

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease characterized by severe, progressive muscle wasting. Cardiomyopathy has emerged as a leading cause of death in patients with DMD. The mechanisms contributing to DMD cardiac disease remain under investigation and specific therapies available are lacking. Our prior work has shown that DMD-iPSC-derived cardiomyocytes (DMD-iCMs) are vulnerable to oxidative stress injury and chronic exposure to DMD-secreted exosomes impaired the cell’s ability to protect against stress. In this study, we sought to examine a mechanism by which DMD cardiac exosomes impair cellular response through altering important stress-responsive genes in the recipient cells. Here, we report that DMD-iCMs secrete exosomes containing altered microRNA (miR) profiles in comparison to healthy controls. In particular, miR-339-5p was upregulated in DMD-iCMs, DMD exosomes and mdx mouse cardiac tissue. Restoring dystrophin in DMD-iCMs improved the cellular response to stress and was associated with downregulation of miR-339-5p, suggesting that it is disease-specific. Knockdown of miR-339-5p was associated with increased expression of MDM2, GSK3A and MAP2K3, which are genes involved in important stress-responsive signaling pathways. Finally, knockdown of miR-339-5p led to mitochondrial protection and a reduction in cell death in DMD-iCMs, indicating miR-339-5p is involved in direct modulation of stress-responsiveness. Together, these findings identify a potential mechanism by which exosomal miR-339-5p may be modulating cell signaling pathways that are important for robust stress responses. Additionally, these exosomal miRs may provide important disease-specific targets for future therapeutic advancements for the management and diagnosis of DMD cardiomyopathy.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease that currently affects approximately 1:5000 boys. DMD is characterized by progressive muscle weakness and loss of function, which typically result in loss of ambulation by age ~13 (1). Often, patients require respiratory ventilator support at age 15. Premature death, due to respiratory or cardiac failure, often occurs when patients are in their late teens or early twenties (2–4). Due to the advancement of novel therapies for skeletal muscle pathology in DMD, cardiomyopathy has emerged as an increasingly significant cause of morbidity and mortality (5). By age 18, 90% of patients demonstrate evidence of cardiac dysfunction (6).
Mutations in the DMD gene result in deficiency of the dystrophin protein, which is an important linkage between the actin cytoskeleton and the extracellular matrix (7). Dystrophin stabilizes the sarcolemma of striated muscle tissue during normal muscle contraction and relaxation, and in its absence, the sarcolemma is weakened and vulnerable to mechanical stress–induced injury (8,9). Studies performed in in vitro and in vivo models of DMD cardiomyopathy have demonstrated increased oxidative stress, disrupted nitric oxide signaling, mitochondrial dysfunction and increased cell death, all contributing to pathology (10–14). Additionally, disruptions in important cell signaling pathways including PI3K-Akt, MAPK and apoptosis have been identified (15–19). However, the mechanisms linking the absence of dystrophin, a membrane protein, to these signaling impairments remain elusive.
MicroRNAs (miRs) are short, non-coding RNAs that have distinct roles in maintaining normal physiologic processes (20,21). Dysregulation of miR expression levels has been associated with pathophysiologic processes in cardiovascular disease (22,23). Because miRs have been implicated in cardiovascular disease processes, they represent interesting targets to evaluate for biomarker or therapeutic potential. Not only do miRs regulate cell processes within the cell, but they may also circulate to affect processes in nearby and distant cells. One route, by which miRs can affect cellular processes in nearby and distant cells, is by being packaged and delivered in exosomes (24,25). Exosomes are small, secreted particles containing proteins, lipids, mRNAs and non-coding RNAs and, as such, have demonstrated roles in intracellular communication and signaling (19,26–28). Exosomes, derived from exogenous cell sources such as cardiospheres or mesenchymal stromal cells, have been examined in DMD for their therapeutic potential (26,29–31). These exosomes modulated inflammatory processes, oxidative injury, mitochondrial respiration, myocyte differentiation and cell death, demonstrating that exosomes are capable of affecting a variety of cellular responses.
In comparison to healthy exosomes, endogenously derived exosomes may have different roles in disease processes (32–34). One study found exosomes derived from DMD-diseased cells promoted pathology via transfer of miR-199 (32). This demonstrated that exosomal content was altered in diseased exosomes and the altered messages the exosomes carried perpetuated the disease phenotype. Proper maintenance of miR levels is essential to maintain homeostasis, as their dysregulation can promote disease processes in DMD (32,35). However, the connection between exosomes, miRs and disease processes underlying DMD cardiomyopathy remains understudied.
In this study, we show several exosomal miRs (exomiRs) are dysregulated as a consequence of dystrophin-deficiency. Comparative analysis of differentially expressed exomiRs, in conjunction with transcriptomic analysis of DMD-iPSC-derived cardiomyocytes (DMD-iCMs) exposed to DMD exosomes, identified altered expression of several miR gene targets involved in stress response and cell death pathways. Upregulation of miR-339-5p was observed in DMD exosomes (DMD-exo) and DMD iPSC-derived cardiomyocytes (DMD-iCMs) in comparison to non-affected exosomes (N-exo) and non-affected iPSC-derived cardiomyocytes (N-iCMs). MiR-339-5p was also upregulated in conditions of stress in DMD-iCMs and mdx mouse cardiac tissue. Knockdown of miR-339-5p was protective against oxidative stress injury by protecting mitochondria and reducing cell death. Knockdown of miR-339-5p was associated with an increase in target genes MDM2 and GSK3A. Together, these findings identify a disease-specific exomiR that may represent a potential therapeutic target in this complex disease.
Experimental Design
Dysregulated exosomal cargo (36–38), including miR cargo, has previously been reported in cardiovascular disease (39–41). Further, dysregulated exosomal miR (exomiR) cargo in DMD has been demonstrated to lead to increased skeletal muscle fibrosis (32) and has failed to protect DMD-iCMs from stress-induced injury (42). Additionally, small RNA sequencing revealed differences in the exomiR cargo of DMD-exo versus N-exo. These differences were linked to regulation of normal physiological pathways stimulated by N-exo including ECM-receptor interaction and metabolism, whereas DMD-exo was associated with pathways impaired in DMD including cGMP-PKG and apoptosis (42). Ultimately, it is unknown whether exomiR alterations occurred as a result of pathological processes occurring in the diseased cardiomyocyte, or whether they were linked to dystrophin-deficiency.
To address whether exomiR alterations were due to dystrophin-deficiency, we designed experiments to express dystrophin in DMD cardiomyocytes and collect exosomes secreted by these cells to evaluate their miR cargo. This was achieved by utilizing a microdystrophin construct ∆R4-R23 (gifted by Dr Jeff Chamberlain, University of Washington), which contains essential domains of the dystrophin protein demonstrated to be expressed in the heart (43,44). The dystrophin construct was engineered into a lentivirus under control of the NCX1 cardiac promotor (Fig. 1) (45), to allow it to be taken up and expressed specifically in iCMs. This group receiving the construct served as a positive control. Once dystrophin expression was confirmed by immunofluorescent staining (4 days after transduction), exosomes were collected from conditioned media, followed by miR isolation and qPCR for miR expression (Fig. 1).
Figure 1 .
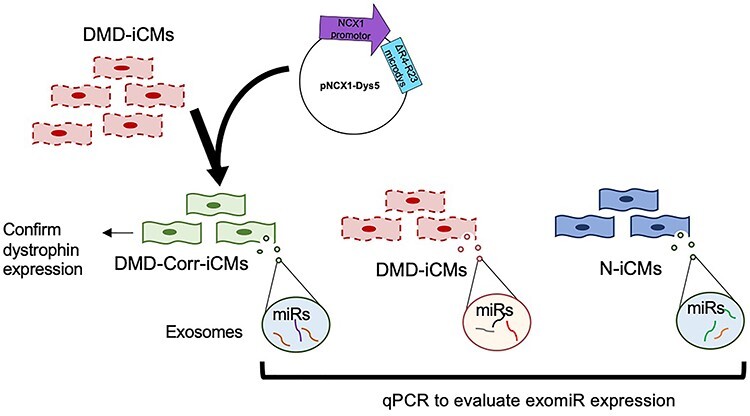
Schematic for dystrophin-restoration and experimental overview. Dystrophin was partially restored with a microdystrophin lentiviral construct containing the ∆R4-R23 domain under control of the cardiac NCX1 promotor. Cardiomyocytes were transduced with the lentivirus and given 4 days recovery for dystrophin to express, followed by harvesting conditioned media after 48 h for exosome isolation, and qPCR for exosomal miRNA expression.
Results
Restoring dystrophin mitigates stress in DMD cardiomyocytes and is associated with altered expression of exosomal miRs
We first confirmed dystrophin was expressed in DMD-iCMs by confirming >95% transduction efficiency via immunostaining for dystrophin (Fig. 2A and B). Induction of metabolic stress in DMD-iCMs causes injury in DMD-iCMs, and restoration of dystrophin with microdystrophin diminished stress-induced ROS levels, mitochondrial membrane potential preservation and decreased cell death (Fig. 2C–E). Therefore dystrophin-deficiency appeared to be responsible for the vulnerability of the cells to stress-induced injury.
Figure 2 .
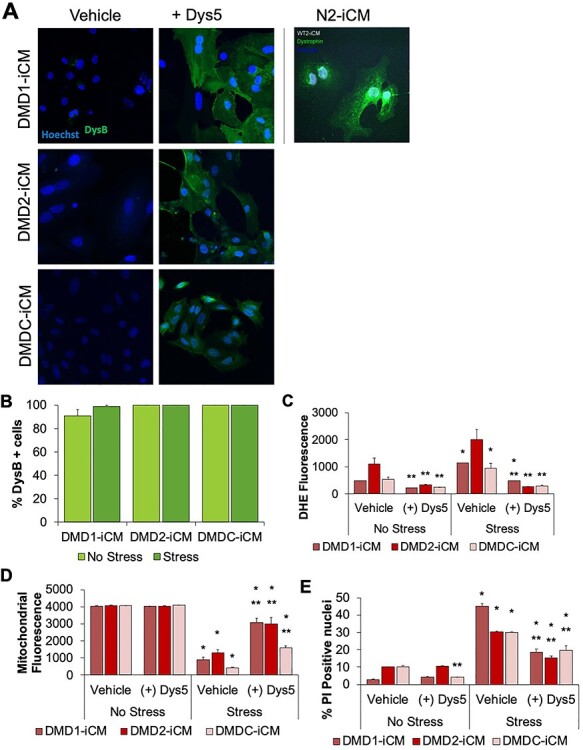
Restoring dystrophin ameliorates stress-induced phenotypes in DMD-iCMs. (A) Four days after transduction, DMD-iCMs were fixed and stained with 1:100 NCL-DysB antibody (Leica) to assess dystrophin expression. (B) Quantification of percentage of dystrophin-positive cells under non-stressed or stressed conditions. Four days after transduction of microdystrophin into three separate DMD-iCM lines, cardioprotective effects are observed with respect to (C) reduction of stress-induced ROS levels, (D) protection of mitochondrial membrane potential and (E) reduction of cell death. These data demonstrate that the stress phenotype is associated with dystrophin-deficiency, as restoring dystrophin reverses the effects of stress injury. N = 3 biological triplicates; *P < 0.05 versus No Stress; **P < 0.05 versus. Vehicle.
To examine whether dystrophin-deficiency of DMD-iCMs could alter expression of exomiRs, exosomes were collected from conditioned media for isolation of exomiRs and experiments were performed as described in Figure 1. Expression of 84 cardiovascular miRs was evaluated by qPCR array to examine whether any DMD exomiRs were normalized to N-exo levels following dystrophin restoration. Since these exosomes are isolated from isogenic control cell lines, altered exomiR levels are predicted to be attributed to dystrophin-deficiency. Controls included N-exo and DMD-exo for comparison and DMD-NCX1 as a transduction control. ExomiRs were dysregulated in DMD-exo in comparison to N-exo (Fig. 3A). Taking into account groups with miR alteration due to transduction (DMD-NCX1) and with dystrophin restoration (DMD-Corr), 10 out of 84 DMD exomiRs were normalized to N-exo levels. Of these exomiRs, let-7b-5p, miR-298, miR-338-3p and miR-485-3p were low in DMD-exo and high in N-exo, and dystrophin restoration led to increased exomiR levels. ExomiRs miR-124-3p, miR-135b-5p, miR-339-5p, miR-346, miR-431 and miR-98-5p were high in DMD-exo and low in N-exo, and dystrophin restoration decreased these exomiR levels. In particular, miR-339-5p was upregulated 35-fold in comparison to N-exo, and this was diminished almost 30-fold in DMD-Corr exosomes (Fig. 3B). Subsequent qPCR analysis confirmed 16-fold upregulation of miR-339-5p in DMD-exo (Fig. 3C). Since restoring dystrophin in iPSC-derived cardiomyocytes altered the profile of several DMD exomiRs to N-exo levels, these findings suggest dysregulation of exomiRs was due, in part, to dystrophin-deficiency.
Figure 3 .
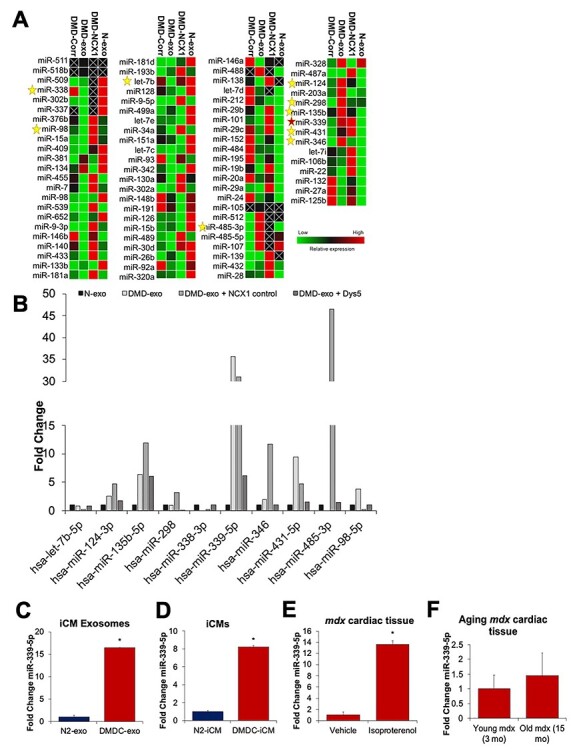
Dystrophin-deficiency is associated with alteration of exosomal miR expression. DMD-iCMs were transduced with a microdystrophin construct to restore dystrophin, followed by collection of conditioned media to harvest and isolate exosomes from DMD-iCMs, DMD-exo from microdystrophin transduced cardiomyocytes (DMD-Corr), DMD-exo from a transduction control (DMD-NCX1) or N-exo from N-iCMs. (A) Heatmap generated from expression of 84 cardiac disease–associated miRNAs in exosomes. The stars denote 10 exo-miRs from DMD-exo normalized to N-exo levels when dystrophin was restored in DMD-iCMs. (B) Graph of 10 exo-miRs altered with dystrophin restoration. (C) qPCR confirmation of miR-339-5p overexpression in DMD-exo in comparison to N-exo. N = 3 independent experiments; *P < 0.05 versus N-exo. (D) MiR-339-5p is also overexpressed in DMD-iCMs. N = 3 independent experiments; *P < 0.05 versus N-iCMs. (E) MiR-339-5p is overexpressed in Isoproterenol-injured mdx cardiac tissue in comparison to vehicle. N = 6 animals; *P < 0.05 versus Vehicle. (F) MiR-339-5p levels are elevated in 15 months mdx cardiac tissue in comparison to 3 months mdx cardiac tissue.
Increased levels of both cellular and exosomal miRs have been observed, suggestive of an association among miR levels from the matching cell-exosome type (46). It is not yet known whether a similar phenomenon exists in cardiovascular disease, and if so, whether cellular and exosomal miR levels are expressed to the same degree. Both cellular and exosomal miRs are responsible for regulation of important processes in cardiovascular disease (47,48). Therefore, we investigated cellular levels of miR-339-5p to establish an association between cellular and exosomal levels. Assessment of miR-339-5p in DMD-iCMs revealed an 8-fold upregulation of miR-339-5p in DMD-iCMs versus N-iCMs (Fig. 3D) and a 15-fold upregulation in DMD-exo versus N-exo. Together, upregulation of miR-339-5p in both DMD-iCMs and DMD-exo suggests a correlation between cellular levels of miRs and the levels resulting in secreted exosomes.
To establish the potential of miR-339-5p as a disease-specific marker for cardiomyopathy in DMD, we assessed miR-339-5p levels in mdx hearts, a mouse model of DMD cardiomyopathy (49). Since the cardiac defect of mdx mice is often not revealed until age 12 months (50,51), we utilized a subacute cardiac injury model with 5× daily isoproterenol intraperitoneal (i.p.) injections in 12-week-old mice to induce cardiac fibrosis (50–53). Upregulation of miR-339-5p was observed in isoproterenol-injured mdx mouse cardiac tissue compared with vehicle control, as well as in aging (15 months) mdx cardiac tissue (Fig. 3E and F). These data indicate that miR-339-5p increases with cardiac injury as well as age in mdx mice. Corresponding expression patterns of DMD-iCM and DMD-exo miRs indicate the disease status of the releasing cell type is sufficient to affect the loading of the exomiR cargo. Further, upregulation of miR-339-5p in DMD-iCMs and mdx hearts reveals its potential to be a cardiomyopathy-sensitive target in DMD.
Identifying stress-responsive gene targets of miR-339-5p in DMD-iCMs
Once we established miR-339-5p as a disease-specific miR in DMD, we next wanted to explore potential gene targets of miR-339-5p, which may be associated with the deleterious stress-responsive role of DMD exosomal miR cargo (42). In DMD, cardiomyocytes are extremely vulnerable to stress, which leads to cellular injury and death (13,14,19). Signaling mechanisms underlying these pathological processes are not well understood. To identify targets of miR-339-5p, which may alter stress responses, the TargetScan 7.2 database was used (54). This search identified MDM2, MAP2K3, GSK3A and MAP3K10 as known and predicted targets of miR-339-5p. Alterations in these targets have been associated with pathological processes in cardiovascular disease (54–58). However, these targets have not yet been investigated in DMD.
Following miR-339-5p target analysis, we wanted to confirm miR-339-5p regulation of these genes in DMD-iCMs. To assess target gene levels, DMD-iCMs were exposed to miR-339-5p-depleted DMD-exo, followed by qPCR analysis. To produce miR-339-5p-depleted exosomes, cellular knockdown was performed, followed by exosome harvest. Transfection of miR-339-5p in DMD-iCMs was observed within 24 h (Supplementary Material, Figure S1). Analysis of miR-339-5p in DMD-iCMs demonstrated a significant reduction in cellular miR-339-5p levels after 48 h (Supplementary Material, Figure S1). Therefore, we proceeded with harvesting DMD-exo to confirm inhibition of exosomal levels of miR-339-5p. Exosomes depleted of miR-339-5p were harvested from DMD-iCM media conditioned for 48 h where DMD-iCMs had >95% efficiency of anti-miR transfection and >60% reduction of cellular miR-339 levels (Fig. 4A). Following these confirmatory steps, exosomal levels of miR-339-5p were verified. Using these quality control steps yielded exosomes depleted of miR-339 by 40% (Fig. 4B).
Figure 4 .
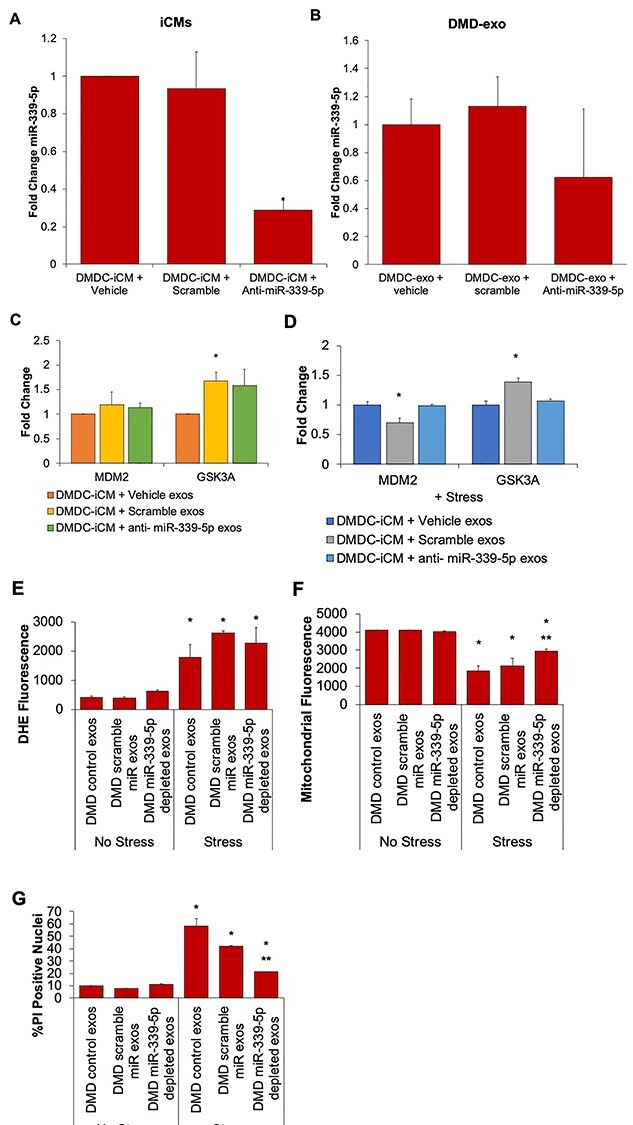
Exposure to miR-339-5p-depleted exosomes alters mitochondrial stress response, but not target gene levels. Following 48 h knockdown with anti-miR-339-5p, miR-339-5p is depleted in (A) DMD-iCMs and (B) DMD-exos. N = 3 independent experiments; *P < 0.05 versus Vehicle. Following 48 h exposure to miR-339-5p-depleted exosomes, levels of target genes MDM2 and GSK3A (C) remain unchanged at baseline and (D) remain unchanged following stress. (E) Stress-induced ROS levels also remain the same following 48 h exposure to miR-339-5p-depleted exosomes. (F) Exposure to miR-339-5p-depleted exosomes results in protection against stress-induced mitochondrial injury and (G) increases in cell death, indicating miR-339-5p may be involved in modulating mitochondrial responses to stress. N = 3 independent experiments; *P < 0.05 versus Vehicle.
Once miR-339-5p depletion in DMD-exo was confirmed, these exosomes were subsequently used in exposure assays to query whether exosomal miR-339-5p altered expression of target genes involved in responding to stress stimuli. Exosomes depleted of miR-339-5p content were added to DMD-iCMs for 48 h, followed by harvesting cellular RNA to assess target gene expression. Since MDM2 and GSK3A are two known targets of miR-339-5p, these were evaluated by qPCR (54,55,59). Exosomes harvested from vehicle-treated DMD-iCMs or scramble-treated DMD-iCMs served as vehicle and negative controls for comparison, respectively. Exposure to miR-339-5p-depleted exosomes for 48 h was not associated with increased MDM2 expression but was associated with increased GSK3A in comparison to controls (Fig. 4C). Since no demonstrable increase in multiple targets was seen with exposure to miR-339-5p exosomes at baseline, a stress protocol involving 1 h of stress media and 4 h recovery in normal media was added to reveal the stress phenotype previously observed in DMD-iCMs (14,19). MDM2 and GSK3A are associated with stimulating protective pathways to respond to cellular stress (56,60); therefore, we predicted adding stress may induce gene expression. Following exposure to miR-339-5p-depleted DMD-exo and stress protocol, RNA was harvested to check gene expression. Interestingly, when stress was added, levels of MDM2 and GSK3A remained unchanged (Fig. 4D). However, there seemed to be an effect with the scramble control exosomes, making it difficult to interpret the results.
While experiments involving miR-339-5p-depleted exosomes did not demonstrate alteration of expression of the two genes assayed, we still wanted to know whether exosomal miR-339-5p was associated with the stress response in DMD-iCMs. It is possible miR-339-5p modulates a cell stress response independently of the two genes assayed. To investigate whether exosomal miR-339-5p was altering the stress response, DMD-iCMs were exposed to miR-339-5p-depleted exosomes for 48 h prior to the stress protocol described above. DMD control exosomes and DMD scramble anti-miR exosomes served as controls for comparison. The stress phenotyping assays involve live cell imaging to measure reactive oxygen species (ROS) levels and mitochondrial membrane potential. Following a 24-h recovery period, cells are fixed for propidium iodide (PI) staining to evaluate the proportion of dead cells versus total cells. Exposure to miR-339-5p-depleted exosomes did not alter stress-induced ROS levels in DMD-iCMs but did protect mitochondrial membrane potential and reduce stress-induced cell death in DMD-iCMs (Fig. 4E–G). These findings suggest exosomal delivery of miR-339-5p is involved in stress responses in DMD-iCMs in a mitochondrial and cell death–dependent manner.
Knockdown of miR-339-5p decreases cell injury in DMD-iCMs by modulating expression of stress-responsive genes
Since exosomal depletion of miR-339-5p improved the cellular response to stress in DMD-iCMs but did not downregulate expression of known targets, we instead looked at how miR-339-5p may be directly modulating a stress response. To assess direct effects of miR-339-5p levels, knockdown cellular expression of miR-339-5p in DMD-iCMs was performed, followed by qPCR and western blotting to check resulting target gene expression and protein levels. Knockdown of miR-339-5p was associated with increased levels of MDM2, GSK3A and MAP2K3, but not MAP3K10 in DMD-iCMs (Fig. 5A). Knockdown of miR-339-5p was also found in conjunction with increased MDM2 protein levels, as well as increases in GSK3A and MAP2K3 proteins (Fig. 5B–G).
Figure 5 .
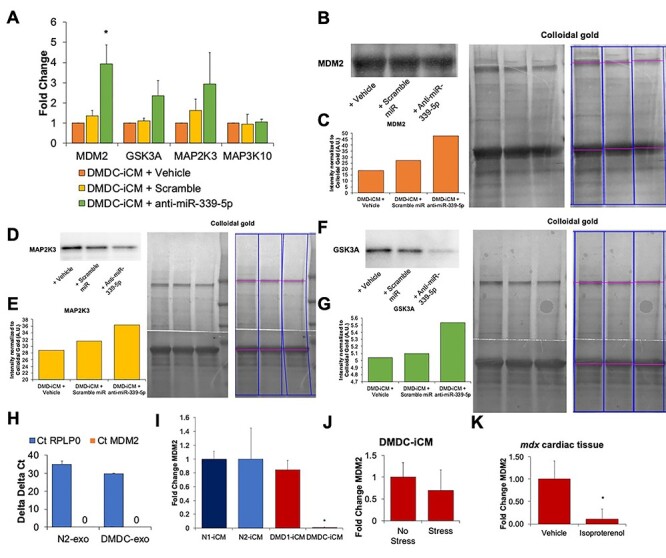
Knockdown of miR-339-5p in DMD-iCMs leads to elevated target gene expression. MiR-339-5p was knocked down in DMD-iCMs, followed by qPCR for target gene expression or western blotting for protein expression. (A) Knockdown of miR-339-5p is associated with increased expression of MDM2, GSK3A and MAP2K3 in DMD-iCMs. N = 3 independent experiments; *P < 0.05 versus Vehicle. For blotting, colloidal gold whole lane staining was used for normalization of proteins. Representative blots indicating lanes analyzed are shown here. (B) and (C) Inhibiting miR-339-5p was associated with an increase in MDM2 protein levels in comparison to vehicle control. (D) and (E) Inhibiting miR-339-5p was associated with a modest increase in MAP2K3 protein levels in comparison to vehicle control. (F) and (G) Similarly, inhibiting miR-339-5p was also associated with increased GSK3A in comparison to vehicle. Biological triplicate samples were pooled and run in technical triplicate on a single blot to serve as N = 1. (H) MDM2 is not found in N- or DMD-exo. N = 3 biological triplicates. (I) MDM2 mRNA levels are downregulated in 1 of 2 DMD-iCM lines tested. N = 3 biological triplicates; *P < 0.05 versus N-iCM. (J) MDM2 mRNA expression is downregulated in DMD-iCMs following exposure to metabolic stress. N = 3 biological triplicates. (K) In mdx mouse cardiac tissue, MDM2 mRNA levels are decreased following exposure to isoproterenol stress. N = 6 animals/group; *P < 0.05 versus Vehicle.
We focused more closely on examining MDM2, since it is associated with regulation of p53-mediated apoptosis (61) and stimulation of pro-survival signaling (62). Increased apoptosis and impaired survival signaling have been previously described in models of DMD (13–15,17–19,51). However, the role of MDM2 in DMD cardiomyopathy has not yet been investigated. Using qPCR, we confirmed knockdown of miR-339-5p was directly responsible for increases in MDM2, rather than exosomal delivery of the gene (Fig. 5H). MiR-339-5p is known to directly bind to MDM2, leading to its downregulation (59). Baseline expression of cellular levels of MDM2 was evaluated in two N-iCM lines and two DMD-iCM lines. MDM2 levels were significantly downregulated in DMD-iCMs in comparison to N-iCMs (Fig. 5I), and therefore DMDC-iCMs were used for evaluating MDM2 further.
To investigate whether MDM2 was important for the stress-phenotype in DMD, expression was characterized in DMD-iCMs subject to the stress protocol and mdx mice hearts subject to isoproterenol-induced injury. MDM2 was downregulated in stressed DMD-iCMs and in isoproterenol-stressed mdx mouse cardiac tissue (Fig. 5J and K). These data support MDM2 as an important stress-responsive target in DMD.
Next, we interrogated whether miR-339-5p was directly involved with modulating the stress response in DMD-iCMs. To assess this, we knocked down cellular levels miR-339-5p in DMD-iCMs, followed by the stress protocol and the stress-phenotype imaging protocols. Knockdown of miR-339-5p was not associated with any change in stress-induced ROS levels (Fig. 6A). However, miR-339-5p depletion was associated with preservation of mitochondrial membrane potential and reduction in stress-induced cell death when compared with vehicle conditions (Fig. 6B and C). These data indicate a role for miR-339-5p in modulating the cellular response to stress in DMD-iCMs.
Figure 6 .
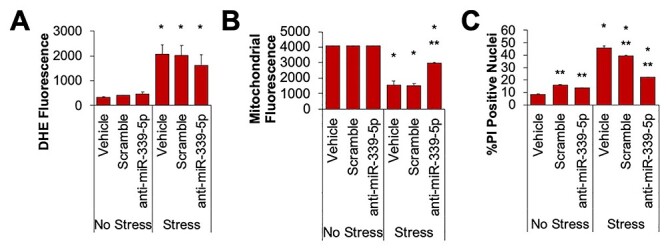
Inhibiting miR-339-5p protects mitochondria and decreases cell death in DMD-iCMs. (A) Knockdown of miR-339-5p does not alter stress-induced ROS levels. (B) Knockdown of miR-339-5p protects mitochondria from stress injury and (C) reduces stress-induced cell death. N = 3 independent experiments; *P < 0.05 versus No Stress; **P < 0.05 versus Vehicle.
Discussion
In our present study, we demonstrated diseased DMD cardiomyocytes secrete exosomes with dysregulated miR content in comparison to healthy controls. Further, dysregulated exomiRs were due in part to dystrophin-deficiency. Both cellular and exosomal miR levels were elevated, demonstrating a relationship between their expression levels. The increased levels in cells and exosomes indicate that the disease status of the releasing cell was sufficient to alter the miR content of exosomes. Analysis of 84 cardiac disease–associated miRs revealed 10 DMD exomiRs that were normalized to N-exo levels following dystrophin restoration. MiR-339-5p was significantly upregulated in DMD-exo, DMD-iCMs and isoproterenol-injured mdx mouse hearts, indicating miR-339-5p is a DMD cardiomyopathy-specific miR. Suppressing miR-339-5p in DMD-exo and DMD-iCMs improved cellular response to stress-induced mitochondrial injury and cell death, suggestive of its role in modulating important stress-responsive pathways. One mechanism cellular miR-339-5p appeared to work through is by modulating expression of gene targets MDM2, GSK3A and MAP2K3, which are genes associated with mounting an effective stress response and downregulating cell death processes (54,55,59,63). These findings identify disease-specific miR-339-5p, and highlight one mechanism by which dysregulated miRs, found in exosomes and in cardiomyocytes, may promote pathological processes in DMD cardiomyopathy.
At present, few studies have identified miR biomarkers in DMD cardiomyopathy (64–66). While these biomarkers are found in aberrant levels in comparison to healthy controls, it is unknown whether they are associated with the disease status of the releasing cell (dystrophin-deficiency) or whether they are consequences of pathological processes occurring in the cell (64–66). Our present study shows restoring dystrophin with microdystrophin in DMD-iCMs alters the expression of 10 DMD exomiRs back to non-affected levels, demonstrating dystrophin-deficiency is sufficient to alter the miR cargo of exosomes. Although cellular conditions, such as oxidative stress, have been shown to alter miR expression (67), whether such conditions subsequently affect their loading of exosomes are still under investigation. It is unknown whether other processes known to contribute to DMD pathology (such as impaired calcium handling, oxidative stress or inflammation) may also be contributing to altering DMD exomiR content (10,14,51,68–70).
Exosomal miR cargo has previously been shown to affect both protective pathways and injurious pathways in the context of cardiovascular disease (48,71–76). Prior work in the lab demonstrated removing DMD exomiR cargo improved the stress response in DMD-iCMs (42). Here, we identify a potential mechanism by which DMD exomiR cargo alters cellular phenotypes. Specifically, exosomal miR-339-5p modulates the impaired stress response in DMD-iCMs. This was shown when decreased miR-339-5p was associated with preservation of mitochondrial membrane potential and reduced stress-induced cell death in DMD-iCMs (Fig. 4). MiR-339-5p is important for regulation of cellular processes such as cell death, and its dysregulation has been associated with ischemic injury and heart failure (77–80). Elevated cell death has been observed in animal and cell-based models of DMD cardiomyopathy (13,14,19,51). Taken together, these findings suggest the exosomal delivery of miR cargo is capable of altering the cell’s response to stress, and miR-339-5p is one candidate for the dysregulation of these processes in DMD-iCMs.
Depletion of miR-339-5p in exosomes did not lead to any alteration in target gene expression in DMD-iCMs (Fig. 4). The DMD-iCMs are continuously releasing non-modified exosomes that may mix with the miR-339-5p-depleted exosomes to mask small changes of mRNA target levels. To address this possibility, we bypassed the indirect effects of exosomal miRs by directly knocking down miR-339-5p in DMD-iCMs. Examination of GSK3A, MAP2K3 and MDM2 mRNA levels revealed a reciprocal increase in mRNA levels with the cellular decrease in miR-339-5p in DMD-iCMs (Fig. 5). GSK3A is a multi-functional kinase known to modulate a pro-survival cellular response to stress via regulation of mitochondrial function and cell death (81). Similarly, MDM2 and MAP2K3 are also known to modulate mitochondrial activity and regulate cell death–associated processes (74,82–85). Focusing on MDM2, a negative regulator of p53-mediated cell death (59,63) and pro-survival signaling (62), we found it was downregulated in in vitro and in vivo models of DMD following stress. MDM2 may be a novel target in DMD cardiomyopathy and is downregulated by overexpression of miR-339-5p.
With knockdown of miR-339-5p, DMD-iCMs were better able to respond to stress conditions by preserving mitochondrial membrane potential and ultimately suppress cell death (Fig. 6). Exposure to miR-339-5p-depleted exosomes or miR-339-5p knockdown was associated with no change in stress-induced ROS levels (Figs 4 and 6), indicating the absence of miR-339-5p may not be sufficient to modulate non-mitochondrial, alternative sources of ROS, such as xanthine oxidase (86). Since knockdown of miR-339-5p was associated with elevated MDM2 levels and an improved response to the effects of stress on mitochondrial injury and cell death, these findings indicate the disease specificity of miR-339-5p in modulating responses to stressful stimuli in DMD-iCMs. In particular, this mechanism may be through modulation of MDM2. Importantly, miR-339-5p is DMD-specific and impairs the ability of diseased DMD cardiomyocytes to stimulate cardioprotective gene expression. Together, these data indicate miR-339-5p may be suppressing a proper response to stress in DMD-iCMs by downregulating important regulators of mitochondrial and cell death.
Here, we provide evidence of miR-339-5p as a novel disease-specific marker of DMD cardiomyopathy. Mechanistically, cellular levels of miR-339-5p appear to downregulate important regulatory genes involved in mitochondrial function and cell death. Upregulation of miR-339-5p is a consequence of dystrophin-deficiency, and its exosomal delivery impairs the ability of DMD cardiomyocytes to protect against stress injury. Identification of this disease-sensitive miR may lead to better understanding of the cellular disruptions occurring during DMD cardiomyopathy. More work is needed to elucidate the cellular pathways and processes miR-339-5p may be altering. These findings may lead to improved assay development to track and monitor disease progress, as well as shed light on the cellular consequences of dystrophin-deficiency, so more targeted therapies can be designed to treat cardiomyopathy in DMD.
Materials and Methods
Culture and cardiac differentiations of iPSCs
For the experiments in this study, one non-affected, control cell line (N2-iPSC) and an isogenic, gene-edited DMD-iPSC (DMDC-iPSC) line were used. Briefly, DMDC-iPSCs were generated from N2-iPSC by CRISPR-Cas9, which generated a 6 bp deletion in exon 1, leading to dystrophin-deficiency (19). In select experiments, two patient-derived DMD-iPSC lines were used (DMD1- and DMD2-). Both of these DMD lines contain exon 3–6 deletions. The pluripotency and genotypes of iPSC lines have been previously characterized (19). Cells were routinely karyotyped. The karyotypes of N2 and DMD1 and DMD2-iPSCs were previously published (19,87). The karyotype of DMDC-iPSCs is normal (Supplementary Material, Figure S2). Cells were maintained in Mtesr1 media (Stem Cell Technologies) on Matrigel (Corning). Cardiac differentiations were performed as previously described (14,19). Cardiomyocytes contracting for 30± 5 days were selected for experiments. Cardiac differentiations of all iCM lines have been characterized previously (14,19).
Exosome exposure assays
Exosomes were isolated from iCMs as previously described (19,42). Briefly, 200 k cells were plated and media was harvested after 48 h. Exosomes were isolated from cell culture media using total exosome isolation reagent from cell culture media (Thermo Fisher) per the manufacturer’s instruction. Exosomes were harvested from cardiac differentiations with >90% cardiomyocytes as confirmed by immunostaining for alpha-actinin (Abcam #9465). Multiple exosome batches were used in experiments to confirm consistency in paracrine effects. For exosome exposure experiments, 5 μl of isolated exosomes were added to cardiomyocytes for 48 h prior to metabolic stress assays as previously described (42). These exosomes have been previously quantified by nanosight and exosome quantitation assays. The dosage of 5 ul was previously optimized and found to elicit phenotypic responses in DMD-iCMs (42).
Metabolic stress injury protocol
For metabolic stress assays, cardiomyocytes were exposed to stress as previously described (19). Briefly, cells were exposed to stress media (100 μm H2O2 in 10 mm deoxyglucose (Sigma) in RPMi—glucose) (Thermo Fisher) for 1 h, followed by a 4-h recovery in normal media (RPMi/B27 + glucose, Thermo Fisher). After 4-h recovery, cells were subject to staining for live imaging.
Live imaging of cardiomyocytes
Cardiomyocytes were dissociated as previously described (19). To identify live cardiomyocytes for imaging, cells were transduced with NCX1-eGFP lentivirus 3–4 days prior to imaging (45,88). This construct contains an enhanced green fluorescent protein (GFP) under the control of cardiac promotor NCX1. Transduction with this construct allowed for selection of cardiomyocytes in subsequent image analysis. In mitochondrial membrane potential assays, cells were stained for 20 min with 50 nm tetramethylrhodamine ethyl ester (TMRE, Sigma). In reactive oxygen species (ROS) assays, cells were stained for 20 min with 10 μm dihydroethidium (DHE, Sigma) to measure cytosolic superoxide levels. Nuclei were counter-stained with Hoechst 33342 (Sigma). Live cell images were taken by laser-scanning confocal fluorescent microscopy (Nikon A1-R) utilizing NIS Elements software (Version 5.11.00, 64 bit). Imaging conditions such as gain levels, frames per second and aperture size were held constant. Three images were taken per coverslip from randomly selected areas, for subsequent analysis. Analysis of superoxide and membrane potential staining was performed in ImageJ software (Version 1.52p, Java 1.8.0_172, National Institutes of Health) as previously described (19,42).
Propidium iodide staining for cell death
Evaluation of cell death was performed by propidium iodide (PI) staining. Twenty-four hours after stress, cells were fixed with 4% paraformaldehyde (Alfa Aesar) for 15 min. Staining with 500 nm PI was performed per the manufacturer’s protocol (Thermo Fisher). Coverslips of fixed cells were mounted to slides for imaging with Fluoroshield containing DAPI (Sigma). Imaging and subsequent analysis of cells was performed as previously described (19,42).
Dystrophin restoration with microdystrophin
To partially restore dystrophin in DMD-iCMs, a microdystrophin construct called Dys5 ∆R4-R23 (kind gift of Dr Jeff Chamberlain, University of Washington) was utilized (43,44). The dystrophin construct was engineered into a lentivirus under control of the NCX1 cardiac promotor (45), to allow it to be taken up and expressed specifically in iCMs. Lentiviral assembly was performed by Versiti Blood Research Institute Viral Vector Core (Milwaukee, WI). Cardiomyocytes were transduced with then NCX1-microdystrophin construct, followed by a media change 24 h post-transduction. Four days after transduction, efficiency of transduction was confirmed by immunofluorescent detection of dystrophin using NCL-DYSB (Leica Novocastra, Cat. NCL-DYSB) as previously described (19). Efficiency of transduction required >90% dystrophin-positive iCMs to be used for downstream experiments. For all subsequent analyses with dystrophin restoration, cells were allowed 4 days for dystrophin to express, followed by phenotypic assays (exosome collection or stress assays).
RNA isolation
RNA was harvested from cardiomyocytes and mouse tissue using the PureLink RNA isolation kit (Thermo Fisher) per the manufacturer’s instruction. Following exosome isolation, RNA was collected using Total Exosome RNA and Protein Isolation kit (Thermo Fisher) per manufacturer’s instruction. The concentration of RNA was measured on a Nanodrop 2000 Spectrophotometer (Thermo Fisher).
qPCR of mRNA
Reverse transcription of iCM RNA was performed using the iScript CDNA synthesis kit (Bio-Rad), and the reaction included 5 min at 25°C, 20 min at 46°C, 1 min at 95°C and holding at 4°C. At least 100 ng RNA was used in qPCR experiments, as determined by Nanodrop readings. Consistent RNA concentrations were used. qPCR was performed using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on a Bio-Rad CFX96 Real-Time System (Bio-Rad) and the reactions were carried out with an initial step of 30 s at 95°C, followed by 40 cycles of denaturation (15 s at 95°C) and annealing (30 s at 60°C), followed by fluorescence capture and holding at 4°C. Primers are listed in Table 1. Data were analyzed using the ∆∆Ct method in CFX Manager software (Bio-Rad) and RPLPO was used as a normalization control. Other controls included a no template control, a no reverse transcriptase control and a genomic DNA contamination control.
Table 1.
qPCR primers used in this study
| Gene target | Primer |
|---|---|
| RPLPO | Qiagen QuantiTect Primer #QT00075012 |
| MDM2 | F: TGGCGTGCCAAGCTTCTCTGT |
| R: ACCTGAGTCCGATGATTCCTGCT | |
| GSK3A | F: GCAGATCATGCGTAAGCTGGAC |
| R: GGTACACTGTCTCGGGCACATA | |
| MAP2K3 | F: CTTGGTGACCATCTCAGAACTGG |
| R: CTTCTGCTCCTGTGAGTTCACG | |
| MAP3K10 | F: GTTTGATGACCTTCGGACCAAGG |
| R: CGATGTCCATCTCACGTTCTGC | |
| RNU6 | HS RNU6–2 miScript Primer Assay # MS00033740 |
| miR-130a-3p | Hs miR-130a-3p miScript Primer Assay # MS00003444 |
| miR-339-5p | Hs miR-339-5p miScript Primer Assay # MS00003997 |
qPCR of miRNA
Reverse transcription of microRNA (miRs) was performed with the miScript RT II kit, per manufacturer’s instruction for mature miRs, on a Bio-Rad C1000 Touch Thermal Cycler (Bio-Rad). qPCR for RNU6 and hsa-miR-130a was performed using the miScript SYBR Green PCR Kit (Qiagen) on a Bio-Rad CFX96 Real-Time System (Bio-Rad) which involved an initial activation step of 15 min at 95°C, followed by 40 cycles of denaturation (15 s at 94°C), annealing (30 s at 55°C) and extension (30 s at 70°C), followed by fluorescent data capture and holding at 4°C. Primers used (Table 1) were specific to miRNA sequences and did not target any known mRNA sequences. Data was analyzed using the ∆∆Ct method and CFX Manager software and RNU6 served as a normalization control. Other controls included a no template control, a no reverse transcriptase control and a genomic DNA contamination control.
qPCR disease pathway miRNA arrays and analysis
To assess changes in exosomal miR expression due to dystrophin-deficiency, DMD-iCMs were transduced with NCX1-microdystrophin (or NCX1-eGFP as a transduction control). Four days after transduction, media was collected from iCMs for exosome isolation and exosomal miRs isolation as described above. Reverse transcription of miRNA was performed as described above, followed by qPCR with miScript miRNA Cardiovascular Disease Pathway Focused Arrays (Qiagen) on a Bio-Rad CFX-96 (Bio-Rad), which involved an initial activation step of 15 min at 95°C, followed by 40 cycles of denaturation (15 s at 94°C), annealing (30 s at 55°C) and extension (30 s at 70°C), followed by fluorescent data capture and holding at 4°C. Data were analyzed using the ∆∆Ct method and CFX Manager software (Bio-Rad) and RNU6 served as a normalization control. Additional controls included a no template control, a no reverse transcriptase control and a genomic DNA contamination control. Comparative analyses of miR expression utilized non-affected and DMD exosomal miR levels for comparison. Altered expression of miRs due to transduction, using the NCX1-eGFP transduction control groups, was eliminated from further consideration.
Identification of miR-339-5p gene targets
To identify interesting gene targets of miR-339-5p in our RNA sequencing dataset, Qiagen’s Ingenuity Pathway Analysis software was used. The RNA sequencing dataset containing gene expression information in DMD-iCMs following DMD exosome exposure was put through a MicroRNA Target Filter analysis including the top three differentially expressed DMD exosome miRs: miR-338-3p, miR-339-5p and miR-431. The target filter analysis allowed us to select for miRs that were upregulated and identified known and predicted gene targets of these miRs in the sequencing dataset. Genes were subsequently filtered down to those that were downregulated and reciprocally the miR was upregulated. Pathway filtering allowed selection for pathways known to regulate stress responses in either DMD or cardiovascular disease: HIF1-alpha, mitochondrial dysfunction, hypoxia signaling, apoptosis signaling. From there, MDM2 was identified as a putative target altered during exposure to DMD exosomes, and it is targeted by miR-339-5p. Therefore, miR-339-5p and MDM2 were chosen for further mechanistic evaluation.
Additional targets of miR-339-5p were chosen by using TargetScan miR database (54). This database revealed GSK3A, MAP2K3 and MAP3K10 as additional targets for examination in mechanistic studies.
Knockdown of miR-339-5p in vitro
To knockdown expression of miR-339-5p in iCMs and exosomes, commercially designed miR-339-5p inhibitors were obtained (Qiagen) and optimized in DMD-iCMs. Cells were transduced with 40 nm anti-miR-339-5p, miRNA scramble or anti-130a-3p inhibitors 48 h prior to experiments. Lipofectamine 3000 (1.5× concentration) served as a vehicle control, anti-miR-130a-3p served as an internal control for knockdown efficiency, and scramble miRNA inhibitors served as an additional control. The inhibitors contained a FAM tag and green fluorescence was observed 24 h post-transduction and served as a positive transduction control. Efficiency of knockdown was confirmed by qPCR for miR-339-5p in iCMs and exosomes, and experiments with >40% miR-339-5p were used for subsequent analyses. In experiments using miR-339-5p-depleted exosomes, cellular knockdown was performed and exosomes were harvested for assays.
Western blotting
Lysates were collected from DMD-iCMs using RIPA buffer (Thermo Fisher) and Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher) on ice. Protein concentration was measured using a BCA Assay (Thermo Fisher). For western blotting, MDM2 (Abcam ab259275) primary antibody was used at 1:200 dilution, GSK3A (Abcam ab40870) was used at 1:2500 dilution and MEK3 (Abcam ab195037) was used at 1:2500 dilution. Blots were incubated for 2 h at room temperature with agitation. Secondary antibody HRP-linked antirabbit IgG (Jackson Labs, #711–035-152) was used for MDM2, GSK3A and MEK3 membranes at a dilution of 1:5000 for 1 h at room temperature with agitation. Protein bands were visualized by Clarity Western ECL Substrate (GE Life Sciences, #RPN2236). Images were taken on ChemiDoc MP System (Bio-Rad) and densitometric analysis was performed using Image Lab Software (Bio-Rad). For normalization of total protein loaded per lane, colloidal gold total protein staining (Bio-Rad) was performed according to Bio-Rad’s protocol. Normalized protein values were calculated by dividing the primary antibody adjusted volume by the colloidal gold total protein adjusted volume (89). In these experiments, biological triplicate samples were pooled and run in technical triplicates on a single blot to serve as N = 1.
Animal models
Animals used in this study received humane care in compliance with the Guide for the Care and Use of Laboratory Animals formulated by the National Research Council (2015) and approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin. All mice in this study were Female 11 to 12 weeks of age C57BL/10ScSn-Dmdmdx/J mice (Jackson Laboratories). In select experiments, female mice were aged to 15 months.
Statistical analysis
All in vitro experiments were performed in biological triplicate (N = 3) per experimental group and condition, unless otherwise noted. Experimental results are presented as mean ± SEM. Statistical analyses including Student’s t-test and one-way ANOVAs were performed as appropriate, using GraphPad Prism software and significance was determined as P ≤ 0.05.
Supplementary Material
Acknowledgements
We would like to acknowledge Margaret Haberman and Leah Thomas for their technical assistance.
Conflicts of Interest statement. M.W.L. is or was recently a scientific advisory board member for Solid Biosciences and Audentes Therapeutics. M.W.L. receives research support from Solid Biosciences and Audentes Therapeutics. M.W.L. is a consultant for Audentes Therapeutics, Encoded Therapeutics, AGADA Biosciences, Prothelia, Biomarin, Kate Therapeutics, Lacerta Therapeutics, Affinia Therapeutics, Modis Therapeutics and Dynacure. All other authors declare no potential conflicts of interest exist.
Contributor Information
Melanie Gartz, Department of Cell Biology, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Research Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Neuroscience Research Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Margaret Beatka, Neuroscience Research Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Department of Pathology, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Mariah J Prom, Neuroscience Research Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Department of Pathology, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Jennifer L Strande, Department of Cell Biology, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Cardiovascular Research Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Department of Medicine, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Michael W Lawlor, Department of Cell Biology, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Neuroscience Research Center, Medical College of Wisconsin, Milwaukee, WI 53226, USA; Department of Pathology, Medical College of Wisconsin, Milwaukee, WI 53226, USA.
Funding
National Institutes of Health (K08HL11148, R01134932).
References
- 1. Hoffman, E.P., Brown, R.H., Jr. and Kunkel, L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 6, 919–928. [DOI] [PubMed] [Google Scholar]
- 2. Mendell, J.R., Rodino-Klapac, L.R., Sahenk, Z., Roush, K., Bird, L., Lowes, L.P., Alfano, L., Gomez, A.M., Lewis, S., Kota, J.et al. (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol., 5, 637–647. [DOI] [PubMed] [Google Scholar]
- 3. Roberto, R., Fritz, A., Hagar, Y., Boice, B., Skalsky, A., Hwang, H., Beckett, L., McDonald, C. and Gupta, M. (2011) The natural history of cardiac and pulmonary function decline in patients with duchenne muscular dystrophy. Spine, 15, E1009–E1017. Epub 2011/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bushby, K., Finkel, R., Birnkrant, D.J., Case, L.E., Clemens, P.R., Cripe, L., Kaul, A., Kinnett, K., McDonald, C., Pandya, S.et al. (2010) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol., 1, 77–93. [DOI] [PubMed] [Google Scholar]
- 5. McNally, E.M. (2007) New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu. Rev. Med., 58, 75–88. [DOI] [PubMed] [Google Scholar]
- 6. Nigro, G., Comi, L.I., Politano, L. and Bain, R.J. (1990) The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol., 3, 271–277. [DOI] [PubMed] [Google Scholar]
- 7. Ervasti, J.M. and Campbell, K.P. (1991) Membrane organization of the dystrophin-glycoprotein complex. Cell, 66, 1121–1131. [DOI] [PubMed] [Google Scholar]
- 8. Rybakova, I.N., Patel, J.R. and Ervasti, J.M. (2000) The dystrophin complex forms a mechanically strong link between the sarcolemma and Costameric actin. J. Cell Bio., 5, 1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petrof, B.J., Shrager, J.B., Stedman, H.H., Kelly, A.M. and Sweeney, H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. PNAS, 8, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jung, C., Martins, A.S., Niggli, E. and Shirokova, N. (2008) Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res., 4, 766–773. [DOI] [PubMed] [Google Scholar]
- 11. Kyrychenko, S., Polakova, E., Kang, C., Pocsai, K., Ullrich, N.D., Niggli, E. and Shirokova, N. (2013) Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res., 4, 666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wehling-Henricks, M., Jordan, M.C., Roos, K.P., Deng, B. and Tidball, J.G. (2005) Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet., 14, 1921–1933. [DOI] [PubMed] [Google Scholar]
- 13. Lin, B., Li, Y., Han, L., Kaplan, A.D., Ao, Y., Kalra, S., Bett, G.C., Rasmusson, R.L., Denning, C. and Yang, L. (2015) Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis. Model. Mech., 5, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Afzal, M.Z., Reiter, M., Gastonguay, C., McGivern, J.V., Guan, X., Ge, Z.D., Mack, D.L., Childers, M.K., Ebert, A.D. and Strande, J.L. (2016) Nicorandil, a nitric oxide donor and ATP-sensitive Potassium Channel opener, protects against dystrophin-deficient cardiomyopathy. J. Cardiovasc. Pharmacol. Ther., 6, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dahiya, S., Givvimani, S., Bhatnagar, S., Qipshidze, N., Tyagi, S.C. and Kumar, A. (2011) Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol., 187, 2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peter, A.K. and Crosbie, R.H. (2006) Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp. Cell Res., 312, 2580–2591. [DOI] [PubMed] [Google Scholar]
- 17. Nakamura, A., Harrod, G.V. and Davies, K.E. (2001) Activation of calcineurin and stress activated protein kinase/p38-mitogen activated protein kinase in hearts of utrophin-dystrophin knockout mice. Neuromuscul. Disord., 11, 251–259. [DOI] [PubMed] [Google Scholar]
- 18. Mourkioti, F., Kustan, J., Kraft, P., Day, J.W., Zhao, M.M., Kost-Alimova, M., Protopopov, A., DePinho, R.A., Bernstein, D., Meeker, A.K.et al. (2013) Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat. Cell Biol., 8, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gartz, M., Darlington, A., Afzal, M.Z. and Strande, J.L. (2018) Exosomes exert cardioprotection in dystrophin-deficient cardiomyocytes via ERK1/2-p38/MAPK signaling. Sci. Rep., 8, 16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu, N. and Olson, E.N. (2010) MicroRNA regulatory networks in cardiovascular development. Dev. Cell, 18, 510–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang, T.C. and Mendell, J.T. (2007) microRNAs in vertebrate physiology and human disease. Annu. Rev. Genomics Hum. Genet., 8, 215–239. [DOI] [PubMed] [Google Scholar]
- 22. Rao, P.K., Toyama, Y., Chiang, H.R., Gupta, S., Bauer, M., Medvid, R., Reinhardt, F., Liao, R., Krieger, M., Jaenisch, R.et al. (2009) Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res., 6, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wojciechowska, A., Braniewska, A. and Kozar-Kamińska, K. (2017) MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med., 26, 865–874. [DOI] [PubMed] [Google Scholar]
- 24. Moghaddam, A.S., Afshari, J.T., Esmaeili, S.A., Saburi, E., Joneidi, Z. and Momtazi-Borojeni, A.A. (2019) Cardioprotective microRNAs: lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis, 285, 1–9. [DOI] [PubMed] [Google Scholar]
- 25. Shurtleff, M.J., Temoche-Diaz, M.M., Karfilis, K.V., Ri, S. and Schekman, R. (2016) Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife, 5, e19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ibrahim, A.G., Cheng, K. and Marbán, E. (2014) Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports., 2, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stahl, P.D. and Raposo, G. (2018) Exosomes and extracellular vesicles: the path forward. Essays Biochem., 62, 199–124. [DOI] [PubMed] [Google Scholar]
- 28. Corrado, C., Raimondo, S., Chiesi, A., Ciccia, F., De Leo, G. and Alessandro, R. (2013) Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int. J. Mol. Sci., 14, 5338–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bier, A., Berenstein, P., Kronfeld, N., Morgoulis, D., Ziv-Av, A., Goldstein, H., Kazimirsky, G., Cazacu, S., Meir, R., Popovtzer, R.et al. (2018) Placenta-derived mesenchymal stromal cells and their exosomes exert therapeutic effects in Duchenne muscular dystrophy. Biomaterials, 174, 67–78. [DOI] [PubMed] [Google Scholar]
- 30. Aminzadeh, M.A., Rogers, R.G., Fournier, M., Tobin, R.E., Guan, X., Childers, M.K., Andres, A.M., Taylor, D.J., Ibrahim, A., Ding, X.et al. (2018) Exosome-mediated benefits of cell therapy in mouse and human models of Duchenne muscular dystrophy. Stem Cell Reports., 10, 942–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogers, R.G., Fournier, M., Sanchez, L., Ibrahim, A.G., Aminzadeh, M.A., Lewis, M.I. and Marbán, E. (2019) Disease-modifying bioactivity of intravenous cardiosphere-derived cells and exosomes in mdx mice. JCI Insight., 4, e130202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zanotti, S., Gibertini, S., Blasevich, F., Bragato, C., Ruggieri, A., Saredi, S., Fabbri, M., Bernasconi, P., Maggi, L., Mantegazza, R.et al. (2018) Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol., 74, 77–100. [DOI] [PubMed] [Google Scholar]
- 33. Shi, R., Zhao, L., Cai, W., Wei, M., Zhou, X., Yang, G. and Yuan, L. (2017) Maternal exosomes in diabetes contribute to the cardiac development deficiency. Biochem. Biophys. Res. Commun., 483, 602–608. [DOI] [PubMed] [Google Scholar]
- 34. Davidson, S.M., Riquelme, J.A., Takov, K., Vicencio, J.M., Boi-Doku, C., Khoo, V., Doreth, C., Radenkovic, D., Lavandero, S. and Yellon, D.M. (2018) Cardioprotection mediated by exosomes is impaired in the setting of type II diabetes but can be rescued by the use of non-diabetic exosomes in vitro. J. Cell. Mol. Med., 1, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexander, M.S., Casar, J.C., Motohashi, N., Vieira, N.M., Eisenberg, I., Marshall, J.L., Gasperini, M.J., Lek, A., Myers, J.A., Estrella, E.A.et al. (2014) MicroRNA-486-dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy-associated symptoms. JCI., 6, 2651–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pironti, G., Strachan, R.T., Abraham, D., Mon-Wei Yu, S., Chen, M., Chen, W., Hanada, K., Mao, L., Watson, L.J. and Rockman, H.A. (2015) Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II type 1 receptors. Circulation, 131, 2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strauss, K.W., Goebel, C., Runz, H., Möbius, W., Weiss, S.L., Feussner, I., Simons, M. and Schneider, A. (2010) Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-pick type C disease. J. Biol. Chem., 285, 26279–26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berrondo, C., Flax, J., Kucherov, V., Siebert, A., Osinski, T., Rosenberg, A., Fucile, C., Richheimer, S. and Beckham, C.J. (2016) Expression of the long non-coding RNA HOTAIR correlates with disease progression in bladder cancer and is contained in bladder cancer patient urinary exosomes. PLoS One, 11, e0147236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gartz, M. and Strande, J.L. (2018) Examining the paracrine effects of exosomes in cardiovascular disease and repair. JAHA., 11, e007954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bang, C., Batkai, S., Dangwal, S., Gupta, S.K., Foinquinos, A., Holzmann, A., Just, A., Remke, J., Zimmer, K., Zeug, A.et al. (2014) Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Invest., 5, 2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Halkein, J., Tabruyn, S.P., Ricke-Hoch, M., Haghikia, A., Nguyen, N., Scherr, M., Castermans, K., Malvaux, L., Lambert, V., Thiry, M.et al. (2013) MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. JCI., 5, 2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gartz, M., Lin, C.W., Sussman, M.A., Lawlor, M.W. and Strande, J.L. (2020) Duchenne muscular dystrophy (DMD) cardiomyocyte-secreted exosomes promote the pathogenesis of DMD-associated cardiomyopathy. Dis. Model. Mech., 13, dmm045559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yue, Y., Li, Z., Harper, S.Q., Davisson, R.L., Chamberlain, J.S. and Duan, D. (2003) Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation, 108, 1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harper, S.Q., Hauser, M.A., Dello Russo, C., Duan, D., Crawford, R.W., Phelps, S.F., Harper, H.A., Robinson, A.S., Engelhardt, J.F., Brooks, S.V.et al. (2002) Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med., 8, 253–261. [DOI] [PubMed] [Google Scholar]
- 45. Barth, A.S., Kizana, E., Smith, R.R., Terrovitis, J., Dong, P., Leppo, M.K., Zhang, Y., Miake, J., Olson, E.N., Schneider, J.W.et al. (2008) Lentiviral vectors bearing the cardiac promoter of the Na+ Ca2+ exchanger report cardiogenic differentiation in stem cells. Mol. Ther., 5, 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warnecke-Eberz, U., Chon, S.H., Hölscher, A.H., Drebber, U. and Bollschweiler, E. (2015) Exosomal onco-miRs from serum of patients with adenocarcinoma of the esophagus: comparison of miRNA profiles of exosomes and matching tumor. Tumour Biol., 36, 4643–4653. [DOI] [PubMed] [Google Scholar]
- 47. Cheng, Y. and Zhang, C. (2010) MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Transl. Res., 3, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiao, J., Pan, Y., Li, X.H., Yang, X.Y., Feng, Y.L., Tan, H.H., Jiang, L., Feng, J. and Yu, X.Y. (2016) Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis., 7, e2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bulfield, G., Siller, W.G., Wight, P.A. and Moore, K.J. (1984) X chromosome-linked muscular dystrophy (mdx) in the mouse. PNAS, 81, 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adamo, C.M., Dai, D.F., Percival, J.M., Minami, E., Willis, M.S., Patrucco, E., Froehner, S.C. and Beavo, J.A. (2010) Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. PNAS, 44, 19079–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Quinlan, J.G., Hahn, H.S., Wong, B.L., Lorenz, J.N., Wenisch, A.S. and Levin, L.S. (2004) Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul. Disord., 14, 491–496. [DOI] [PubMed] [Google Scholar]
- 52. Kamdar, F., Das, S., Gong, W., Klaassen-Kamdar, A., Meyers, T.A., Shah, P., Ervasti, J.M., Townsend, D., Kamp, T.J., Wu, J.C.et al. (2020) Stem cell-derived Cardiomyocytes and Beta-adrenergic receptor blockade in Duchenne muscular dystrophy cardiomyopathy. J. Am. Coll. Cardiol., 75, 1159–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yue, Y., Skimming, J.W., Liu, M., Strawn, T. and Duan, D. (2004) Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Hum. Mol. Genet., 15, 1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Agarwal, V., Bell, G.W., Nam, J.W. and Bartel, D.P. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife, 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Betel, D., Wilson, M., Gabow, A., Marks, D.S. and Sander, C. (2008) The microRNA.Org resource: targets and expression. Nucleic Acids Res., 36, D149–D153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Markou, T., Cullingford, T.E., Giraldo, A., Weiss, S.C., Alsafi, A., Fuller, S.J., Clerk, A. and Sugden, P.H. (2008) Glycogen synthase kinases 3alpha and 3beta in cardiac myocytes: regulation and consequences of their inhibition. Cell. Signal., 20, 206–218. [DOI] [PubMed] [Google Scholar]
- 57. Li, Y., Li, Z., Zhang, C., Li, P., Wu, Y., Wang, C., Bond Lau, W., Ma, X.L. and Du, J. (2017) Cardiac fibroblast-specific activating transcription factor 3 protects against heart failure by suppressing MAP2K3-p38 Signaling. Circulation, 135, 2041–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhu, J., Chen, T., Yang, L., Li, Z., Wong, M.M., Zheng, X., Pan, X., Zhang, L. and Yan, H. (2012) Regulation of microRNA-155 in atherosclerotic inflammatory responses by targeting MAP3K10. PLoS One, 7, e46551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jansson, M.D., Damas, N.D., Lees, M., Jacobsen, A.N. and Lund, A.H. (2015) miR-339-5p regulates the p53 tumor-suppressor pathway by targeting MDM2. Oncogene, 34, 1908–1918. [DOI] [PubMed] [Google Scholar]
- 60. Pikkarainen S, Kennedy RA, Marshall AK, Tham el L, Lay K, Kriz TA, Handa BS, Clerk A, Sugden PH. Regulation of expression of the rat orthologue of mouse double minute 2 (MDM2) by H(2)O(2)-induced oxidative stress in neonatal rat cardiac myocytes. J. Biol. Chem. 2009. (284) 27195–27210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de Rozieres, S., Maya, R., Oren, M. and Lozano, G. (2000) The loss of mdm2 induces p53-mediated apoptosis. Oncogene, 19, 1691–1697. [DOI] [PubMed] [Google Scholar]
- 62. Singh, S., Ramamoorthy, M., Vaughan, C., Yeudall, W.A., Deb, S. and Palit, D.S. (2013) Human oncoprotein MDM2 activates the Akt signaling pathway through an interaction with the repressor element 1 silencing transcription factor conferring a survival advantage to cancer cells. Cell Death Differ., 20, 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang, C., Liu, J., Wang, X., Wu, R., Lin, M.S., Laddha, S.V., Yang, Q., Chan, C.S. and Feng, Z.H. (2014) MicroRNA-339-5p inhibits colorectal tumorigenesis through regulation of the MDM2/p53 signaling. Oncotarget, 5, 9106–9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jeanson-Leh, L., Lameth, J., Krimi, S., Buisset, J., Amor, F., Le Guiner, C., Barthelemy, I., Servais, L., Blot, S., Voit, T.et al. (2014) Serum profiling identifies novel muscle miRNA and cardiomyopathy-related miRNA biomarkers in golden retriever muscular dystrophy dogs and Duchenne muscular dystrophy patients. Am. J. Pathol., 11, 2885–2898. [DOI] [PubMed] [Google Scholar]
- 65. Becker, S., Florian, A., Patrascu, A., Rösch, S., Waltenberger, J., Sechtem, U., Schwab, M., Schaeffeler, E. and Yilmaz, A. (2016) Identification of cardiomyopathy associated circulating miRNA biomarkers in patients with muscular dystrophy using a complementary cardiovascular magnetic resonance and plasma profiling approach. J. Cardiovasc. Magn. Reson., 18, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Florian, A., Patrascu, A., Tremmel, R., Rösch, S., Sechtem, U., Schwab, M., Schaeffeler, E. and Yilmaz, A. (2018) Identification of cardiomyopathy-associated circulating miRNA biomarkers in muscular dystrophy female carriers using a complementary cardiac imaging and plasma profiling approach. Front. Physiol., 9, 1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Simone, N.L., Soule, B.P., Ly, D., Saleh, A.D., Savage, J.E., DeGraff, W., Cook, J.A., Harris, C.C., Gius, D. and Mitchell, J.B. (2009) Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One, 4, e6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lohan, J. and Ohlendieck, K. (2004) Drastic reduction in the luminal Ca2+ binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim. Biophys. Acta, 1689, 252–258. [DOI] [PubMed] [Google Scholar]
- 69. Kyrychenko, V., Poláková, E., Janíček, R. and Shirokova, N. (2015) Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium, 58, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mavrogeni, S., Papavasiliou, A., Spargias, K., Constandoulakis, P., Papadopoulos, G., Karanasios, E., Georgakopoulos, D., Kolovou, G., Demerouti, E., Polymeros, S.et al. (2010) Myocardial inflammation in Duchenne muscular dystrophy as a precipitating factor for heart failure: a prospective study. BMC Neurol, 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Feng, Y., Huang, W., Wani, M., Yu, X. and Ashraf, M. (2014) Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS One, 2, e88685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen, L., Wang, Y., Pan, Y., Zhang, L., Shen, C., Qin, G., Ashraf, M., Weintraub, N., Ma, G. and Tang, Y. (2013) Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem. Biophys. Res. Commun., 431, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang, X., Gu, H., Qin, D., Yang, L., Huang, W., Essandoh, K., Wang, Y., Caldwell, C.C., Peng, T., Zingarelli, B.et al. (2015) Exosomal miR-223 contributes to mesenchymal stem cell-elicited Cardioprotection in Polymicrobial sepsis. Sci. Rep., 5, 13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wang, K., Jiang, Z., Webster, K.A., Chen, J., Hu, H., Zhou, Y., Zhao, J., Wang, L., Wang, Y., Zhong, Z.et al. (2017) Enhanced Cardioprotection by human endometrium mesenchymal stem cells driven by Exosomal MicroRNA-21. Stem Cells Transl. Med., 6, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang, X., Huang, W., Liu, G., Cai, W., Millard, R.W., Wang, Y., Chang, J., Peng, T. and Fan, G.C. (2014) Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol., 74, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zheng, B., Yin, W.N., Suzuki, T., Zhang, X.H., Zhang, Y., Song, L.L., Jin, L.S., Zhan, H., Zhang, H., Li, J.S.et al. (2017) Exosome-mediated miR-155 transfer from smooth muscle cells to endothelial cells induces endothelial injury and promotes atherosclerosis. Mol. Ther., 25, 1279–1294. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77. Dhiraj, D.K., Chrysanthou, E., Mallucci, G.R. and Bushell, M. (2013) miRNAs-19b, − 29b-2* and −339-5p show an early and sustained up-regulation in ischemic models of stroke. PLoS One, 8, e83717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hu, T., Chong, Y., Lu, S., Wang, R., Qin, H., Silva, J., Kitamura, E., Chang, C.S., Hawthorn, L. and Cowell, J.K. (2018) miR-339 promotes development of stem cell Leukemia/lymphoma syndrome via downregulation of the BCL2L11 and BAX Proapoptotic genes. Cancer Res., 78, 3522–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mukhopadhyay, P., Mukherjee, S., Ahsan, K., Bagchi, A., Pacher, P. and Das, D.K. (2010) Restoration of altered microRNA expression in the ischemic heart with resveratrol. PLoS One, 5, e15705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Abu-Halima, M., Meese, E., Keller, A., Abdul-Khaliq, H. and Rädle-Hurst, T. (2017) Analysis of circulating microRNAs in patients with repaired tetralogy of Fallot with and without heart failure. J. Transl. Med., 15, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chiara, F. and Rasola, A. (2013) GSK-3 and mitochondria in cancer cells. Front. Oncol., 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Arena, G., Cissé, M.Y., Pyrdziak, S., Chatre, L., Riscal, R., Fuentes, M., Arnold, J.J., Kastner, M., Gayte, L., Bertrand-Gaday, C.et al. (2018) Mitochondrial MDM2 regulates respiratory complex I activity independently of p53. Mol. Cell, 69, 594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Elkholi, R., Abraham-Enachescu, I., Trotta, A.P., Rubio-Patiño, C., Mohammed, J.N., Luna-Vargas, M.P.A., Gelles, J.D., Kaminetsky, J.R., Serasinghe, M.N., Zou, C.et al. (2019) MDM2 integrates cellular respiration and apoptotic Signaling through NDUFS1 and the mitochondrial network. Mol. Cell, 74, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mannam, P., Rauniyar, N., Lam, T.T., Luo, R., Lee, P.J. and Srivastava, A. (2016) MKK3 influences mitophagy and is involved in cigarette smoke-induced inflammation. Free Radic. Biol. Med., 101, 102–115. [DOI] [PubMed] [Google Scholar]
- 85. Baldari, S., Ubertini, V., Garufi, A., D'Orazi, G. and Bossi, G. (2015) Targeting MKK3 as a novel anticancer strategy: molecular mechanisms and therapeutical implications. Cell Death Dis., 6, e1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lindsay, A., McCourt, P.M., Karachunski, P., Lowe, D.A. and Ervasti, J.M. (2018) Xanthine oxidase is hyper-active in Duchenne muscular dystrophy. Free Radic. Biol. Med., 129, 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mitzelfelt, K.A., McDermott-Roe, C., Grzybowski, M.N., Marquez, M., Kuo, C.T., Riedel, M., Lai, S., Choi, M.J., Kolander, K.D., Helbling, D.et al. (2017) Efficient precision genome editing in iPSCs via genetic co-targeting with selection. Stem Cell Reports., 3, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Canfield, S.G., Sepac, A., Sedlic, F., Muravyeva, M.Y., Bai, X. and Bosnjak, Z.J. (2012) Marked hyperglycemia attenuates anesthetic preconditioning in human-induced pluripotent stem cell-derived cardiomyocytes. Anesthesiology, 4, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Egger, D. and Bienz, K. (1992) Colloidal gold staining and immunoprobing on the same western blot. Methods Mol. Biol., 80, 247–253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.