


Abstract
The human pseudouridine synthase PUS7 is a versatile RNA modification enzyme targeting many RNAs thereby playing a critical role in development and brain function. Whereas all target RNAs of PUS7 share a consensus sequence, additional recognition elements are likely required, and the structural basis for RNA binding by PUS7 is unknown. Here, we characterize the structure–function relationship of human PUS7 reporting its X-ray crystal structure at 2.26 Å resolution. Compared to its bacterial homolog, human PUS7 possesses two additional subdomains, and structural modeling studies suggest that these subdomains contribute to tRNA recognition through increased interactions along the tRNA substrate. Consistent with our modeling, we find that all structural elements of tRNA are required for productive interaction with PUS7 as the consensus sequence of target RNA alone is not sufficient for pseudouridylation by human PUS7. Moreover, PUS7 binds several, non-modifiable RNAs with medium affinity which likely enables PUS7 to screen for productive RNA substrates. Following tRNA modification, the product tRNA has a significantly lower affinity for PUS7 facilitating its dissociation. Taken together our studies suggest a combination of structure-specific and sequence-specific RNA recognition by PUS7 and provide mechanistic insight into its function.
INTRODUCTION
Every cellular RNA is covalently modified to influence maturation, stability or function by increasing their chemical diversity beyond the four canonical nucleotides (1). More than 150 chemical modifications occur through the action of numerous RNA-modifying enzymes in all cells from bacteria to archaea to eukaryotes (2). These modifications to the four standard RNA nucleotides range in chemical diversity from small structural changes such as isomerization and methylation to large, multi-step modifications that are implicated in various important roles, e.g. for RNA stability, structure and protein binding (3). The single most abundant RNA modification is pseudouridine found in non-coding RNA (tRNA, snRNA, rRNA) and in coding RNA (4–7). Pseudouridine is a C5-glycosidic isomer of uridine that contains a C-C bond between the C1′ of the ribose sugar and the C5 of uracil rather than the usual C1′-N1 bond found in uridine.
The enzymes responsible for this modification are called pseudouridine synthases and fall in two categories, the guide RNA-dependent enzymes and the stand-alone enzymes. Thirteen enzymes possessing a pseudouridine synthase (PUS) domain have been identified in humans, among them dyskerin (Cbf5 in yeast) that assembles into H/ACA small ribonucleoproteins together with H/ACA RNAs responsible for target RNA recognition (8). All other pseudouridine synthases are called ‘stand-alone’ because they can modify their target RNA without a guide RNA by directly recognizing a sequence and/or an RNA structure (9). Twelve stand-alone enzymes have been described in humans and are related to their bacterial counterparts which are classified into six families: TruA, TruB, TruD, RsuA, RluA and Pus1 (9,10).
PUS7 is a member of the TruD family which is present in eubacteria, archaea and eukaryotes (11–13). TruD shows little sequence similarity to other pseudouridine synthases, but structural determination of Escherichia coli TruD revealed that it shares the conserved catalytic domain with all other pseudouridine synthases (12,14,15). Interestingly, the discrepancy between the low sequence similarity but conserved catalytic domain between TruD and other pseudouridine synthases can be explained by a circular permutation of TruD’s fold which likely arose from a gene rearrangement early in evolution (15). Besides the conserved catalytic domain, E. coli TruD and its homologs all contain a unique TruD insertion domain which has been speculated to contribute to RNA binding. Sequence alignments show that the eukaryotic homologs of TruD contain an additional insertion close to the N-terminus which is not found in eubacteria or archaea (16). Moreover, the TruD insertion domain is also larger in eukaryotes. Like other pseudouridine synthases, TruD and its homologs contain a conserved aspartate residue (D294 in human PUS7) that is essential for catalysis by attacking the ribose of the target uridine causing separation of the uracil base and formation of a glycal intermediate (17). The catalytic aspartate is stabilized by a conserved arginine also found in all other pseudouridine synthases. Notably, the third conserved active site residue in TruD is a phenylalanine whereas all other pseudouridine synthase families contain a tyrosine in this position. The exact role of this residue remains under debate (9).
Many pseudouridines in different types of RNA are produced by PUS7, and in several cases pseudouridine formation by PUS7 is modulated by the cellular condition. In addition to the pseudouridine synthase PUS4/TRUB1, PUS7 is the only other known pseudouridine synthase that utilizes a consensus sequence for substrate recognition namely UNUAR (6,18). In Saccharomyces cerevisiae, PUS7 is responsible for modifying position U35 of U2 snRNA (16), and absence of pseudouridine 35 impairs the function of U2 snRNA in mRNA splicing (19,20). PUS7 also modifies the position U56 of U2 snRNA in yeast upon heat shock or nutrient deprivation (21). Moreover, the pseudouridine synthase PUS7 is long known to target certain cytoplasmic tRNAs at position 13 and pre-tRNATyr at position 35 as well as position 50 of 5S rRNA in S. cerevisiae (18,22). Interestingly, the modification of the 5S rRNA is not stoichiometric as the level of modification increases dependent on the temperature (6). Transcriptome-wide pseudouridine sequencing demonstrated that PUS7 also targets many mRNAs in S. cerevisiae and humans, and the number of modification sites depends on the cellular growth conditions (4–6,23). Upon heat shock, yeast PUS7 protein and RNA levels decrease and PUS7 is re-localized from the nucleus to the cytoplasm (6). The re-localization of PUS7 may induce the modification of new target RNAs, possibly playing a stabilizing role for these transcripts and conferring a survival advantage. Similarly, pseudouridylation of mRNA increases in humans upon heat shock at 42°C (23); while it is currently not known which pseudouridine synthase is responsible for this phenomenon, human PUS7 likely contributes to heat-induced mRNA pseudouridylation.
By pseudouridylating different RNAs, PUS7 fulfills a variety of cellular functions. In Candida albicans, PUS7 is required for normal growth, filamentation and virulence (24). In humans, multiple reports have shown that mutations and deletions within the PUS7 gene cause intellectual disabilities, microcephaly and short stature suggesting a role in brain development (25–27). Even single amino acid substitutions can have deleterious effects such as the substitution Gly128Arg in human PUS7 which was identified in patients with intellectual disability, speech delay, motor impairments and aggressive behavior (25). Similarly, the substitution Asp503Tyr in the insertion domain of human PUS7 has been described to be associated with intellectual disability and progressive microcephaly, and patients display reduced formation of pseudouridine 13 in tRNAs. (26). More recently, mutations within PUS7 have also been implicated in age-related macular degeneration in multiple unrelated families (28). In addition, PUS7 is critical for stem cell differentiation by controlling protein synthesis. In the absence of PUS7, stem cells display increased protein synthesis and defective differentiation which is common to certain types of cancers (29). The control of protein synthesis has been reported to occur through a special class of small, 18-mer tRNA-derived fragments (tRF) containing a 5′ terminal oligoguanine (TOG) motif such as fragments derived from tRNAAla called mini TOGs (mTOGs) that require a pseudouridine at position 8. Since the role of PUS7 for embryonic stem cell differentiation depends on its catalytic activity and since U8 in the mTOG is embedded in a PUS7 consensus sequence (UNUAR), it was proposed that PUS7 is responsible for modifying this site although it does not reside at the canonical tRNA position 13 usually targeted by PUS7.
With the goal of describing the structure–function relationship of human PUS7 as a critical step to understand its cellular role, we report here the crystal structure of the human pseudouridine synthase PUS7 at 2.26 Å resolution, which reveals the presence of two additional subdomains compared to the bacterial homolog TruD. Furthermore, we biochemically characterize tRNA binding and modification by human PUS7 uncovering critical features of this enzyme. Together, our structural and functional insights into human PUS7 suggest that this pseudouridine synthase employs a structure-specific mechanism for tRNA recognition interacting with the entire tRNA structure using an extended RNA binding surface along all domains of PUS7.
MATERIALS AND METHODS
PUS7 expression and purification
A group of constructs encoding the PUS7 gene (Homo sapiens pseudouridylate synthase 7) were subcloned into the pET28-MHL vector (Addgene plasmid # 26096) using ligation independent cloning (LIC) to generate full-length PUS7 and N-terminally truncation (Δ1–98). Additional variants were generated in these constructs containing point mutations and a truncation within residues 99–661 referred to as D294N, G128R, D503Y and Δ398–459, respectively. The pairs of primers used for PCR amplification or mutagenesis are listed in the supplementary information (Supplementary Table S1). All PUS7 constructs described in this manuscript were expressed in E. coli BL21(DE3)-V3R-pRARE2 in regular TB media. Protein expression was induced at OD600 of 1.2–1.4 with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and cells were harvested after 16–18 h incubation at 20°C. The recombinant PUS7(Δ1–98) and PUS7(Δ1–98) D294N proteins were first affinity purified with Ni-NTA resin followed by size-exclusion chromatography using S200 column pre-equilibrated with 20 mM PIPES pH 6.5 and 250 mM NaCl. The N-terminal His6 tag was then cleaved overnight by incubating the protein with TEV protease at 4°C. The protein was further purified to homogeneity using ion-exchange Source S column pre-equilibrated with 20 mM PIPES pH 6.5 (buffer A) and eluted with 20 mM PIPES pH 6.5 and concentration gradient of 1 M NaCl (buffer B). For crystallographic phasing purposes, the recombinant PUS7(Δ1–98) protein was also expressed in selenomethionine (SeMet) media and purified same as TB-expressed PUS7(Δ1–98) protein described above. The incorporation of SeMet in PUS7(Δ1–98) was confirmed by mass spectrometry; the measured mass after TEV cleavage was 65,214 Da. The remaining G128R and Δ398–459 variants of PUS7 were affinity purified with Ni-NTA resin followed by size-exclusion chromatography using S200 column pre-equilibrated with 20 mM PIPES pH 6.5 and 250 mM NaCl, without TEV cleavage.
The recombinant full-length PUS7 protein was first affinity purified with Ni-NTA resin followed by size-exclusion chromatography using S200 column pre-equilibrated with 20 mM Tris-HCl pH 8.0 and 150 mM NaCl. The N-terminal His6 tag was then cleaved overnight by incubating the protein with TEV protease at 4°C. The protein was further purified to homogeneity using ion-exchange Source 30Q column pre-equilibrated with 20 mM Tris pH 7.5 (buffer A) and eluted with 20 mM Tris pH 7.5 and concentration gradient of 1 M NaCl (buffer B). Measured mass for full-length PUS7 after TEV cleavage was 75095.2 Da measured by mass spectrometry. Protein concentrations were determined using Nanodrop, taking into consideration the extinction coefficients calculated in Expasy, ProtParam.
PUS7 crystallization and structural determination
Diffraction quality crystals for PUS7(Δ1–98) (SeMet) were obtained in vapor-diffusion sitting drops by mixing equal volumes of PUS7 at 10.1 mg/ml and precipitant solution containing 23% w/v PEG 3350, 0.2 M sodium formate, pH 7.0. Crystals were then cryo-protected using reservoir solution supplemented with 10% (v/v) ethylene glycol and cryo-cooled in liquid nitrogen.
X-ray diffraction data of PUS7(Δ1–98) (SeMet) were collected at 100 K at the 22ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory. The raw data were processed and scaled using the HKL-3000 suite (30). The PUS7 structure was solved by single-wavelength anomalous diffraction (SAD) method using SOLVE/RESOLVE (31). Graphics program COOT (32) was used for model building and visualization. Restrained refinement and structure validation was carried out using BUSTER (33) and MOLPROBITY (34), respectively. Images were prepared in PyMol (www.pymol.org).
Computational docking of PUS7 with tRNAGln
The computational docking of PUS7-tRNAGln complex was performed in NPDock (35,36). The coordinates of a previously published co-crystal structure of tRNAGln (PDB ID: 3AKZ) was used as a template for docking experiments. The substrate U13 residue in tRNAGln was manually flipped out in COOT, to mimic the substrate uridyl conformation in the co-crystal structure of TruB bound to a T stem-loop RNA (PDB ID: 1K8W), to enable a U13 conformation in tRNAGln that is amenable for pseudouridylation reaction by pseudouridine synthases. An interface filtering option was added in NPDock to take into consideration the proximity of target residue U13 in tRNAGln to Asp294 of PUS7 (PDB ID: 5KKP). The overall best-scored complex was used for structural characterization of the complex and for generating Figure 1C.
Figure 1.

Overall structure of human PUS7. (A) Crystal structure of PUS7(Δ1−98) is shown in cartoon representation and color-coded according to its domain architecture as labeled. The catalytic D294 residue is shown in sticks and marked with an asterisk. (B) Overlay of PUS7(Δ1−98), color-coded same as (A), and Escherichia coli TruD (gray) (PDB: 1SB7). (C) Overall best-scored predicted model of PUS7-tRNAGln complex. Electrostatic surface potential representation of PUS7(Δ1−98) shown in similar orientation as (A), docked with tRNAGln (yellow). The target U13 in tRNAGln is highlighted in red. PUS7 surface color indicates electrostatic potential ranging from − 10 kT/e (red) to + 10 kT/e (blue). Electrostatic surface potentials were calculated in APBS (36).
In vitro transcription of RNA substrates
Templates used for in vitro transcriptions were generated by PCR extension of two partially overlapping oligonucleotides (Supplementary Table S2) containing the T7 promoter sequence (TAATACGACTCACTATAG) on the 5′ end of each construct.
Each in vitro transcription contained PCR generated template [10% (v/v) which is equivalent to a concentration range of 450−700 ng/μl] in transcription buffer (40 mM TrisHCl pH 7.5, 15 mM MgCl2, 2 mM spermidine, 10 mM NaCl, 10 mM DTT), 3 mM NTPs (ATP, CTP, GTP and UTP), 5 mM GMP, 0.01 U/ml inorganic pyrophosphatase, 0.3 mM T7 RNA Polymerase and 0.12 U/ml RNase inhibitor. In vitro transcriptions were incubated at 37°C for 4 h. Template DNA was digested with DNaseI for 1 h at 37°C. The RNA was then precipitated by addition of 1 volume isopropanol and 1/5 volumes of KOAc pH 5.2 for 16 h at −20°C. RNAs were purified by crush and soak gel extraction and were re-dissolved in H2O and stored at −20°C until further use. The concentration of RNA was determined by A260 measurements using extinction coefficients calculated by OligoAnalyzer 3.1 (IDT) and the purity was assessed by measuring the A260 to A280 ratio.
For the generation of [3H]-labeled tRNA substrate, UTP was substituted with 0.1 mM [5–3H]-UTP (0.46 Ci/mmole) (Moravek Biochemicals Inc.) and in vitro transcriptions and purifications by crush and soak gel extraction were conducted as described. RNA concentration was determined by A260, and the specific activity was determined by scintillation counting.
Tritium release assay
For initial characterization of the enzymes, single turnover tritium release assays were performed using 1000 nM of each enzyme and 100 nM substrate tRNA. For multiple turnover assays, 50 nM of enzyme were incubated in presence of 600 nM of substrate tRNAs. Before starting the reactions, substrate tRNAs were refolded in TAKEM4 buffer (50 mM TrisHCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 1 mM EDTA, 4 mM MgCl2) at 65°C for 5 min followed by cooling at room temperature for 10 min. The modification reaction was performed at 37°C. Samples containing 7.5–25 pmol of RNA (depending on the specific activity) were removed after desired time points and quenched in 1 ml 5% (w/v) activated charcoal (Norit A) in 0.1 M HCl. After centrifugation, 900 μl of the supernatant was mixed with 500 μl 5% Norit A (w/v) in 0.1 M HCl and centrifuged again. The supernatant was filtered through glass wool, and 900 μl of the filtrate was subjected to scintillation counting to determine the amount of tritium released corresponding to the amount of pseudouridine formed. Data were analyzed using GraphPad Prism. Initial velocities were estimated using multiple turnover time courses by linear regression of the initial region (<30% of the measured end-level).
Preparation of cy-tRNAGln (Ψ13)
In vitro transcribed [3H]tRNAGln was refolded in TAKEM4 buffer by heating to 65°C for 5 min and cooled to room temperature for 10 min. To prepare modified [3H]tRNAGln (Ψ13), 1.38 μM of tritium-labeled of cy-tRNAGln was incubated with 4 μM of purified full-length PUS7 (PUS7 FL) enzyme for 2 h at 37°C in a 3 ml reaction. Enzyme was subsequently removed by phenol−chloroform extraction followed by ethanol precipitation of the tRNA. After resuspending modified [3H]tRNAGln (Ψ13) in water, [3H]tRNAGln (Ψ13) concentration was determined by A260 measurements and scintillation counting.
To quantify the level of modification for prepared [3H]tRNAGln (Ψ13), small-scale tritium release assays were performed in parallel and in duplicate to estimate the level of modification within the large-scale preparation. Thereby, it was determined that [3H]tRNAGln (Ψ13) was fully modified.
Nitrocellulose filter binding assay
Substrate tRNAs were refolded in TAKEM4 buffer as described. Increasing concentrations of the enzyme (from 0 to 5 μM) were incubated with 20 nM of tritium-labeled substrate RNAs for 10 min at 37°C. The complete reaction was filtered through a nitrocellulose membrane, followed by washing of the nitrocellulose membrane with 1 ml cold TAKEM4 buffer. The nitrocellulose membrane was then dissolved in 10 ml EcoLite scintillation cocktail (EcoLite (+), MP Biomedical) followed by scintillation counting to determine the amount of radioactivity bound to the membrane corresponding to the enzyme bound to each substrate. Data were analyzed using GraphPad Prism. Dissociation constants (KD) were determined by fitting the binding curves to a hyperbolic function:
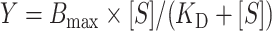 |
where [S] is the substrate concentration and Bmax is the maximum binding.
RESULTS
Structure of human PUS7
PUS7 is a 661-residue protein containing a predicted N-terminal disordered region (residues 1–98) and a pseudouridine synthase domain including several insertions (residues 99–645) (Figure 1A). In order to elucidate the structure–function relationship of human PUS7, we first determined the crystal structure of the pseudouridine synthase domain of PUS7(Δ1–98) refined to 2.26 Å resolution. Data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| PUS7(Δ1–98) (PDB ID: 5KKP) | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 56.4, 73.4, 138.2 |
| α, β, γ (°) | 90.0,90.0,90.0 |
| Resolution (Å) (highest resolution shell) | 50.00–2.26 (2.30–2.26) |
| Measured reflections | 364318 |
| Unique reflections | 27643 |
| R merge | 10.1 (83.2) |
| CC1/2 | 0.996 (0.795) |
| I/σI | 40.2 (1.7) |
| Completeness (%) | 99.6 (95.3) |
| Redundancy | 13.2 (8.8) |
| Refinement | |
| Resolution (Å) | 27.4–2.26 |
| No. of reflections (test set) | 27570 (695) |
| R work/ R free (%) | 21.0/26.3 (22.4/26.8) |
| No. atoms | |
| Protein | 3938 |
| Water | 82 |
| B-factors (Å2) | |
| Protein | 59.7 |
| Water | 52.1 |
| RMSD | |
| Bond lengths (Å) | 0.01 |
| Bond angles (º) | 1.08 |
| Ramachandran plot % residues | |
| Favored | 96.7 |
| Additional allowed | 3.3 |
| Disallowed | 0 |
*Values in parentheses are for highest resolution shell.
PUS7(Δ1–98) has an overall structural fold that is most similar to E. coli TruD (PDB ID: 1SB7) (15) with a root-mean-square deviation (RMSD) of 2.24 Å over 300 aligned Cα atoms (Figure 1A and B, Table 1). As expected, it has a pseudouridine synthase domain (catalytic domain) that is structurally conserved throughout the entire pseudouridine synthase families (Supplementary Figures S1A and S2), followed by a C-terminal insertion (residues 382–577) specific to the TruD family proteins, referred to here as Insertion C (Figure 1A and B). Interestingly, the Insertion C in PUS7 contains an extra Helix-Turn-Helix (HTH) motif (residues 398–457) that is missing in E. coli TruD (Figure 1B; Supplementary Figures S1B and S2). Human PUS7 also contains an insertion toward the N-terminal region of the catalytic domain (residues 163–235) not observed in any other pseudouridine synthase (Supplementary Figures S1C and S2). This N-terminal insertion has structural similarity to the R3H domain family known for nucleic-acid binding to both DNA and RNA (37). The overall structure of PUS7(Δ1–98) resembles a question mark together with its N-terminal R3H-like insertion domain (Figure 1A and C). A large positively charged cleft is formed between the catalytic domain and TruD-like insertion of PUS7, which also harbors the strictly conserved catalytic aspartate 294 (Figure 1A) as well as other conserved aromatic and positively charged residues (F137, F345, K131) forming the PUS7 active-site (Supplementary Figure 1D).
To further investigate the structural aspects of PUS7 interaction with tRNA substrates, we performed computational docking experiments using the NPDock web server (35). We used the Apo-PUS7(Δ1–98) structure as a starting model for docking with a tRNAGln template that was modified from a previously published structure (PDB ID: 3AKZ) by flipping out the target U13 residue to be able to interact with the catalytic pocket of PUS7. The overall best-scored predicted model of PUS7(Δ1–98)-tRNAGln complex showed a large interaction surface between the tRNAGln and PUS7, which enables the protein to contact not only the elbow region containing the target U13 through its catalytic domain but also the anticodon with the R3H-like insertion domain as well as the acceptor arm with the HTH motif in Insertion-C (Figure 1C). Nevertheless, the target U13 of tRNAGln in our predicted model is located outside of the active site pocket and over 14 Å away from the catalytic aspartate 294. This is in contrast to the co-crystal structure of TruB bound to a T stem-loop RNA (PDB ID: 1K8W) in which the target U55 is positioned inside the catalytic pocket and coordinated by conserved aspartate 48. Furthermore, none of the 30 best-scored predicted models showed accurate positioning of the target U13 in the PUS7 catalytic pocket, suggesting that the PUS7 catalytic pocket is inaccessible for U13 of tRNAGln without further structural rearrangements and potentially local tRNA melting. The presence of an extra HTH motif in PUS7 prevents proper positioning of tRNAGln U13 within the catalytic pocket of PUS7 due to steric clashes between the HTH motif of PUS7 and the stem-loop of tRNAGln. We hypothesize that our predicted model may represent an initial recognition state between PUS7 and tRNA, which, upon binding to the correct tRNA substrate, triggers structural rearrangements in PUS7 and tRNA necessary for accurate positioning of target U13 in the PUS7 active pocket for pseudouridylation.
Initial characterization of PUS7 binding and modification of tRNA
To characterize the pseudouridylation activity of human PUS7, different cytoplasmic and mitochondrial tRNAs (Figure 2A and B) were screened as potential substrates for PUS7 whose yeast homolog has been shown to modify U13 in selected tRNAs (22). Pseudouridylation activity was quantified by the amount of tritium released when using [C5-3H]-labeled tRNA as substrate in a single-turnover assay with enzyme present in excess over tRNA (Figure 2C). As expected, PUS7(Δ1–98) efficiently pseudouridylates cytoplasmic tRNAGln which is known to have a pseudouridine at position 13, but PUS7 is not active on cytoplasmic tRNAMet known to lack a pseudouridine at position 13 (2). Based on these results, it seems highly likely that human PUS7 indeed targets uridine 13; however, the tritium release assay is not site-specific. To confirm the target uridine, we therefore prepared [C5-3H]-labeled tRNAGln with a single U13C substitution. This tRNA was not modified by human PUS7 (Figure 2D) verifying U13 as the target uridine of the human enzyme.
Figure 2.

Identification of an RNA target and modification position for human PUS7. (A) Sequence and secondary structure of three cytoplasmic (cy) tRNAs and (B) two mitochondrial (mt) tRNAs. The positions of U13 in tRNAGln and U8 in tRNAAla as predicted PUS7 target sites are highlighted in yellow. (C) Different tRNAs were tested for pseudouridylation by PUS7(Δ1–98) using a tritium release assay. Single turnover pseudouridylation reactions were performed with 100 nM of [3H-C5] uridine-labeled tRNA and 1000 nM PUS7(Δ1–98). Results of tritium release assay are presented in pseudouridine (Ψ) formed in pmol over time. The error bars represent one standard deviation (n = 3−6). (D) To verify that PUS7 pseudouridylates U13 in cytoplasmic tRNAGln, a variant tRNA with a U13C substitution was prepared and tested in single turnover tritium release assays. As positive control, it is shown that human TruB1 can modify this tRNA at position 55 (n = 3). (E) PUS7(Δ1–98) activity was tested in single turnover tritium release assays for non-specific activity using a non-tRNA substrate, corresponding to a fragment of the human mt-16S rRNA (n = 3).
Next, we analyzed whether the disordered N-terminus region of PUS7 is important for its pseudouridylation activity (Figure 1A). Using cytoplasmic tRNAGln as substrate, we show that both full-length PUS7 as well as truncated PUS7(Δ1–98) lacking the first 98 amino acids possess similar pseudouridylation activity (Figure 3A and Table 2). These data support our structural model lacking residues 1–98 as representing a physiologically relevant construct of human PUS7. Since the truncated PUS7(Δ1–98) variant is more stable, it has been used in all subsequent assays unless indicated. Moreover, we confirmed that residue D294 is the catalytic aspartate of human PUS7 as a PUS7(Δ1–98) D294A variant is completely inactive in pseudouridine formation (Figure 3B and Supplementary Figure S1D).
Figure 3.

Functional role of catalytic aspartate D294 and the N-terminus of PUS7. (A) Comparison of truncated PUS7(Δ1–98) and full-length (FL) PUS7 in tritium release assays under multiple-turnover conditions with cytoplasmic (cy) tRNAGln. (B) Analysis of PUS7(Δ1–98)D294A, where the catalytic aspartate is substituted by alanine, in single-turnover tritium pseudouridylation assays compared to PUS7(Δ1–98) and TruB1. The error bars represent one standard deviation from several measurements (n = 3−6).
Table 2.
Initial velocity of multiple-turnover pseudouridine formation in cytoplasmic tRNAGln by PUS7. The initial velocity is calculated for each replicate, and here the average is reported with its standard deviation.
| Protein | Initial velocity, nM/min |
|---|---|
| PUS7FL – 37°C | 135 ± 53 |
| PUS7(Δ1–98) – 37°C | 186 ± 45 |
In addition to the modification activity, we also quantified the affinity of different PUS7 variants for tRNAGln (Figure 4). In brief, nitrocellulose filtration assays determine the amount of radioactively labeled tRNA which is retained on a nitrocellulose filter when bound to protein, but which is washed off the filter when it is not bound to protein. Full-length PUS7 and PUS7(Δ1–98) bind tRNAGln with similar affinity (KD around 600 nM); as both enzymes are active, this affinity reflects the binding of pseudouridylated product tRNA. In contrast, PUS7(Δ1–98) D294A binds tRNA with approximately 5-fold higher affinity (KD of 170 nM) corresponding to binding of unmodified substrate tRNA. Similarly, tRNAGln (U13C), which cannot be modified, binds with a high affinity (KD of 50 nM) to active PUS7(Δ1–98) (Table 3) confirming the difference between substrate and product affinity of PUS7. Notably, this difference in affinity for substrate and product tRNA is not observed when PUS7 binds tRNA already containing Ψ13 (Table 3) suggesting that modification of tRNA by PUS7 results in a different conformation of protein or tRNA than binding of pre-modified tRNA to PUS7. Lastly, we assessed whether PUS7 can bind non-target RNA using cytoplasmic tRNAMet and observed binding with an affinity of about 400 nM (Table 3).
Figure 4.
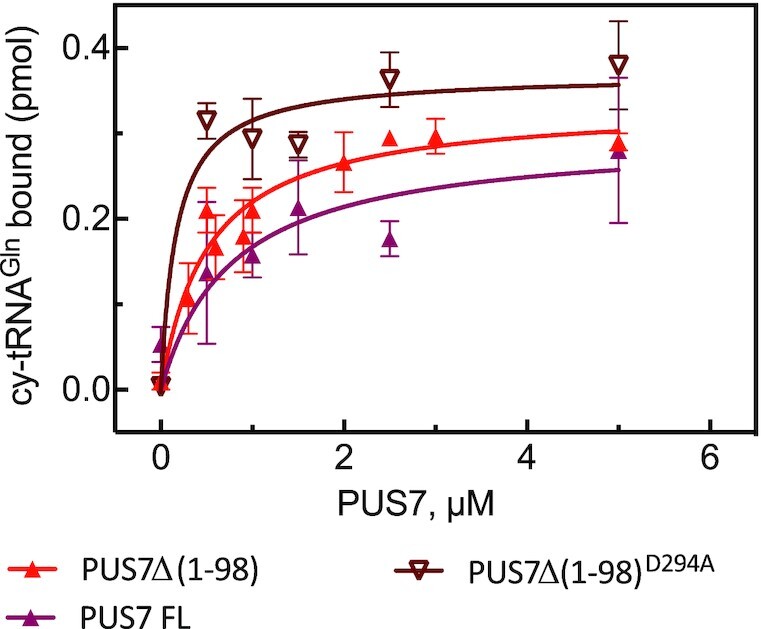
Affinity of PUS7 for cytoplasmic tRNAGln. The affinity of different constructs of PUS7 for cy-tRNAGln was determined using nitrocellulose filtration. About 20 nM of cytoplasmic (cy) tRNAGln was incubated in presence of increasing concentration of protein for 10 min in triplicate. The amount of tRNAGln bound to PUS7 in pmol was determined by nitrocellulose filtration and is represented as average with one standard deviation (n = 3−6). The dissociation constants (KD) were determined by fitting the data to a hyperbolic equation (smooth lines) and are summarized in Table 3.
Table 3.
Affinity of different PUS7 constructs for different RNA substrates and variants thereof. Affinity of PUS7 for different RNA substrates was determined using nitrocellulose filtration. About 20 nM of RNA was incubated in presence of increasing concentration of proteins for 10 min (see Figure 4). Here, the average dissociation constants are reported together with the standard deviation (SD). The number of replicates used for each KD determination is indicated (n). The RNA targets, which will be modified by PUS7 as measured in tritium release assay, are designed with an asterisk (*), and in these cases the reported KD reflects binding of a modified product RNA to PUS7.
| PUS7 variant | RNA substrate | K D, nM | SD | n |
|---|---|---|---|---|
| PUS7 FL | cy-tRNAGln (*) | 621 | 291 | 4 |
| PUS7(Δ1–98) D294A | cy-tRNAGln | 170 | 89 | 3 |
| cy-tRNAGln (Ψ13) | 30 | 15 | 3 | |
| PUS7(Δ1–98) | cy-tRNAGln (*) | 519 | 97 | 3 |
| cy-tRNAGln -( 13) | 92 | 11 | 3 | |
| cy-tRNAGln(U13C) | 50 | 35 | 4 | |
| cy-tRNAGln(G22A) | 47 | 2 | 3 | |
| micro-cy-tRNAGln | 629 | 387 | 7 | |
| 18mer-cy-tRNAGln | 267 | 91 | 8 | |
| cy-tRNAGln Δ(anticodon) | 127 | 83 | 5 | |
| cy-tRNAGln Δ(T arm) | 225 | 100 | 5 | |
| (D arm)-cy-tRNAGln | 58 | 12 | 3 | |
| cy-tRNAMet | 421 | 149 | 3 | |
| mt-16S rRNA | 277 | 97 | 3 | |
| cy-tRNAAla | 203 | 103 | 3 | |
| micro-cy-tRNAAla | 698 | 418 | 5 | |
| PUS7(Δ1–98) G128R | cy-tRNAGln | 71 | 30 | 5 |
| PUS7(Δ398–459) | cy-tRNAGln | 188 | 87 | 5 |
Structural determinants in tRNA required for recognition by PUS7
It was previously shown that the presence of the consensus sequence UGUAR in target RNA is required for S. cerevisiae PUS7 modification activity (22). As expected, this motif is also present in human tRNAGln, the substrate used here for human PUS7. In yeast, the D-arm alone including the consensus sequence is sufficient for pseudouridine formation by PUS7, and the remainder of the tRNA structure seems dispensable (38). In addition to the consensus sequence, PUS7 requires an additional hairpin in order to modify U35 in S. cerevisiae U2 snRNA suggesting that recognition of RNA structural elements contributes to the target recognition by PUS7 (16).
Here, we asked whether other structural elements of the tRNA are needed for recognition by human PUS7(Δ1–98). We prepared tRNA variants lacking selected arms, a micro-tRNA consisting only of the acceptor and T-arms connected by a loop with the PUS7 target sequence, a single-stranded 18-nucleotide RNA comprising the 5′ end of tRNAGln including the PUS7 consensus sequence (18-mer-cy-tRNAGln) as well as a 21-nucleotide RNA comprising the D-arm of tRNAGln (D arm-cy-tRNAGln) (Figure 5A). Interestingly, single turnover pseudouridylation assays revealed that only full-length tRNAGln is modified by human PUS7(Δ1–98) (Figure 5B). The target U13 of PUS7 is typically not in a Watson−Crick base-pairing interaction. To assess the importance of U13 base-pairing interactions, we also prepared a tRNA variant containing a mutation at the position pairing with U13 (cy-tRNAGln (G22A)). Changing the U13 interaction from a U-G to a Watson−Crick U-A base pair abolishes PUS7(Δ1–98) activity (Figure 5B). These results demonstrate that besides the target U13 in the D-arm, also the T-arm and the anticodon arm are required for human PUS7 activity which is in contrast to the reports for S. cerevisiae PUS7 (38). Moreover, the base-pairing interactions of U13 are critical for tRNA recognition by PUS7.
Figure 5.

Dissecting the structural requirements in tRNAGln for modification by human PUS7. (A) Secondary structure of cytoplasmic (cy) tRNAGln derivatives that are lacking arms of the canonical tRNA cloverleaf structure. (B) Single turnover pseudouridylation reactions were performed by mixing 100 nM of [3H-C5] uridine-labeled tRNAGln (full-length and truncated derivatives) with 1000 nM of PUS7(Δ1–98). The error bars represent one standard deviation (n = 3−6).
Subsequently, we used nitrocellulose filtration assays to assess whether PUS7(Δ1–98) can bind the truncated tRNAGln variants. Interestingly, RNA binding was observed for all RNA constructs although they could not be modified (Table 3). Most RNAs bind with similar affinity as full-length, unmodified tRNAGln (KD of 127–267 nM). Only binding of the microRNA consisting of the acceptor and the T-arm connected by a loop is weaker with a KD of about 600 nM. As negative control, we tested binding of PUS7 to a fragment of the mitochondrial 16S rRNA that does not resemble a tRNA and surprisingly observed binding with a KD of 277 nM (Table 3). In conclusion, human PUS7 binds several RNAs quite tightly without modifying these RNAs. As further discussed below, this interaction with many RNAs may help human PUS7 to search for a productive substrate RNA.
Structure−function analysis of human PUS7
As single-residue substitutions in human PUS7 cause intellectual disability, we investigated how such substitutions affect tRNA binding and modification. Toward this goal, the disease variants PUS7(Δ1–98)G128R and PUS7(Δ1–98)D503Y were cloned and expressed in E. coli for biophysical characterization. Contrary to PUS7(Δ1–98)G128R, the PUS7(Δ1–98)D503Y expression was not detectable in induced E. coli cells, suggesting that this substitution in the C-terminal insertion domain causes a structural instability of the protein. The second variant, PUS7(Δ1–98)G128R is located in a loop in the catalytic domain in close vicinity of the active site (Figure 6A). Notably, this variant displays no pseudouridylation activity in single-turnover pseudouridylation assays with tRNAGln (Figure 6B). Nevertheless, nitrocellulose filtration assays reveal that PUS7(Δ1–98)G128R is able to bind tRNAGln with a high affinity of 71 nM (Table 3). Taken together, the biochemical analysis of PUS7 disease variants show that the D503Y substitution likely causes structural instability whereas the G128R substitution impairs the catalytic activity of PUS7.
Figure 6.

tRNA modification activity of PUS7 variants. (A) Substitutions G128R in the catalytic domain and D503Y in the C-terminal insertion domain are indicated in dotted spheres and residues 398–459, the helix-turn-helix motif, which was deleted, are highlighted in (magenta). (B) Single turnover pseudouridylation reactions were performed by mixing 100 nM of [3H-C5] uridine-labeled tRNAGln with 1000 nM of the different PUS7 variants. The error bars represent one standard deviation (n = 3−6).
Next, we used a similar approach to characterize the importance of the unique helix-turn-helix insertion on the C-terminal insertion domain of human PUS7 (Figure 1). The entire helix-turn-helix motif was deleted creating a PUS7 variant lacking residues 398−459 (Figure 6A). This deletion results in complete loss of the tRNA pseudouridylation activity of PUS7 (Figure 6B) although the enzyme variant retains the ability to bind tRNA with a reasonable affinity of 188 nM (Table 3). As further discussed below, these observations suggest that the helix-turn-helix motif does not contribute substantially to the binding energy of PUS7 interacting with tRNA but is critical for correct positioning of the tRNA relative to the active site allowing for productive pseudouridine formation.
Interaction of PUS7 with cytoplasmic tRNAAla and a derived fragment
To explain the role of PUS7 in embryonic stem cell differentiation, it was proposed that PUS7 produces a pseudouridine at position 8 found in small tRNA-derived fragments called mTOGs generated for example from tRNAAla. The modification of these mTOGs could occur before or after tRNA cleavage, and thus either the full-length tRNAAla or the mTOG could be the substrate of PUS7 (29). Therefore, we directly assessed whether human PUS7(Δ1–98) can modify tRNAAla, a truncated micro-variant comprising only the acceptor and T-arm and an 18-nucleotide 5′ fragment of tRNAAla corresponding to the mTOG in vitro (Figure 7 and Supplementary Figure S3). However, in several independent experiments, we have not observed any pseudouridylation signal for PUS7(Δ1–98) or PUS7FL modifying tRNAAla or its shorter derivatives as predicted (29). As observed for other non-target tRNAs, the non-cognate tRNAAla can be bound by PUS7(Δ1–98) with an affinity of about 200 nM (Table 3).
Figure 7.

Comparison of PUS7 activity modifying tRNAGln or tRNAAla. (A) 18-nucleotide 5′ fragment of cytoplasmic (cy) tRNAAla and micro-variants of cytoplasmic tRNAAla that only contain the acceptor and T-arm including a loop with the PUS7 consensus sequence; the putative target uridine is shown in yellow. The secondary structures have been predicted using mFold. (B) Multiple-turnover tritium release assays of PUS7(Δ1–98) with cytoplasmic tRNAGln, tRNAAla, its micro-RNA variant and a short 18-nucleotide 5′ fragment of tRNAAla. The error bars represent one standard deviation (n = 4).
Interestingly, a tRNAAla-derived fragment comprising the first 30 nucleotides has been reported to form an RNA G quadruplex structure (39). Therefore, we asked whether the formation of RG4s in cy-tRNAAla may inhibit PUS7 activity and be responsible for the observed lack of pseudouridine formation. As G quadruplex are differentially stabilized in the presence of different cations (K+ > Na+ > Li+) (40), we incubated in vitro transcribed cytoplasmic tRNAAla and a short, 18-nucleotide fragment of this tRNA corresponding to the mTOG (29) in presence of different salts and within the reaction buffer TAKEM4. G quadruplexes are stable and migrate slower than other species on a denaturing gel allowing us to directly observe G quadruplex formation (Supplementary Figure S4). We confirmed the formation of G quadruplex structures by the 18-nucleotide tRNAAla 5′ fragment; however, the proportion of free RNA that does not form a G quadruplex is still high, suggesting that G quadruplex formation is not responsible for the lack of PUS7 activity. Moreover, a tRNAAla variant with a reduced number of Gs at the 5′ end is also not modified whereas a tRNAGln variant harboring a stretch of five Gs at the 5′ end can be pseudouridylated (Supplementary Figure S5). Together, these observations suggest that G quadruplex formation does explain why PUS7 does not modify tRNAAla or its mTOG in vitro.
DISCUSSION
Combining structural and biochemical studies, we have gained important insight into the structure-function relationship of the human pseudouridine synthase PUS7 which plays critical roles in development and brain function. In comparison to the structure of its bacterial homolog TruD, the structure of human PUS7(Δ1–98) reveals two additional subdomains that are inserted within the catalytic and the so-called insertion domain, respectively. Interestingly, docking studies suggest that all domains of PUS7 may contribute to binding the entire length of the L-shaped tRNA to position the target U13 in proximity to the catalytic aspartate, although further conformational adjustments of the complex are needed to enable a productive enzyme−substrate complex. In agreement with this model, assessing the pseudouridylation activity of PUS7 for different tRNA variants demonstrates that in addition to a consensus sequence, human PUS7 requires all structural features of the tRNA in contrast to its yeast homolog (38). Our analysis also explains the detrimental defects of disease-causing substitutions in PUS7 which either impair structural stability in case of the D503Y substitution or abolish catalytic activity as observed for the G128R substitution. Interestingly, and in accordance with the large RNA-binding surface, PUS7 is able to bind many RNAs without modifying them. Lastly, we find no evidence for in vitro modification by human PUS7 of tRNAAla or a small tRNA fragment thereof at the predicted position U8 (29).
tRNA recognition by human PUS7
The structure of human PUS7(Δ1–98) reveals a large, positively charged surface extending along the catalytic and insertion domain that is likely to electrostatically interact with the target RNA beyond the recognition consensus sequence (Figure 1C). When docking a tRNA onto PUS7 by closely aligning the target U13 with the active site, this model suggests that PUS7 recognizes the entire tRNA utilizing the additional subdomains not found in its bacterial homolog TruD. Specifically, the R3H-like insertion in the catalytic domain, which is a known nucleic acid binding domain, is well positioned to interact with the anticodon loop of the tRNA, in particular if the anticodon undergoes a slight conformational change, e.g. along the tRNA hinge region as observed during tRNA’s interactions with the ribosome (41). The additional helix-turn-helix subdomain found at the tip of the Insertion C domain is in a location suitable to interact with the acceptor arm of tRNA. Importantly, this model of PUS7-tRNA recognition is in agreement with our biochemical data that PUS7 requires the entire tRNA structure for productive pseudouridine formation (Figure 5).
The formation of multiple different contacts between PUS7 and the entire tRNA might be required to trigger structural rearrangement and local tRNA unwinding to correctly position the target uridine within the active site. Accordingly, truncated tRNA variants can still interact with PUS7, but fail to be modified as they will likely not adopt the correct relative position preventing the target U13 from reaching the catalytic center. Similarly, the deletion of the helix-turn-helix motif in PUS7, which eliminates only a small portion of the interaction surface with tRNA, does not impair tRNA binding, but tRNA modification (Figure 6). It is conceivable that the large RNA-binding surface of PUS7 allows it to interact with a broad range of structured RNAs (vide infra). It is interesting that yeast PUS7 has been reported to require only the D-arm to modify U13 which is in contrast to our findings for human PUS7 (38).
While we focused our analysis on the interaction of human PUS7 with tRNA, the extended RNA-binding surface of PUS7 supports the hypothesis that also other target RNAs such as mRNAs form an extended interaction network with PUS7 including several structural elements. For example, pseudouridylation of U2 snRNA requires not only the consensus motif surrounding the target uridine but also an additional stem loop (16). In order to fully elucidate the recognition of other RNAs by PUS7 and the specific interactions with the consensus motif, a structure of PUS7 in complex with RNA will be required.
RNA binding features of human PUS7
The detailed quantitative investigation of human PUS7 binding to a range of different RNAs independent of its pseudouridylation activity reveals several interesting characteristics (Figure 4 and Table 3). First, PUS7 binds tRNA that it has pseudouridylated with 4- to 10-fold lower affinity than unmodified tRNA containing U13 as evident from comparisons of active and inactive PUS7 as well as wild-type and a U13C variant of tRNA (Table 3). This feature will allow PUS7 to rapidly release the tRNA after modification. Notably, tRNA already containing a pseudouridine at position 13 is bound more tightly than tRNA that is modified by PUS7 wild-type during the assay. This observation can be explained by a conformational change induced after pseudouridine formation in the protein and/or the tRNA that facilitates product release similar as proposed for other pseudouridine synthases (42). For example, it can be envisioned that pseudouridine 13 is expelled from the active site after catalysis and re-inserted into the tRNA core facilitating tRNA dissociation whereas binding of pseudouridylated tRNA may allow pseudouridine 13 to flip into the active site of PUS7 causing tight tRNA binding.
Second, we observe significant binding of RNAs to PUS7 that are not being modified such as truncated variants of tRNAGln, a fragment of mitochondrial 16S rRNA, tRNAMet and tRNAAla (Table 3). These RNAs bind to PUS7 with dissociation constants that are higher than the KD for unmodified tRNAGln(U13C) but lower than the KD for modified tRNAGln. Importantly, an 18-nucleotide short variant of tRNAGln, which may fold into a small hairpin and harbors the consensus site, binds with an affinity of only 267 nM. This observation suggests that the consensus sequence itself only partially contributes to the interaction strength. Thus, we conclude that human PUS7 interacts with medium affinity with a variety of RNAs that either are structured similarly to a tRNA or contain the consensus sequence. This broad RNA binding may allow PUS7 to sample all tRNAs in the cell through rapid binding and release in order to search for its correct target RNAs harboring the consensus sequence around U13.
Pseudouridylation of tRNAAla and its fragments at U8
Despite the reports of the importance of PUS7 for embryonic stem cell differentiation and the role of pseudouridylated mTOGs in controlling protein synthesis (29), we have not discovered any evidence for direct modification of U8 by human PUS7 in vitro in either tRNAAla prior to cleavage or in its 18-nucleotide cleavage product called mTOG (Figure 7). This finding can be explained by our observation that human PUS7 recognizes the entire structure of tRNA: U8 is located between the anticodon and D-arm which is in 3D vicinity to U13 and thus possibly close to the active site of PUS7. However, the recognition sequence UNUAR surrounding U8 is likely partially buried in the tRNA tertiary structure and thus not in the correct orientation to be recognized by PUS7. Notably, human tRNAAla has not been reported to contain a pseudouridine at position 8 such that the observations by Guzzi et al. would rather result from a modification of the mTOG after tRNA cleavage (2). Since human PUS7 cannot modify an 18-nucleotide fragment of tRNAGln comprising the consensus sequence, it is not surprising that a fragment of tRNAAla also cannot be pseudouridylated by PUS7 in vitro. The formation of G quadruplexes in tRNAAla and truncations thereof does not prevent binding to PUS7 (Table 3), potentially because G quadruplex formation is only observed in a fraction of RNAs under our buffer conditions (Supplementary Figure S2).
Therefore, our observations cannot explain the presence of pseudouridine 8 in mTOGs observed in embryonic stem cells (29). In this context, it is noteworthy that Guzzi et al. did not directly demonstrate formation of this pseudouridine by PUS7 or its absence in strains lacking PUS7 or carrying a catalytically inactive PUS7 variant. Hence, it cannot be completely ruled out that another pseudouridine synthase is responsible for modifying the mTOG. However, given the function of both PUS7 and the pseudouridylated mTOG in controlling protein synthesis in embryonic stem cells, it may be more likely that PUS7 requires additional factors such as interaction proteins present in the cell to modify the mTOG which are missing from our in vitro reconstituted experimental system.
CONCLUSION
In summary, the structure of human PUS7 and the characterization of tRNA pseudouridylation by PUS7 provide critical insight into the function of this enzyme and its mode of RNA recognition. First, PUS7 searches for a suitable target RNA by binding many cellular target RNAs through interactions with its extended RNA binding surface spanning all domains. Incorrect RNAs likely dissociate rapidly, whereas unmodified substrate RNA is bound most tightly and recognized both by its consensus sequence surrounding the target uridine as well as its structure allowing the target U13 to flip into the active site of PUS7. After catalysis of pseudouridylation, the affinity of PUS7 to the modified tRNA is significantly reduced presumably by conformational changes in the tRNA and/or PUS7 allowing the pseudouridylated RNA to dissociate.
Several questions remain regarding the plethora of cellular RNA targets of PUS7 and its physiological function. In future, it will be insightful to obtain a structure of PUS7 bound to its different target RNAs to fully explain how this enzyme recognizes such a variety of RNAs. Moreover, it will be interesting to characterize other interactions of human PUS7 with factors that could regulate or enhance its activity, e.g. for mTOGs or at elevated temperatures. And most importantly, we still must clearly identify the cellular function of the many pseudouridines in coding and non-coding RNAs that are introduced by PUS7 and other pseudouridine synthases.
DATA AVAILABILITY
Coordinates and structure factors for PUS7 have been deposited in the Protein Data Bank under the accession code 5KKP.
Supplementary Material
ACKNOWLEDGEMENTS
Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. SER-CAT is supported by its member institutions (see www.ser-cat.org/members.html) from the National Institutes of Health.
Contributor Information
Julia Guegueniat, Alberta RNA Research and Training Institute (ARRTI), Department of Chemistry and Biochemistry, University of Lethbridge, AB, T1K 3M4, Canada.
Levon Halabelian, Structural Genomics Consortium, University of Toronto, Toronto, ON, M5G 1L7, Canada; Department of Pharmacology and Toxicology, University of Toronto, Toronto, ON, M5S 1A8, Canada.
Hong Zeng, Structural Genomics Consortium, University of Toronto, Toronto, ON, M5G 1L7, Canada.
Aiping Dong, Structural Genomics Consortium, University of Toronto, Toronto, ON, M5G 1L7, Canada.
Yanjun Li, Structural Genomics Consortium, University of Toronto, Toronto, ON, M5G 1L7, Canada.
Hong Wu, Protein Technologies Center, St Jude Children’s Research Hospital, Memphis, TN 38105, USA.
Cheryl H Arrowsmith, Structural Genomics Consortium, University of Toronto, Toronto, ON, M5G 1L7, Canada; Princess Margaret Cancer Centre and Department of Medical Biophysics, University of Toronto, Toronto, ON, M5G 2M9, Canada.
Ute Kothe, Alberta RNA Research and Training Institute (ARRTI), Department of Chemistry and Biochemistry, University of Lethbridge, AB, T1K 3M4, Canada; Department of Chemistry, University of Manitoba, Winnipeg, Manitoba, R3T 2N2, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Structural Genomics Consortium is a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer and Takeda.
Conflict of interest statement. None declared.
REFERENCES
- 1. Schwartz S. Cracking the epitranscriptome. RNA. 2016; 22:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boccaletto P., Machnicka M.A., Purta E., Piatkowski P., Baginski B., Wirecki T.K., de Crecy-Lagard V., Ross R., Limbach P.A., Kotter A.et al.. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018; 46:D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lewis C.J., Pan T., Kalsotra A.. RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 2017; 18:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V.. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014; 515:143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lovejoy A.F., Riordan D.P., Brown P.O.. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One. 2014; 9:e110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., Leon-Ricardo B.X., Engreitz J.M., Guttman M., Satija R., Lander E.S.et al.. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014; 159:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spenkuch F., Motorin Y., Helm M.. Pseudouridine: still mysterious, but never a fake (uridine)!. RNA Biol. 2014; 11:1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baker D.L., Youssef O.A., Chastkofsky M.I., Dy D.A., Terns R.M., Terns M.P.. RNA-guided RNA modification: functional organization of the archaeal H/ACA RNP. Genes Dev. 2005; 19:1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamma T., Ferre-D’Amare A.R.. Pseudouridine synthases. Chem. Biol. 2006; 13:1125–1135. [DOI] [PubMed] [Google Scholar]
- 10. Rintala-Dempsey A.C., Kothe U.. Eukaryotic stand-alone pseudouridine synthases - RNA modifying enzymes and emerging regulators of gene expression?. RNA Biol. 2017; 14:1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ericsson U.B., Andersson M.E., Engvall B., Nordlund P., Hallberg B.M.. Expression, purification, crystallization and preliminary diffraction studies of the tRNA pseudouridine synthase TruD from Escherichia coli. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:775–776. [DOI] [PubMed] [Google Scholar]
- 12. Ericsson U.B., Nordlund P., Hallberg B.M.. X-ray structure of tRNA pseudouridine synthase TruD reveals an inserted domain with a novel fold. FEBS Lett. 2004; 565:59–64. [DOI] [PubMed] [Google Scholar]
- 13. Kaya Y., Ofengand J.. A novel unanticipated type of pseudouridine synthase with homologs in bacteria, archaea, and eukarya. RNA. 2003; 9:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaya Y., Del Campo M., Ofengand J., Malhotra A.. Crystal structure of TruD, a novel pseudouridine synthase with a new protein fold. J. Biol. Chem. 2004; 279:18107–18110. [DOI] [PubMed] [Google Scholar]
- 15. Hoang C., Ferre-D’Amare A.R.. Crystal structure of the highly divergent pseudouridine synthase TruD reveals a circular permutation of a conserved fold. RNA. 2004; 10:1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma X., Zhao X., Yu Y.T.. Pseudouridylation (Psi) of U2 snRNA in S. cerevisiae is catalyzed by an RNA-independent mechanism. EMBO J. 2003; 22:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Veerareddygari G.R., Singh S.K., Mueller E.G.. The pseudouridine synthases proceed through a glycal intermediate. J. Am. Chem. Soc. 2016; 138:7852–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Decatur W.A., Schnare M.N.. Different mechanisms for pseudouridine formation in yeast 5S and 5.8S rRNAs. Mol. Cell. Biol. 2008; 28:3089–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang C., McPheeters D.S., Yu Y.T.. Psi35 in the branch site recognition region of U2 small nuclear RNA is important for pre-mRNA splicing in Saccharomyces cerevisiae. J. Biol. Chem. 2005; 280:6655–6662. [DOI] [PubMed] [Google Scholar]
- 20. Zhao X., Yu Y.T.. Pseudouridines in and near the branch site recognition region of U2 snRNA are required for snRNP biogenesis and pre-mRNA splicing in Xenopus oocytes. RNA. 2004; 10:681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu G., Xiao M., Yang C., Yu Y.T.. U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J. 2011; 30:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Behm-Ansmant I., Urban A., Ma X., Yu Y.T., Motorin Y., Branlant C.. The Saccharomyces cerevisiae U2 snRNA:pseudouridine-synthase Pus7p is a novel multisite-multisubstrate RNA:Psi-synthase also acting on tRNAs. RNA. 2003; 9:1371–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X., Zhu P., Ma S., Song J., Bai J., Sun F., Yi C.. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 2015; 11:592–597. [DOI] [PubMed] [Google Scholar]
- 24. Pickerill E.S., Kurtz R.P., Tharp A., Guerrero Sanz P., Begum M., Bernstein D.A.. Pseudouridine synthase 7 impacts Candida albicans rRNA processing and morphological plasticity. Yeast. 2019; 36:669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Darvish H., Azcona L.J., Alehabib E., Jamali F., Tafakhori A., Ranji-Burachaloo S., Jen J.C., Paisan-Ruiz C.. A novel PUS7 mutation causes intellectual disability with autistic and aggressive behaviors. Neurol. Genet. 2019; 5:e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaheen R., Tasak M., Maddirevula S., Abdel-Salam G.M.H., Sayed I.S.M., Alazami A.M., Al-Sheddi T., Alobeid E., Phizicky E.M., Alkuraya F.S.. PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum. Genet. 2019; 138:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Brouwer A.P.M., Abou Jamra R., Kortel N., Soyris C., Polla D.L., Safra M., Zisso A., Powell C.A., Rebelo-Guiomar P., Dinges N.et al.. Variants in PUS7 cause intellectual disability with speech delay, microcephaly, short stature, and aggressive behavior. Am. J. Hum. Genet. 2018; 103:1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ratnapriya R., Acar I.E., Geerlings M.J., Branham K., Kwong A., Saksens N.T.M., Pauper M., Corominas J., Kwicklis M., Zipprer D.et al.. Family-based exome sequencing identifies rare coding variants in age-related macular degeneration. Hum. Mol. Genet. 2020; 29:2022–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guzzi N., Ciesla M., Ngoc P.C.T., Lang S., Arora S., Dimitriou M., Pimkova K., Sommarin M.N.E., Munita R., Lubas M.et al.. Pseudouridylation of tRNA-derived fragments steers translational control in stem cells. Cell. 2018; 173:1204–1216. [DOI] [PubMed] [Google Scholar]
- 30. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M.. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 31. Terwilliger T.C. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D. Biol. Crystallogr. 2003; 59:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bricogne G., B.E. Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P, Sharff A., Smart O.S., Vonrhein C., Womack T.O. Buster version 2.10.2. 2017; Cambridge, UK: Global Phasing Ltd. [Google Scholar]
- 34. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B.et al.. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tuszynska I., Magnus M., Jonak K., Dawson W., Bujnicki J.M.. NPDock: a web server for protein-nucleic acid docking. Nucleic Acids Res. 2015; 43:W425–W430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L.E., Brookes D.H., Wilson L., Chen J., Liles K.et al.. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018; 27:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liepinsh E., Leonchiks A., Sharipo A., Guignard L., Otting G.. Solution structure of the R3H domain from human Smubp-2. J. Mol. Biol. 2003; 326:217–223. [DOI] [PubMed] [Google Scholar]
- 38. Urban A., Behm-Ansmant I., Branlant C., Motorin Y.. RNA sequence and two-dimensional structure features required for efficient substrate modification by the Saccharomyces cerevisiae RNA:{Psi}-synthase Pus7p. J. Biol. Chem. 2009; 284:5845–5858. [DOI] [PubMed] [Google Scholar]
- 39. Ivanov P., O’Day E., Emara M.M., Wagner G., Lieberman J., Anderson P.. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc. Natl. Acad. Sci. U. S. A. 2014; 111:18201–18206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bhattacharyya D., Mirihana Arachchilage G., Basu S.. Metal cations in G-quadruplex folding and stability. Front Chem. 2016; 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valle M., Zavialov A., Li W., Stagg S.M., Sengupta J., Nielsen R.C., Nissen P., Harvey S.C., Ehrenberg M., Frank J.. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat. Struct. Biol. 2003; 10:899–906. [DOI] [PubMed] [Google Scholar]
- 42. Duan J., Li L., Lu J., Wang W., Ye K.. Structural mechanism of substrate RNA recruitment in H/ACA RNA-guided pseudouridine synthase. Mol. Cell. 2009; 34:427–439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors for PUS7 have been deposited in the Protein Data Bank under the accession code 5KKP.