Mutant libraries, required for Hsp90 genetic interaction network mapping, have been engineered in C. albicans, C. parapsilosis, C. glabrata, A. fumigatus and the basidiomycete Cryptococcus neoformans with the aim to comprehensively identify and study virulence factors. They also provide the unique opportunity to study Hsp90 network evolution.
Figure 1.
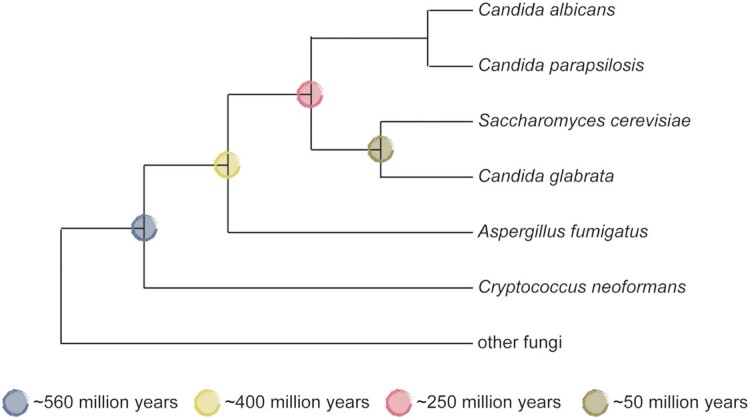
Phylogenetic relationships amongst fungal species with available mutant libraries. Divergence times for branches leading to C. albicans, S. cerevisiae and C. glabrata (all Saccharomycotina), A. fumigatus (Pezizomycotina) and C. neoformans (Basidiomycota) are indicated by colored circles.
Hsp90 in S. cerevisiae
Saccharomyces cerevisiae has been instrumental in understanding eukaryotic Hsp90 function and regulation. Unlike other fungi, S. cerevisiae expresses two isoforms of Hsp90. One, Hsc82, is constitutively expressed at very high levels, and the other, Hsp82, is expressed at much lower levels but is strongly induced in response to heat shock. Expression of either gene is essential for growth, however, each isoform has distinct functions and clients (Borkovich et al. 1989; Girstmair et al. 2019). Crystalizing full-length S. cerevisiae Hsp90 together with an ATP analogue and a co-chaperone revealed the complex architecture of the clamp-like structure of the chaperone and the intricate conformational changes required to execute chaperone function (Ali et al. 2006). Due to its ease of manipulation, S. cerevisiae has been extensively used to identify and characterize the role of post-translational modifications in chaperone regulation. For more specific examples see this review by Mollapour and Neckers (2012). The phosphorylation site essential for survival of high temperatures was initially detected and characterized in S. cerevisiae (Nathan and Lindquist 1995).
Hsp90 in C. albicans
Amongst pathogenic fungi, Hsp90 is best understood in C. albicans. This yeast causes ∼750 000 life-threatening invasive infections world-wide each year with mortality rates of up to 75% (Brown et al. 2012; Bongomin et al. 2017). In addition to this burden on human life and health, candidemia adds substantially to health care costs. The United States alone spent $1.4 billion on over 26 K hospitalizations necessitated by candidemia in 10 years (Benedict et al. 2019). High mortality rates and extended hospital stays are due to the currently available treatments being rather ineffective or the emergence of antifungal drug resistance. It is, thus, imperative to understand C. albicans virulence mechanisms and stress response pathways to develop more efficacious treatment strategies. Hsp90 is a key regulator of C. albicans virulence (Cowen et al. 2009), morphogenesis (Shapiro et al. 2009), drug resistance in planktonic cells (Singh et al. 2009) and biofilms (Robbins et al. 2011) as well as cell cycle progression (Senn, Shapiro and Cowen 2012). Despite promising results in insect models of fungal disease, targeting Hsp90 with inhibitors tested in clinical trials as anticancer drugs resulted in severe host toxicity in mice with candidemia. Due to the high sequence conservation between fungal Hsp90 and the mammalian ortholog, both othologs were inhibited, which caused complications for the host (Cowen et al. 2009). Hsp90 itself is therefore not suitable as an antifungal drug target until fungal-specific inhibitors of Hsp90 exist. Hence, alternative strategies need to be explored and Hsp90 interactors could prove useful as future drug targets. They could either be targeted by monotherapy or in combination therapy with already available Hsp90 inhibitors.
Hsp90 in C. parapsilosis
Candida albicans’ relative C. parapsilosis causes 33% of candidemia infections in pre-term infants with mortality rates of 10% (Pammi et al. 2013). Candida parapsilosis infections can be difficult to treat due to reduced susceptibility to the echinocandin class of antifungals. This is caused by a naturally occurring amino acid substitution in the protein encoding the echinocandin target Fks1 relative to other Candida species (Garcia-Effron et al. 2008). Similar to C. albicans, Hsp90 represses filamentation in C. parapsilosis (Hossain, Veri and Cowen 2020) and the combination of triazoles and the Hsp90 inhibitor, geldanamycin, acts synergistically, reducing minimum inhibitory concentrations to triazoles (Mahmoudi et al. 2019). Beyond this, Hsp90’s role in virulence in this pathogen of premature infants remains uncharacterized.
Hsp90 in C. glabrata
Although closely related to S. cerevisiae, comparatively little is known about Hsp90 in C. glabrata. This yeast is the leading cause of non-albicans candidiasis in Northern Europe and the United States of America, and the third or fourth in Asia (Kumar et al. 2019). Commensal strains found in the oral cavity or gut are often the causative agents in clinical C. glabrata infections (Pfaller et al. 2002; Wang et al. 2013; Guinea 2014; Khatib et al. 2016; Nash et al. 2017). Candida glabrata employs a suite of virulence traits that facilitate infection of humans including surface adhesion, biofilm production, tissue invasion, macrophage survival, immune dampening and drug resistance. Echinocandin resistance requires the environmentally responsive phosphatase, calcineurin, in C. glabrata, as in C. albicans. Inhibition or genetic repression of Hsp90 phenocopies that of Cnb1, a subunit of calcineurin, when measuring echinocandin resistance in C. glabrata (Singh-Babak et al. 2012). In addition to rapid development of drug resistance, C. glabrata is more intrinsically resistant to many drugs, especially the azole anti-fungals. Fluconazole became fungicidal instead of fungistatic when C. glabrata Hsp90 was inhibited by geldanamycin (Borah, Shivarathri and Kaur 2011). Apart from these studies, the role of Hsp90 in C. glabrata virulence remains unstudied.
Hsp90 in A. fumigatus
The Pezizomycotina are a sister group to the Saccharomycotina yeasts. Amongst the Pezizomycotina, the ‘deadly mould’ A. fumigatus causes >300 000 invasive infections world-wide each year with mortality rates of up to 95% (Brown et al. 2012; Bongomin et al. 2017). In addition to causing life-threatening invasive infections, mainly of the lung, A. fumigatus causes chronic and allergic pulmonary disease in ∼8 million patients world-wide (Bongomin et al. 2017). The United States alone spent ∼$1.2 billion on hospitalizations necessitated by invasive aspergillosis over 10 years (Benedict et al. 2019). These staggering numbers are due to a suite of host- and fungal-specific factors. Aspergillus fumigatus is highly prevalent in the environmental and its small spore size allows the fungus to reach the bronchoalveolar space. Once inside the host, A. fumigatus effectively adheres to the human lung lumen and extensive secretion of galactosaminogalactan and extracellular proteases facilitate persistence in the human lung (Gago, Denning and Bowyer 2019). This is further confounded by a broad and increasing patient demographic that includes leukemic patients and those that received hematopoietic stem cell or solid organ transplants (Kontoyiennis et al. 2010; Pappas et al. 2010). Hsp90’s role in A. fumigatus virulence, drug resistance and morphogenesis has been reviewed here (Lamoth, Juvvadi and Steinbach 2016). More recently, Hsp90 expression, controlled by the transcription factor (TF) HsfA (Fabri et al. 2021), has been shown to be up-regulated in response to heat-shock and azole treatment (Tu, Yin and Li 2020). Resembling findings in C. albicans (Lafayette et al. 2010; Caplan et al. 2018), A. fumigatus Hsp90 governs the cell wall integrity pathway (CWIP) by stabilizing key kinases of this pathway (Rocha et al. 2021). While specific Hsp90 interactors, such as the CWIP kinases have been identified, a global view of Hsp90 genetic and physical interactors is yet to be obtained.
Hsp90 in C. neoformans
The basidiomycetous yeast C. neoformans, the causative agent of the AIDS-defining illness cryptococcal meningitis, causes >1 million cases world-wide each year with mortality rates of up to 70% (Brown et al. 2012). From its environmental reservoirs, mainly pigeon guano but also Eucalyptus trees (Edwards et al. 2021), C. neoformans spores enter the host via inhalation and then move to the brain (Lin and Heitman 2006). Important virulence factors supporting host colonization include an antiphagocytic capsule (Kozel et al. 1988) and melanin production, which provides protection from UV light, oxidative stress, microbicidal peptides and phagocytic cells (Casadevall, Steenbergen and Nosanchuk 2003). Upon entry of C. neoformans into the mammalian lung, Hsp90 is up-regulated (Hu et al. 2008). Pharmacological inhibition of Hsp90 reduced C. neoformans tolerance to thermal stress, antifungal drugs and virulence in an invertebrate model of fungal virulence (Cordeiro et al. 2016). It has, furthermore, been observed that Hsp90 is physically associated with the C. neoformans cell wall and regulates capsule induction and maintenance (Chatterjee and Tatu 2017). Hsp90 interactors are yet to be identified and characterized in C. neoformans.
To date, ten fungal Hsp90 interaction networks have been mapped. Due to limited availability of essential tools, such as genome-scale mutant libraries, Hsp90 networks are currently restricted to the eukaryotic model system S. cerevisiae and the major fungal pathogen C. albicans. Here, we will review the tools and technologies that made Hsp90 network mapping possible and how they may be extended into other fungal species.
Mutant libraries available in fungi
To get a global view of the genes and proteins that either directly or indirectly interact with Hsp90, a suite of genetic and molecular biological manipulations is required. These include genome-scale collections of loss-of-function mutants and epitope-tagged strains.
Genetic interactions are defined as two genes that together produce an unexpected phenotype (Costanzo et al. 2019; Fig. 2). To map genome-scale Hsp90 genetic interaction networks, suitable mutant collections are required. These mutant libraries, containing hundreds or thousands of loss-of-function mutants, were initially developed to study eukaryotic gene function in S. cerevisiae or to identify virulence genes in pathogenic fungi. To generate these collections (Table 1), the majority of which were made possible by community efforts, different genetic approaches were deployed.
Figure 2.

Synthetic lethality identifies genes acting in the same pathway or complex. Yeast cells are viable when experiencing either sub-lethal depletion of Hsp90 function or loss of function of ‘your favourite gene’ (YFG). The combination of both, however, is not tolerated and yeast cells are either ‘sick’ (reduced growth) or dead. Hsp90 function can be reduced by either pharmacological inhibition or the use of hypomorphic alleles and loss-of-function mutations can be achieved as described in the text.
Table 1.
Fungal mutant libraries.
| Species | # Mutants/# genes | Background strain | Mutant auxotrophic and resistance markers | Reference | Availability |
|---|---|---|---|---|---|
| S. cerevisiae | 4815/4815 MATa barcoded haploid deletionmutants | BY4730 | leu, met, ura, kanR | Giaever et al. (2002) | Horizon Discovery Ltd.(https://bit.ly/3xqwJTR) |
| S. cerevisiae | 4803/4803 MATalpha barcoded haploid deletion mutants1 | BY4739 | leu, lys, ura, kanR | Giaever et al. (2002) | Horizon Discovery Ltd.(https://bit.ly/3xqwJTR) |
| S. cerevisiae | 4757/4757 barcoded homozygous diploid mutants2 | BY4743 | his, leu, ura, kanR | Giaever et al. (2002) | Horizon Discovery Ltd.(https://bit.ly/3xqwJTR) |
| C. albicans | 1248/703 transposon insertion (Tn7) mutants of transcription factor, kinase and random genes | BWP17(Wilson, Davis and Mitchell 1999) | his | Davis et al. (2002) | Fungal Genetics Stock Centre (https://bit.ly/3xqmI8Z) |
| C. albicans | 365/166 transcriptional regulator knockouts | SN152 (SC5314)(Noble and Johnson 2005) | arg | Homann et al. (2009) | Fungal Genetics Stock Centre (https://bit.ly/3xqmI8Z) |
| C. albicans | 3000/674 barcoded gene deletions | SN152 (SC5314; Noble and Johnson 2005) | arg | Noble et al. (2010) | Fungal Genetics Stock Centre (https://bit.ly/3xqmI8Z) |
| C. albicans | 2357/2357 Gene Replacement and Conditional Expression (GRACE) mutants (Merck-Frosst library)3 | CaSS1 (CAI4) | natR | Roemer et al. (2003) | National Research Council of Canada (https://bit.ly/3yyPX9J) |
| C. albicans | 5099 ORF clones using Invitrogen Gateway technology (ORFeome collection for C2H) | pDONR207 (Brand, MacCallum and Walker 2012) | Legrand et al. (2018) | http://candidaorfeome.eu/ https://www6.inrae.fr/cirm_eng/ | |
| C. parapsilosis | 200/100 barcoded gene deletions of transcription factors, kinases, species-specific genes | CLIB214 | Holland et al. (2014) | Please contact the authors | |
| C. glabrata | 1601/619 barcoded gene deletions | HTL | natR, his, trp, leu | Schwarzmüller et al. (2014) | Please contact the authors |
| A. fumigatus | 484/484 transcription factor null mutants | MFIG001 (A1160; Fraczek et al. 2013) | ku80-, pyrG+, hphR | Furukawa et al. (2020) | National Collection of Pathogenic Fungi(https://bit.ly/3AxPCWH) |
| C. neoformans | 322/155 barcoded transcription factor deletion mutants | H99S | natR | Jung et al. (2015) | Fungal Genetics Stock Centre (http://www.fgsc.net/crypto/crypto.htm) |
| C. neoformans | 264/129 barcoded kinase deletion mutants | H99S | natR | Lee et al. (2016) | Fungal Genetics Stock Centre (http://www.fgsc.net/crypto/crypto.htm) |
| C. neoformans | 230/114 barcoded phosphatase deletion mutants | H99S | natR | Jin et al. (2020) | Fungal Genetics Stock Centre (http://www.fgsc.net/crypto/crypto.htm) |
| C. neoformans | 2112/2112 barcoded gene deletion mutants (2015 set) | KN99alpha (Nielsen et al. 2003) | natR | Chun and Madhani (2010) | Fungal Genetics Stock Centre (http://www.fgsc.net/crypto/crypto.htm) |
| C. neoformans | 1919/1919 barcoded gene deletion mutants (2016 set) | KN99alpha (Nielsen et al. 2003) | natR | Chun and Madhani (2010) | Fungal Genetics Stock Centre (http://www.fgsc.net/crypto/crypto.htm) |
| C. neoformans | 662/662 barcoded gene deletion mutants (2020 set) | KN99alpha (Nielsen et al. 2003) | natR | Chun and Madhani (2010) | Fungal Genetics Stock Centre (http://www.fgsc.net/crypto/crypto.htm) |
Barcoded haploid MATa and MATalpha deletion libraries in the BY4741 (MATa, his3∆, leu2∆, met15∆, ura3∆ andKanMX) and BY4742 (MATalpha, his3∆, leu2∆, lys2∆ andura3∆) are also available.
A barcoded heterozygous diploid mutant library in the BY4743 background containing 5916 mutants exists as well.
Merck Sharp and Dohme Corp has also produced a heterozygous double barcoded library with 5467 mutants (Xu et al. 2007). This library is available from the National Research Council of Canada (https://bit.ly/3yyPX9J).
Gene deletion libraries are easier to assemble in haploid organisms, as only one allele requires manipulation. Cryptococcus neoformans, C. glabrata and A. fumigatus are haploid, while C. albicans and C. parapsilosis are diploid. In contrast, non-laboratory S. cerevisiae strains are usually diploid (Diezmann and Dietrich 2009), while the S. cerevisiae laboratory strain S288c is an artificial haploid (Mortimer and Johnston 1986). This is due to mutations in the HO-endonuclease, whose wild-type facilitates mating-type switching and consequently selfing (Meiron, Nahon and Raveh 1995).
Saccharomyces cerevisiae mutant libraries
Saccharomyces cerevisiae has been a trailblazer in building interaction networks due to ease of genetic manipulation and a readily executable sexual cycle. Various libraries, differing by mating type, ploidy and auxotrophic markers have been generated (Table 1). To delete target genes in a haploid background, wild-type alleles of non-essential genes were replaced with the KanMX cassette, conferring resistance to the antibiotic geneticin (G418) to select for successful transformation events. These haploid deletion strains were then mated and selected for based on their auxotrophies to create diploid homozygous deletion strains (Giaever et al. 2002). Additionally, in these libraries the KanMX cassette is flanked 3’ and 5’ by 20-mer oligonucleotides, which serve as unique barcodes. Barcode frequencies can either be quantified via microarray technology or next-generation sequencing, allowing pooling of libraries and screening of thousands of mutants in a single vial under the exact same conditions.
Candida albicans mutant libraries
Research on pathogenic fungi is often hampered by low homologous integration rates and the absence of a canonical sexual cycle. Consequently, different strategies have been implemented to manipulate gene function. The situation is further confounded by C. albicans being diploid, which means that depletion of gene function requires two rounds of transformation. This is usually done using a modified S. cerevisiae lithium acetate protocol, which can inadvertently lead to changes in chromosome copy numbers (Bouchonville et al. 2009).
The first publicly available mutant library deployed random transposon insertion mutagenesis (Davis et al. 2002). Transposon mutagenesis is a widely used technique through which genetic material, such as auxotrophic markers, are distributed throughout the host genome via a mobile DNA element, the transposon. This process is mostly random but can lead to gene inactivation, should the transposon insert into coding DNA. Here, the Tn7 transposon, carrying the UAU1 cassette (Tn7-UAU1), was transformed into the C. albicans genome. The UAU1 cassette contains a complete copy of ARG4 flanked by the 3’ and the 5’ regions of URA3. Transforming the Tn7-UAU1 construct into C. albicans strain BWP17 (arg, his and ura) yields Arg+ transformants upon successful integration into allele one. Should recombination of the UAU1 cassette occur, the URA3 marker reconstitutes while being inserted into allele two, therefore, homozygous mutants are Arg+ Ura+. This strategy delivered >1200 histidine–auxotroph transposon insertion mutants representing 703 genes. It should be noted that the progenitor strain used to generate this library, BWP17, is missing a part of the right arm of chromosome 5B (Forche et al. 2004; Selmecki, Bergmann and Berman 2005)
Conversely, a TF library was produced using a clean gene deletion approach to remove both alleles of 166 non-essential TF genes (Homann et al. 2009). Auxotrophic markers HIS1 and LEU2 replace each wild-type TF allele in the progenitor strain SN152 (Noble and Johnson 2005). This approach was then expanded to create a homozygous gene deletion library containing 3000 mutants representing 674 genes (Noble et al. 2010). These mutants are also tagged with one of 48 different oligonucleotide barcodes. This 20-mer, adjacent to the selectable marker, allows for mutants to be pooled in groups of 48, thereby drastically reducing the experimental load.
In addition to transposon insertions and clean gene deletions, C. albicans loss-of-function mutants have also been created using a gene replacement and conditional expression (GRACE) strategy (Roemer et al. 2003). To produce each mutant, the progenitor strain CaSS1, a histidine–auxotroph CAI derivative, has one allele replaced with the HIS3 auxotrophic marker, flanked by two distinct barcodes, and the second allele's promoter is replaced by a SAT1-marked tetracycline promoter. Transformants were then selected for nourseothricin resistance and expression of allele two can be repressed by culturing with doxycycline or tetracycline. It was successfully used to identify essential genes in C. albicans. The library design allows for the prototrophic and nourseothricin resistant mutants to be pooled but limits further genetic manipulations.
Of the four C. albicans libraries described above, there is very little overlap in genes represented (Fig. 3). The largest portion of genes, 221, is shared by the Noble and the GRACE library. Only 13 genes are covered by all four libraries. Thanks to these efforts, the C. albicans community has access to four mutant libraries that together cover ∼50% of the genome.
Figure 3.

Candida albicans mutant libraries sizes and overlaps. Upset R-plot depicting the size of each library on the left (set size) and the overlap between different libraries on the right. The Homann library covers 166 TF gene deletions (Homann et al. 2009). The Mitchell library consists of 703 genes disrupted by transposon insertions (Davis et al. 2002). The Noble library comprises 674 clean gene deletion mutants (Noble et al. 2010). The GRACE library provides repressible mutants for 2357 genes (Roemer et al. 2003). Vertical bars represent the number of genes shared between each of the libraries, the libraries sharing these genes are indicated by the connected dots. There is little overlap in genes represented between libraries, together these libraries allow disruption of 2603 genes, covering 42% of the C. albicans genome.
Candida parapsilosis and C. glabrata libraries
Homologous integration rates are also extremely low in the other Candida species, including C. parapsilosis and C. glabrata. To counter-act this and improve transformation rates, wild-type alleles were replaced in both species with constructs flanked by 500 bp homology arms made using fusion PCR (Noble and Johnson 2005). To create the C. parapsilosis library, the first wild-type allele was replaced with the C. maltosa LEU2 gene and the second allele with the C. dubliniensis HIS1 gene (Holland et al. 2014). Each of the 200 mutants, two per gene, is also barcoded with a 20-mer signature DNA tag permitting pooling of otherwise prototrophic mutants. To replace wild-type alleles in haploid C. glabrata, the NAT1 marker (Shen, Guo and Köhler 2005), conferring resistance to nourseothricin, flanked by two barcodes, was deployed. The library, containing 1601 mutants, covers ∼10% of the genome and mutants can be pooled in groups of 96 (Schwarzmüller et al. 2014). Mutants are nourseothricin resistant and auxotrophic for histidine only or for histidine, tryptophan and leucine, permitting further manipulations if required.
The A. fumigatus library
To create the A. fumigatus gene deletion library, the hygromycin B phosphotransferase cassette (hph) was amplified with 1 kb flanking regions using a fusion PCR protocol (Szewczyk et al. 2006). This construct was transformed into the progenitor strain MFIG001 (Fraczek et al. 2013). MFIG001 is deficient for homologous end joining (akuBKU80; Kress et al. 2006) and carries the pyrG gene, which encodes the orotidine-5’-phosphate decarboxylase, complementing for uracil and uridine auxotrophy (Osmani, Oakley and Osmani 2006). As a consequence, the almost 500 TF gene deletion mutants are prototrophic and resistant to hygromycin B, precluding further genetic modifications (Furukawa et al. 2020).
Different sets of mutant libraries exist for C. neoformans. In a targeted approach utilizing database predictions of gene function, wild-type alleles of TFs, kinases and phosphatases were deleted (Jung et al. 2015; Lee et al. 2016; Jin et al. 2020) in the wild-type strain C. neoformans H99S (Janbon et al. 2014). To do so, the nourseothricin-resistance marker NAT1 was amplified together with a barcode using either an overlap PCR (Davidson et al. 2002) or a double-joint PCR approach (Kim et al. 2009) and transformed into H99S. Mutants, usually multiple per gene, are prototrophic and barcoded for ease of handling. A second, much larger, set of C. neoformans mutants was generated by transforming wild-type strain KN99alpha with overlap fusion PCR products (Chun, Liu and Madhani 2007). The transformation constructs harbor the NAT1 marker, together with 48 unique barcodes (Liu et al. 2008) flanked by 1 kb regions of homology to the up- and down-stream regions of the target gene. In this library, each gene is represented by one prototrophic, nourseothricin resistant, barcoded mutant. Note that H99S and KN99 are derivatives of H99, which was collected from a patient at Duke Medical Center in 1978. For genetic relationships amongst these and other H99 derivatives see here (Janbon et al. 2014).
Together, these mutant libraries enable the large-scale studies into genetic interactions, including that of Hsp90 across evolutionary time in species with different life history trajectories.
Useful techniques for mapping Hsp90 networks in fungi
Several genetic and biochemical approaches have been developed to map Hsp90 interaction networks. Screens are conducted to catalog genes or proteins that are part of the same cellular pathways as Hsp90 or depend on Hsp90 for regulation, stability and/or activation.
Perturbing Hsp90 function
Knock-out mutants have long provided a fundamental technique to investigate the function and interactions of genes, however, this is confined only to non-essential genes. Since Hsp90 is essential, deletion mutants are not viable, so pharmacological inhibition or genetic depletion have been used to carry out synthetic lethality screens to build Hsp90 interaction networks. Pharmacological inhibition of Hsp90 can be achieved by addition of one of several commercially available Hsp90 inhibitors, such as geldanamycin and macbecin II, which would be the ones most commonly used in Hsp90 screens. It should be noted that while both drugs are commonly considered to inhibit Hsp90 by binding to its ATP-binding pocket (Prodromou et al. 1997; Martin et al. 2008), they appear to exert multiple subtle effects that in combination reduce Hsp90 function and may cause small off-target effects (Schmid, Götz and Hugel 2018). Genetic depletion of Hsp90 is possible by producing mutants where Hsp90 is under a repressible promoter such as the tetracycline promoter (Gari et al. 1997; Nakayama et al. 2000). Both, chemical and genetic Hsp90 perturbation, allow fine tuning of Hsp90 function to elicit synthetic lethality in loss-of-function libraries, a prerequisite for the mapping of genetic interaction networks.
Synthetic lethality screens
Genetic interactions occur between genes in the same pathways or molecular complexes, and the functional associations between genes throughout the genome form a genetic interaction network. Genetic interactions can be investigated using the concept of synthetic lethality. Synthetic lethality states that if two non-essential genes genetically interact, the viability of a double mutant is significantly affected, causing reduced fitness (synthetic sickness) or death (synthetic lethality; Dobzhansky 1946; Bendert and Pringle 1991). Organisms have inherent redundancy in their cellular pathways, thereby if one component is not functional, a second route exists that can by-pass the non-functional component. However, when two interacting genes are not functioning, the pathway is no longer functional, and the organism's survival is affected (Fig. 2). Gene deletion libraries can be screened for inviability in response to Hsp90 inhibition in studies termed chemical genetic synthetic lethality (CGSL) screens.
Synthetic genetic arrays (SGA) also use the premise of synthetic lethality but exploit sexual recombination to cross haploid S. cerevisiae gene deletion strains (Tong et al. 2001). Again, since Hsp90 is essential, a hypomorphic Hsp90 allele must be used. Inviable crosses indicate that the non-functional genes in the haploid parents genetically interact (Novick, Osmond and Botstein 1989).
Y2H and C2H protein–protein interaction screens
Hsp90 forms numerous protein complexes with clients and co-chaperones through its role as a molecular chaperone. Some co-chaperones contain a conserved Hsp90-binding sequence, however, client proteins lack such a motif (Scheufler et al. 2000). This hinders bioinformatic prediction of Hsp90 clients, necessitating proteomic and physical interaction screens.
A canonical, large-scale method to identify protein–protein interactions (PPIs) is the yeast two-hybrid system (Y2H; Fields and Song 1989). This system employs a reporter gene such as HIS3, ADE2, LEU2 or Escherichia coli LacZ down-stream of an inducible promoter such as GAL1p (Vojtek, Hollenberg and Cooper 1993; Estojak, Brent and Golemis 1995; James, Halladay and Craig 1996). The original and most utilized system uses the S. cerevisiae TF, Gal4 to bind to GAL1p and induce expression of LacZ. Gal4 comprises an N-terminal DNA binding domain (DBD) and a C-terminal activator domain (AD). When both Gal4 domains come together, expression of β-galactosidase from LacZ is induced. On addition of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), β-galactosidase cleaves the X-gal to form a blue-colored molecule. In Y2H screens the Gal4 binding domain is fused to one protein (termed the ‘bait’) and the activating domain to another protein (‘prey’). If these proteins interact, Gal4 is reconstituted, allowing expression of β-galactosidase and cleavage of X-gal, causing the colony to turn blue (Fig. 4A). To enable high-throughput screening of the S. cerevisiae proteome, a haploid MATa bait library containing the Gal4 DBD attached to each protein in the S. cerevisiae proteome can be mated with a MATalpha prey strain containing a gene of interest attached to the Gal4 activating domain (Uetz et al. 2000). When crossed, selected for diploid cells and grown with X-gal, blue colonies indicate that the Gal4-domain tagged proteins in the haploid parental strains directly and physically interact.
Figure 4.
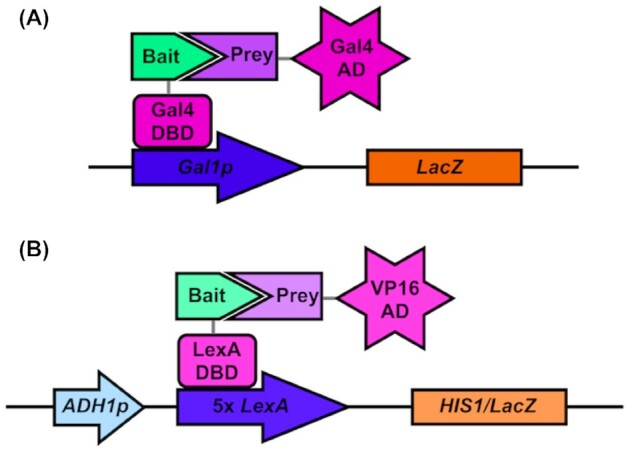
Set-up of the original yeast two-hybrid system for use in S. cerevisiae and its adaption to Candida two-hybrid. (A) The original yeast two-hybrid system uses LacZ as the reporter gene (Fields and Song 1989). When the Gal4 DBD-tagged bait protein interacts with the Gal4 AD-tagged prey protein, Gal4 induces the expression of LacZ via the GAL1 promoter. Colonies where bait and prey proteins interact will appear blue when grown on X-gal media. (B) The Candida two-hybrid system uses C. albicans optimized genes (Stynen, van Dijck and Tournu 2010; Legrand et al. 2018; Schoeters et al. 2018). The background strain, SC2H3, has two reporter genes, Streptococcus thermophilus LacZ and C. albicans HIS1. Each reporter gene is under the C. albicans ADH1 promoter and five copies of the Staphylococcus aureus LexA operon. The LacZ reporter cassette is integrated into chromosome 1 and the HIS1 reporter cassette is integrated into chromosome 4. When S. aureus LexA DBD-tagged bait interacts with viral VP16 AD-tagged prey, expression of LacZ and HIS1 is induced. Strains where bait and prey proteins interact will grow on histidine deficient media and have increased β-galactosidase activity, measurable via assay.
Due to C. albicans’ alternative codon usage (CUG coding for leucine instead of serine) and this species’ inability to maintain plasmids, Y2H systems designed in S. cerevisiae are not reliable in this pathogenic yeast. The first 2H system designed for C. albicans (C2H) comprises plasmids that are integrated into the genome (Stynen, van Dijck and Tournu 2010). A total of two reporter genes, C. albicans HIS1 and Staphylococcus thermophilus LacZ were used, up-stream of five copies of the Staphylococcus aureus LexA operon and the C. albicans ADH1 promoter. An artificial TF comprised of S. aureus LacZ DBD and the viral activating domain VP16 activate the LexA operon and induce expression of LacZ and HIS1. The bait and prey plasmids allow the tagging of genes of interest with the LexA DBD and VP16 activating domain under the MET3 repressible promoter. Reporter gene, bait and prey plasmids are then linearized and integrated into sections of the C. albicans genome where their affects were predicted to be minimal. If the bait and prey proteins interact, strains grown in methionine deficient media are able to grow on histidine selection media and have high activity in a β-galactosidase assay (Fig. 4B).
This C2H system was developed by making the vector plasmids Gateway compatible and mating-inducible C. albicans strains were constructed to allow crossing of bait and prey strains rather than triple-transforming the same strain with three plasmids (Legrand et al. 2018). Again, further improvements to allow high-throughput screens were made by optimizing a mating protocol on agar rather than in broth (Schoeters et al. 2018). Furthermore, the ORFeome library (each C. albicans ORF in a Gateway vector, Table 1) allows the cloning of each C. albicans ORF into the bait or prey C2H vectors for high-throughput interaction studies. Now, the mating-inducible strain containing the reporter gene cassette can be transformed with either the bait or prey plasmids, mated and screened for histidine prototrophy, which indicates the bait and prey proteins interact.
Tandem affinity purification—mass spectrometry proteomics
The Y2H and C2H systems involve several transformations, which is not an insignificant task on a high-throughput level, especially in C. albicans which has low rates of homologous integration. An alternative proteomic approach to investigate PPIs is that of tandem affinity purification (TAP) coupled with mass spectrometry (MS; Rigaut et al. 1999). The TAP tag is formed of calmodulin binding peptide (CBP) and protein A (ProtA) linked with a Tobacco Etch Virus protease (TEV) cleavage site. Tagging a protein with this epitope allows stringent purification first using ProtA's strong affinity to immunoglobulin G (IgG), the protein is then released by TEV protease cleavage and purified again using calmodulin in the presence of Ca2+. Chelating the calcium ions by egtazic acid (EGTA) releases the TAP-tagged protein from calmodulin. Any proteins that form stable complexes with the TAP-tagged protein will co-purify and can be identified by MS. This technique needs only one transformation of a yeast, tagging a gene of interest with the TAP epitope, of which S. cerevisiae and C. albicans optimized sequences exist (Gavin et al. 2002; Lavoie et al. 2008).
Multiplexed quantitative proteomics
Another approach is to utilize multiplexed quantitative proteomics to identify both direct and indirect interactions of Hsp90. The most used technique, stable isotope labeling by amino acids in cell culture (SILAC), involves growing cells in the presence of heavy or light carbon-labeled amino acids, usually arginine and lysine, while inhibiting or repressing Hsp90 (Ong et al. 2002; Gopinath et al. 2014; O'Meara et al. 2019). The cells assimilate these isotopic amino acids into newly synthesized proteins. When subjected to MS, the differently labeled samples can be quantified individually, allowing comparisons between up to three samples at once: those with natural isotopes, light isotopes and heavy isotopes. A more powerful quantitative proteomic approach is Tandem Mass Tagging (TMT; Thompson et al. 2003). This technique covalently attaches one of up to ten different isobaric tags of the same mass to a cell protein sample. During MS, the tags fragment into reporter ions with differing masses, allowing comparison of up to 11 samples simultaneously. Since many samples can be compared concurrently and because tagging occurs after protein extraction, TMT allows robust quantitative proteomic studies without extremely high costs or specialist cell-culture techniques. Although TMT proteomics is yet to be applied to fungal Hsp90 studies, this technique has been used on Hsp90-inhibited human lung cancer and squamous cell carcinoma cell lines (Grimes et al. 2018; Mehta et al. 2020).
Quantitative proteomics on Hsp90-impaired cells can identify interactors up and down-stream of Hsp90 in molecular pathways. Interactors up-stream of Hsp90 are likely to increase in abundance to mitigate loss of Hsp90 function, while clients which are dependent upon Hsp90 for their stability and folding will decrease in abundance. Since clients lack a conserved Hsp90 binding motif to allow identification bioinformatically, quantitative proteomics provides a unique, proteome-wide view to predict novel Hsp90 clients.
The use of these techniques provides a comprehensive toolbox to investigate genetic, proteomic and PPIs.
Fungal Hsp90 interaction networks
Inhibiting Hsp90 function results in a multitude of phenotypes indicative of the central role this molecular chaperone plays. Genetic, physical and proteomic interaction networks allow identification of the molecular pathways and complexes through which Hsp90 exerts its control. To date, ten networks have been mapped in two fungi, yielding insights into Hsp90 function and regulation (Table 2).
Table 2.
Hsp90 interaction networks at a glance.
| Organism | Experimental technique | # of interactors | Key discoveries | Reference |
|---|---|---|---|---|
| S. cerevisiae | Yeast two-hybrid (Y2H) using the Hsp82E33A mutant as bait with a library of ∼6000 prey strains. The E33A allele stabilizes transient Hsp90 interactions. | 177 |
|
Millson et al. (2005) |
| S. cerevisiae | Y2H using different Hsp82 domains and full-length Hsp82 as bait against the entire S. cerevisiae genome.TAP-MS using N-terminal tagged Hsp82 and ∼4000 C-terminal TAP-tagged single gene constructs.Synthetic Genetic Array (SGA) on a MATα hsc82∆ HSP82ts strain mated with the MATa deletion library of ∼4700 non-essential genes. Nonviable or slow growing diploids at 35°C indicative of Hsp90 genetic interaction.Chemical genetic synthetic lethality (CGSL) screen of barcoded haploid deletion library grown in the presence of Hsp90 inhibitor geldanamycin (GdA). | Y2H = 90TAP-MS = 118SGA = 300CGSL = 200Total = 627 |
|
Zhao et al. (2005) |
| S. cerevisiae | CGSL screen on a barcoded homozygous diploid deletion library using the Hsp90 inhibitor macbecin II at 30°C and 37°C.CGSL screen on barcoded heterozygous diploid deletion library at 30 and 37°C. | Homozygous screen:102 at 30°C118 at 37°C90 sharedTotal = 310Heterozygous screen:235 at 30°C241 at 37°C 40 sharedTotal = 516 |
|
McClellan et al. (2007) |
| S. cerevisiae | Mass-spectrometric identification of interactions between 63 TAP-tagged chaperones and 4562 TAP-tagged individual genes. | 259 chaperone–chaperone interactions4340 chaperone–protein interactions |
|
Gong et al. (2009) |
| S. cerevisiae | CGSL screen on barcoded heterozygous diploid mutant library at 15°C.Combined analysis with the published heterozygous screens at 30 and 37°C (McClellan et al. 2007). | Total at 15°C = 27356 shared at 15 and 30˚C20 shared at 15 and 37˚C10 shared at all temperatures |
|
Franzosa et al. (2011) |
| S. cerevisiae | SILAC proteomics on hsp82∆ TETp-HSC82 ura3::tTA strain coupled with transcriptomics.Combined analysis with publicly available data from S. cerevisiae and human. | Total = 90466% of interactors displayed no change in transcript levels. 74% of post-transcriptionally regulated proteins displayed decreased abundance, putative clients. |
|
Gopinath et al. (2014) |
| C. albicans | CGSL screen on transposon insertion library covering 10% of the genome (Davis et al. 2002) using GdA in six environmental conditions (37°C, 41°C, NaCl, tunicamycin, caspofungin and fluconazole). | Total = 226 |
|
Diezmann et al. (2012) |
| C. albicans | CGSL screen of two homozygous deletion mutant libraries covering 13% of the genome (Homann et al. 2009; Noble et al. 2010) using GdA and the same conditions as before (Diezmann et al. 2012). | Total = 158 |
|
O'Meara et al. (2016a) |
| C. albicans | Affinity purification (AP-MS) on HSP90E36A-GFP, HSP90E36A-TAP and C-terminally TAP-tagged cochaperones (Aha1, Cdc37, Cpr7, Cpr7, Cns1, Hch1, Sba1, Sti1 and Sgt1) grown at 30˚C.The E33A homologous allele E36A stabilizes transient Hsp90 interactions.SILAC to compare Hsp90 competent to Hsp90 inhibited (GdA) or Hsp90-repressed (tetO-HSP90) cells. | AP-MS = 188SILAC on GdA treated cells = 505SILAC on tetracycline-repressed cells = 629overlap = 400 proteins found in GdA and tet-repressed cells. |
|
O'Meara et al. (2019) |
| Yarrowia lipolytica | Saccharomyces cerevisiae strain hsp82∆ hsc82∆ heterologously expressing HSP90 from Yarrowia lipolytica, Naumovozyma castellii, Kluyveromyces lactis.Phenotypic characterization, experimental evolution, sequence analysis of evolved strains. | 51 genetic interactions in strain expressing Ylip-HSP90 |
|
Koubkova-Yu, Chao and Leu (2018) |
Hsp90 networks in S. cerevisiae
Probably the most thorough examination of the fungal Hsp90 network landscape has been achieved in S. cerevisiae. Deploying this yeast's extensive toolbox, which includes loss-of-function mutant libraries (Table 1), epitope-tagged libraries (Janke et al. 2004; Howson et al. 2005) and genome-scale collections suitable for Y2H screens (Uetz et al. 2000), provided a comprehensive overview of the eukaryotic Hsp90 interaction landscape. Key findings of the six screens conducted in S. cerevisiae are highlighted in Table 2. Based on these screens, several general features of Hsp90 biology can be concluded.
Hsp90 is a network hub that interacts with at least 10% of the proteome (Zhao et al. 2005). Genome-scale Hsp90 network data have not just yielded lists of genes and proteins that function in the same pathways and complexes as Hsp90, but further analyses of these extensive lists of Hsp90 clients provided important insights into Hsp90’s role in genome evolution (Zhao et al. 2005; Gong et al. 2009). Comparative analyses of evolutionary rates of S. cerevisiae Hsp90 clients with their homologs in the close relative S. paradoxus showed that Hsp90 clients diverged faster due to Hsp90’s ability to buffer destabilizing mutations (Koubkova-Yu, Chao and Leu 2018; Alvarez-Ponce et al. 2019). Furthermore, gene/genome duplication is an important pillar of genetic diversification as it produces new genetic material that can take on a new function, share the function of the predecessor, or be lost. The molecular mechanisms involved in these processes are not yet fully understood. The ancestor of S. cerevisiae underwent whole genome duplication (Langkjær et al. 2003) and S. cerevisiae thus provides an ideal testing ground to determine Hsp90’s role in the fate of duplicated genes. Indeed, comparing the evolutionary rates of Hsp90 clients with those of their non-client paralogs revealed that Hsp90 clients evolved faster and Hsp90 thus facilitates the divergence of gene duplicates (Lachowiec et al. 2013). Hence, Hsp90 is an important contributor to genome diversification.
Hsp90 interactors are involved in many fundamental cellular processes, therefore, inhibition or depletion of Hsp90 results in a multitude of phenotypes. The most extensive S. cerevisiae screen to date, combining Y2H, TAP-MS, SGA and CGSL screens, showed that at 30˚C >10% of interactors function in transcription, cellular fate, protein post-translational modifications, metabolism, cellular transport and the cell cycle and DNA processing (Zhao et al. 2005). This finding was supported by a CGSL screen comparing Hsp90 genetic interactors from cells grown at 30 and 37˚C (McClellan et al. 2007). Here, Hsp90 interactors are 2-fold enriched for genes involved in nuclear organization, protein binding, signal transduction and pseudohyphal growth. The second screen, furthermore showed that Hsp90 genetic interactors differ with the test environment (McClellan et al. 2007) and interactor profiles in cells that were grown in more similar temperatures are more similar to each other (Franzosa et al. 2011). Thus, the Hsp90 interaction network is environmentally responsive.
Further, genome-scale S. cerevisiae Hsp90 network screens have identified novel Hsp90 co-chaperones, which differ from traditional co-chaperones that are essential for Hsp90 activity, client specificity and directionality of the chaperone cycle (Zuehlke and Johnson 2010). Co-chaperones identified as part of genomic screens for Hsp90 interactors have established links between Hsp90 and specific cellular pathways, such as epigenetic gene regulation (Zhao et al. 2005). While rich in discovery, these findings are limited to one species and may not be necessarily transferable to species with other evolutionary trajectories and ecologies.
Hsp90 networks in C. albicans comprise novel regulators of Hsp90 and fungal virulence
To reveal more detail of Hsp90’s role as a regulator of virulence traits in C. albicans, the Hsp90 genetic interaction network was mapped using the transposon insertion mutant library (Davis et al. 2002). In a CGSL screen, mutants were screened for loss of viability in response to Hsp90 inhibition in six environmental conditions: two different temperatures, mild osmotic stress and three common antifungal drugs (Diezmann et al. 2012). Most interactions were detected under just one or two experimental conditions. Only very few interactors were detected during exposure to five or six different conditions. Further analyses showed that high-connectivity interactors, such as Ckb1, are required for Hsp90 phosphorylation and expression, while various low-connectivity interactors were shown to be Hsp90 clients. The experimental set-up of the initial screen was applied to two more C. albicans libraries, the Homann TF library and the Noble deletion mutant library (Table 1), whose overlap with the transposon insertion mutant library is limited to 25 mutants (Fig. 3), bringing coverage of the C. albicans genome to ∼20%. Here, a similar pattern of high- and low-connectivity interactors was observed (O'Meara et al. 2016a). A total of two high-connectivity interactors, ERG5 and STT4, were further characterized. Lack of ERG5 resulted in hypersensitivity to Hsp90 inhibitors, due to the additional cell stress of a destabilized cell wall and loss of STT4 increases cellular demand for Hsp90 due to defects in actin organization. Mapping Hsp90 networks in C. albicans not only contributed to a better understanding of how the chaperone network modulates C. albicans virulence traits (O'Meara, Robbins and Cowen 2017) but revealed another feature of Hsp90 interactors. High-connectivity interactors, those that are detected as essential for growth during most test conditions, affect Hsp90 expression, phosphorylation and function (Diezmann et al. 2012; O'Meara et al. 2016b). Low-connectivity interactors, those detected at specific environmental conditions, depend on Hsp90 for stability and function (Diezmann et al. 2012).
Given the limitations associated with targeting fungal Hsp90, its interactors could provide novel avenues to reducing fungal virulence. To demonstrate just how broadly Hsp90 interactors are involved in different C. albicans virulence factors, three were selected for a more detailed review, two of which, AHR1 and ERG5, were identified as high connectivity interactors. The third, CKA2, was initially identified as a low-connectivity interactor but was later found to be critical for phosphorylation of a Hsp90 serine residue and modulating Hsp90 is a hallmark of a high-connectivity interactor.
The Candida-specific zinc cluster TF Ahr1 (Table 2) activates numerous genes required for fundamental processes of virulence, including adhesion, hyphal growth and biofilm formation (Askew et al. 2011) as well as HSP90 expression (Diezmann et al. 2012). Ahr1 furthermore acts as repressor of the white-to-opaque transition (Wang et al. 2011) by being one of three core regulators of the white cell regulatory network. This network comprises 179 genes, 93 of which are activated by Ahr1 (Hernday et al. 2013). Most recently, it was shown that Ahr1 also activates expression of ECE1 in hyphae (Ruben et al. 2020). ECE1 is the most abundant transcript in hyphae and the precursor of Candidalysin, the first fungal cytolytic toxin to be identified. Candidalysin is critical for mucosal pathogenesis (Moyes et al. 2016). Given how many aspects of C. albicans virulence are controlled by Ahr1, it is not surprising that the ahr1∆/∆ mutant strain displayed attenuated virulence in a murine model of systemic infection (Askew et al. 2011).
Erg5 (Table 2) is part of the ergosterol biosynthetic pathway, which is a prominent drug target making Erg5 itself a prominent component of antifungal drug resistance. Deletion of this C-22 sterol desaturase (P450 cytochrome) results in accumulation and integration of different sterol intermediates into the cell membrane, which causes Hsp90 stress (O'Meara et al. 2016b). Deletion of ERG5 also renders S. cerevisiae cells resistant to polyene antifungals, such as nystatin (Parks et al. 1985). Mutations in ERG5 and ERG11, as identified in a clinical isolate of C. albicans from a patient with recurrent oral candidosis, resulted in multi-drug resistance. The strain was reported to be resistant against the most commonly deployed class of antifungals, the azoles and the last-line antimycotic Amphotericin B, severely compromising the antifungal armamentarium available to treat this patient (Martel et al. 2010).
The tetrameric kinase Ck2 is not well-characterized in C. albicans, but plays a central role in mammalian regulation of cell proliferation and DNA damage repair (Filhol and Cochet 2009). Ck2’s catalytic subunit Cka2 is required for invasion of oral epithelial cells (Chiang et al. 2007) and a mutant lacking CKA2 displays increased resistance to fluconazole (Bruno and Mitchell 2005). The latter phenotype could be explained by the lack of phosphorylation of serine residue 530 in C. albicans Hsp90 (Alaalm et al. 2021). This phospho-switch regulates Hsp90 stability and the expression of various virulence traits, including drug resistance. Phosphorylation of S530 results in a loss-of-function phenotype, as exemplified by susceptibility to fluconazole, filamentous growth and increased susceptibility to thermal stress. Thus, Cka2 is a repressor of Hsp90 function that requires Hsp90 chaperoning for stability (Diezmann et al. 2012).
Further dissecting the functions of Hsp90 interactors identified in networking mapping efforts, will provide new insights into the molecular pathways through which this protein hub governs fungal virulence.
Core network of Hsp90 interactors comprises key regulators of the environmental stress response
Comparing Hsp90 networks from different species has revealed limited overlap between Hsp90 genetic interactors. Only ∼17% of interactors are conserved between S. cerevisiae and C. albicans (Zhao et al. 2005; Diezmann et al. 2012). However, a core of conserved Hsp90 interactors is beginning to emerge. Unsurprisingly, with Hsp90 being a stress-responsive chaperone, core interactors are also involved in stress-response pathways, more specifically several are stress-responsive kinases. In total, two examples of core Hsp90 client kinases are components of the CWIP, which is required for survival during thermal stress. Protein kinase C (Pkc1), which activates the mitogen-activated protein kinase (MAPK) cascade that is integral to the CWIP, is stabilized by Hsp90 in C. albicans (Caplan et al. 2018), as is the A. fumigatus homolog PkcA (Rocha et al. 2021). Mkc1, a MAPK of the CWIP, is stabilized by Hsp90 in C. albicans (Lafayette et al. 2010), in S. cerevisiae (Slt2; Millson et al. 2005) and A. fumigatus (MpkA; Rocha et al. 2021). Yet, Hsp90 core interactors are not restricted to CWIP kinases. Hog1, which regulates the osmolarity signaling pathway (Schüller et al. 1994) is stabilized by Hsp90 in S. cerevisiae (Millson et al. 2005) and C. albicans (Diezmann et al. 2012). Interestingly, even the human homolog, p38, is stabilized and activated by Hsp90 in murine cardiomyocytes (Ota et al. 2010) and human sperm (Sun et al. 2021). Understanding core Hsp90 interactors may shed new light onto the early days of evolution of this intricate and divergent network.
Beyond experimental mapping and validation of Hsp90 interactions, existing PPI databases, such as the STRING and BioGRID (https://string-db.org/; https://thebiogrid.org/; von Mering et al. 2005; Oughtred et al. 2021) can be mined for Hsp90 interactions. Querying the STRING database for PPIs between different molecular chaperones in S. cerevisiae, C. albicans, A. fumigatus and C. neoformans, revealed that protein interactors have diverged between the different species (Horianopoulos and Kronstad 2021). This is supported by a comparison between experimental data of Hsp90 interactors from two different studies in S. cerevisiae (Zhao et al. 2005) and C. albicans (Diezmann et al. 2012), which showed that less than 20% of interactors are shared between these two species. Yet, to fully understand the degree of divergence between Hsp90 chaperone networks in different species, network assays need to be done under comparable conditions.
CONCLUSIONS AND OUTLOOK
Being able to map Hsp90 interaction networks in diverse fungal species facilitates detection of signatures of evolution. Comparing Hsp90 networks mapped in S. cerevisiae that commonly lives on fruit and in the soil, with those gleaned from C. albicans, a common human commensal and opportunistic pathogen, will identify common interactors due to shared ancestry and those that evolved in response to selection exerted by the environmental niche. Comparisons between Hsp90 networks in different species will furthermore provide novel insights into general features of network biology, allowing network dynamics and properties, such as degrees of connectivity, to be established more robustly. Also, experimental identification of Hsp90 interactors has the potential to improve existing databases that allow in silico investigations of PPI networks, such as STRING and BioGRID databases (https://string-db.org/; https://thebiogrid.org/; von Mering et al. 2005; Stark et al. 2006; Oughtred et al. 2021). While powerful with regards to the fungal kingdom, these databases are often limited to experimentally validated interactions in S. cerevisiae, which are then used to predict interactions of orthologs in other species. With the arrival of experimental data in species other than S. cerevisiae, predictions can be refined and improved.
Lastly, research on non-model fungal pathogens, which is hampered by low rates of homologous recombination, is currently being revolutionized by CRISPR-Cas technology (Jinek et al. 2012). To date, CRISPR-Cas has been adapted to operate in C. albicans (Vyas, Barrasa and Fink 2015; Min et al. 2016), C. parapsilosis (Lombardi et al. 2017), C. glabrata (Enkler et al. 2016), C. neoformans (Arras et al. 2016) and A. fumigatus (van Rhijn et al. 2020). Being able to selectively and efficiently manipulate specific loci, will not only enable expansion of existing libraries, but also facilitate creation of libraries in differing genetic backgrounds with varying environmental origins. Being able to mine population-scale mutant collections will further increase resolution of Hsp90 interaction networks.
The model eukaryote S. cerevisiae has been instrumental in extracting fundamental knowledge of the nature of Hsp90 networks. Fungal Hsp90 networks can provide critical insights into the evolution of complex chaperone networks and emergence of pathogenesis.
ACKNOWLEDGEMENTS
The authors would like to thank Heath O'Brien for comparing Candida library sizes.
Contributor Information
Julia L Crunden, School of Cellular and Molecular Medicine, University of Bristol, University Walk, Bristol BS8 1TD, UK.
Stephanie Diezmann, School of Cellular and Molecular Medicine, University of Bristol, University Walk, Bristol BS8 1TD, UK.
FUNDING
Work on fungal Hsp90 interaction networks in the Diezmann lab is supported by the BBSRC and a studentship from the GW4 MRC Doctoral Training Program to JLC.
Conflicts of Interest
None declared.
REFERENCES
- Alaalm L, Crunden JL, Butcher Met al. Identification and phenotypic characterization of Hsp90 phosphorylation sites that modulate virulence traits in the major human fungal pathogen Candida albicans. Front Cell Infect Microbiol. 2021;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MMU, Roe SM, Vaughan CKet al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D, Aguilar-Rodríguez J, Fares MAet al. Molecular chaperones accelerate the evolution of their protein clients in yeast. Genome Biol Evol. 2019;11:2360–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arras SDM, Chua SMH, Wizrah MSIet al. Targeted genome editing via CRISPR in the pathogen Cryptococcus neoformans. PLoS ONE. 2016;11:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew C, Sellam A, Epp Eet al. The zinc cluster transcription factor Ahr1p directs Mcm1p regulation of Candida albicans adhesion. Mol Microbiol. 2011;79:940–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beimforde C, Feldberg K, Nylinder Set al. Estimating the Phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol Phylogenet Evol. 2014;78:386–98. [DOI] [PubMed] [Google Scholar]
- Bendert A, Pringle JR.. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:1295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict K, Jackson BR, Chiller Tet al. Estimation of direct healthcare costs of fungal diseases in the United States. Clin Infect Dis. 2019;68:1791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongomin F, Gago S, Oladele ROet al. Global and multi-national prevalence of fungal diseases—estimate precision. J Fungi. 2017;3:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah S, Shivarathri R, Kaur R. The Rho1 GTPase-activating protein CgBem2 is required for survival of azole stress in Candida glabrata. J Biol Chem. 2011;286:34311–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Farrelly FW, Finkelstein DBet al. hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol. 1989;9:3919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchonville K, Forche A, Tang KESet al. Aneuploid chromosomes are highly unstable during DNA transformation of Candida albicans. Eukaryot Cell. 2009;8:1554–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AC, MacCallum DM, Walker JM. (Ed.) Methods in Molecular Biology. Humana Press, New York, NY, 2012. [Google Scholar]
- Brown GD, Denning DW, Gow NARet al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4:165rv13. [DOI] [PubMed] [Google Scholar]
- Bruno VM, Mitchell AP. Regulation of azole drug susceptibility by Candida albicans protein kinase CK2. Mol Microbiol. 2005;56:559–73. [DOI] [PubMed] [Google Scholar]
- Caplan T, Polvi EJ, Xie JLet al. Functional genomic screening reveals core modulators of echinocandin stress responses in Candida albicans. Cell Rep. 2018;23:2292–8. [DOI] [PubMed] [Google Scholar]
- Casadevall A, Steenbergen JN, Nosanchuk JD.“Ready made” virulence and “dual use” virulence factors in pathogenic environmental fungi - the Cryptococcus neoformans paradigm. Curr Opin Microbiol. 2003;6:332–7. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Tatu U.. Heat shock protein 90 localizes to the surface and augments virulence factors of Cryptococcus neoformans. PLoS NeglTrop Dis. 2017;11:e0005836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang LY, Sheppard DC, Bruno VM. et al. Candida albicans protein kinase CK2 governs virulence during oropharyngeal candidiasis. Cell Microbiol. 2007;9:233–45. [DOI] [PubMed] [Google Scholar]
- Chun CD, Liu OW, Madhani HD.. A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans. PLoS Pathog. 2007;3:0225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun CD, Madhani HD.. Applying genetics and molecular biology to the study of the human pathogen Cryptococcus neoformans. Methods Enzymol. 2010;470:797–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro R de A, Evangelista AJ de J, Serpa Ret al. Inhibition of heat-shock protein 90 enhances the susceptibility to antifungals and reduces the virulence of Cryptococcus neoformans/Cryptococcus gattii species complex. Microbiology. 2016;162:309–17. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Kuzmin E, van Leeuwen Jet al. Global genetic networks and the genotype-to-phenotype relationship. Cell. 2019;177:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE, Lindquist S. Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science. 2005;309:2185–9. [DOI] [PubMed] [Google Scholar]
- Cowen LE, Singh SD, Köhler B JRet al. Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc Natl Acad Sci. 2009;106:2818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RC, Blankenship JR, Kraus PRet al. A PCR-based strategy to generate integrative targeting alleles with large regions of homology. Microbiology. 2002;148:2607–15. [DOI] [PubMed] [Google Scholar]
- Davis DA, Bruno VM, Loza L. et al. Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics. 2002;162:1573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezmann S, Dietrich FS.. Saccharomyces cerevisiae: population divergence and resistance to oxidative stress in clinical, domesticated and wild isolates. PLoS ONE. 2009;4:e0005317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezmann S, Michaut M, Shapiro RSet al. Mapping the Hsp90 genetic interaction network in Candida albicans reveals environmental contingency and rewired circuitry. PLos Genet. 2012;8:e1002562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky TH. Genetics of natural populations. XIII. Recombination and variabiity in populations of Drosophila pseudoobscura. Genetics. 1946;31:269–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Morris JL, Richardson JBet al. Cryptospores and cryptophytes reveal hidden diversity in early land floras. New Phytol. 2014;202:50–78. [DOI] [PubMed] [Google Scholar]
- Edwards HM, Cogliati M, Kwenda Get al. The need for environmental surveillance to understand the ecology, epidemiology and impact of Cryptococcus infection in Africa. FEMS Microbiol Ecol. 2021;97:fiab093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. [DOI] [PubMed] [Google Scholar]
- Enkler L, Richer D, Marchand ALet al. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci Rep. 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estojak J, Brent R, Golemis EA.. Correlation of two-hybrid affinity data with in vitro measurements. Mol Cell Biol. 1995;15:5820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabri JHTM, Rocha MC, Fernandes CMet al. The heat shock transcription factor HsfA is essential for thermotolerance and regulates cell wall integrity in Aspergillus fumigatus. Front Microbiol. 2021;12. DOI: 10.3389/fmicb.2021.656548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Song OK. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–6. [DOI] [PubMed] [Google Scholar]
- Filhol O, Cochet C. Cellular functions of protein kinase CK2: a dynamic affair. Cell Mol Life Sci. 2009;66:1830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Magee PT, Magee BBet al. Genome-wide single-nucleotide polymorphism map for Candida albicans. Eukaryot Cell. 2004;3:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraczek MG, Bromley M, Buied Aet al. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother. 2013;68:1486–96. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, Albanèse V, Frydman Jet al. Heterozygous yeast deletion collection screens reveal essential targets of Hsp90. PLoS ONE. 2011;6:e28211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, van Rhijn N, Fraczek Met al. The negative cofactor 2 complex is a key regulator of drug resistance in Aspergillus fumigatus. Nat Commun. 2020;11:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gago S, Denning DW, Bowyer P.. Pathophysiological aspects of Aspergillus colonization in disease. Med Mycol. 2019;57:S219–27. [DOI] [PubMed] [Google Scholar]
- Garcia-Effron G, Katiyar SK, Park Set al. A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob Agents Chemother. 2008;52:2305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari E, Piedrafita L, Aldea MUet al. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast. 1997;13:837–48. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bösche M, Krause Ret al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–7. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower Ket al. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni Let al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–91. [DOI] [PubMed] [Google Scholar]
- Girstmair H, Tippel F, Lopez Aet al. The Hsp90 isoforms from S. cerevisiae differ in structure, function and client range. Nat Commun. 2019;10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Kakihara Y, Krogan Net al. An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol Syst Biol. 2009;5:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath RK, You S-T, Chien K-Yet al. The Hsp90-dependent -proteome is conserved and enriched for hub proteins with high levels of protein-protein connectivity. Genome Biol Evol. 2014;6:2851–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes M, Hall B, Foltz Let al. Integration of protein phosphorylation, acetylation, and methylation data sets to outline lung cancer signaling networks. Sci Signal. 2018;11:eaaq1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinea J. Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infect. 2014;20:5–10. [DOI] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Fordyce PMet al. Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol Microbiol. 2013;90:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LM, Schröder MS, Turner SAet al. Comparative phenotypic analysis of the major fungal pathogens Candida parapsilosis and Candida albicans. PLoS Pathog. 2014;10:e1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann OR, Dea J, Noble SMet al. A phenotypic profile of the Candida albicans regulatory network. PLos Genet. 2009;5:e1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horianopoulos LC, Kronstad JW. Chaperone networks in fungal pathogens of humans. J Fungi. 2021;7:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain S, Veri AO, Cowen LE.. The proteasome governs fungal morphogenesis via functional connections with Hsp90 and cAMP-protein kinase a signaling. MBio. 2020;11:e00290–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howson R, Huh WK, Ghaemmaghami Set al. Construction, verification and experimental use of two epitope-tagged collections of budding yeast strains. Comp Funct Genomics. 2005;6:2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Cheng PY, Sham Aet al. Metabolic adaptation in Cryptococcus neoformans during early murine pulmonary infection. Mol Microbiol. 2008;69:1456–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA.. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janbon G, Ormerod KL, Paulet Det al. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLos Genet. 2014;10:e1004261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder Net al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–62. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Lindquist S. Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science. 2010;330:1820–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JH, Lee KT, Hong Jet al. Genome-wide functional analysis of phosphatases in the pathogenic fungus Cryptococcus neoformans. Nat Commun. 2020;11:4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara Iet al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KW, Yang DH, Maeng Set al. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat Commun. 2015;6:6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatib R, Johnson LB, Fakih MGet al. Current trends in candidemia and species distribution among adults: Candida glabrata surpasses C. albicans in diabetic patients and abdominal sources. Mycoses. 2016;59:781–6. [DOI] [PubMed] [Google Scholar]
- Kim MS, Kim SY, Yoon JKet al. An efficient gene-disruption method in Cryptococcus neoformans by double-joint PCR with NAT-split markers. Biochem Biophys Res Commun. 2009;390:983–8. [DOI] [PubMed] [Google Scholar]
- Kontoyiennis DP, Marr KA, Park BJet al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001-2006: overview of the transplant- associated infection surveillance network (TRANSNET) database. Clin Infect Dis. 2010;50:1091–100. [DOI] [PubMed] [Google Scholar]
- Koubkova-Yu TCT, Chao JC, Leu JY. Heterologous Hsp90 promotes phenotypic diversity through network evolution. PLoS Biol. 2018;16:e2006450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozel TR, Pfrommer GST, Guerlain ASet al. Role of the capsule in phagocytosis of Cryptococcus neoformans. Clin Infect Dis. 1988;10:436–9. [DOI] [PubMed] [Google Scholar]
- Kress MRVZ, Savoldi M, Goldman MHSet al. The akuBKU80 mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot Cell. 2006;5:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K, Askari F, Sahu MS. et al. Candida glabrata: a lot more than meets the eye. Microorganisms. 2019;7:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachowiec J, Lemus T, Thomas JHet al. The protein chaperone HSP90 can facilitate the divergence of gene duplicates. Genetics. 2013;193:1269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafayette SL, Collins C, Zaas AKet al. PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010;6:79–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoth F, Juvvadi PR, Steinbach WJ. Heat shock protein 90 (Hsp90): a novel antifungal target against Aspergillus fumigatus. Crit Rev Microbiol. 2016;42:310–21. [DOI] [PubMed] [Google Scholar]
- Langkjær RB, Cliften PF, Johnston Met al. Yeast genome duplication was followed by asynchronous differentiation of duplicated genes. Nature. 2003;421:848–52. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Sellam A, Askew Cet al. A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans. BMC Genomics. 2008;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KT, So YS, Yang DHet al. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat Commun. 2016;7:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand M, Bachellier-Bassi S, Lee KKet al. Generating genomic platforms to study Candida albicans pathogenesis. Nucleic Acids Res. 2018;46:6935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Heitman J. The biology of the Cryptococcus neoformans species complex. Annu Rev Microbiol. 2006;60:69–105. [DOI] [PubMed] [Google Scholar]
- Liu OW, Chun CD, Chow EDet al. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135:174–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi L, Turner SA, Zhao Fet al. Gene editing in clinical isolates of Candida parapsilosis using CRISPR/Cas9. Sci Rep. 2017;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loron CC, François C, Rainbird RHet al. Early fungi from the Proterozoic era in Arctic Canada. Nature. 2019;570:232–5. [DOI] [PubMed] [Google Scholar]
- Mahmoudi S, Rezaie S, Ghazvini DaieR et al. In vitro interaction of geldanamycin with triazoles and echinocandins against common and emerging Candida species. Mycopathologia. 2019;184:607–13. [DOI] [PubMed] [Google Scholar]
- Martel CM, Parker JE, Bader Oet al. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14α-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob Agents Chemother. 2010;54:3578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CJ, Gaisser S, Challis IRet al. Molecular characterisation of macbecin as an Hsp90 inhibitor. J Med Chem. 2008;51:2853–7. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Xia Y, Deutschbauer AMet al. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–35. [DOI] [PubMed] [Google Scholar]
- Mehta RK, Pal S, Kondapi Ket al. Low dose Hsp90 inhibitor selectively radiosensitizes HNSCC and pancreatic xenografts. Clin Cancer Res. 2020;26:5246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiron H, Nahon E, Raveh D. Identification of the heterothallic mutation in HO-endonuclease of S. cerevisiae using HO/ho chimeric genes. Curr Genet. 1995;28:367–73. [DOI] [PubMed] [Google Scholar]
- Millson SH, Truman AW, King Vet al. A two-hybrid screen of the yeast proteome for Hsp90 interactors uncovers a novel Hsp90 chaperone requirement in the activity of a stress-activated mitogen-activated protein kinase, Slt2p (Mpk1p). Eukaryot Cell. 2005;4:849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min K, Ichikawa Y, Woolford CA. et al. Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere. 2016;1:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami Y, Kimura Y, Kawasaki Het al. The carboxy-terminal region of mammalian HSP90 is required for its dimerization and function in vivo. Mol Cell Biol. 1994;14:1459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta Mol Cell Res. 2012;1823:648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran L, Mirault ME, Arrigo APet al. Heat shock of Drosophila melanogaster induces the synthesis of new messenger RNAs and proteins. Philos Trans R Soc London. 1978;283:391–406. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR. Genealogy of principal strains of the yeast genetic stock center. Genetics. 1986;113:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes DL, Wilson D, Richardson JPet al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016;532:64–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Mio T, Nagahashi Set al. Tetracycline-regulatable system to tightly control gene expression in the pathogenic fungus Candida albicans. Infect Immun. 2000;68:6712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash AK, Auchtung TA, Wong MCet al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. 2017;5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Harju Vos M, Lindquist S. Identification of SSF1, CNS1, and HCH1 as multicopy suppressors of a Saccharomyces cerevisiae Hsp90 loss-of-function mutation. Proc Natl Acad Sci. 1999;96:1409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen K, Cox GM, Wang Pet al. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect Immun. 2003;71:4831–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, French S, Kohn LAet al. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat Genet. 2010;42:590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell. 2005;4:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Osmond BC, Botstein D. Suppressors of yeast actin mutations. Genetics. 1989;121:659–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara TR, O'Meara MJ, Polvi EJet al. Global proteomic analyses define an environmentally contingent Hsp90 interactome and reveal chaperone-dependent regulation of stress granule proteins and the R2TP complex in a fungal pathogen. PLoS Biol. 2019;17:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara TR, Robbins N, Cowen LE. The Hsp90 chaperone network modulates Candida virulence traits. Trends Microbiol. 2017;25:809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara TR, Veri AO, Polvi EJet al. Mapping the Hsp90 genetic network reveals ergosterol biosynthesis and phosphatidylinositol-4-kinase signaling as core circuitry governing cellular stress. PLos Genet. 2016a;12:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara TR, Veri AO, Polvi EJet al. Mapping the Hsp90 genetic network reveals ergosterol biosynthesis and phosphatidylinositol-4-kinase signaling as core circuitry governing cellular stress. PLos Genet. 2016b;12:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova Iet al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. [DOI] [PubMed] [Google Scholar]
- Osmani AH, Oakley BR, Osmani SA.. Identification and analysis of essential Aspergillus nidulans genes using the heterokaryon rescue technique. Nat Protoc. 2006;1:2517–26. [DOI] [PubMed] [Google Scholar]
- Ota A, Zhang J, Ping Pet al. Specific regulation of non-canonical p38 activation by Hsp90-Cdc37 chaperone complex in cardiomyocyte. Circ Res. 2010;29:997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oughtred R, Rust J, Chang Cet al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021;30:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pammi M, Holland L, Butler G. et al. Candida parapsilosis is a significant neonatal pathogen a systematic review and meta-analysis. Pediatr Infect Dis J. 2013;32:e206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas PG, Alexander BD, Andes DRet al. Invasive fungal infections among organ transplant recipients: results of the transplant-associated infection surveillance network (Transnet). Clin Infect Dis. 2010;50:1101–11. [DOI] [PubMed] [Google Scholar]
- Parks L, Bottema C, Rodriguez Ret al. Yeast sterols: yeast mutants as tools for the study of sterol metabolism. Methods Enzymol. 1985;111:333–46. [DOI] [PubMed] [Google Scholar]
- Pfaller MA, Messer SA, Hollis RJet al. In vitro activities of ravuconazole and voriconazole compared with those of four approved systemic antifungal agents against 6,970 clinical isolates of Candida spp. Antimicrob Agents Chemother. 2002;46:1723–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien Ret al. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. [DOI] [PubMed] [Google Scholar]
- Queitsch C, Sangster TA, Lindquist S. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417:618–24. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz Bet al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–2. [DOI] [PubMed] [Google Scholar]
- Robbins N, Uppuluri P, Nett Jet al. Hsp90 governs dispersion and drug resistance of fungal biofilms. PLoS Pathog. 2011;7:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha MC, Minari K, Fabri JHTM. et al. Aspergillus fumigatus Hsp90 interacts with the main components of the cell wall integrity pathway and cooperates in heat shock and cell wall stress adaptation. Cell Microbiol. 2021;23:e13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Jiang B, Davison Jet al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol. 2003;50:167–81. [DOI] [PubMed] [Google Scholar]
- Rohner N, Jarosz DF, Kowalko JEet al. Cryptic variation in morphological evolution: HSP90 as a capacitor for loss of eyes in cavefish. Science. 2013;342:1372–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben S, Garbe E, Mogavero S. et al. AHR1 and TUP1 contribute to the transcriptional control of virulence-associated genes in Candida albicans. MBio. 2020;11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–42. [DOI] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov Get al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. [DOI] [PubMed] [Google Scholar]
- Schmid S, Götz M, Hugel T. Effects of inhibitors on Hsp90′s conformational dynamics, cochaperone and client interactions. ChemPhysChem. 2018;19:1716–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Linka RM, Reinke H. HSP90 affects the stability of BMAL1 and circadian gene expression. J Biol Rhythms. 2014;29:87–96. [DOI] [PubMed] [Google Scholar]
- Schoeters F, Munro CA, D'enfert Cet al. A high-throughput Candida albicans two-hybrid system. mSphere. 2018;3:e00391–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller C, Brewster JL, Alexander MRet al. The HOG pathway controls osmotic regulation of transcription via the stress response element (STRE) of the Saccharomyces cerevisiae CTT1 gene. EMBO J. 1994;13:4382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzmüller T, Ma B, Hiller Eet al. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 2014;10:e1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Bergmann S, Berman J.. Comparative genome hybridization reveals widespread aneuploidy in Candida albicans laboratory strains. Mol Microbiol. 2005;55:1553–65. [DOI] [PubMed] [Google Scholar]
- Senn H, Shapiro RS, Cowen LE.. Cdc28 provides a molecular link between Hsp90, morphogenesis, and cell cycle progression in Candida albicans. Mol Biol Cell. 2012;23:268–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RS, Uppuluri P, Zaas AKet al. Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr Biol. 2009;19:621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Guo W, Köhler JR.. CaNAT1, a heterologous dominant selectable marker for transformation of Candida albicans and other pathogenic Candida species. Infect Immun. 2005;73:1239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X-X, Steenwyk JL, Labella ALet al. Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci Adv. 2020;6:eabd0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeta T, Zaizen Y, Sugimoto Yet al. Heat shock protein 90 acts in brassinosteroid signaling through interaction with BES1/BZR1 transcription factor. J Plant Physiol. 2015;178:69–73. [DOI] [PubMed] [Google Scholar]
- Singh SD, Robbins N, Zaas AKet al. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 2009;5:e000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Babak SD, Babak T, Diezmann Set al. Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Pathog. 2012;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly Tet al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stechmann A, Cavalier-Smith T.. Evolutionary origin of Hsp90 chaperones and a deep paralogy in their bacterial ancestors. J Eukaryot Microbiol. 2004;51:364–73. [DOI] [PubMed] [Google Scholar]
- Stynen B, van Dijck P, Tournu H.. A CUG codon adapted two-hybrid system for the pathogenic fungus Candida albicans. Nucleic Acids Res. 2010;38:gkq725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Wang Y, Gao Tet al. Hsp90 modulates human sperm capacitation via the Erk1/2 and p38 MAPK signaling pathways. Reprod Biol Endocrinol. 2021;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk E, Nayak T, Oakley ECet al. Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc. 2006;1:3111–20. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–28. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva Met al. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn Ket al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–904. [DOI] [PubMed] [Google Scholar]
- Tong AHY, Evangelista M, Parsons ABet al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–8. [DOI] [PubMed] [Google Scholar]
- Tu B, Yin G, Li H. Synergistic effects of vorinostat (SAHA) and azoles against Aspergillus species and their biofilms. BMC Microbiol. 2020;20:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney Get al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–7. [DOI] [PubMed] [Google Scholar]
- van Rhijn N, Furukawa T, Zhao Cet al. Development of a marker-free mutagenesis system using CRISPR-Cas9 in the pathogenic mould Aspergillus fumigatus. Fungal Genet Biol. 2020;145:103479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase raf. Cell. 1993;74:205–14. [DOI] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Snel Bet al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33:433–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas VK, Barrasa MI, Fink GR. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv. 2015;1:e1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Song W, Huang G. et al. Candida albicans Zcf37, a zinc finger protein, is required for stabilization of the white state. FEBS Lett. 2011;585:797–802. [DOI] [PubMed] [Google Scholar]
- Wang H, Xu J, Guo Het al. Patterns of human oral yeast species distribution on Hainan Island in China. Mycopathologia. 2013;176:359–68. [DOI] [PubMed] [Google Scholar]
- Wilson RB, Davis D, Mitchell AP.. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol. 1999;181:1868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Jiang B, Ketela Tet al. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog. 2007;3:0835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Davey M, Hsu YCet al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell. 2005;120:715–27. [DOI] [PubMed] [Google Scholar]
- Zuehlke A, Johnson JL.. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers. 2010;93:211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]