


Abstract
Direct sequencing of single, native RNA molecules through nanopores has a strong potential to transform research in all aspects of RNA biology and clinical diagnostics. The existing platform from Oxford Nanopore Technologies is unable to sequence the very 5′ ends of RNAs and is limited to polyadenylated molecules. Here, we develop True End-to-end RNA Sequencing (TERA-Seq), a platform that addresses these limitations, permitting more thorough transcriptome characterization. TERA-Seq describes both poly- and non-polyadenylated RNA molecules and accurately identifies their native 5′ and 3′ ends by ligating uniquely designed adapters that are sequenced along with the transcript. We find that capped, full-length mRNAs in human cells show marked variation of poly(A) tail lengths at the single molecule level. We report prevalent capping downstream of canonical transcriptional start sites in otherwise fully spliced and polyadenylated molecules. We reveal RNA processing and decay at single molecule level and find that mRNAs decay cotranslationally, often from their 5′ ends, while frequently retaining poly(A) tails. TERA-Seq will prove useful in many applications where true end-to-end direct sequencing of single, native RNA molecules and their isoforms is desirable.
INTRODUCTION
Rather than a single RNA species, the transcriptional output of a eukaryotic gene is an assortment of RNA molecules whose sequences differ through processes such as alternative transcription initiation (ATI), generating 5′ ends of various lengths; splicing, generating isoforms with different exon compositions; and alternative cleavage and polyadenylation (APA), generating 3′ ends of various lengths (1). Such variations affect translation, RNA stability and localization. At any time, full-length RNA isoforms coexist with their decay fragments, which are generated by exo- and endonucleolysis (2). Initially thought to be restricted to problematic messenger RNAs (mRNAs) (3–5), cotranslational mRNA degradation is now known to be the governing pathway of general mRNA decay in eukaryotes (6–10). Cytoplasmic recapping of processed or decay RNA fragments (11,12) adds to the heterogeneity of gene output. Ribonucleoside modifications such as adenine methylation and cytidine acetylation further expand the complexity of individual RNA molecules (13–15).
Short-read RNA-Seq is currently the most established method for analyzing transcriptomes used in countless applications for both biological research and clinical diagnostics (16). Short-read RNA-Seq involves sequencing of complementary DNA (cDNA) molecules derived after RNA fragmentation, reverse transcription and polymerase chain reaction (PCR) amplification (16). Although widely used and useful, RNA-Seq cannot accurately identify the true transcriptome complexity or its dynamics as short reads cannot be precisely assigned to individual, longer molecules. The most widely used long-read sequencing platforms are Single-Molecule Real-Time (SMRT) sequencing of DNA molecules, a technology used by Pacific Biosciences (PacBio); and nanopore sequencing by Oxford Nanopore Technology (ONT) (17). PacBio sequencing is based on direct observation of DNA polymerization during the replication process of the target DNA molecule through the use of fluorescently labeled deoxyribonucleoside triphosphates and an engineered DNA polymerase (17,18). RNA sequencing with PacBio (Iso-Seq) involves cDNA generation from polyadenylated –poly(A)– RNA, PCR amplification and SMRT sequencing (17,19–22). Recently, a detailed description of poly(A) tails with PacBio has been achieved with FLAM-Seq (23). The authors showed high accuracy of PacBio consensus reads as well as individual nucleotides other than adenosines embedded within poly(A) tails (23). However, Iso-Seq cannot determine the chemical nature of the 5′ end (whether capped; 5′-monophosphate, –5P–; or 5′-hydroxyl, –5OH–), as template switching occurs when reverse transcriptase (RT) reaches the 5′ end of the RNA molecule (20–22). PacBio requires expensive sequencing instruments that are not widely available. Importantly, PacBio is unable to sequence RNA molecules directly.
ONT is rapidly emerging as a user-friendly platform for DNA and RNA sequencing with a unique method for direct sequencing of native, single RNA molecules of any length preserving their modifications (17,24). ONT is poised to transform the way transcriptomes are sequenced and analyzed as the sequencing devices are affordable, portable, can be installed in any lab and both unamplified and amplified cDNA or RNA can be sequenced (17). ONT direct RNA sequencing protocol involves sequential ligation of double-stranded DNA adapters to the poly(A) tail of each RNA molecule. The first adapter contains a stretch of ten thymidines that base-pair with the poly(A) tail while its complementary strand (termed RTA; reverse transcriptase adapter) is ligated directly to the 3′-terminal adenosine of the RNA molecule. This is followed by reverse transcription to create a cDNA strand that alleviates RNA intramolecular secondary structures to improve the sequencing process. Subsequently, a sequencing adapter is attached and the strand ligated to the RTA is equipped with a motor protein, which threads the ligated RNA molecule through a protein nanopore in a 3′ to 5′ direction for sequencing (25). However, as currently designed, ONT cannot perform true end-to-end sequencing as the 5′ end of the RNA molecule is never fully sequenced due to the protein pore's inability to read the terminal 10–15 nucleotides (nt) ((24,26,27), and see below). Moreover, molecules may appear ‘truncated’ because they are incompletely sequenced (27,28) often as a result of signal artifacts due to motor protein stalling, extraneous voltage spikes, stalled pore unblocking (27,29), or other unknown reasons (29).
Identifying the chemical nature of the 5′ end of mRNAs can provide valuable biological insights into mRNA biogenesis, processing and decay. Capped 5′ ends typically signify Transcription Start Sites (TSS) and the very beginning of mRNA molecules (30–32) but in some cases may also indicate cytoplasmic recapping of mRNA decay fragments (11,12,32). 5P ends are typically generated by endogenous nucleolytic processes, including those associated with cotranslational mRNA decay (7–9,33–36). mRNAs with 5P ends are the primary substrates for Xrn1, a conserved exoribonuclease that targets cytoplasmic RNA substrates marked by 5P for processive 5′-to-3′ degradation (37). 5OH ends may be generated by many endogenous nucleases (36,38) but may also reflect exogenous RNA fragmentation. The latter may be due to contamination by RNases secreted by our epidermis and mucosal surfaces (36,39,40), or by commercial RNAses (such as RNAse A, RNAse T1, RNAse I). All these RNAses use acid-base catalysis, taking advantage of the 2′ OH group of RNA as a reactive species and generate 5OH termini (36,40). Another source of exogenous degradation is due to the inherent instability of RNA caused be spontaneous cleavage of phosphodiester bonds by intramolecular, in-line nucleophilic attacks that also generate 5OH termini (41,42).
Recently, cap-dependent ligation and capture of RNAs allowed direct RNA sequencing of the capped ends of transcripts in the migratory locust (26) and in Arabidopsis thaliana (24). However, ONT methods to precisely identify RNAs bearing 5P or 5OH ends, are not available. Finally, as currently constructed, ONT library kits are limited to sequencing of polyadenylated RNAs thus completely missing molecules without poly(A) tail or with different 3′ ends. Such molecules, if identified, would illuminate biological aspects of RNA processing and decay.
Here, we present a new and straightforward method for True End-to-end RNA Sequencing (TERA-Seq), by ligating uniquely designed adapters to the 5′ and 3′ ends of RNA molecules, which can be combined with various treatments. We employ TERA-Seq to accurately define the actual status of RNAs and characterize the human HeLa protein-coding transcriptome. We also introduce a modified ONT direct RNA sequencing protocol for use with a 3′ adapter that allows sequencing of RNAs regardless of the presence of poly(A) tails. With 5TERA, we analyze capped polyadenylated RNAs (cap), as well as processing and decay intermediates containing 5P, or 5OH termini. With TERA3, we analyze 3′ ends of all native transcripts including those lacking poly(A) tails. By attaching both 5′ and 3′ adapters on the same RNA molecule with 5TERA3, we analyze full-length, decay, and processing intermediates from end-to-end, with or without poly(A) tail. 5TERA reveals, in addition to full-length transcripts, prevalent capping downstream of canonical TSSs in fully spliced, polyadenylated mRNAs. We find that full-length mRNAs show marked variation of poly(A) tail lengths. 5TERA, TERA3 and 5TERA3 reveal RNA processing and decay in unprecedented detail at the level of single molecules. We find that mRNAs decay cotranslationally, often while retaining poly(A) tails. We also find that mRNAs primarily decay from their 5′ ends, and this process is not strictly dependent upon prior deadenylation.
MATERIALS AND METHODS
Cell culture
HeLa cells were obtained from ATCC (CCL-2.1) and maintained in DMEM (Gibco) supplemented with 10% FBS (Sigma), at 37°C in 5% CO2. The cells were free of mycoplasma.
RNA preparation
Total RNA was extracted from cells after immediate cell lysis with Trizol, and was treated with DNase as previously described (43). The integrity of total RNA was assessed on a Bioanalyzer prior to each library preparation (Agilent, Supplementary Figure S1a). Depending on downstream application, total RNA was subjected to either poly(A) selection (∼75 μg) using oligo-dT dynabeads (Thermo Fisher) according to manufacturer's protocol, or to ribosomal RNAs (rRNAs) and abundant small, non-coding RNAs subtraction (∼100–130 μg) using custom antisense biotinylated DNA oligonucleotides as previously described (9,43). After the initial processing, the RNA was subjected to various enzymatic treatments and the native ends were marked by adapter ligation to the 5′ end (5TERA), 3′ end (TERA3), or both ends (5TERA3). All downstream protocols converge to the same Oxford Nanopore Technologies (ONT) steps.
TERA-Seq library preparation and sequencing
Libraries were prepared using nanopore direct RNA sequencing kits (SQK-RNA001 and SQK-RNA002, ONT) following manufacturer's instructions for the CTRL-Poly(A) library or with modifications as follows. To identify 5′ ends of RNAs (5TERA), several enzymatic treatments were performed. To select 5′ monophosphate (5P)-containing RNAs, poly(A)-enriched RNA was ligated on beads to a 5′ biotinylated adapter (5′ adapter; Supplementary Table S1) using T4 RNA ligase 1 (NEB) plus 12.5% polyethylene glycol (PEG) in a 50 μl reaction at 37°C for 3 h with gentle agitation in a thermomixer. In our initial experiments, we used a 16 nt 5′ adapter, but we were unable to detect it in the nanopore reads. We then designed a 58 nt adapter, the longest possible that could be synthesized, with a sequence to avoid formation of hairpin structure that might impede ligation, of self- or heterodimers with the 3′ adapter used in TERA3 (Supplementary Table S1). Beads were washed three times with Washing buffer B (10 mM Tris-Cl pH 7.5, 150 mM LiCl, 1 mM EDTA pH 8.0). RNA was eluted off the beads in 50 μl RNase-free water at 75°C for 2 min. The eluate was rebound to 1.8x Agencourt RNAClean XP beads (Beckman Coulter). RNA was then eluted in 9.5 μl RNase-free water and ligated to the RTA using T4 DNA ligase (2M, NEB), 1x Quick ligase buffer (NEB) at 30°C for 15 min. First-strand cDNA was synthesized by SuperScript III Reverse Transcriptase (Thermo Fisher). The RNA-cDNA was purified using RNAClean XP beads. RNA was eluted in 20 μl RNase-free water as directed by ONT protocol. The sequencing adapter was ligated using T4 DNA ligase, 1x Quick ligase buffer at 30°C for 15 min. Ligated-RNA was cleaned up with RNAClean XP beads and eluted in 21 μl Elution Buffer. To identify 5′ hydroxyl (5OH)-containing RNAs, poly(A)-enriched RNA was ligated on beads to the 5′ adapter that specifically ligates to 5OH ends (Supplementary Table S1) using RtcB ligase (NEB) according to manufacturer's protocol. Beads were washed three times with Washing buffer B. RNA was eluted off the beads in 50 μl RNase-free water at 75°C for 2 min. The eluate was rebound to RNAClean XP beads, RNA was eluted in RNase-free water, and ligated to the RTA and the sequencing adapter as described above.
For selection of only capped RNA molecules, poly(A)-enriched RNA was dephosphorylated on beads using Quick Calf Intestinal Phosphatase (CIP, NEB) according to manufacturer's protocol. Beads were washed three times with Washing Buffer B, and once with 1× Thermopol buffer (NEB). The 5′ cap of RNA was removed on beads using 20 μl of RNA 5′ Pyrophosphohydrolase (RppH, NEB), 1× Thermopol buffer, in a 200 μl reaction at 37°C for 1 h 10 min. The reaction was stopped by the addition of 1 μl of 500 mM EDTA pH 8.0. Beads were washed three times with Washing Buffer B and once with 1× Reaction Buffer provided with T4 RNA ligase 1. Previously capped RNAs were ligated on beads to the 5′ adapter using T4 RNA ligase 1 plus 12.5% PEG in a 50 μl reaction at 37°C for 3 h. Beads were washed three times with Washing Buffer B. RNA was eluted from the oligo-dT beads, rebound to RNAClean XP beads, and eluted in RNase-free water for library preparation (as described above). To identify both capped and 5P-containing RNAs, poly(A)-enriched RNA was only treated with RppH followed by 5′ adapter ligation prior to library generation.
To select native 3′ ends of RNA (TERA3), total RNA depleted of rRNAs and abundant small, non-coding RNAs, was ligated to the 3′ adapter (Supplementary Table S1) using T4 RNA ligase 1 plus 7.5% PEG in a 50 μl reaction at 37°C for 3 h in a thermomixer. RNA was cleaned up using 1x RNAClean XP beads and eluted in 9.5 μl RNase-free water. RNA was then ligated to a custom RTA (Supplementary Table S1) followed by reverse transcription, and ligation of the sequencing adapter (as described above). The custom RTA was prepared by mixing equal volume of top strand that contains the RTA sequence and the bottom strand that contains a complementary sequence to the 3′ adapter (Supplementary Table S1) in a 1× Annealing Buffer (10 mM Tris–HCl pH 8.0, 25 mM NaCl, 0.1 mM EDTA) at 95°C for 10 min and cooling down slowly to room temperature. Annealed adapter was stored in aliquots at –20°C.
To simultaneously identify the 5′ and 3′ ends of RNAs (5TERA3), total RNA depleted of rRNAs and abundant small, non-coding RNAs, was first subjected to decapping using 35 μl of RppH in a 350 μl reaction at 37°C for 1 h 15 min. One microliter of 500 mM EDTA pH 8.0 was added, and the RNA was then cleaned up using 1x RNAClean XP beads and eluted in 20 μl of RNase-free water. RNA was then ligated to the 5′ adapter using T4 RNA ligase 1 plus 7.5% PEG, in a 50 μl reaction at 37°C for 3 h. To enrich for ligated RNAs, the reaction was incubated with MyOne C1 Streptavidin beads (Thermo Fisher) (43). Beads were washed three times with 1× BW Buffer (5 mM Tris–Cl pH 7.5, 0.5 mM EDTA pH 8.0, 1 M NaCl) and once with 1× Reaction Buffer. RNA was then ligated on beads to the 3′ adapter using T4 RNA ligase 1 plus 12.5% PEG in a 50 μl reaction at 37°C for 3 h. Beads were washed three times with 1× BW Buffer. RNA was eluted in 50 μl Formamide Elution Buffer (95% formamide, 5 mM EDTA pH 8.0) at 65°C for 5 min. RNA was cleaned up using 1.8× RNAClean XP beads and eluted in 9.5 μl RNase-free water. The RNA was then ligated to the custom RTA followed by reverse transcription, and ligation of the sequencing adapter, as described earlier.
One microliter of each library was quantified using Qubit fluorometer with the DNA HS assay (Thermo Fisher) according to manufacturer's instruction. Sequencing was performed on a MinION device using R9.4 flow cell (FLO-MIN106), and the standard MinKNOW settings recommended by ONT for 48 h or 72 h run.
Data analysis
Basecalling and adapter detection
The raw fast5 files were basecalled using Guppy (v3.3.2–1) (https://community.nanoporetech.com/downloads) in the GPU mode with parameters guppy_basecaller--flowcell FLO-MIN106--kit SQK-RNA002--hp_correct on--trim_strategy none. Cutadapt (v2.5) (44), seqkit (v0.11.0) (45), and seqtk (v1.3-r106) (https://github.com/lh3/seqtk) were used for adapter detection and trimming. To reduce the possibility of false-positive (FP) adapter detection, we tested the adapter trimming with different Cutadapt settings on the reference transcript sequences. The maximum FP detection rate (determined as a random match of adapter and reference sequence) was set to 0.2% (∼300 transcripts). Parameters passing the set FP rate were then tested on the reads and the combination of settings with the highest number of trimmed reads was selected. Additionally, three rounds of randomized adapter sequence trimming with the same settings had to result in 0% trimmed reads. If more than one settings combination resulted in the same number of trimmed reads the highest error rate and smallest minimal overlap combination was preferred. For the 5′ adapter detection, the resulting parameters were --overlap 31--error-rate 0.29 (see also Supplementary Note), and detection of internal adapters was disabled (XADAPTER). For the 3′ adapter detection, the final parameters were --overlap 16--error-rate 0.18, and the detection was limited to the last 200 nt of the read. All reads shorter than 25 nt after the adapter trimming were discarded. Of note, libraries generated using 3′ adapter cannot be sequenced unless the 3′ adapter is ligated to the RNA molecule because the custom RTA will not bind molecules without this adapter.
Alignment and postprocessing
The basecalled reads were aligned to both genome and transcriptome reference sequences (GRCh38; Ensembl 91) (46) using minimap2 (v2.17-r941) (47). All pseudogenes sequences were removed from the reference transcriptome for all alignments. For poly(A) libraries mapping, only transcripts with poly(A) tail (protein_coding, lincRNA, antisense_RNA, bidirectional_promoter_lncRNA, processed_transcript, retained_intron, sense_intronic, sense_overlapping) were subset from the reference transcriptome. For genomic alignments, splicing and secondary alignments were allowed (minimap2 -a -x splice -k 12 -p 1 -u b--secondary=yes). Transcriptome alignment was run with the recommended ONT settings (minimap2 -a -x map-ont -k 12 -p 1 -u f--secondary=yes). Samtools (v1.9) (48), samutils (commit 3e9da2b) (https://github.com/mnsmar/samutils), and CLIPSeqTools (commit 8b8a7b7) (49) were used in alignment postprocessing steps. For most of the analyses, only unambiguous alignments were kept. Unambiguous alignments were determined as alignments with a single highest alignment score (minimap2 ms tag).
Transcriptome completeness
To better determine molecule completeness, we corrected the general Ensembl transcriptome annotation to fit HeLa cell line sequencing results. The re-defined start and end of a transcript were determined by the highest read coverage position outside the annotated CDS region using reads with 5′ adapter from Cap-Poly(A) library. A minimum of 2 reads was required to re-define the new end. The corrected annotation was used for analyses in Figures 1B (right), C (right), D, 3B, 4B and 5C, and Supplementary Figures S2b, c and d. The transcripts were then divided into 20 equal length bins (0–19). The percentages of 5′ and 3′ ends in the individual bins were calculated separately. Furthermore, the transcripts were grouped by length (500 nt increments). Mapped bins for both 5′ and 3′ ends were additionally summarized in a heatmap.
Figure 1.

True end-to-end sequencing of single native polyadenylated RNA molecules with 5′ adapter ligation (5TERA). (A) Method schematic. Enzymatic treatments to identify indicated 5′ ends by adapter ligation are shown in box. ONT, Oxford Nanopore Technologies; RTA, reverse transcriptase adapter; CIP, Calf Intestinal Phosphatase; RppH, RNA 5′ Pyrophosphohydrolase; 5P, 5′ monophosphate; 5OH, 5′ hydroxyl, Gppp, 5′ cap; A(n), poly(A) tail; T, thymidine. (B) Heatmap of read density of the 5′ ends close to the annotated transcription start site based on Ensembl annotation (left) and on re-annotated transcripts (right) from Cap-Poly(A) library. Only molecules with 5′ adapter are used for the analysis. Y-axis corresponds to individual transcripts. Positions up to 150 nucleotides from transcription start site are shown on the x-axis. Z-scores are calculated per row and scale is depicted on top. Number of reads corresponding to each transcript is shown on the right. Only top 30% most expressed transcripts are shown. (C) Correlation of the completeness of CDS and mRNA with expression levels based on Ensembl annotation (left) and on re-annotated transcripts (right) from Cap-Poly(A) library. Only molecules with 5′ adapter are used for the analysis. Each point represents an individual transcript. Color represents transcript expression level, calculated as the log2 of reads per million (RPM). Pearson's correlation (R) and associated P-value are shown on top. CDS, Coding Sequence; mRNA, messenger RNA. (D) Distribution of molecule ends per transcript length from indicated 5TERA libraries on HeLa re-annotated transcripts. Distribution of reads is calculated for individual transcripts and then averaged for visualizing (green line). Shaded area (green) represents the standard deviation. Only molecules with 5′ adapter are used for the analysis. Meta-coordinates are defined by splitting each transcript into 20 equal bins. Transcript lengths, grouped by 500 nucleotides are shown on the right.
Figure 3.

Identification of native 3′ ends of single RNA molecules by direct sequencing with 3′ adapter ligation (TERA3). (A) Method schematic. rRNAs, ribosomal RNAs; snRNAs, small nuclear RNAs; Gppp, 5′ cap; A(n), poly(A) tail; custom RTA, reverse transcriptase adapter with custom bottom sequence. (B) Heatmap of molecule ends from a TERA3 library on transcript meta-coordinates. Each dot (black) corresponds to a single molecule and its meta-coordinates are defined according to the 5′ and 3′ end position along 20 bins on the corresponding transcript. The shade of a square represents the total sum (log10(count)) of ends mapped to the indicated meta-coordinate. The scale is shown on the top right. The total distribution for each meta-coordinate is summarized independently (5′, top; 3′, right). Mol., molecules. (C) Visualization of coverage and alignment of sequenced molecules to Ferritin Light Chain gene from FTL-201 (ENST00000331825) transcript. Genomic coordinates and gene model (orange) with numbered introns are shown on top. Arrowhead and arrow show molecules with retained intron 3, and introns 2 and 3, respectively. Only molecules with 3′ adapter are visualized. Mb, megabase. (D) Distance of 3′ ends of RNAs identified by TERA3 (representative library, blue line), and control (grey), to APA sites. Control was created by generating 100 000 random positions within transcripts detected in at least one library. Only positions with a direct overlap or downstream to Alternative Polyadenylation (APA) sites were considered. Y-axis represents the cumulative percentage of overlaps within the visualized range section. nt, nucleotides.
Figure 4.

Poly(A) tail characterization with TERA-Seq and its relation to 5′ end decay. (A) Histogram of poly(A) tail lengths from indicated 5TERA libraries and a representative TERA3 library; dashed lines, median values of adapter-ligated molecules. Lengths are binned by 10 nucleotides. Tails longer than 300 nucleotides are merged to the 300 nt bin. (B) Distribution of 5′ (green) and 3′ (blue) ends of Ferritin Heavy Chain 1 transcript (FTH1-201) RNA molecules from indicated 5TERA libraries. Meta-coordinates are defined by splitting each transcript into 20 equal bins. Each horizontal line represents single RNA molecule aligned to FTH1-201. Poly(A) tail length of each molecule (grey line) is shown on the right. Tails longer than 300 nucleotides (nt) are capped to 300 nt. (C, D) Relation between 5′ end meta-coordinates and poly(A) tail length of all molecules (C) and transcripts (D) from 5P-Poly(A) library. Meta-coordinates are defined by splitting each transcript into 20 equal bins. Each point represents a single RNA molecule (C) or transcript (D). Only molecules with 5′ adapter are visualized. Kendall's Tau correlation value, associated p-value, and linear regression (red) are also shown. Poly(A) tail length was capped at 600 nucleotides (nt).
Figure 5.

True, end-to-end sequencing of single, native RNA processing and decay intermediates with concurrent 5′ and 3′ adapter ligation (5TERA3). (A) Method schematic. 5P, 5′ monophosphate; A(n), poly(A) tail; B, biotin. Ligated molecules are enriched on streptavidin beads. (B) Histogram of poly(A) tail lengths; dashed line, median value of adapter-ligated molecules from 5TERA3. Lengths are binned by 10 nucleotides (nt). Tails longer than 300 nucleotides are merged to the 300 nt bin. (C) Average read density distribution of molecule ends across re-annotated HeLa transcripts in 5TERA3. Meta-coordinates are defined by splitting each transcript into 20 equal bins. Only top 30% expressed transcripts are shown. Shaded area (green) represents the standard deviation. (D) Visualization of coverage and alignment of sequenced molecules to Thymosin Beta 10 gene from TMSB10-201 (ENST00000233143) transcript with 5TERA3 (top; dark grey); poly(A) tails shown in blue. Non-polyadenylated molecules are shown with red arrowheads. Illumina (short-read; data obtained from (41,42)) coverage and read alignment (fuchsia) are shown on bottom. Genomic coordinates and Ensemble exons (orange) with numbered introns are shown on top. Mb, megabase.
CAGE and APA overlaps
Genomic positions of 5′ and 3′ mapped TERA-Seq read ends were obtained using bedtools bamtobed (v2.29.0) (50) and the distances to the nearest CAGE/APA tag with bedtools closest. HeLa CAGE signals were converted from hg19 to hg38 coordinates using liftOver (v385) (51,52). For CAGE analysis, we considered features as overlapping if the CAGE signal directly overlapped the mapped 5′ end or was located upstream. The same rule applied for the APA (53) overlaps but using the mapped 3′ end instead. Since the ONT direct RNA sequencing is strand-specific (forward/sense), the same strand overlap was required. Controls were created by generating 100 000 random positions within transcripts detected in at least one 5TERA library.
Promoter region heatmaps
ENCODE SCREEN (54) promoter-like signature (PLS) regions were downloaded from UCSC (52)
(http://hgdownload.soe.ucsc.edu/gbdb/hg38/encode3/ccre/encodeCcreCombined.bb) in bigBed format. BigBed files were converted to bigWig using kentUtils (v385) (52) and used as score file (-S) to deepTools computeMatrix reference-point (v3.5.0) (55). Genomic positions of 5′ ends were extracted the same way as in the CAGE and APA overlaps section above. The mapped 5′ ends were collapsed to avoid duplicated positions and used as region files (-R). Window of 500 bp upstream to 500 bp downstream is visualized.
Transcript and gene coverage
Transcript coverage (Figure 2B) was calculated from transcriptome alignments using unambiguously mapped reads. For this visualization, genomic coordinates of exon boundaries, CAGE, NET-CAGE (56), and APA sites were converted to transcriptomic by GenomicFeatures (v1.32.7) (57) and rtracklayer (v1.40.6) (58) R/Bioconductor (v3.8) (59) packages. The mapped positions were binned by 5 nt. Gene coverage (Figures 2C, 3C, and 5D, and Supplementary Figures S2b and S5) was visualized by Gviz (v1.24.0) (60) using genome mapped reads. The Illumina coverage was calculated from STAR (v2.7.2b) (61) mapped reads (STAR --outMultimapperOrder Random--outFilterMultimapScoreRange 1--alignMatesGapMax 1000000--alignIntronMax 1000000--outFilterIntronMotifs RemoveNoncanonicalUnannotated--alignIntronMin 20--alignSJoverhangMin 5--alignSJDBoverhangMin 3--twopassMode Basic--outFilterType BySJout--outFilterMatchNmin 10--outFilterMultimapNmax 20--outFilterMismatchNmax 999--outFilterMismatchNoverReadLmax 1.0--outFilterMismatchNoverLmax 0.05--outFilterScoreMinOverLread 0.66--outFilterMatchNminOverLread 0.66--sjdbOverhang 100).
Figure 2.

Relation of 5′ ends of native RNA molecules to transcription start sites and to active promoters. (A) Distance of capped 5′ adapter-ligated ends of polyadenylated RNAs identified with 5TERA (green line), and control (grey), to CAGE sites. Control was created by generating 100 000 random positions within transcripts detected in at least one library. Only positions with a direct overlap or downstream to CAGE sites were considered. Y-axis represents the cumulative percentage of overlaps within the visualized range section. CAGE, Cap Analysis of Gene Expression. (B) Coverage of Ferritin Heavy Chain transcript 1 (FTH1-201) from indicated libraries and positions of Native Elongating Transcript–Cap Analysis of Gene Expression (NET-CAGE) signals (purple), CAGE signals (black), and Alternative Polyadenylation (APA) sites (blue). Only molecules with 5′ adapter are analyzed. Dashed orange lines indicate exon-exon boundaries. All visualized positions are binned by 5 nucleotides. CDS, Coding Sequence. (C) Visualization of coverage and alignment of sequenced molecules to FTH1 gene from FTH1-201 transcript, from indicated 5TERA libraries. Genomic coordinates, Ensembl transcript (orange) with numbered introns, NET-CAGE signals (purple), and CAGE signals (black) are shown on top. Only molecules with 5′ adapter are analyzed. Mb, megabase. (D) Summary plots and heatmaps of ENCODE-annotated, promoter-like signals (PLS) around 5′ ends of mRNAs identified by 5TERA. The summary profile plot (top panel) indicates enrichment around the 5′ ends (position 0) for reads with adapter (green) and without adapter (black). Heatmaps show distribution of PLS for each 5′ end separately. Each line in the heatmap represents a single 5′ end/read. Reads with adapter (middle panels) and reads without adapter (bottom panels) are visualized separately. Scale is shown at the bottom; low signal, dark blue; strong signal, yellow). All 5′ ends were collapsed prior to analysis. 500 base pair (bp) region upstream and downstream from 5TERA 5′ ends is visualized.
Poly(A) tail
Nanopolish (v0.11.1) (27) was used to estimate poly(A) tail lengths in all 5TERA, TERA3 and 5TERA3 libraries. As recommended by the authors, we used transcriptome alignments of RTA untrimmed reads for the estimates. Estimating poly(A) length directly from the basecalled sequence is not reliable and was not used in any of the analyses (see also Supplementary Note). As indicated in figures and their legends, for some visualizations, poly(A) tail lengths were capped at 300 nt and tails longer than 300 nt were merged to the last bin; or at 600 nt.
Ribosome profiling and Akron5 overlaps
Ribosome profiling and Akron5 overlaps with the mapped TERA-Seq 5′ ends were done as described previously (9). Briefly, 5′ end coordinates of mapped reads were overlapped with coordinates of 5′ mapped ribosome-protected fragment (RPF) (STAR --outFilterMultimapScoreRange 0--alignIntronMax 50000--outFilterIntronMotifs RemoveNoncanonicalUnannotated--outFilterMatchNmin 15--outFilterMatchNminOverLread 0.9). RPF mapped positions were collapsed and used as reference points to which the TERA-Seq reads were related. Only 29 nt long RPFs were selected for the analysis as they show the highest periodicity. A window of +/- 45 nt around the RPF 5′ ends was visualized. Discrete Fourier transformation and density calculation was used to determine the 3 nt periodicity of the overlaps. Akron5 5′ ends overlaps (STAR --outFilterMultimapScoreRange 0--alignIntronMax 50000--outFilterMatchNmin 8--outFilterMatchNminOverLread 0.7--sjdbOverhang 80--alignSJDBoverhangMin 1--seedSearchStartLmax 15) were done in the same manner as RPFs.
Evolutionary conservation
Conservation analysis (62) was done as previously described (9). 5′ ends of mapped reads with and without 5TERA adapter were used as the central positions for the analysis.
Transcript expression and correlation
Transcript expression was calculated as RPM (reads per million) ‘ number of mapped reads to the transcript/(number of mapped reads to all the transcripts/1 000 000). The correlation was calculated from log2(RPM + minimum RPM from all transcripts) counts using Pearson's correlation (r) with corrplot (v0.84) (https://github.com/taiyun/corrplot).
RESULTS
To develop TERA-Seq, we chose the human HeLa cell line based on its extensive characterization and numerous, publicly available experimental datasets. As starting material for all experiments, we extracted total RNA after immediate cell lysis with Trizol and assessed its integrity with capillary electrophoresis on a Bioanalyzer (Supplementary Figure S1a) prior to each library preparation. Depending on the downstream application, we then isolated poly(A) molecules by oligo-dT magnetic beads; or subtracted ribosomal RNAs (rRNAs) and abundant small, non-coding RNAs from total RNA with custom biotinylated oligonucleotides (oligos) (9,43).
True end-to-end sequencing of single, native polyadenylated RNA molecules with 5′ adapter ligation (5TERA)
In our first trials, we attached short, 16 nt 5′ adapter to poly(A) RNA 5′ ends with T4 RNA ligase 1, either before (5P short-Poly(A) library) or after T4 Polynucleotide Kinase (PNK) treatment (5P short-PNK-Poly(A) library) to identify RNAs with 5P, or with both 5P and 5OH respectively. However, we were not able to identify the adapter sequence (<0.01% of molecules) of the RNA molecules in neither library, indicating that ONT was unable to read the very 5′ ends. To verify this observation, we increased the adapter length to 58 nt. We were able to efficiently detect the adapter (see below for more details), and used it for generation of all subsequent libraries (5′ adapter; sequences of all adapters are shown in Supplementary Table S1). We also added biotin to the 5′ end of the longer adapter to allow, if desired, selective capture of ligated molecules.
To identify molecules with 5P, we ligated the 5′ adapter to poly(A) RNA with T4 RNA ligase 1 (5P-Poly(A) library; Figure 1A) under conditions that minimize ligation biases (9,63). A 5P is obligatory for adapter ligation with T4 RNA ligase 1 (64–66). To identify capped molecules, we first treated poly(A) RNA with Calf Intestinal Phosphatase (CIP) to dephosphorylate 5P-containing RNAs preventing them from ligating the 5′ adapter. Then, we used RNA 5′ Pyrophosphohydrolase (RppH) to hydrolyze the cap triphosphate linkage, leaving a 5P in its place (67). This renders only previously capped molecules amenable to 5′ adapter ligation with T4 RNA ligase (Cap-Poly(A) library; Figure 1A). To identify both capped and 5P molecules, we treated poly(A) RNA with RppH followed by 5′ adapter ligation with T4 RNA ligase (Cap & 5P-Poly(A) library; Figure 1A). The levels of endogenous mRNAs bearing 5OH termini are very low and attempts by us (9) and others (7) to capture them had not been successful. The Hesselberth lab developed a sensitive method that specifically identifies 5OH termini of RNA for sequencing, by using the RtcB ligase (38). Thus, to identify molecules with 5OH, we ligated a 5′ adapter bearing a 3′ phosphate (Supplementary Table S1) to poly(A) RNA with RtcB ligase (5OH-Poly(A) library; Figure 1A). We also generated a library without any adapter ligation (CTRL-Poly(A) library). We implemented a rigorous scheme to detect the TERA adapters, minimizing false-positive detection and maximizing accurate identification of the nucleotides at both ends of adapter-ligated RNA molecules (see Supplementary Note). We successfully identified the long 5′ adapter in all libraries and confirmed that ONT indeed misses ∼13 nt from the 5′ ends of molecules (Supplementary Figure S1b), as previously observed (24,26,27). These findings indicate that ligation of the long 5′ adapter can bypass the systematic ONT error and permits sequencing of exact 5′ ends of input polynucleotides.
Our initial experiments showed higher yield of short molecules relative to long molecules. For this reason, we next considered a preferential sequencing of short molecules as another potential ONT aspect. The speed of the motor protein translocating RNA molecule through the nanopore is fixed, as is the number of nanopores reused during the sequencing run (25), potentially leading to smaller molecules being sequenced more readily. Furthermore, incomplete sequencing of longer molecules (27,29) may artificially inflate the number of shorter reads. However, a large number of short molecules may also be an accurate reflection of the sample composition as libraries are loaded on the flow cell based on total sample mass and without any prior size selection. Since the number of molecules in a given RNA mass is inversely proportional to their length, the molarity differences between short and long molecules vary greatly within a sample as complex as a human cell transcriptome. To investigate whether ONT favors short RNA molecules over long ones, we examined its performance with synthetic transcripts. We re-analyzed direct RNA sequencing reads from datasets that included Spike-In RNA Variant Control Mix E2 (SIRV) (68). SIRVs are 69 isoform transcripts (from 7 SIRV genes), ranging in length from 191 to 2 528 nt, that mimic complexity of the human transcriptome. They are generated by in vitro transcription with T7 RNA polymerase, and all contain a poly(A30) tail and a 5′ triphosphate, the latter as expected of all RNA polymerase products (69). SIRV E2 mix contains four expression groups each with equimolar concentration of the transcripts and was previously used to show that ONT RNA sequencing is highly accurate in quantifying gene expression (68). The advantage of this dataset is that SIRVs were added directly to the samples and sequenced along the cellular transcripts (68). Our analysis shows that the expression level of SIRVs is not dependent on transcript length (Supplementary Figure S1c) indicating that ONT does not have length selection bias.
Accurate characterization of the HeLa protein-coding transcriptome using 5TERA
Next, we analyzed our HeLa ONT libraries prepared from poly(A) RNA using 5TERA. As expected, we find that ∼98% of molecules in all 5TERA libraries contain poly(A) tails attesting to the efficiency of the ONT platform to sequence poly(A) molecules. It also shows that TERA-Seq did not disrupt the capture of polyadenylated molecules. We also examined the specificity of the oligo-dT beads and of the ONT platform by spiking in a non-polyadenylated synthetic transcript to total RNA preparations before oligo-dT purification and library generation. This synthetic RNA was not detected upon ONT sequencing (data not shown). We found similar size distribution of reads in 5P-Poly(A) and CTRL-Poly(A) libraries (Supplementary Figure S1d), indicating that adapter ligation did not have adverse effects.
For analyses detailed below, we focus only on protein-coding transcripts (70). In 5TERA libraries, we do not selectively enrich for adapter-ligated RNA molecules by streptavidin bead capture (Figure 1A), therefore we expect to observe both molecules with the 5′ adapter (adapted) and without (non-adapted). Thus, we are able to compare properties of adapted and non-adapted molecules from the same RNA extraction and all subsequent steps including the library preparation and sequencing. When examining non-adapted reads in the 5P-Poly(A) library, we anticipate seeing molecules both with and without 5P (such as capped molecules). In Cap-Poly(A) and Cap & 5P-Poly(A) libraries, we expect to see even distributions in adapted and non-adapted molecules. We detect the 5′ adapter in ∼7% of molecules in the 5P-Poly(A) library, and this ratio increases to ∼20% and to ∼25% of molecules in the Cap-Poly(A) and Cap & 5P-Poly(A) libraries, respectively. The difference in percentage of the detected adapters between 5P-Poly(A) and Cap & 5P-Poly(A) libraries indicates that capped poly(A) RNAs are roughly three and a half times more abundant than 5P-containing RNAs. We detect the 5′ adapter in less than 1% of molecules in the 5OH-Poly(A) library, consistent with the low level of 5OH mRNAs in cells (7,9), and the absence of any appreciable, exogenous, RNA degradation. To explore the differences between adapted and non-adapted molecules, we first examined the 5′ and 3′ ends distributions of RNA molecules without the 5′ adapter on meta-mRNA plots based on Ensembl transcript annotation. Roughly 61% of molecules per transcript reach the annotated 3′ end (last 5% of the meta-mRNA) in all libraries while approximately 26% reach the annotated 5′ end (first 5% of the meta-mRNA) (Supplementary Figure S2a). Reads whose 5′ ends are within the first 5% are considered to represent full- or close to full-length molecules.
To investigate the low levels of complete molecules, we analyzed the distribution of read 5′ ends around the annotated mRNA TSSs. We focused on molecules with 5′ adapter from the Cap-Poly(A) library to ensure the 5′ end was read accurately and that we only consider capped molecules. Interestingly, we find that the most covered position at the 5′ end for the majority of transcripts does not coincide with the Ensembl annotated mRNA TSSs (position 0; Figure 1B; left). This reflects the limitations of broad gene annotation databases as they are curated from a variety of biological sources that do not necessarily reflect the individual cell-type or tissue of interest. To adapt the general annotation for the HeLa protein-coding transcriptome, we re-annotated the transcripts by adjusting their transcript start and end sites to positions with the highest read coverage outside the annotated coding sequence (CDS) region (Figure 1B; right). We find that the coincidence of the most covered position with the re-annotated TSSs increased significantly as expected (Figure 1B). To confirm our adjustment, we compared the completeness of mRNAs and their CDSs under the hypothesis that a complete CDS should originate from a complete mRNA. The ratio of incomplete mRNAs with complete CDS is significantly lower after our HeLa-specific re-annotation (Figure 1C; right) compared to the original Ensembl annotation (Figure 1C; left) highlighting the importance of precise 5′ identification for accurate transcriptome representation. An example of the effect of re-annotation is shown in Supplementary Figure S2b. Having the re-annotated HeLa transcriptome, we plotted the distribution of 5′ and 3′ ends of RNA molecules without the 5′ adapter on meta-mRNA for all the libraries and found that the percentage of RNA molecules reaching the 3′ and 5′ ends increases to ∼80% and ∼32%, respectively (Supplementary Figure S2c). We used the adjusted transcript annotation in all subsequent analyses.
Next, we extracted reads containing the 5′ adapter from each 5TERA library to investigate the native 5′ ends of RNA molecules. The vast majority of molecules with 5′ adapter (∼81%) reach the 3′ end in all libraries. As expected, we find only ∼6% of 5P molecule ends (5P-Poly(A) library) reaching the transcript 5′ end while the remainder (∼94%) is distributed along the mRNA body indicating they are mostly decay intermediates of polyadenylated mRNAs. Similarly, we find ∼5% of 5OH-containing molecules (5OH-Poly(A) library) reach the transcript 5′ end. We find that ∼29% of the 5′ ends of capped molecules per transcript (Cap-Poly(A) library) reach the 5′ ends indicative of full-length molecules. The remainder (∼71%), whose 5′ ends are downstream of the TSS, are indicative of mRNA molecules derived from alternative TSSs (51), un-annotated isoforms, or recapped transcripts (11,12,71). In the Cap & 5P-Poly(A) library, which identifies the 5′ ends of both capped and 5P-containing molecules, we find that ∼37% of molecules per transcript reach the 5′ end (Supplementary Figure S2d). The differences in 5′ end distributions between adapted and non-adapted molecules in Cap & 5P-Poly(A) libraries might be partially explained by technical errors during ONT sequencing. To corroborate our findings, we analyzed HeLa datasets obtained by an orthogonal approach, Cap-analysis of gene expression (CAGE)-Seq (31). CAGE is a highly specific method for identifying capped ends of RNA molecules (72) and widely used for TSS annotation (51,73). Application of CAGE to nascent elongating transcripts (NET-CAGE) has further enriched annotation and characterization of 5′ ends of RNAs (56). We detect a similar distribution of full-lengthiness with ∼36% of all CAGE signals found in close proximity to 5′ ends of transcripts.
To investigate TERA-Seq performance in sequencing RNA molecules of various sizes, we examined the positional distribution of 5′ ends of molecules on meta-mRNAs grouped by the transcript length. To maintain accurate 5′ end identification, for all subsequent analyses, we use only RNA molecules containing the 5′ adapter from each library. We find that in transcripts shorter than 1 kilobases (kb), ∼60% of the 5′ ends of capped molecules per transcript reach the 5′ ends while this percentage drops to ∼30% for transcripts longer than 1.5 kb (Cap-Poly(A) library; Figure 1D). We detect similar distribution of the 5′ ends of molecules from 5P & Cap-Poly(A) library (Figure 1D). The 5′ ends of molecules with 5P (5P-Poly(A) library) and 5OH (5OH-Poly(A) library) are more evenly distributed along the meta-mRNA for the various transcript lengths, although we see a higher percentage of molecules reaching the 5′ ends in shorter 5P- bearing transcripts (Figure 1D). Collectively, these findings reflect both the inherent challenges of quantifying complex transcriptomes composed of RNAs of varying sizes as well as the underlying biology of mRNA production and decay. Longer genes are more likely to generate shorter capped isoforms from ATI or from recapping of decay fragments, than shorter genes. Once initiated, decay of a short transcript to completion is expected to proceed faster than that of a long transcript, resulting in fewer lingering decay fragments.
Identification of single, capped RNA molecules using 5TERA
We then compared the 5′ ends of mRNAs from all 5TERA libraries, to those found by CAGE-Seq (31), and by NET-CAGE (56). We find that ∼52% and ∼61% of the 5′ ends of molecules in Cap-Poly(A) and Cap & 5P-Poly(A) libraries, respectively, overlap CAGE signals (51) within a 10 nt window (Figure 2A and Supplementary Figure S3a). As expected, we observe less overlap between CAGE signals and 5P ends (∼35%, Supplementary Figure S3b) or 5OH ends (∼38%, plot not shown) of molecules. Overlap of random positions (control) within the same 10 nt window is ∼12%. Cumulative plots with an extended, 500 nt window, are shown in Supplementary Figure S3c. A representative example shows the cap- and 5P-containing molecules for FTH1-201 transcript along with annotated transcript landmarks (exon-exon boundaries, CAGE (73), NET-CAGE (56) and APA sites (53)) both as cumulative coverage (Figure 2B) and at the level of individual molecules (Figure 2C), enabling detailed examination of individual mRNAs. Next, we examined the proximity of the 5′ ends of mRNAs from all 5TERA libraries to transcriptionally active promoters based on the latest ENCODE-annotated, Promoter-Like Signatures (PLS) (54). As shown in Figure 2D, adapted 5′ ends from the Cap-Poly(A) and Cap & 5P-Poly(A) libraries show a strong and tight signal just downstream of active promoters, which is weaker in adapted 5′ ends from 5P-Poly(A) library, and essentially absent in adapted 5′ ends from the 5OH-poly(A) library. The 5′ ends of non-adapted molecules in all libraries are expected to represent many full-length molecules and consequently, show strong and tight signal just downstream of active promoters in all libraries (Figure 2D).
Identification of native 3′ ends of single RNA molecules by direct sequencing with 3′ adapter ligation (TERA3)
Sequencing of single RNA molecules from total RNA, without oligo-dT selection, offers distinct advantages not afforded by the existing ONT platform. First is the sequencing of non-polyadenylated transcripts. Second is the ability to quantify polyadenylated versus non-polyadenylated molecules for each transcript isoform, enabling comparisons between the presence and the length of poly(A) tail to other transcript features at the single molecule level. This can illuminate biological aspects of RNA processing and decay.
We used custom antisense biotinylated DNA oligos to subtract rRNAs and abundant small non-coding RNAs from total RNA by streptavidin beads capture (9,43). We retained the eluate, containing RNAs with and without poly(A) tails, for the library preparation. As an alternative method of rRNA depletion, we applied RNase H degradation of rRNAs, guided by tiled antisense DNA oligos, as described in (74). However, we found that although the depletion of rRNAs was nearly complete, there was a prominent off-target digestion of almost all transcripts leading to marked overrepresentation of ∼300 nt RNA fragments in the ONT libraries (data not shown). Although such off-target fragmentation may not be of consequence for short-read RNA-Seq, it precludes long-read RNA sequencing. Recently, similar off-target effects of RNase H that is detrimental for ribosome profiling were reported (75). Therefore, we used our original depletion strategy based on biotinylated antisense oligos (43), followed by ligation of an RNA adapter to the 3′ ends of eluted RNA molecules (3′ adapter; Supplementary Table S1). We used a custom DNA duplex adapter whose top strand contains the RTA and the bottom contains a complementary sequence to the 3′ adapter (Figure 3A and Supplementary Table S1). In total, we prepared three TERA3 biological replicate libraries.
We find that TERA3 detects full-length as well as shorter molecules, representing processing and decay intermediates, as revealed by analysis on meta-mRNAs (Figure 3B), and by visualizing individual molecules mapping to transcripts (representative example shown in Figure 3C). We find that the largest variation involves the 5′ end of molecules while the 3′ end is more well-defined (Figures 3B and C), even though TERA3 does not enrich for poly(A) tailed transcripts and the input is total RNA. To confirm the efficiency of the TERA3 protocol to correctly identify molecules without a poly(A) tail, we extracted reads mapping to histone transcripts, as they are not expected to have poly(A) tails (76). Indeed, ∼99% of histone mapped reads did not bear any poly(A) tail. We next examined the distribution of 3′ ends of all sequenced molecules in relationship to annotated APA sites regardless of the poly(A) tail status. We find an ∼82% overlap between 3′ ends identified by TERA3 and human APA sites (53) within a 10 nt window (Figure 3D and Supplementary Figure S4a). Overlap of random positions (control) within the same window is ∼4%. We find that ∼75% of molecules in the TERA3 libraries contain a poly(A) tail; ∼4% do not contain a poly(A) tail and represent histone mRNAs; and the remainder ∼21% of molecules are de-adenylated protein-coding transcripts. Of the non-histone mRNA molecules without poly(A) tail, we find that ∼41% have 3′ ends mapped more than 10 nt upstream from APA sites, representing mostly decay intermediates. The remainder, have 3′ ends mapped within 10 nt upstream from APA sites and likely represent deadenylated transcripts. Collectively, these findings indicate that at steady state, the majority of mRNAs have intact 3′ ends.
Poly(A) tail characterization with TERA-Seq
To examine the association of poly(A) tails with mRNAs and their decay fragments, we used Nanopolish (27) to compute the poly(A) tail lengths of molecules in all libraries. Addition of the 3′ adapter does not disrupt the ability of Nanopolish to detect poly(A) tails (representative example shown in Supplementary Figure S4b, and see Supplementary Note). We observe a median tail length between 101 to 113 nt in 5TERA and CTRL libraries, and 72 to 96 nt in TERA3 libraries (Figure 4A and Supplementary Figures S4c, d). These values are in broad agreement with poly(A) tail lengths identified with short-read based methods (77) and other long-read human sequencing datasets (23,27). The smaller median value of poly(A) length in some of the TERA3 libraries is likely a reflection of the oligo-dT-enrichment step, which can introduce selection bias for molecules with longer tails (77) in 5TERA libraries (Supplementary Figure S4e).
We next examined poly(A) tails of individual RNA molecules. For each transcript, we plotted the distribution of 5′ and 3′ ends of the corresponding molecules binned across the transcript length (20 equal length bins), followed by the length of their poly(A) tail (representative example is shown in Figure 4B). Interestingly, this reveals marked variation of the poly(A) tail of single molecules in both Cap-Poly(A) and 5P-Poly(A) libraries. Notably, poly(A) tails from capped FTH1-201 molecules reach up to 546 nt (median 84 nt). We find no relationship between the level of 5′ end decay and the poly(A) tail length, when examining individual molecules (Figure 4C) or transcripts (Figure 4D) from the 5P-Poly(A) library. This indicates that mRNA degradation at the single molecule level is not strictly dependent upon prior deadenylation. Although we show a wide range of poly(A) tail lengths, we note that the precision of Nanopolish to call poly(A) tails has been tested on synthetic tails with lengths of up to 150 nt (27,78).
True, end-to-end sequencing of single, native RNA molecules with concurrent 5′ and 3′ adapter ligation (5TERA3)
To simultaneously identify both ends of single RNA molecules independent of the presence of poly(A) tails, we ligated adapters at both 5′ and 3′ ends of transcripts isolated from total RNA after depletion of rRNAs and abundant small non-coding RNAs (Figure 5A). We converted the 5′ cap of transcripts to a 5P, ligated the biotinylated 5′ adapter and enriched ligated molecules on streptavidin beads, capturing both capped molecules and decay or processing intermediates with pre-existing 5P. After washes to remove non-ligated transcripts without the 5′ adapter, we ligated the 3′ adapter and proceeded with downstream steps for ONT sequencing (Figure 5A). We prepared three 5TERA3 biological replicate libraries, which were later combined into one for subsequent analyses. The sequencing depth of the 5TERA3 libraries was heavily affected by adapters-only reads, a byproduct of using adapters without blocked 3′ end, which was necessary for ligating the 5′ adapter to the RNA molecule, and the 3′ adapter to the RTA adapter.
We find that ∼60% of molecules in the 5TERA3 libraries contain a poly(A) tail, with the median tail length of ∼84 nt (Figure 5B). To explore the difference between transcripts with and without poly(A) tail, we first examined the distribution of 5′ and 3′ ends of RNA molecules on meta-mRNA plots. About 60% of molecules both with or without poly(A) tails reach the annotated 5′ ends. Approximately 90% of molecules with poly(A) tails (Figure 5C; top) reach the annotated 3′ end compared to only ∼30% of molecules without poly(A) tails (Figure 5C; bottom). A representative example for the TMSB10 gene is shown in Figure 5D and reveals the full spectrum of RNA molecules. In addition to full-length polyadenylated molecules, we detect numerous decay intermediates where most of them retain complete 3′ ends and have poly(A) tails of various lengths (Figure 5D), congruent with findings from 5TERA and TERA3. Decay intermediates with 3′ truncations lacking poly(A) tail are also identified but they are a notable minority (arrowheads, Figure 5D). For comparison, alignment of sequenced molecules from the CTRL-Poly(A) library, to TMSB10-201, is shown in Supplementary Figure S5. In contrast to TERA-Seq, Illumina RNA-Seq (79,80), is unable to identify the extreme ends of molecules or to unambiguously assign short-reads to individual RNA molecules (Figure 5D).
Collectively, findings from all TERA-Seq libraries indicate that at steady state, the majority of transcripts have intact 3′ ends, and are composed of full-length molecules along with numerous, often shorter, decay and processing products. We also find that human mRNAs often decay from their 5′ ends.
Cotranslational mRNA decay identified with TERA-Seq
By analyzing the relationship between native ends of RNA decay intermediates identified by Akron-Seq, a technique based on short-read sequencing (43), to the position of translating ribosomes, identified by ribosome-protected fragments (RPFs), we previously described ribothrypsis, a process that degrades canonical human mRNAs as they are being translated (9). To assess if TERA-Seq is capable of capturing ribothrypsis signatures, we plotted the distribution density of 5P ends of RNA molecules as identified by 5TERA, upstream and downstream of 5′ ends of RPFs from HeLa Ribo-Seq datasets (81) (Figure 6A). For direct comparison, we performed the same analysis using 5′ ends of polyadenylated RNAs previously identified with Akron-Seq (9). We detect clear 3 nt periodicity (Figure 6B), which we verify on the frequency domain with discrete Fourier transformation (Figure 6C), indicating that TERA-Seq, like Akron-Seq, identifies the cotranslational generation of 5′ ends of mRNA fragments. Likewise, the shape of the plots is very similar, with an increase in density starting at position 0 and decrease towards the end of the RPF at position ∼26, one of the characteristics of ribothrypsis (Figure 6B) (9). We also find significant overlap between the 5P ends of mRNA fragments identified by TERA-Seq and Akron-Seq (Figure 6D). Finally, just as we observed with Akron-Seq (9), we find that the regions around 5′ ends of mRNAs identified from adapted molecules in the 5P-Poly(A) library, are conserved in vertebrates (Figure 6E). Notably, the conservation is higher for reads with 5′ adapter compared to reads without (Figure 6E), further supporting that adapter ligation enables accurate identification of native 5′ ends of RNAs. Together, these findings indicate that TERA-Seq can also be employed to study mRNA translation and decay
Figure 6.
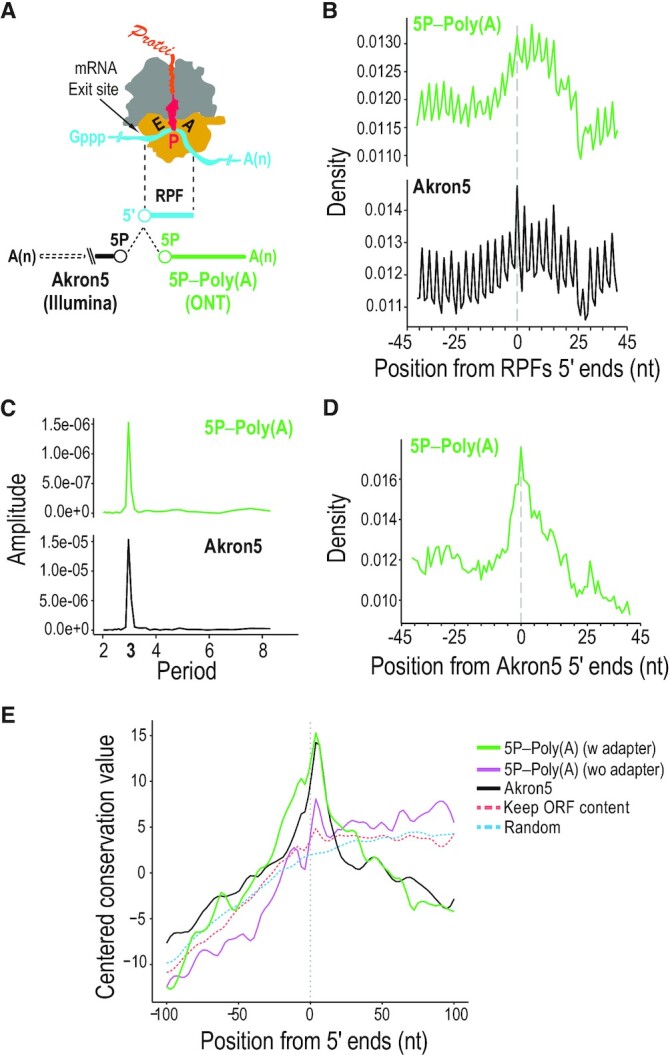
Cotranslational mRNA decay identified with TERA-Seq. (A) Schematic of elongating ribosome with ribosome-protected fragment (RPF) and relative position analyses of 5′ ends from TERA-Seq (5P–Poly(A); ONT) and Akron-Seq (Akron5; Illumina) to the 5′ ends of RPFs. E, tRNA-exit site; P, peptidyl-tRNA site; A, aminoacyl-tRNA-binding site; yellow, 40S subunit with mRNA channel; grey, 60S subunit with polypeptide channel; red, peptidyl-tRNA attached to nascent protein; Gppp, 5′ cap; A(n), poly(A) tail; 5P, 5′ monophosphate. (B) Density plots of 5′ ends distances from indicated libraries relative to RPF 5′ ends (centered at position 0) in coding regions. RPF, ribosome-protected fragment. nt, nucleotide. (C) Discrete Fourier transformation of read density around RPFs for indicated libraries. (D) Density plot of 5′ ends distances from TERA-Seq (5P–Poly(A)) relative to Akron-Seq (Akron5). (E) Evolutionary conservation for 100 vertebrates (PhastCons) upstream and downstream of 5′ ends of mRNAs identified from the 5P-Poly(A) TERA-Seq library; adapted (green) and non-adapted (purple) reads and reads from Akron5 Illumina library (black) are shown. A random control maintaining the nucleotide and open reading frame (ORF) context of 5P-Poly(A) 5′ ends (dashed orange) and a completely random control (dashed blue) are shown.
Altogether, we generated 12 TERA-Seq libraries and one conventional ONT library (CTRL-Poly(A)) of HeLa transcriptome and sequenced ∼24 million direct RNA reads (Supplementary Table S2). To generate each 5TERA library, poly(A) RNA isolated from ∼75 μg of total RNA was used. To generate each TERA3 and 5TERA3 libraries, ∼100 and ∼130 μg of total RNA was used respectively and rRNAs and abundant small, non-coding RNAs were subtracted. Each TERA-Seq library was run in one R9.4 flow cell on a MinION device. Time considerations and resource requirements for all TERA-Seq protocol variants are shown in Table 1. We find high correlation of transcript expression across all libraries (Supplementary Figure S6). On average, ∼85% of all raw reads from 5TERA, 76% from TERA3 and 39% from 5TERA3 libraries used for the main analyses map to the human genome. From those, ∼84%, 11% and 10% mapped to the protein-coding transcriptome respectively (Supplementary Table S2). The reads not mapping to the genome in the 5TERA3 libraries are almost exclusively shorter than 200 nt and primarily represent adapter-dimers ligated to the custom RTA. If we remove these short reads, the genome mapping rate increases to ∼86%.
Table 1.
TERA-Seq: reagents, major steps and time considerations
| Reagents (company, catalogue number) | 5TERA | TERA3 | 5TERA3 |
|---|---|---|---|
| Trizol (Thermo Fisher, 15596026) | Y | Y | Y |
| Acid Phenol/Chloroform, pH 4.5 (Ambion, AM9722) | Y | Y | Y |
| Chloroform (Sigma-Aldrich, C2432) | Y | Y | Y |
| RQ1 nuclease-free DNase enzyme (Promega, M6101) | Y | Y | Y |
| RNasin Ribonuclease Inhibitor (Promega, N2515) | Y | Y | Y |
| 10 mM dNTP solution (NEB, N0447) | Y | Y | Y |
| T4 RNA Ligase I (NEB, M0204S) | Y | Y | Y |
| RtcB (NEB, M0458S) | Y | ||
| Quick CIP (NEB, M0525S) | Y | ||
| RppH (NEB, M0356S) | Y | Y | |
| 10x Thermopol buffer (NEB, B9004S) | Y | Y | |
| Quick Ligation Reaction Buffer (NEB, B6058) | Y | Y | Y |
| T4 DNA Ligase 2M (NEB, M0202) | Y | Y | Y |
| SuperScript III Reverse Transcriptase (Thermo Fisher, 18080044) | Y | Y | Y |
| Dynabeads Oligo(dT)25 (Thermo Fisher, 61002) | Y | ||
| Dynabeads MyOne streptavidin C1 (Thermo Fisher, 65001) | Y | ||
| RNAClean XP (Beckman Coulter, A63987) | Y | Y | Y |
| Qubit dsDNA HS assay kit (Thermo Fisher, Q32851) | Y | Y | Y |
| Total amount of input RNA (μg) | 75 | 100 | 130 |
| Buffers | 5TERA | TERA3 | 5TERA3 |
| Washing buffer B (10 mM Tris–Cl pH 7.5, 150 mM LiCl, 1 mM EDTA pH 8.0) | Y | ||
| 1× Annealing Buffer (10 mM Tris–HCl pH 8.0, 25 mM NaCl, 0.1 mM EDTA) | Y | Y | |
| 1× BW Buffer (5 mM Tris–Cl pH 7.5, 0.5 mM EDTA pH 8.0, 1 M NaCl) | Y | ||
| Formamide Elution Buffer (95% formamide, 5 mM EDTA pH 8.0) | Y | ||
| Major Steps | 5TERA | TERA3 | 5TERA3 |
| Total RNA isolation, DNase treatment, Agilent/agarose assessment | 1 | 1 | 1 |
| Poly(A) mRNA enrichment, enzymatic treatment, and library generation | 1 | ||
| Depletion #, enzymatic treatment, and library generation | 2 | 3 | |
| MinION sequencing run | 2 | 2 | 2 |
| Total numbers of days | 4 | 5 | 6 |
Y, yes; empty cells, not applicable; #, after depletion of ribosomal RNAs and snRNAs in TERA3 and 5TERA3, RNA can be stored at –80°C or processed immediately for library generation.
The longest alignment was 11 092 nt to the reference genome, and 8 194 nt to the reference transcriptome. The average median mapped read length was ∼549 nt to the genome and ∼515 nt to the transcriptome.
DISCUSSION
Advantages of TERA-Seq
By identifying endogenous RNA ends, TERA-Seq can harness the full potential of ONT to sequence native, single RNA molecules directly. Existing direct RNA sequencing methods can only identify the 5′ ends of capped, polyadenylated RNAs (24,26), In addition to capped ends, TERA-Seq can identify monophosphorylated and hydroxylated 5′ ends of RNAs, allowing studies of full-length, processing and decay intermediates at the single molecule level. Furthermore, TERA-Seq can identify native 3′ ends of all RNAs by capturing both molecules with and without poly(A), and preserve the original poly(A) tail length. Additional benefits of direct sequencing of RNA molecules are identification of base modifications, and the avoidance of biases associated with reverse transcription and amplification, steps that are required for other commonly used cDNA-based, RNA-Seq platforms, such as Illumina and PacBio. TERA-Seq uses common molecular biological methods and reagents, and is simple to use, generating sequencing results with a short turnaround time (Table 1).
Although Illumina short-read sequencing-based studies of RNA ends have led to many important discoveries, they are inherently limited as the sequenced fragments cannot be precisely assigned to individual molecules (7,9,31,33–35,77,81,82). To address this impediment, analyses involving short reads are often aggregated to the gene level, missing molecular events that take place at the level of individual RNA molecules. For example, metabolic labeling combined with Illumina-based sequencing of RNA 3′ ends found that shortening of the poly(A) tail is the initiating step in canonical mRNA decay in mouse 3T3 cells, and that most mRNA molecules degrade only after their tail lengths shorten below 25 nt (83). Yet, TERA-Seq reveals that many decay fragments have poly(A) tails as long as their parental, full-length molecules, indicating that in human HeLa cells, decay can initiate from the 5′ end or from within the mRNA body and is not strictly dependent on prior deadenylation. Very recently, a study of mRNA dynamics with nano-ID, an ONT-based method, revealed a large variation between the stability of individual RNA isoforms in human K562 cells, which cannot be identified by aggregating analysis to the gene level (84).
We envision that combining TERA-Seq with metabolic labeling, such as nano-ID, may constitute a powerful platform to investigate RNA synthesis and turnover at the single-molecule and single-isoform level in future studies. Because TERA-Seq captures the precise 5′ ends of capped and decay intermediates, coupling TERA-Seq with metabolic labeling and gene knockouts of relevant pathways, may identify the temporal sequence of events, such as decapping, deadenylation, exo- and endonucleolysis, and the contribution of these processes to the decay of each transcript.
Illumina-based CAGE-Seq, identified numerous cap signals downstream of canonical TSSs (51,73). Without knowledge of the downstream sequence, it is difficult to infer how these capped ends arise. TERA-Seq supports recapping origin for many of these molecules, consistent with previously described cytoplasmic recapping of decay fragments (11,12). It also shows that some of the internally capped molecules might have originated from alternative TSSs. The biological significance of recapping is largely unknown (12). We speculate that cytoplasmic recapping may facilitate translation-dependent decay of mRNA fragments that escaped 5′-to-3′ exonucleolysis and we expect that TERA-Seq will facilitate future studies to address the biology of recapping (12).
Studies often discard shorter and incomplete molecules as potential RNA degradation products created during library preparation. TERA-Seq shows that many such molecules are biologically relevant and should be considered during downstream analyses. It is also important to note that studies focusing on novel isoform identification have to be carefully interpreted. For example, novel shorter RNA isoforms that share the exon structure with the parent transcript may, in many cases, represent biologically degraded and re-capped molecules, or artificial products created by ONT sequencing errors. TERA-Seq is able to distinguish the latter technical errors from the actual novel biological isoforms, paving the way for true isoform detection and discovery.
TERA-Seq may also prove useful for the study of RNA modifications and their functions in splicing and mRNA turnover. For example, by manipulating the levels of RNA modification enzymes, readers and erasers, and by employing TERA-Seq, changes in capped, processed or decay mRNA intermediates can be assessed at the single-molecule level. More generally, TERA-Seq can identify changes in capped, processed or decay intermediates at the single RNA molecule level under any experimental manipulation.
Limitations of TERA-Seq
While TERA-Seq resolves many of the ONT direct RNA sequencing limitations, several challenges still remain. A relatively large amount of sample RNA is required. Such amount is easily obtained from cultured cells, or from animal or human tissues but may be a limiting factor for clinical samples. TERA-Seq cannot be used for single-cell RNA-Seq. The basecalling accuracy of ONT overall is lower than Illumina or PacBio. PacBio has superior accuracy for homopolymers and the PacBio-based FLAM-Seq can identify non-A nucleotides within poly(A) tails (23), which is not currently possible with ONT. TERA-Seq and ONT in general, has lower throughput than Illumina. Moreover, electronic signal noise from nanopores often results in incomplete sequencing (27), reducing the final number of usable fully sequenced reads. The latter is, at least in part, addressed by TERA-Seq since endogenous ends are marked by adapter ligation. We anticipate that improvements of ONT flow cells and software may address such limitations.
In summary, we regard that TERA-Seq is a robust method for accurate and improved transcriptome characterization, eminently suitable for applications where end-to-end sequencing of single, native RNA molecules and their modifications is desirable.
DATA AVAILABILITY
All datasets have been deposited in the Sequence Read Archive under the BioProject accession number PRJNA673166. Source code utilized for all analyses and fast5 files are available from GitHub at https://github.com/mourelatos-lab/TERA-Seq_manuscript.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: F.I., M.M. and Z.M. conceived the study. F.I. and Z.M. led the study. F.I. developed TERA-Seq, modified the nanopore direct RNA sequencing protocol, performed and interpreted all wet lab experiments, and library generation and sequencing. J.O. and M.M. designed, performed and interpreted all computational analyses with contributions by F.I. All authors analyzed the data. F.I. and Z.M. wrote the manuscript with input and edits from J.O. and M.M.
Contributor Information
Fadia Ibrahim, Department of Pathology and Laboratory Medicine, Division of Neuropathology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; Department of Biochemistry and Molecular Biology, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA 19107, USA.
Jan Oppelt, Department of Pathology and Laboratory Medicine, Division of Neuropathology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
Manolis Maragkakis, Laboratory of Genetics and Genomics, National Institute on Aging, Intramural Research Program, National Institutes of Health, Baltimore, MD 21224, USA.
Zissimos Mourelatos, Department of Pathology and Laboratory Medicine, Division of Neuropathology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [GM133154 to Z.M.]; Intramural Research Program of the National Institute on Aging, NIH [to M.M.]. Funding for open access charge: Intramural Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Krebs J.E., Goldstein E.S., Kilpatrick S.T.. Lewin's Genes. 2018; 12th edn.Jones & Bartlett Learning. [Google Scholar]
- 2. Schoenberg D.R., Maquat L.E.. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012; 13:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Isken O., Maquat L.E.. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007; 21:1833–1856. [DOI] [PubMed] [Google Scholar]
- 4. Shoemaker C.J., Green R.. Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 2012; 19:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inada T. Quality controls induced by aberrant translation. Nucleic. Acids. Res. 2020; 48:1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu W., Sweet T.J., Chamnongpol S., Baker K.E., Coller J.. Co-translational mRNA decay in Saccharomyces cerevisiae. Nature. 2009; 461:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pelechano V., Wei W., Steinmetz L.M.. Widespread Co-translational RNA decay reveals ribosome dynamics. Cell. 2015; 161:1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu X., Willmann M.R., Anderson S.J., Gregory B.D.. Genome-wide mapping of uncapped and cleaved transcripts reveals a role for the nuclear mRNA Cap-binding complex in cotranslational RNA decay in arabidopsis. Plant Cell. 2016; 28:2385–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ibrahim F., Maragkakis M., Alexiou P., Mourelatos Z.. Ribothrypsis, a novel process of canonical mRNA decay, mediates ribosome-phased mRNA endonucleolysis. Nat. Struct. Mol. Biol. 2018; 25:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuck A.C., Rankova A., Arpat A.B., Liechti L.A., Hess D., Iesmantavicius V., Castelo-Szekely V., Gatfield D., Buhler M.. Mammalian RNA decay pathways are highly specialized and widely linked to translation. Mol. Cell. 2020; 77:1222–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiss D.L., Oman K., Bundschuh R., Schoenberg D.R.. Uncapped 5′ ends of mRNAs targeted by cytoplasmic capping map to the vicinity of downstream CAGE tags. FEBS Lett. 2015; 589:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trotman J.B., Schoenberg D.R.. A recap of RNA recapping. Wiley Interdiscip. Rev. RNA. 2019; 10:e1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arango D., Sturgill D., Alhusaini N., Dillman A.A., Sweet T.J., Hanson G., Hosogane M., Sinclair W.R., Nanan K.K., Mandler M.D.et al.. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018; 175:1872–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaccara S., Ries R.J., Jaffrey S.R.. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019; 20:608–624. [DOI] [PubMed] [Google Scholar]
- 16. Wang Z., Gerstein M., Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oikonomopoulos S., Bayega A., Fahiminiya S., Djambazian H., Berube P., Ragoussis J.. Methodologies for transcript profiling using long-read technologies. Front. Genet. 2020; 11:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B.et al.. Real-time DNA sequencing from single polymerase molecules. Science. 2009; 323:133–138. [DOI] [PubMed] [Google Scholar]
- 19. Au K.F., Sebastiano V., Afshar P.T., Durruthy J.D., Lee L., Williams B.A., van Bakel H., Schadt E.E., Reijo-Pera R.A., Underwood J.G.et al.. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. U. S. A. 2013; 110:E4821–E4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kulpa D., Topping R., Telesnitsky A.. Determination of the site of first strand transfer during Moloney murine leukemia virus reverse transcription and identification of strand transfer-associated reverse transcriptase errors. EMBO J. 1997; 16:856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu Y.Y., Machleder E.M., Chenchik A., Li R., Siebert P.D.. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Bio. Tech. 2001; 30:892–897. [DOI] [PubMed] [Google Scholar]
- 22. Ramskold D., Luo S., Wang Y.C., Li R., Deng Q., Faridani O.R., Daniels G.A., Khrebtukova I., Loring J.F., Laurent L.C.et al.. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012; 30:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Legnini I., Alles J., Karaiskos N., Ayoub S., Rajewsky N.. FLAM-seq: full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods. 2019; 16:879–886. [DOI] [PubMed] [Google Scholar]
- 24. Parker M.T., Knop K., Sherwood A.V., Schurch N.J., Mackinnon K., Gould P.D., Hall A.J., Barton G.J., Simpson G.G.. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m(6)A modification. Elife. 2020; 9:e49658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garalde D.R., Snell E.A., Jachimowicz D., Sipos B., Lloyd J.H., Bruce M., Pantic N., Admassu T., James P., Warland A.et al.. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods. 2018; 15:201–206. [DOI] [PubMed] [Google Scholar]
- 26. Jiang F., Zhang J., Liu Q., Liu X., Wang H., He J., Kang L.. Long-read direct RNA sequencing by 5′-Cap capturing reveals the impact of Piwi on the widespread exonization of transposable elements in locusts. RNA Biol. 2019; 16:950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J.et al.. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019; 16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soneson C., Yao Y., Bratus-Neuenschwander A., Patrignani A., Robinson M.D., Hussain S.. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 2019; 10:3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Payne A., Holmes N., Rakyan V., Loose M.. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics. 2019; 35:2193–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shatkin A.J., Manley J.L.. The ends of the affair: capping and polyadenylation. Nat. Struct. Biol. 2000; 7:838–842. [DOI] [PubMed] [Google Scholar]
- 31. Shiraki T., Kondo S., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T.et al.. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15776–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramanathan A., Robb G.B., Chan S.H.. mRNA capping: biological functions and applications. Nucleic. Acids. Res. 2016; 44:7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. German M.A., Pillay M., Jeong D.H., Hetawal A., Luo S., Janardhanan P., Kannan V., Rymarquis L.A., Nobuta K., German R.et al.. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008; 26:941–946. [DOI] [PubMed] [Google Scholar]
- 34. Gregory B.D., O’Malley R.C., Lister R., Urich M.A., Tonti-Filippini J., Chen H., Millar A.H., Ecker J.R.. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev. Cell. 2008; 14:854–866. [DOI] [PubMed] [Google Scholar]
- 35. Addo-Quaye C., Eshoo T.W., Bartel D.P., Axtell M.J.. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008; 18:758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011; 44:1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jinek M., Coyle S.M., Doudna J.A.. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol. Cell. 2011; 41:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peach S.E., York K., Hesselberth J.R.. Global analysis of RNA cleavage by 5′-hydroxyl RNA sequencing. Nucleic Acids Res. 2015; 43:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sorrentino S. Human extracellular ribonucleases: multiplicity, molecular diversity and catalytic properties of the major RNase types. Cell. Mol. Life Sci. 1998; 54:785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sorrentino S. The eight human “canonical” ribonucleases: molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010; 584:2194–2200. [DOI] [PubMed] [Google Scholar]
- 41. Soukup G.A., Breaker R.R.. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999; 5:1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Regulski E.E., Breaker R.R.. In-line probing analysis of riboswitches. Methods Mol. Biol. 2008; 419:53–67. [DOI] [PubMed] [Google Scholar]
- 43. Ibrahim F., Mourelatos Z.. Capturing 5′ and 3′ native ends of mRNAs concurrently with Akron sequencing. Nat. Protoc. 2019; 14:1578–1602. [DOI] [PubMed] [Google Scholar]
- 44. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011. 2011; 17:3. [Google Scholar]
- 45. Shen W., Le S., Li Y., Hu F.. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016; 11:e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R.et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maragkakis M., Alexiou P., Nakaya T., Mourelatos Z.. CLIPSeqTools—a novel bioinformatics CLIP-seq analysis suite. RNA. 2016; 22:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lizio M., Harshbarger J., Shimoji H., Severin J., Kasukawa T., Sahin S., Abugessaisa I., Fukuda S., Hori F., Ishikawa-Kato S.et al.. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015; 16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haeussler M., Zweig A.S., Tyner C., Speir M.L., Rosenbloom K.R., Raney B.J., Lee C.M., Lee B.T., Hinrichs A.S., Gonzalez J.N.et al.. The UCSC Genome Browser database: 2019 update. Nucleic. Acids Res. 2019; 47:D853–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Herrmann C.J., Schmidt R., Kanitz A., Artimo P., Gruber A.J., Zavolan M.. PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 2010; 48:D174–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Consortium E.P., Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A.et al.. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hirabayashi S., Bhagat S., Matsuki Y., Takegami Y., Uehata T., Kanemaru A., Itoh M., Shirakawa K., Takaori-Kondo A., Takeuchi O.et al.. NET-CAGE characterizes the dynamics and topology of human transcribed cis-regulatory elements. Nat. Genet. 2019; 51:1369–1379. [DOI] [PubMed] [Google Scholar]
- 57. Lawrence M., Huber W., Pages H., Aboyoun P., Carlson M., Gentleman R., Morgan M.T., Carey V.J.. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lawrence M., Gentleman R., Carey V.. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009; 25:1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T.et al.. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015; 12:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hahne F., Ivanek R.. Visualizing genomic data using gviz and Bioconductor. Methods Mol. Biol. 2016; 1418:335–351. [DOI] [PubMed] [Google Scholar]
- 61. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S.et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Song Y., Liu K.J., Wang T.H.. Elimination of ligation dependent artifacts in T4 RNA ligase to achieve high efficiency and low bias microRNA capture. PLoS One. 2014; 9:e94619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Silber R., Malathi V.G., Hurwitz J.. Purification and properties of bacteriophage T4-induced RNA ligase. Proc. Natl. Acad. Sci. U.S.A. 1972; 69:3009–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cranston J.W., Silber R., Malathi V.G., Hurwitz J.. Studies on ribonucleic acid ligase. Characterization of an adenosine triphosphate-inorganic pyrophosphate exchange reaction and demonstration of an enzyme-adenylate complex with T4 bacteriophage-induced enzyme. J. Biol. Chem. 1974; 249:7447–7456. [PubMed] [Google Scholar]
- 66. Sugino A., Snoper T.J., Cozzarelli N.R.. Bacteriophage T4 RNA ligase. Reaction intermediates and interaction of substrates. J. Biol. Chem. 1977; 252:1732–1738. [PubMed] [Google Scholar]
- 67. Almeida M.V., de Jesus Domingues A.M., Lukas H., Mendez-Lago M., Ketting R.F.. RppH can faithfully replace TAP to allow cloning of 5′-triphosphate carrying small RNAs. MethodsX. 2019; 6:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sessegolo C., Cruaud C., Da Silva C., Cologne A., Dubarry M., Derrien T., Lacroix V., Aury J.M.. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 2018; 9:14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Paul L., Kubala P., Horner G., Ante M., Holländer I., Alexander S., Reda T.. SIRVs: spike-In RNA variants as external isoform controls in RNA-sequencing. 2016; bioRxiv doi:13 October 2016, preprint: not peer reviewed 10.1101/080747. [DOI]
- 70. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S.et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kiss D.L., Oman K.M., Dougherty J.A., Mukherjee C., Bundschuh R., Schoenberg D.R.. Cap homeostasis is independent of poly(A) tail length. Nucleic Acids Res. 2016; 44:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Adiconis X., Haber A.L., Simmons S.K., Levy Moonshine A., Ji Z., Busby M.A., Shi X., Jacques J., Lancaster M.A., Pan J.Q.et al.. Comprehensive comparative analysis of 5′-end RNA-sequencing methods. Nat. Methods. 2018; 15:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Forrest A.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J., Haberle V., Lassmann T., Kulakovskiy I.V., Lizio M., Itoh M.et al.. A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morlan J.D., Qu K., Sinicropi D.V.. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One. 2012; 7:e42882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zinshteyn B., Wangen J.R., Hua B., Green R.. Nuclease-mediated depletion biases in ribosome footprint profiling libraries. RNA. 2020; 26:1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Marzluff W.F., Koreski K.P.. Birth and death of histone mRNAs. Trends Genet. 2017; 33:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chang H., Lim J., Ha M., Kim V.N.. TAIL-seq: genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell. 2014; 53:1044–1052. [DOI] [PubMed] [Google Scholar]
- 78. Krause M., Niazi A.M., Labun K., Torres Cleuren Y.N., Muller F.S., Valen E.. tailfindr: alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA. 2019; 25:1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Park J.E., Yi H., Kim Y., Chang H., Kim V.N.. Regulation of Poly(A) tail and translation during the somatic cell cycle. Mol. Cell. 2016; 62:462–471. [DOI] [PubMed] [Google Scholar]
- 82. Subtelny A.O., Eichhorn S.W., Chen G.R., Sive H., Bartel D.P.. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014; 508:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Eisen T.J., Eichhorn S.W., Subtelny A.O., Lin K.S., McGeary S.E., Gupta S., Bartel D.P.. The dynamics of cytoplasmic mRNA metabolism. Mol. Cell. 2020; 77:786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Maier K.C., Gressel S., Cramer P., Schwalb B.. Native molecule sequencing by nano-ID reveals synthesis and stability of RNA isoforms. Genome Res. 2020; 30:1492–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets have been deposited in the Sequence Read Archive under the BioProject accession number PRJNA673166. Source code utilized for all analyses and fast5 files are available from GitHub at https://github.com/mourelatos-lab/TERA-Seq_manuscript.