

Abstract
Epidemiological studies have long recognized risky behaviors as potentially modifiable factors for the onset and flares of inflammatory bowel disease (IBD); yet, the underlying mechanisms are largely unknown. Recently, the genetic susceptibilities to cigarette smoking, alcohol and cannabis use [i.e. substance use (SU)] have been characterized by well-powered genome-wide association studies (GWASs). We aimed to assess the impact of genetic determinants of SU on IBD risk. Using Mount Sinai Crohn’s and Colitis Registry (MSCCR) cohort of 1058 IBD cases and 188 healthy controls, we computed the polygenic risk score (PRS) for SU and correlated them with the observed IBD diagnoses, while adjusting for genetic ancestry, PRS for IBD and SU behavior at enrollment. The results were validated in a pediatric cohort with no SU exposure. PRS of alcohol consumption (DrnkWk), smoking cessation and age of smoking initiation, were associated with IBD risk in MSCCR even after adjustment for PRSIBD and actual smoking status. One interquartile range decrease in PRSDrnkWk was significantly associated to higher IBD risk (i.e. inverse association) (with odds ratio = 1.65 and 95% confidence interval: 1.32, 2.06). The association was replicated in a pediatric Crohn’s disease cohort. Colocalization analysis identified a locus on chromosome 16 with polymorphisms in IL27, SULT1A2 and SH2B1, which reached genome-wide statistical significance in GWAS (P < 7.7e-9) for both alcohol consumption and IBD risk. This study demonstrated that the genetic predisposition to SU was associated with IBD risk, independent of PRSIBD and in the absence of SU behaviors. Our study may help further stratify individuals at risk of IBD.
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the digestive tract with two major subtypes, ulcerative colitis (UC) and Crohn’s disease (CD). In the past decades, the incidence and prevalence of IBD were increasing worldwide, especially among children (1,2). It is estimated that 568 per 100 000 people in the USA are affected by IBD (249 by UC and 319 by CD) (1). The risk factors of IBD include genetic predisposition, environmental exposure, unbalanced gut microbiome and their complex interplays. Cigarette smoking has long been recognized as an environmental factor modifying IBD risk (3). Interestingly, smoking behavior is reported to have a distinct effect on UC and CD. Specifically, UC is mostly a disease of non-smokers and former smokers, and smoking is protective against UC, whereas a detrimental effect of smoking on the CD risk has been reported (3,4), with interventional studies showing the positive effects of smoking cessation in CD patients. Moreover, alcohol consumption has been considered as a potential trigger for IBD flares (5) and worsening of the IBD symptoms (6); however, its effect on IBD onset has been inconclusive (7,8). Furthermore, over the last decade, interest in the therapeutic potential of cannabis and its constituents in the management of IBD has escalated (9); yet, there are no studies examining the role of cannabinoid use to IBD risk.
It is now recognized that genetic risk of many complex traits and diseases, including IBD and substance use (SU), for example, alcohol, cannabis and cigarettes smoking, is driven by multiple common genetic polymorphisms (10,11) (Table 1). A genome-wide association study (GWAS) of nearly 60 000 individuals has identified 240 IBD susceptibility loci (Table 1) (12,13). Moreover, the recent GWAS & Sequencing Consortium of Alcohol and Nicotine use (GSCAN) study, that aggregated genetic association findings across scores of studies with up to 1.2 million individuals (14), identified 406 loci associated with multiple phenotypes of cigarette use as well as alcohol consumption. Furthermore, the largest cannabis use GWAS (15) based on the analysis of 184 765 subjects revealed 35 significant susceptibility loci.
Table 1.
Study data sets
| Study name | Traits | N | Data type | Reference |
|---|---|---|---|---|
| IBD data sets | ||||
| MSCCR |
|
1246 | Sample-level genotypes and phenotypes | (38) |
| RISK |
|
873 | Sample-level genotypes and phenotypes | (19,40) |
| de Lange et al. |
|
59 957 | Summary GWAS stats | (12) |
| Substance use data sets | ||||
| GSCAN |
|
1 200 000 | Summary GWAS stats | (14) |
| PGC-Alcdep |
|
46 568 | Summary GWAS stats | (36) |
| Cannabis-ICC-UKB |
|
184 765 | Summary GWAS stats | (15) |
aOnly results from the European ancestry cohort were used in the present study; ICC: International Cannabis Consortium; UKB: UK Biobank.
Given an epidemiological link between IBD onset and disease course and risky behaviors, and the strong polygenic predisposition to IBD and various substances use, the goal of this study was to determine whether genetic susceptibility to SU could affect the risk of IBD in independent cohorts of adult and pediatric IBD cases and controls. To capture the polygenic nature of IBD and SU risk, we calculated a series of polygenic risk scores (PRS), which aggregate the effects of disease-associating variants across multiple loci (16,17). PRS derived from large GWASs tend to be more powerful in predicting disease outcomes (16). Identifying genetic correlations between IBD and SU can provide useful etiological insights into IBD pathogenesis (18). Also, given an emerging interest in applying PRS to improve clinical decision-making, this study may help risk stratify individuals at a high risk of IBD.
Results
Association between IBD outcomes and SU PRS in MSCCR
The overall study design is illustrated in Figure 1. We computed PRS in the 1246 Mount Sinai Crohn’s and Colitis Registry (MSCCR) subjects (Supplementary Material, Table S1), using linear combinations of the imputed genotype dosages and regression coefficients from the respective summary association statistics retrieved from previously published GWAS (Table 1), for seven SU traits: alcohol dependency (PRSAlcdep), alcohol drinks per week (PRSDrnkWk), smoking initiation (PRSSmkInit), smoking cessation (PRSSmkCes), age of smoking initiation (PRSAgeSmk), cigarettes per day (PRSCPD) and cannabis use (PRScannabis); and three IBD endpoint variables: IBD (PRSIBD), UC (PRSUC) and CD (PRSCD, see Materials and Methods). For each disease and trait, we calculated eight sets of PRS using GWAS P-value thresholds of 1e-1, 1e-2, 1e-3, 1e-4, 1e-5, 1e-6, 1e-7 and 1e-8 for including Single Nucleotide Polymorphisms (SNPs) in the PRS derivation.
Figure 1 .
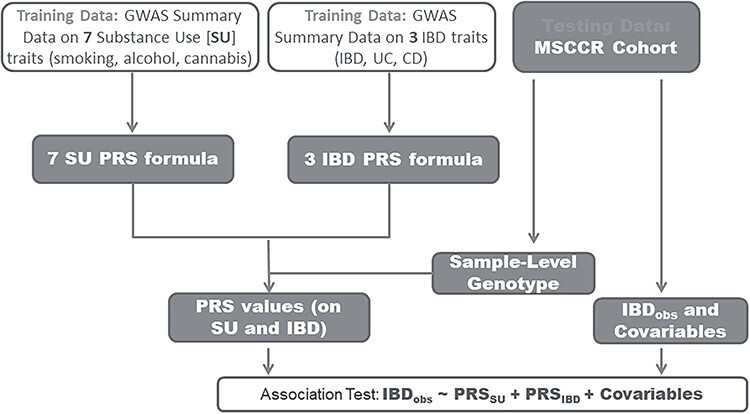
Analysis workflow. Seven SU GWAS: alcohol dependency, alcohol drinks per week, smoking initiation, smoking cessation, smoking age of initiation, cigarettes per day and cannabis use. Three IBD GWASs: IBD, UC and CD.
Marginal associations were tested between each SU PRS and each observed IBD diagnosis (Fig. 2 and Supplementary Material, Table S2). Bonferroni correction was applied to account for multiple testing and we presented adjusted P-value (i.e. Padj). As expected, the associations between observed IBD endpoints and PRSIBD were highly significant (Supplementary Material, Table S3). PRSDrnkWk was inversely associated with IBD risk. One interquartile range (IQR) increase of PRSDrnkWk led to lower IBD (OR = 0.61, Padj = 1.68e-3), CD (OR = 0.58, Padj = 9.25e-4) and UC risks [odds ratio (OR) = 0.64, Padj = 5.37e-2] (see Supplementary Material, Table S2). PRS for smoking behavior traits were correlated with the risk of IBD, UC and CD (Fig. 2). Specifically, higher PRSAgeSmk, representing an older age of smoking initiation, was associated with higher risk of UC (Padj = 4.39e-3), but not CD. One IQR increase of PRSSmkCes was significantly associated with higher IBD (OR = 1.73, Padj = 2.42e-5), CD (OR = 1.73, Padj = 1.41e-4) and UC (OR = 1.70, Padj = 8.29e-4) risks. Of note, the smoking cessation variable was coded as 1 for current smoker and 0 otherwise (14); therefore, our results indicate that genetic determinants of inability to stop smoking significantly increase the risk of IBD, CD and UC.
Figure 2 .
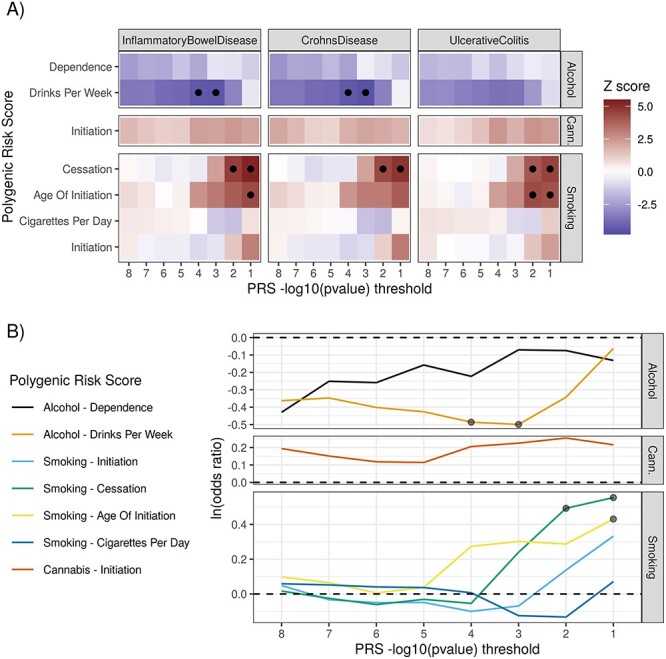
PRSs for SU traits were associated with IBD endpoints. (A) Z-scores of marginal association, estimated through logistic regression on the IBD case/control cohort (MSCCR). Circles mark associations passing the 1% FDR cutoff. (B) Estimated effect sizes of genetically determined SU traits on IBD risk (log-OR). Circles mark associations passing the 1% FDR cutoff. PRSs for three SU traits (alcohol drinks per week, smoking cessation and smoking age of initiation) are significantly associated with IBD risk. We observe similar association patterns for IBD, CD and UC. Large effect sizes are observed for alcohol drinks per week and smoking cessation. Cann., cannabis.
Given PRSDrnkWk, PRSSmkCes and PRSAgeSmk showed significant association with the observed IBD diagnosis, we further examined the associations after controlling for PRSIBD and observed smoking status at enrollment (Models (1–3), see Materials and Methods). To reduce the number of multivariate models, we focused on the PRS that yielded the most significant association in the univariate analysis. The multivariate analysis revealed two key observations. First, the association between SU PRS (i.e. PRSDrnkWk, PRSSmkCes and PRSAgeSmk) and IBD endpoints had consistent direction as in univariate analysis, but of slightly smaller effect sizes (Fig. 3 and Table 2). For example, one IQR increase in PRSDrnkWk was associated with lower IBD (OR = 0.76, P = 0.038) and CD (OR = 0.73, P = 0.031) risk, a smaller effect than in the univariate model (Table 2). Second, the effect size of SU PRS on IBD risk (even when significant) was 4–7 times smaller than that of PRSIBD (Table 2). PRScannabis were not significantly associated with the IBD endpoints (Supplementary Material, Table S2).
Figure 3 .
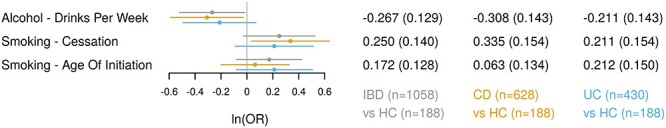
Multivariate regression analysis between IBD endpoints and PRSs for SU traits in MSCCR cohort. Three disease endpoints: IBD, CD and UC. The regression model was adjusted for age, sex, observed smoking status at disease diagnosis, first 10 genetic principal components and PRS of disease endpoint (i.e. IBD, CD and UC). The forest plot presents the OR per interquartile increase in the PRS. Circles: point estimate of log-odds ratio; horizontal error bars: 95% confidence intervals.
Table 2.
Association between IBD/CD/UC diagnosis and PRS in univariate and multivariate analyses
| Dependent variable |
N
a
(cases + controls) |
Regressor | GWAS P-value threshold | Univariate regression | Multivariate regression | ||||
|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P-value | OR | 95% CI | P-value | ||||
| IBD | 1246 | PRSDrnkWk | 1E-3 | 0.61 | 0.49–0.76 | 1.00E-05 | 0.76 | 0.59–0.98 | 0.0375 |
| 1246 | PRSSmkCes | 1E-1 | 1.73 | 1.41–2.13 | 1.44E-07 | 1.28 | 0.98–1.69 | 0.075 | |
| 1246 | PRSAgeSmk | 1E-1 | 1.54 | 1.27–1.87 | 1.31E-05 | 1.19 | 0.92–1.52 | 0.179 | |
| 1246 | PRSIBD | 1E-3 | 4.01 | 3.11–5.18 | 1.43E-26 | 3.63 | 2.79–4.73 | 6.84E-22 | |
| CD | 816 | PRSDrnkWk | 1E-3 | 0.58 | 0.46–0.73 | 5.50E-06 | 0.73 | 0.55–0.97 | 0.0312 |
| 816 | PRSSmkCes | 1E-1 | 1.73 | 1.39–2.15 | 8.39E-07 | 1.40 | 1.04–1.90 | 0.0291 | |
| 816 | PRSAgeSmk | 1E-1 | 1.48 | 1.21–1.80 | 1.02E-04 | 1.06 | 0.82–1.38 | 0.636 | |
| 816 | PRSCD | 1E-3 | 4.48 | 3.37–5.95 | 4.74E-25 | 4.22 | 3.10–5.75 | 9.21E-20 | |
| UC | 618 | PRSDrnkWk | 1E-3 | 0.64 | 0.51–0.82 | 3.19E-04 | 0.81 | 0.61–1.07 | 0.141 |
| 618 | PRSSmkCes | 1E-1 | 1.70 | 1.35–2.13 | 4.93E-06 | 1.23 | 0.91–1.67 | 0.17 | |
| 618 | PRSAgeSmk | 1E-1 | 1.63 | 1.30–2.05 | 2.61E-05 | 1.23 | 0.92–1.66 | 0.159 | |
| 618 | PRSUC | 1E-3 | 3.35 | 2.51–4.47 | 2.56E-16 | 3.63 | 2.68–4.93 | 1.58E-16 | |
The multivariate regression models (Models (1–3)) were specified in the Materials and Methods section. All PRSs were centered and scaled to have median = 0 and IQR = 1. In constructing PRS, the GWAS P-value thresholds were selected to maximize significance of the association between the PRS and IBD diagnosis in univariate regression analysis.
aAnalyses for all 3 dependent variables share the same 188 HC.
Predictive value of PRS for alcohol consumption for IBD risk
To assess the additive predictive value of SU PRS on IBD risk, we used the multivariate model (Models (4–6), see Materials and Methods), to simultaneously examine the effect of PRSIBD and SU PRS, comparing the top/bottom 10th percentiles of each PRS with the rest of the cohort. As expected, the PRSIBD was significantly associated with IBD case status, with subjects in the top 10% of PRSIBD showing a 5.61-fold [95% confidence interval (CI): 2.04–15.41] higher IBD risk than the remaining 95% of subjects (Table 3). Importantly, PRSDrnkWk provided an additional prediction value toward IBD risk independent of PRSIBD. Since the PRSDrnkWk and IBD risk were inversely associated, we focused on subjects within the bottom 10% of PRSDrnkWk, who turned out to have 1.84-fold (95% CI: 1.17–2.89) higher IBD risk when comparing with the rest of the cohort. Similarly, conditional on PRSCD, subjects within the bottom 10% of PRSDrnkWk had 1.94-fold (95% CI: 1.18–3.19) higher CD risk when comparing with the rest of the cohort (Table 3).
Table 3.
Alcohol use PRS provides independent prediction power toward IBD diagnosis
| Endpoint | Dichotomized PRS | OR | 95% CI | |
|---|---|---|---|---|
| IBD | DrnkWk (bottom 10% vs. rest) | 1.83 | 1.17 | 2.87 |
| SmkCes (top 10% vs. rest) | 1.58 | 0.85 | 2.94 | |
| AgeSmk (top 10% vs. rest) | 1.59 | 0.85 | 2.97 | |
| IBD (top 10% vs. rest) | 5.72 | 2.08 | 15.72 | |
| CD | DrnkWk (bottom 10% vs. rest) | 1.92 | 1.17 | 3.15 |
| SmkCes (top 10% vs. rest) | 1.57 | 0.82 | 3.02 | |
| AgeSmk (top 10% vs. rest) | 1.73 | 0.90 | 3.33 | |
| CD (top 10% vs. rest) | 10.15 | 3.16 | 32.55 | |
| UC | DrnkWk (bottom 10% vs. rest) | 1.65 | 0.99 | 2.76 |
| SmkCes (top 10% vs. rest) | 1.74 | 0.89 | 3.39 | |
| AgeSmk (top 10% vs. rest) | 1.51 | 0.76 | 2.97 | |
| UC (top 10% vs. rest) | 4.40 | 1.85 | 10.47 | |
We dichotomized each continuous PRS into a binary variable, for example, PRSDrnkWk to IDrnkWk. In PRSIBD, PRSCD, PRSUC, PRSSmkCes and PRSAgeSmk, if a given subject was within the top 10% of the PRS, we assigned the corresponding binary variable as 1, otherwise 0. If a subject was within the bottom 10% of the PRSDrnkWk, we assigned IDrnkWk = 1, otherwise 0. OR, computed using multivariate model (Models (4–6), see Materials and Methods).
Replication in pediatric Crohn’s disease cohort
We validated the marginal association (i.e. without covariates adjustment) between SU PRS and CD diagnosis in a cohort of 769 pediatric CD cases and 104 age-matched healthy controls (HC) of Caucasian ancestry (RISK cohort, Table 1 and Supplementary Material, Table S4) (19,20). The range and median age in cases were 1.8–17.3 and 12.6 years and in controls were 2.3–16.9 and 12.5 years, respectively, which is below the age of alcohol exposure. We applied the same regression model and parameter setting as in the MSCCR cohort. We confirmed a significant, negative effect of PRSDrnkWk on the risk of CD (OR = 0.78; P = 0.038). The PRS for smoking traits were not significantly associated with CD risk.
Shared risk variants between IBD and alcohol consumption
Comparison of IBD (12) and DrnkWk (14) GWASs revealed a region on chromosome 16 (28.2–29.2 Mbp, hg19) with shared, genome-wide significant variants between the two traits (Supplementary Material, Fig. S1). Colocalization analysis (see Materials and Methods) supported a shared causal variant driving both DrnkWk and IBD (H4 = 89.1%, Supplementary Material, Table S5) and DrnkWk and CD (H4 = 87.6%). Three genes in this region contained non-synonymous variants associated with both IBD and DrnkWk at the genome-wide significance (Table 4): IL27 (rs181206), SULT1A2 (rs1059491) and SH2B1 (rs7498665). IL27 has been extensively studied in relation to IBD, and it plays an important role in the immune response. SULT1A2 is involved in ethanol metabolism and appears to be a plausible gene regulating alcohol consumption. Importantly, these three non-synonymous SNPs are in tight linkage disequilibrium (LD) (rs181206 vs. rs1059491: r2 = 0.74; rs7498665 vs. rs181206: r2 = 0.55; rs7498665 vs. rs1059491: r2 = 0.66), and further functional studies are necessary to dissect their functional roles.
Table 4.
Shared non-synonymous SNPs between GWAS on IBD and GWAS on alcohol drinks per week
| rsID | chr | Position (hg19) | Gene symbol | Alleles | Amino acid change | IBD | Alcohol drinks per week | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ref | alt | ref/alt | Beta | P-value | Beta | P-value | ||||
| rs181206 | 16 | 28 513 403 | IL27 | A | G | L/P | 0.0945 | 9.07e-12 | --0.0121 | 7.67e-09 |
| rs1059491 | 16 | 28 603 655 | SULT1A2 | T | G | N/T | 0.0886 | 4.22e-12 | --0.0124 | 7.9e-10 |
| rs7498665 | 16 | 28 883 241 | SH2B1 | A | G | T/A | 0.0965 | 3.5e-14 | --0.0132 | 2.4e-11 |
ref: Reference allele; alt: Alternative allele
Discussion
IBD is believed to result from the complex interactions between the patient’s genome and environmental exposures (21). While the associations of smoking and alcohol consumption with IBD diagnosis and activity have been extensively reported, the contribution of genetic predisposition to SU, which is now well established (14), to IBD pathogenesis has not been explored. In this study, we demonstrated that genetic determinants of alcohol consumption and smoking behavior, summarized as PRS, are predictors of IBD risk independent of PRSIBD. Specifically, individuals at the bottom 10% of PRSDrnkWk had 1.8-fold higher IBD risk when compared with the remaining 90% individuals after accounting for all covariates. The association between CD and PRSDrnkWk was replicated in a pediatric IBD cohort where the subjects were below the age of alcohol exposure. The data sets consistently indicate that SU PRS influence IBD risk without necessarily modifying SU behavior, which is also supported by a Mendelian randomization study demonstrating that smoking behavior does not affect the risk of developing IBD (22).
Both IBD and SU are polygenic phenotypes, whereby numerous common genetic variants with small effect sizes located in or near various genes contribute to the disease risk or trait variability. PRS assesses the cumulative genetic burden across these susceptibility loci with potential clinical applications for a number of traits (11,23,24). As expected, our study demonstrated a very strong correlation between PRS and a respective diagnosis across various GWAS P-value thresholds (P < 4.9e-16 between PRSIBD and IBD, P < 5.0e-17 between PRSCD and CD, and P < 2.9e-9 between PRSUC and UC; Supplementary Material, Table S3). Individuals at the top 10% of PRSIBD, PRSCD or PRSUC had at least a 4-fold increased risk of IBD, CD or UC, respectively, further supporting the predictive value of PRS. Moreover, lower PRSDrnkWk was associated with a higher risk of IBD, CD and UC at certain GWAS P-value thresholds (i.e. inverse association), with being at the bottom 10% PRSDrnkWk increased the risk of the disease by >80% (statistically significant for IBD and CD, Table 3). Importantly, the association remained significant for IBD and CD after the adjustment for respective PRSIBD or PRSCD. Same direction of the effect, though with lower magnitude, was detected in the pediatric cohort of CD patients who did not reach the age of alcohol or nicotine exposure, further indicating that genetic determinants of alcohol use are negatively associated with CD risk in the absence of actual alcohol consumption.
Also, a positive association was found between PRSSmkCes and risk of IBD, CD or UC, but the significance only remained for CD after adjustment for PRSCD. Smoking cessation was a binary variable contrasting current versus former smoking (14), therefore, indicating a higher risk of the disease associated with the inability to quit smoking, which is consistent with the epidemiological data linking CD risk to nicotine exposure (3). Moreover, higher PRSAgeSmk, representing an older age of smoking initiation, was associated with higher risk of IBD, CD or UC. However, among all smoking PRS, only those of the associations between CD and PRSSmkCes survived the adjustment for the respective disease PRS (Table 2). Further, PRScannabis was not in association with IBD risk.
Our results suggest that SU PRS possess effects on IBD risk, which are both independent of and overlapping with PRSIBD. PRSDrnkWk and PRSSmkCes explained an additional genetic risk associated with IBD diagnosis not captured by PRSIBD, likely because the GWAS that informed the SU PRS derivations (GSCAN study, sample size up to 1.2 million subjects) (14) was much larger than the GWAS used for IBD PRS calculation (sample size ~ 60 000 subjects) (12). It has been previously shown that combining PRS and environment/life-style parameters achieves better prediction than PRS alone (11,24). Our findings showcase the ability to improve disease risk prediction by leveraging PRS of predisposition to life-style behaviors (see Fig. 3 and Table 3), which has been studied using a much larger sample. The effect of alcohol use and smoking on IBD incidence should be systematically investigated in future prospective studies since incorporating alcohol use and/or SU PRS may improve the performance of prediction models toward IBD risk.
There are two possible mechanisms that can explain the genetic overlap between SU and IBD risk. One is a mediation model where the genetic determinants of SU lead to SU behavior which in turn modified the IBD risk. Second is an independent effect model when the genetic determinants of SU affect the IBD risk independent of influencing smoking or drinking behavior. In the MSCCR cohort, smoking status at the time of cohort enrollment was available, and we found that controlling for the observed smoking status did not change the association between smoking PRS and IBD endpoints. Moreover, the RISK cohort subjects (Supplementary Material, Table S4) were below the age of smoking/drinking exposure; however, PRSDrnkWk was still significantly associated with CD diagnosis. Taken together, this evidence supports the independent effect of SU PRS on IBD risk without necessarily modifying SU behavior. Our conclusion is also supported by Mendelian randomization study that smoking behavior does not affect the risk of developing IBD (22).
The pathophysiological mechanisms underlying the connection of SU and IBD are of great importance. In the SU GWAS, Liu et al. (14), in addition to reporting enrichment for nervous system cell types, and more specifically pathways involved into dopaminergic and glutamatergic transmission, among the top SU susceptibility loci, also found enrichment for the immune system and liver cells, though those findings were not confirmed across different analytical methods. Our genetic colocalization analysis identified the Chr16 locus with multiple variants, in moderate or high LD with each other, which played a causal role in both predisposition to alcohol drinking and IBD risk. Polymorphisms in the three genes in this locus, IL27, SULT1A2 and SH2B1, reached genome-wide statistical significance for both phenotypes (Table 4). Variations in IL27, which encodes an immunomodulatory cytokine that regulates adaptive immunity responses, and in SULT1A2, which encodes sulfotransferases that catalyze sulfate conjugation of catecholamines, phenolic drugs and neurotransmitters, have been linked to early onset IBD (25). Deleterious mutations in SH2B1 have been detected using whole exome resequencing of candidate gene in IBD patients with severe early onset IBD (26). At the same time, SULT1A2 is involved in ethanol metabolism and appears to be a plausible gene associated with alcohol consumption, while SH2B1 regulates liver lipid metabolism, which is significantly perturbed by ethanol (27). Moreover, significant implications of the innate immune and complement systems in responses to alcohol have been reported (28–32). Strong links can be also drawn between the variations in these loci and obesity, as all three variations reported here in Table 4 also show a strong, positive association with body mass index (BMI) in a large GWAS meta-analysis of about 700 thousands individuals of European ancestry (P-values all smaller than 1E-34) (32). This last observation might help explain the findings of a positive association between obesity and IBD risk from a recently published meta-analysis of 15.6 million individuals (HR = 1.20, 95% CI: 1.08–1.34) (33). These Chr16 locus genes could be the high-priority candidate genes in future functional studies to determine their role as potential drug targets to treat IBD, especially in individuals with genetic predisposition to SU or obesity.
This study has several limitations. Only individuals of European ancestry were included in our analysis limiting the generalizability of our results. However, the largest GWAS have been conducted in Europeans, and PRS derived using regression coefficients from these GWAS do not perform well in non-Europeans (17,34). We argue the reason that PRSDrnkWk and PRSSmkCes explained an additional genetic risk associated with IBD diagnosis not captured by PRSIBD is because the SU GWAS is much larger (i.e. up to 1.2 million subjects) (14) than the IBD GWAS to date (sample size~60 000 subjects) (12). This rationale should be examined in the future when the size and power of IBD GWASs further increase.
In summary, in this study, we found that genetic determinants of alcohol consumption and smoking behavior strongly correlated with IBD risk independent of genetic predictors of IBD. Individuals at the bottom 10% of PRSDrnkWk having their IBD risk increased by over 80%. We also identified shared genetic effects between SU and IBD diagnosis through the chromosome 16 locus, which encodes genes involved in the IBD pathogenesis and ethanol-related metabolism. Our study demonstrates the value of SU PRS to better understand disease etiology and to help stratify individuals at high risk of IBD.
Materials and Methods
Summary-level GWAS data
We curated three sets of GWAS results: IBD GWAS reported by de Lange et al. (deLange-IBD) (12), GSCAN (14) and Psychiatrics Genomics Consortium-Alcohol Dependency Study (PGC-Alcdep) (35) (Table 1). In total, summary-level data for nine traits were used, including three IBD traits: IBD diagnosis (12), UC diagnosis (12) and CD diagnosis (12); and six SU traits: alcohol use, defined as drinks per week (14); alcohol dependence, defined as a binary trait (35), smoking initiation, smoking cessation, age of smoking initiation, smoking cigarettes per day (14,36) and lifetime cannabis use (15) (Table 1).
Individual-level IBD genotype and phenotype data sets
Patients UC or CD and unaffected controls (ages 18 and older) were recruited into the MSCCR between December 2013 and September 2016 via a protocol approved by the Icahn School of Medicine at the Mount Sinai Institutional Review Board. Study participants were enrolled during an office visit at the Susan and Leonard Feinstein IBD Center for a routine colonoscopy, while the majority of controls were undergoing endoscopy as part of colon cancer screening. All patients gave written informed consent to participate in the study. The clinical and demographic information was obtained through an extensive questionnaire. UC or CD diagnoses were confirmed by chart reviews of electronic medical records. Blood samples were obtained, DNA were extracted and genotype data were generated using the high-density Illumina Multi-Ethnic Global Array (MEGAEX) and Infinium ImmunoArray-24 v2 BeadChip arrays, as reported elsewhere (37). Genotypes were further imputed using the Michigan Imputation Server (38) and using the 1000 Genomes reference panel. A summary of the study cohort demographics, clinical variables and smoking behavior is reported in Supplementary Material, Table S1.
The Crohn’s and Colitis Foundation-sponsored RISK study is a prospective inception cohort study, which enrolled pediatric patients with IBD from 28 sites in North America between 2008 and 2012 (20,39). We used the CD cases and controls of Caucasian ancestry (Supplementary Material, Table S4) to replicate the association between SU PRS and CD diagnosis.
PRS construction
The overall study design is illustrated in Figure 1 and is further described in the Results section. The PRS construction was carried out in two steps:
(1) We assembled the PRS formula based on regression coefficients from the respective summary association statistics retrieved from previously published GWAS (Table 1) and thresholding-pruning workflow (40). The GWAS summary data were filtered using eight predefined GWAS P-value thresholds for variant inclusion in the formula (<1e-1, <1e-2, <1e-3, <1e-4, <1e-5, <1e-6, <1e-7 and < 1e-8). Then, we pruned the SNPs to reduce the inter-correlations (using LD threshold of r2 ≥ 0.1) as implemented in PLINK software (41), using the 1000 Genomes European reference. In total, we constructed PRS formulas for 10 traits: PRSAlcdep (alcohol dependency), PRSDrnkWk (alcohol drinking per week), PRSCPD (cigarettes per day), PRSSmkCes (smoking cessation, where higher score indicates current smoking), PRSAgeSmk (age of smoking initiation), PRSSmkInit (smoking initiation, which denotes ever smoking), PRScannabis, PRSIBD, PRSUC and PRSCD. We created eight sets of PRS for each trait, corresponding to eight GWAS P-value thresholds:
(2) Individual-level PRS values for the 10 traits (seven SU traits and three IBD outcome traits) were computed for the MSCCR and RISK subjects using linear combinations of the imputed genotype dosages and regression coefficients from the respective GWASs; each PRS value was standardized to median = 0 and IQR = 1.
Marginal association between SU PRS and observed IBD outcomes
Marginal association tests (i.e. without adjusting for covariates) were conducted in MSCCR between each pair of SU PRS and observed IBD endpoints (IBD, CD and UC). In total, 168 tests were performed and Bonferroni correction was applied. Associations of adjusted P < 0.01 were considered significant and used in further downstream analysis (Fig. 2). As a positive control, we also tested associations between the observed IBD diagnoses and the respective PRSIBD, PRSUC, and PRSCD.
Multivariate analysis between SU PRS and observed IBD diagnoses
In the multivariate analysis, we focused on PRSDrnkWk, PRSSmkCes and PRSAgeSmk, which showed significant associations in univariate analysis after Bonferroni correction. The genetic determinants of IBD, UC and CD were represented by PRSIBD, PRSUC and PRSCD, respectively. From the eight sets of PRS corresponding to various GWAS P-value thresholds, we only used those that yielded the most significant association in univariate analysis (Supplementary Material, Tables S2 and S3). We fit regression models as follows:
logit(IBD) ~ PRSDrnkWk + PRSSmkCes + PRSAgeSmk + PRSIBD + Smkobs + Z (1)
logit(CD) ~ PRSDrnkWk + PRSSmkCes + PRSAgeSmk + PRSCD + Smkobs + Z (2)
logit(UC) ~ PRSDrnkWk + PRSSmkCes + PRSAgeSmk + PRSUC + Smkobs + Z (3)
where Z represents other covariates, including age at enrollment, sex and the first five genetic principal components. Importantly, Smkobs denotes observed smoking status at the time of enrollment. We fit Models (1–3) on the MSCCR data to dissect the independent effect of SU PRS, while conditioning on PRSIBD as well as on observed smoking behavior. Observed drinking behavior was not available for the MSCCR data set and thus could not be included in the regression models.
It is of clinical significance to quantify the IBD risk for subjects with extreme SU PRS values. We dichotomized each continuous PRS into a binary variable, for example, PRSDrnkWk to IDrnkWk, PRSSmkCes to ISmkCes, PRSAgeSmk to IAgeSmk, PRSIBD to IIBD, PRSCD to ICD, and PRSUC to IUC. In PRSIBD, PRSCD, PRSUC, PRSSmkCes and PRSAgeSmk, if a given subject was within the top 10% of the PRS, we assigned the corresponding binary variable as 1, otherwise 0. Since PRSDrnkWk was negatively associated with IBD risk, subjects within the bottom 10 percentile of the PRSDrnkWk were assigned IDrnkWk = 1, otherwise 0. We then fit multivariate logistic regression model as follows:
logit(IBD) ~ IDrnkWk + ISmkCes + IAgeSmk + IIBD + Smkobs + Z, (4)
logit(CD) ~ IDrnkWk + ISmkCes + IAgeSmk + ICD + Smkobs + Z, (5)
logit(UC) ~ IDrnkWk + ISmkCes + IAgeSmk + IUC + Smkobs + Z. (6)
Genetic colocalization analysis
Genetic colocalization between IBD and SU endpoints was tested using coloc, version 3.1 (42), using publicly available summary GWAS statistics in a region with genome-wide significant loci shared by alcohol dependence and IBD, CD and UC, in turn: Chr16:28.2Mbp-29.2Mbp (hg19). Default prior parameter settings were used, as provided by the software package. In total, five hypotheses were evaluated: H0: no genetic association with either trait; H1: association with trait 1 only; H2: association with trait 2 only; H3: association with both trait 1 and trait 2, but with different causal loci and H4: association with both trait 1 and trait 2, with a shared underlying causal variant.
Conflict of interest statement. None declared.
Supplementary Material
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institute of Health (NIDDK/NIH) grants R01DK106593, U24DK062429 and 1R01ES029212-01.
Authors’ contribution
Conception and design were by A.F.D.N., A.H. and K.H.; analysis and interpretation were by A.F.D.N., A.H. and K.H.; contributing data sets were provided by A.H. and K.H. Drafting the manuscript for important intellectual content was managed by A.F.D.N., A.H., R.K., C.A., L.P., A.S., H.C., Z.Z., M.S., J.C., I.P., A.K., E.E.S. and KH.
References
- 1. Molodecky, N.A., Soon, I.S., Rabi, D.M., Ghali, W.A., Ferris, M., Chernoff, G., Benchimol, E.I., Panaccione, R., Ghosh, S., Barkema, H.W.et al. (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology, 142, 46–54.e42quiz e30. [DOI] [PubMed] [Google Scholar]
- 2. Coward, S., Clement, F., Benchimol, E.I., Bernstein, C.N., Avina-Zubieta, J.A., Bitton, A., Carroll, M.W., Hazlewood, G., Jacobson, K., Jelinski, S.et al. (2019) Past and future burden of inflammatory bowel diseases based on modeling of population-based data. Gastroenterology, 156, 1345–1353.e1344. [DOI] [PubMed] [Google Scholar]
- 3. Birrenbach, T. and Böcker, U. (2004) Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm. Bowel Dis., 10, 848–859. [DOI] [PubMed] [Google Scholar]
- 4. Somerville, K.W., Logan, R., Edmond, M. and Langman, M. (1984) Smoking and Crohn’s disease. Br. Med. J. (Clin. Res. Ed.), 289, 954–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swanson, G.R., Sedghi, S., Farhadi, A. and Keshavarzian, A. (2010) Pattern of alcohol consumption and its effect on gastrointestinal symptoms in inflammatory bowel disease. Alcohol, 44, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mantzouranis, G., Fafliora, E., Saridi, M., Tatsioni, A., Glanztounis, G., Albani, E., Katsanos, K.H. and Christodoulou, D.K. (2018) Alcohol and narcotics use in inflammatory bowel disease. Ann. Gastroenterol., 31, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang, Y.-F., Ou-Yang, Q., Xia, B., Liu, L.-N., Gu, F., Zhou, K.-F., Mei, Q., Shi, R.-H., Ran, Z.-H., Wang, X.-D.et al. (2013) Multicenter case-control study of the risk factors for ulcerative colitis in China. World J. Gastroenterol., 19, 1827–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porter, C.K., Welsh, M., Riddle, M.S., Nieh, C., Boyko, E.J., Gackstetter, G. and Hooper, T.I. (2017) Epidemiology of inflammatory bowel disease among participants of the Millennium Cohort: incidence, deployment-related risk factors, and antecedent episodes of infectious gastroenteritis. Aliment. Pharmacol. Ther., 45, 1115–1127. [DOI] [PubMed] [Google Scholar]
- 9. Picardo, S., Kaplan, G.G., Sharkey, K.A. and Seow, C.H. (2019) Insights into the role of cannabis in the management of inflammatory bowel disease. Ther. Adv. Gastroenterol., 12, https://pubmed.ncbi.nlm.nih.gov/31523278/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McGovern, D.P., Kugathasan, S. and Cho, J.H. (2015) Genetics of inflammatory bowel diseases. Gastroenterology, 149, 1163–1176.e1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khera, A.V., Chaffin, M., Aragam, K.G., Haas, M.E., Roselli, C., Choi, S.H., Natarajan, P., Lander, E.S., Lubitz, S.A., Ellinor, P.T.et al. (2018) Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet., 50, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Lange, K.M., Moutsianas, L., Lee, J.C., Lamb, C.A., Luo, Y., Kennedy, N.A., Jostins, L., Rice, D.L., Gutierrez-Achury, J., Ji, S.G.et al. (2017) Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet., 49, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramos, G.P. and Papadakis, K.A. (2019) Mechanisms of disease: inflammatory bowel diseases. Mayo Clin. Proc., 94, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu, M., Jiang, Y., Wedow, R., Li, Y., Brazel, D.M., Chen, F., Datta, G., Davila-Velderrain, J., McGuire, D., Tian, C.et al. (2019) Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet., 51, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pasman, J.A., Verweij, K.J., Gerring, Z., Stringer, S., Sanchez-Roige, S., Treur, J.L., Abdellaoui, A., Nivard, M.G., Baselmans, B.M. and Ong, J.-S. (2018) GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal effect of schizophrenia liability. Nat. Neurosci., 21, 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin, A.R., Kanai, M., Kamatani, Y., Okada, Y., Neale, B.M. and Daly, M.J. (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet., 51, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marquez-Luna, C., Loh, P.R. and Price, A.L. (2017) Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol., 41, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li, C., Yang, C., Gelernter, J. and Zhao, H. (2014) Improving genetic risk prediction by leveraging pleiotropy. Hum. Genet., 133, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kugathasan, S., Baldassano, R.N., Bradfield, J.P., Sleiman, P.M., Imielinski, M., Guthery, S.L., Cucchiara, S., Kim, C.E., Frackelton, E.C. and Annaiah, K. (2008) Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat. Genet., 40, 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cutler, D.J., Zwick, M.E., Okou, D.T., Prahalad, S., Walters, T., Guthery, S.L., Dubinsky, M., Baldassano, R., Crandall, W.V. and Rosh, J. (2015) Dissecting allele architecture of early onset IBD using high-density genotyping. PLoS One, 10, 1–12.e0128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loddo, I. and Romano, C. (2015) Inflammatory bowel disease: genetics, epigenetics, and pathogenesis. Front. Immunol., 6, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones, D.P., Richardson, T.G., Davey Smith, G., Gunnell, D., Munafò, M.R. and Wootton, R.E. (2020) Exploring the effects of cigarette smoking on inflammatory bowel disease using Mendelian randomization. Crohns Colitis 360, 2, 1–7.otaa018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khera, A.V., Chaffin, M., Wade, K.H., Zahid, S., Brancale, J., Xia, R., Distefano, M., Senol-Cosar, O., Haas, M.E., Bick, A.et al. (2019) Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell, 177, 587–596.e589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chaudhury, S., Brookes, K.J., Patel, T., Fallows, A., Guetta-Baranes, T., Turton, J.C., Guerreiro, R., Bras, J., Hardy, J., Francis, P.T.et al. (2019) Alzheimer’s disease polygenic risk score as a predictor of conversion from mild-cognitive impairment. Transl. Psychiatry, 9, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Imielinski, M., Baldassano, R.N., Griffiths, A., Russell, R.K., Annese, V., Dubinsky, M., Kugathasan, S., Bradfield, J.P., Walters, T.D., Sleiman, P.et al. (2009) Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet., 41, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christodoulou, K., Wiskin, A.E., Gibson, J., Tapper, W., Willis, C., Afzal, N.A., Upstill-Goddard, R., Holloway, J.W., Simpson, M.A., Beattie, R.M.et al. (2013) Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut, 62, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. You, M. and Arteel, G.E. (2019) Effect of ethanol on lipid metabolism. J. Hepatol., 70, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szabo, G. and Saha, B. (2015) Alcohol’s effect on host defense. Alcohol Res., 37, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou, Z., Xu, M.J. and Gao, B. (2016) Hepatocytes: a key cell type for innate immunity. Cell. Mol. Immunol., 13, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frank, J., Witte, K., Schrodl, W. and Schutt, C. (2004) Chronic alcoholism causes deleterious conditioning of innate immunity. Alcohol Alcohol, 39, 386–392. [DOI] [PubMed] [Google Scholar]
- 31. Li, S., Tan, H.Y., Wang, N., Feng, Y., Wang, X. and Feng, Y. (2019) Recent insights into the role of immune cells in alcoholic liver disease. Front. Immunol., 10, 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pulit, S.L., Stoneman, C., Morris, A.P., Wood, A.R., Glastonbury, C.A., Tyrrell, J., Yengo, L., Ferreira, T., Marouli, E. and Ji, Y. (2019) Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet., 28, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhagavathula, A.S., Clark, C.C., Rahmani, J. and Chattu, V.K. (2021) Healthcare. Multidisciplinary Digital Publishing Institute, Basel, Switzerland, Vol. 9, p. 35.33401588 [Google Scholar]
- 34. Duncan, L., Shen, H., Gelaye, B., Meijsen, J., Ressler, K., Feldman, M., Peterson, R. and Domingue, B. (2019) Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun., 10, 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walters, R.K., Polimanti, R., Johnson, E.C., McClintick, J.N., Adams, M.J., Adkins, A.E., Aliev, F., Bacanu, S.-A., Batzler, A., Bertelsen, S.et al. (2018) Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci., 21, 1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Furberg, H., Kim, Y., Dackor, J., Boerwinkle, E., Franceschini, N., Ardissino, D., Bernardinelli, L., Mannucci, P.M., Mauri, F. and Merlini, P.A. (2010) Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat. Genet., 42, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mayte, S.-F., Tokuyama, M., Wei, G., Huang, R., Livanos, A., Jha, D., Levescot, A., et al. (2021) Intestinal Inflammation Modulates the Expression of ACE2 and TMPRSS2 and Potentially Overlaps With the Pathogenesis of SARS-CoV-2-related Disease. Gastroenterology, 160, 287-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Das, S., Forer, L., Schonherr, S., Sidore, C., Locke, A.E., Kwong, A., Vrieze, S.I., Chew, E.Y., Levy, S., McGue, M.et al. (2016) Next-generation genotype imputation service and methods. Nat. Genet., 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haberman, Y., Tickle, T.L., Dexheimer, P.J., Kim, M.O., Tang, D., Karns, R., Baldassano, R.N., Noe, J.D., Rosh, J., Markowitz, J.et al. (2014) Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest., 124, 3617–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Choi, S.W., Shin-Heng Mak, T. and O’Reilly, P.F. (2020) Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc., 15, 2759–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J., Sklar, P., De Bakker, P.I. and Daly, M.J. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giambartolomei, C., Vukcevic, D., Schadt, E.E., Franke, L., Hingorani, A.D., Wallace, C. and Plagnol, V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, 1–15.e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.