

Abstract
Aortic aneurysms (AAs) are pathological dilatations of the aorta. Pathogenic variants in genes encoding for proteins of the contractile machinery of vascular smooth muscle cells (VSMCs), genes encoding proteins of the transforming growth factor beta signaling pathway and extracellular matrix (ECM) homeostasis play a role in the weakening of the aortic wall. These variants affect the functioning of VSMC, the predominant cell type in the aorta. Many variants have unknown clinical significance, with unknown consequences on VSMC function and AA development. Our goal was to develop functional assays that show the effects of pathogenic variants in aneurysm-related genes. We used a previously developed fibroblast transdifferentiation protocol to induce VSMC-like cells, which are used for all assays. We compared transdifferentiated VSMC-like cells of patients with a pathogenic variant in genes encoding for components of VSMC contraction (ACTA2, MYH11), transforming growth factor beta (TGFβ) signaling (SMAD3) and a dominant negative (DN) and two haploinsufficient variants in the ECM elastic laminae (FBN1) to those of healthy controls. The transdifferentiation efficiency, structural integrity of the cytoskeleton, TGFβ signaling profile, migration velocity and maximum contraction were measured. Transdifferentiation efficiency was strongly reduced in SMAD3 and FBN1 DN patients. ACTA2 and FBN1 DN cells showed a decrease in SMAD2 phosphorylation. Migration velocity was impaired for ACTA2 and MYH11 cells. ACTA2 cells showed reduced contractility. In conclusion, these assays for showing effects of pathogenic variants may be promising tools to help reclassification of variants of unknown clinical significance in AA-related genes.
Graphical Abstract
Graphical Abstract.
Introduction
Aortic aneurysms (AA) are pathological dilatations of the aorta. AA can be life threatening in cases of untreated aneurysm growth leading to rupture, with a mortality rate of up to 80% (1). AA can occur in both segments of the aorta and are referred to accordingly as thoracic and abdominal AA. Genetic causes for thoracic and abdominal AA underlie syndromic and non- syndromic familial disease (2,3). AA may be caused by a variety of pathogenic variants in genes encoding structural components of the extracellular matrix (ECM), cytoskeletal/smooth muscle contraction proteins and proteins associated with the TGFβ signaling pathway. Pathogenic variants in the genes encoding the cytoskeleton/contractile machinery of vascular smooth muscle cells (VSMCs), such as ACTA2 (α-smooth muscle actin), MYH11 (myosin heavy chain 11), MYLK (myosin light chain kinase) and PRKG1 (Cyclic guanosine monophosphate (cGMP)-dependent protein kinase 1 alpha isozyme) (4–7) may lead to altered contractility of VSMCs. Aneurysm-related genes having an effect on the TGFβ signaling pathway are TGFβ receptor 1 (TGFBR1), TGFBR2, TGFB2, TGFB3, SMAD2 and SMAD3 (8–17). Pathogenic variants in aneurysm-related genes encode for components of the ECM and lead to disruption of the ECM homeostasis, resulting in a weakening of the aortic wall, loss of mechanical resilience, elasticity and altered ability to retain latent TGFβ. Genes in this group include FBN1 (encoding fibrilin-1), COL3A1 (collagen type 3 α1), ELN (elastin), EFEMP2 (fibulin-4) and LOX (lysyl oxidase) (18–29).
The pathogenic mechanism underlying aneurysm formation is not fully understood. Mutations in the aforementioned genes affect the normal functioning of VSMC, such as their contractile properties, interaction with the ECM and TGFβ signaling. These mutations affect the ability of VSCMs to adapt to mechanical stimuli and subsequent expression of aneurysm-related genes (30). To study the effects of pathogenic variants in aneurysm-related genes affecting VSMC function in vitro, VSMCs can be isolated from aortic tissue retrieved from open aneurysm surgery. Alternatively, dermal fibroblasts grown on a scaffold containing collagen and elastin or coverslips can be transdifferentiated into VSMC-like cells within 2 weeks using TGFβ1. This model has previously been tested and showed that VSMC-like cells were comparable with primary human aortic smooth muscle cell (SMC) with regard to messenger RNA expression of SMC markers ACTA2, SM22 and CNN1 (31). The purpose of this study was to investigate and develop assays that can identify the molecular phenotype and measure effects of pathogenic variants, in order to have a panel of pathogenic signatures of aneurysm genes that can be used as reference for reclassification of variants of unknown clinical significance (VUS) in the aneurysm-related genes. To develop and validate these novel robust functional assays, we used VSMC-like cells of healthy controls and AA patients with a pathogenic variant in the AA-related genes [ACTA2, MYH11, SMAD3, FBN1 haploinsufficient (HI) and dominant negative (DN)] and assessed their potential to differentiate, TGFβ signaling, migratory capacity and contractility.
Results
Assessment of baseline values for control VSMC-like cell transdifferentiation efficiency
Human dermal fibroblasts from three healthy donors were transdifferentiated into VSMC. A schematic overview of the transdifferentiation protocol is shown in Figure 1A. Transdifferentiation efficiency was determined by quantifying VSMC marker proteins. Western blot analysis of smooth muscle actin (SMA) and transgelin (SM22) expression in control VSMC-like cells showed the presence of both proteins in all controls after transdifferentiation (Fig. 1B). Quantification of SMA and SM22 protein levels also revealed variability in expression between the controls. The variability in relative protein expression of SMA in the controls ranged from 0.46 to 1.54 (Fig. 1C) and 0.54 to 1.4 for SM22 (Fig. 1D). Western blot analysis was performed to demonstrate baseline values for control fibroblasts (Supplementary Material, Fig. S5).
Figure 2 .
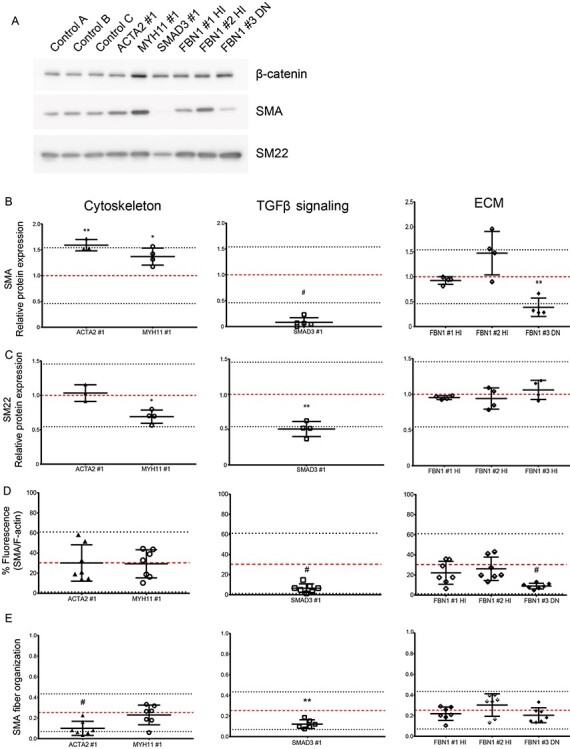
Transdifferentiation efficiency of VSMC-like cells of aneurysm patients with a pathogenic mutation. (A) Western blot detecting SMA and SM22 protein expression and β-catenin expression as loading control in three control cell and six patient VSMC-like cells. (B) Quantification of western blot SMA expression normalized by β-catenin expression. (C) Quantification of western blot SM22 expression normalized by β-catenin expression. Each data point represents a separate experiment, n = 3. (D) Quantification of immunofluorescent staining for SMA, normalized by total F-actin fluorescence. (E) SMA fiber anisotropy. Each data point represents a separate image, n = 7. Data are presented as boxplots with mean ± 2SD; red dotted line indicates mean value of the control cells, black dotted lines indicate ±2SD.
Figure 1 .
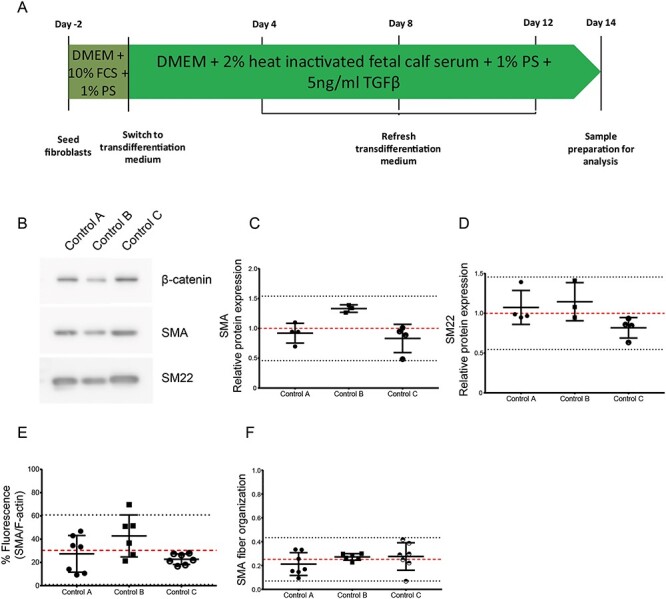
Baseline values for control VSMC-like cell transdifferentiation efficiency. (A) Schematic representation of the transdifferentiation protocol. (B) Western blot detecting SMA and SM22 protein expression and β-catenin expression as loading control. (C) Quantification of western blot SMA normalized by β-catenin expression. (D) Quantification of western blot SMA normalized by β-catenin expression. Each data point represents a separate experiment, n = 3. (E) Quantification of immunofluorescent staining for SMA, normalized by total F-actin fluorescence. (F) SMA fiber anisotropy. Each data point represents a separate image, n = 7. Data are presented as boxplots with mean ± 2SD; red dotted line indicates mean value, black dotted lines indicate ±2SD.
Subsequently, we determined the transdifferentiation efficiency by analyzing the protein expression level of SMA and the organization of the newly expressed SMA fibers by immunofluorescence. Representative images are shown in Supplementary Material, Figure S1. Quantification of the immunofluorescent staining is shown in Supplementary Material, Figure S3. All cells were positive for both markers, although marker expression was variable. To quantify the amount of VSMC-specific SMA fibers, we determined the ratio between SMA fibers and total F-actin content in the cytoplasm. The transdifferentiated controls showed an overlay of ~30% of SMA fiber fluorescence with total F-actin, with the 2 × standard deviation (SD) range running from 0 to 61% (Fig. 1E). SMA fibers anisotropy, as an indicator of fiber organization, was quantified. Control VSMC-like cells showed a mean anisotropy coefficient of 0.25, with a range from 0.07 to 0.43 (Fig. 1F).
Taken together, these data show that all controls can be transdifferentiated to VSMC-like cells upon stimulation with TGFβ, as confirmed by SMA and SM22 marker expression and SMA fiber organization.
Transdifferentiation efficiency of VSMC-like cells of aneurysm patients
The goal of our study is to develop functional assays to determine aberrant phenotypes in AA patient cells. Since these assays have not been previously used to uncover the consequences of the mutations on VSMC function, there is no quantitative reference available for normal cell output. We thus used the mean and 2 × SD range of the control cell lines as a reference and compared the output of patient cells to determine if they were considered significantly aberrant from the range of the controls.
Transdifferentiation efficiency was determined by quantitative western blot analysis of SMA and SM22 protein levels (Fig. 2A). SMA and SM22 were present after transdifferentiation in both cell lines with a mutation in cytoskeleton encoding genes. Quantification revealed increased SMA protein levels (1.59) in ACTA2 #1 VSMC-like cells (Fig. 2B, P < 0.01), whereas SM22 protein levels (1.03) were comparable with controls (Fig. 2C). MYH11 #1 VSMC-like cells showed increased SMA protein levels (1.37) compared with controls, suggesting increased transdifferentiation potential (Fig. 2B, P < 0.05). SM22 was significantly decreased (0.69) in MYH11 #1 VSMC-like cells compared with controls, but was also remained in the control range (Fig. 2C, P < 0.05). SMA was almost undetectable (0.08) in SMAD3 #1 VSMC-like cells compared with controls, and SM22 protein levels (0.51) were decreased (Fig. 2B and C, P < 0.001). FBN1 #1 HI and FBN1 #2 HI VSMC-like cells showed similar SMA (0.92 and 1.47, respectively) and SM22 (0.94 and 0.95, respectively) protein levels to controls after transdifferentiation (Fig. 2B and C). FBN1 #3 DN VSMC-like cells showed decreased SMA protein levels (0.39) compared with controls (Fig. 2B, P < 0.01), whereas having comparable SM22 protein levels (1.06) (Fig. 2C).
Figure 5 .
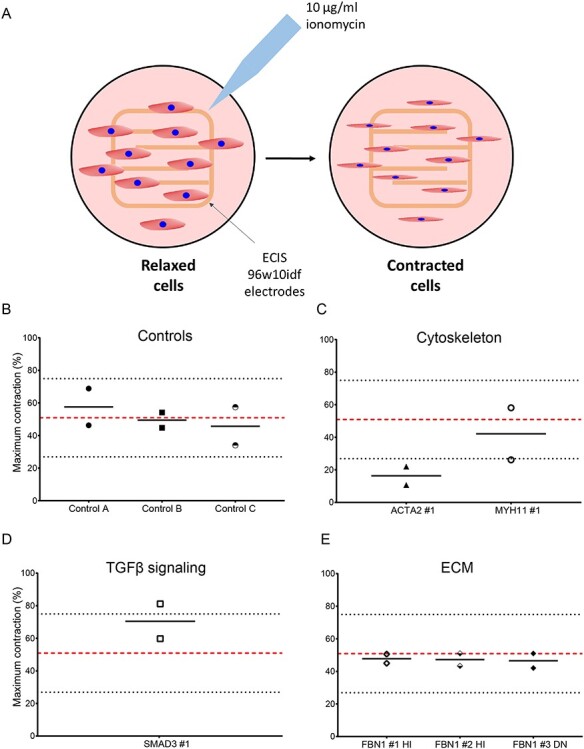
Maximum contraction of control and VSMC-like cells of patients with a pathogenic mutation. (A) Schematic representation of measuring VSMC-like cell contractility using ECIS. Cells are seeded on electrodes which record the baseline resistance of relaxed cells as a function of their surface. Upon ionomycin stimulation, cells contract, which reduces their surface and subsequently resistance. (A) Maximum contraction of controls VSMC-like cells. (B) Maximum contraction of ACTA2 and MYH11 variant VSMC-like cells. (C) Maximum contraction of SMAD3 variant VSMC-like cells. (D) Maximum contraction of FBN1 variant VSMC-like cells, n = 2. Data are presented as boxplots with mean ± 2SD; red dotted line indicates mean value of the control cells, black dotted lines indicate ±2SD.
Transdifferentiation efficiency was further assessed by immunofluorescence (representative images are shown in Supplementary Material, Figure S2). Quantification of the immunofluorescent staining is shown in Supplementary Material, Figure S3. Baseline values for all non-transdifferentiated control and patient fibroblast are quantified and shown in Supplementary Material, Figure S4. Quantification of SMA fiber fluorescence, normalized for total F-actin, showed that ACTA2 #1 and MYH11 #1 VSMC-like cells have a similar SMA fluorescence as controls, 30 and 29%, respectively (Fig. 2D). Analysis of anisotropy confirmed decreased organization of SMA (0.099) in ACTA2 #1 VSMC-like cells (Fig. 2E, P < 0.001). MYH11 #1 VSMC-like cells showed a similar anisotropy for SMA (0.23) as controls (Fig. 2E). Quantification of SMA fluorescence levels revealed that SMA fluorescence levels (6.5%) were significantly reduced in SMAD3 #1 VSMC-like cells (Fig. 2D, P < 0.001). SMA fibers that were present in SMAD3 #1 VSMC-like cells after TGFβ stimulation furthermore showed a significantly decreased SMA anisotropy (0.12) compared with controls (Fig. 2E, P < 0.01). FBN1 #1 HI and FBN1 #2 HI VSMC-like cells showed similar SMA fluorescence, 22 and 26%, respectively, compared with controls (Fig. 2D). Quantification of anisotropy for SMA (0.22 and 0.30) in FBN1 #1 HI and FBN1 #2 HI VSMC-like cells revealed no aberrations in the fiber organization compared with controls (Fig. 2E). In contrast, after stimulation with TGFβ DN FBN1 #3 DN VSMC-like cells showed a very low presence of SMA-positive cells with a significant decrease in intensity in SMA fluorescence (8.9%), suggesting transdifferentiation potential (Fig. 2D, P < 0.001). The SMA fibers in FBN1 #3 DN VSMC-like cells showed no aberrations in SMA anisotropy (0.20) compared with controls (Fig. 2E).
In conclusion, SMA and SM22 western blot and immunostaining indicated that SMAD3 #1 and FBN1 #3 DN cells did not develop SMA fibers and have a decreased transdifferentiation potential. ACTA2 #1 VSMC-like cells showed disorganized SMA fibers.
Kinetics of pSMAD2 activation in control and AA patients’ fibroblasts
To investigate the effects of TGFβ stimulation over time on TGFβ signaling activity, the C-terminal phosphorylation of SMAD2 was analyzed, as illustrated in Figure 3A. The response of non-transdifferentiated control fibroblasts to TGFβ over time was calculated from the ratio of receptor-activated (phosphorylated) SMAD2 compared with total SMAD2 protein following TGFβ stimulation (Fig. 3B). Overall, phosphorylated SMAD2 was detected 15 min after stimulation with TGFβ. The amount of phosphorylated SMAD2 peaked after 30 min in the controls and the expression level at this time point was used as a reference for TGFβ signaling activity. At the 30 min time point, the mean pSMAD2/SMAD2 ratio was ~1 and ranged from 0.32 to 1.72 (Fig. 3B).
Figure 3 .
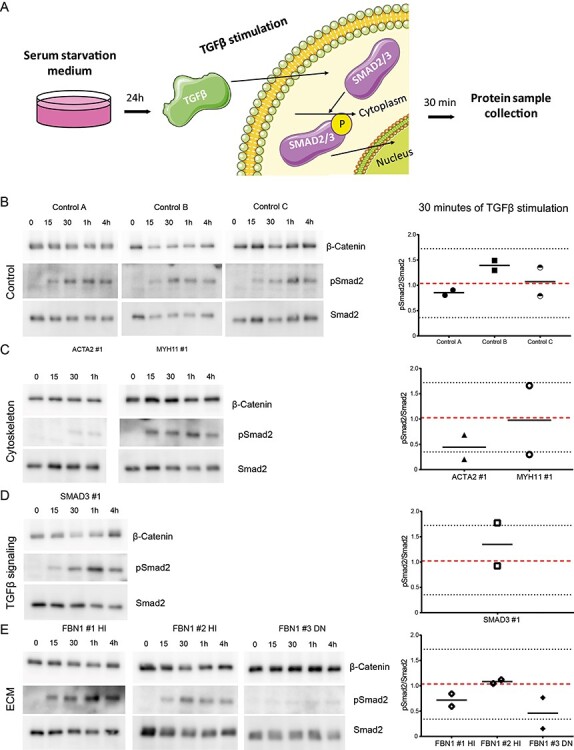
TGFβ responsiveness of control and fibroblasts of patients with a pathogenic mutation. (A) Schematic depicting TGFβ stimulation of control and patient fibroblasts. Cells are serum starved for 24 h, upon which they are stimulated with TGFβ, which induces SMAD2 phosphorylation. Samples are harvested for western blot after 30 min. (B) Western blots detecting pSMAD2 and SMAD2 in control fibroblasts before and upon stimulation with TGFβ (15, 30 min, 1 and 4 h) and quantification of the pSMAD2/SMAD2 ratio. (C) Western blots detecting pSMAD2 and SMAD2 in fibroblasts with a mutation in a cytoskeleton encoding gene before and upon stimulation with TGFβ (15, 30 min, 1 and 4 h) and quantification of the pSMAD2/SMAD2 ratio. (D) Western blots detecting pSMAD2 and SMAD2 in fibroblasts with a mutation in a TGFβ signaling pathway encoding gene before and upon stimulation with TGFβ (15, 30 min, 1 and 4 h) and quantification of the pSMAD2/SMAD2 ratio. (E) Western blots detecting pSMAD2 and SMAD2 in fibroblasts with a mutation in an ECM encoding gene before and upon stimulation with TGFβ (15, 30 min, 1 and 4 h) and quantification of the pSMAD2/SMAD2 ratio. β-catenin levels serve as a loading control. Data are presented as boxplots with mean ± 2SD; red dotted line indicates mean value of the control cells, black dotted lines indicate ±2SD.
TGFβ signaling activity in patient fibroblasts was compared with the ratio obtained from the mean and 2 × SD ratio of the controls. ACTA2 #1 fibroblasts showed a pSMAD2/SMAD2 ratio of 0.44 after 30 min of TGFβ stimulation, which was just above the 2 × SD range of the controls (Fig. 3C). MYH11 #1 fibroblasts showed comparable phosphorylation of SMAD2 to control fibroblasts (Fig. 3C). Quantification also indicated a normal pSMAD2/SMAD2 ratio of 0.98 for MYH11 #1 fibroblasts compared with controls (Fig. 3C). Phosphorylation of SMAD2 was comparable with controls for SMAD3 #1 fibroblasts, the pSMAD2/SMAD2 ratio at 30 min of 1.35 and was therefore similar to the controls (Fig. 3D). SMAD2 phosphorylation in FBN1 #1 HI and FBN1 #2 HI fibroblasts showed similar pSMAD2/SMAD2 ratios, 0.72 and 1.08, respectively, compared with controls after 30 min of TGFβ stimulation (Fig. 3E). FBN1 #3 DN fibroblasts showed reduced phosphorylation of SMAD2 after TGFβ stimulation (Fig. 3E). The pSMAD2/SMAD2 ratio of 0.46 for FBN1 #3 DN fibroblasts after 30 min of TGFβ stimulation remained just above the 2 × SD range of the controls (Fig. 3E).
Taken together, these data show that pSMAD2 activation is highly dynamic. In non-transdifferentiated cells, we detected differences in ACTA2 #1 and FBN1 #3 DN cells compared with controls, although with considerable inter-experimental variability.
Migration capacity of control and VSMC-like cells with a pathogenic variant
The migration capacity of the transdifferentiated VSMC-like cells was investigated with the use of a ring barrier migration assay (Fig. 4A). Migration velocity of ACTA2 #1 VSMC-like cells was decreased (5.0 versus 9.0 μm/h in controls) (P < 0.001, Fig. 4B). Likewise, MYH11 #1 VSMC-like cells showed decreased migration velocity (3.5 versus 9.4 μm/h in controls) (P < 0.001, Fig. 4C). SMAD3 #1, FBN1 #1 HI and #3 DN VSMC-like cells showed similar migration velocity as controls (Fig. 4D–F).
Figure 4 .
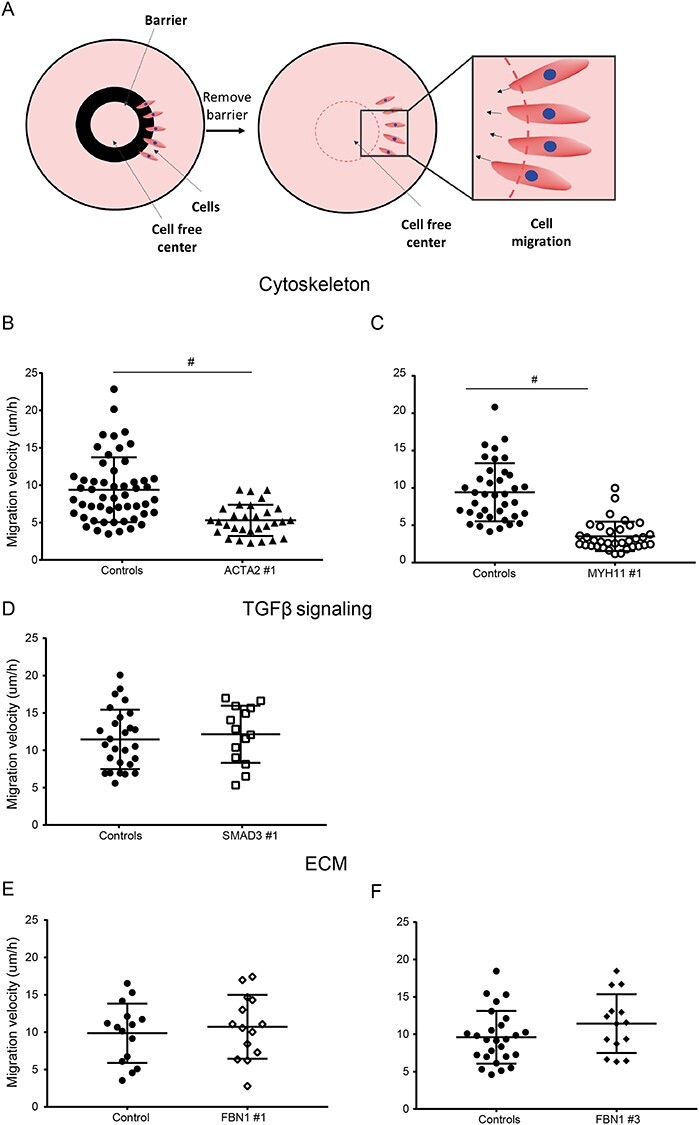
Migration potential of control and VSMC-like cells of patients with a pathogenic mutation. (A) Schematic representation of the ring barrier assay to measure migration. Cells are seeded on coverslips in the outside area of the wall. A ring is blocking the migration of the cells, preserving a cell-free are in the center. Once the ring is removed, cells start to migrate towards the center and the migration velocity is quantified. (B–F) Migration velocity of pathogenic variant VSMC-like cells compared with controls. (A) ACTA2, (B) MYH11, (C) SMAD3, (D) FBN1 #1 and (E) FBN1 #3 VSMC-like cells, n = 2, #P = <0.001. Data are presented as boxplots with mean ± 2SD; red dotted line indicates mean value of the control cells, black dotted lines indicate ±2SD.
These data indicate that the migration assay was informative for ACTA2 #1 and MYH11 #1 VSMC-like cells, showing reduced migration after transdifferentiation.
Contractility of pathogenic variant and control VSMC-like cells
Contractility of VSMC-like cells was determined upon ionomycin stimulation using the electric cell-substrate impedance sensing (ECIS) (Fig. 5A). The maximum contraction of control VSMC-like cells was on average 49.7% with a 2 × SD ranging from 23.3 to 76.1% (Fig. 5B). The maximum contraction of ACTA2 #1 VSMC-like cells was 16.3% and thereby below the 2 × SD range of the controls. MYH11 #1 VSMC-like cells showed a maximum contraction of 42.1% and did not differ from controls (Fig. 5C). SMAD3 #1 VSMC-like cells displayed a maximum contraction of 70.4%, indicating a slight, but not significant, increase in contractility (Fig. 5D). FBN1 #1 HI and FBN1 #2 HI VSMC-like cells had a maximum contractility of 47.8 and 47.2%, respectively, and were thereby comparable with the controls. Likewise FBN1 #3 DN VSMC-like cells were similar to controls with a maximum contraction of 46.5% (Fig. 5E).
From these data, we can conclude that the contractility assay was informative for ACTA2 #1 VSMC-like cells showing a reduced contractility. In the other patients, contractility was unimpaired.
Discussion
Genetic causes for syndromal and non-syndromal AA include a growing number of variants in genes with a wide range of interacting functions. It is important to find ways to distinguish and quantify the detrimental effects of variants in these genes into pathogenic and non-pathogenic, in particular due to the wide implementation of next-generation sequencing for molecular diagnosis of genetic aneurysms and in vitro classification of variants, resulting in a growing number of VUS. Functional assays with robust readouts for pathogenic effect will help to improve the diagnostic yield of aneurysm testing.
In this study, we used VSMC-like cells of AA patients to evaluate the clinical relevance of novel functional assays that could detect the pathologic molecular phenotype and measure the effect of different pathogenic variants in aneurysm-related genes. We measured a strong decrease in SMA expression and thereby reduced transdifferentiation potential in SMAD3 and FBN1 dominant negative (FBN1 #3 DN) patient cells. ACTA2 and FBN1 DN patients also showed a decrease in intracellular TGFβ signaling. Migration velocity was impaired for ACTA2 and MYH11, whereas only ACTA2 showed reduced contractility. Below, we will discuss these results per mutation. A summary of the data can also be found in Table 2. VMCs can uniquely be distinguished by the expression of VSMC-specific markers, such as SMA, calponin, SM22 and MYH11. As we demonstrated in our previous publication (31) where we first described and validated the method, our cells phenocopy VSMCs as they express calponin and smoothelin on protein level, markers of mature contractile SMC. We also demonstrated that in VSMC-like cells of AA patients, there is a significant upregulation of ACTA2, CNN1, MYH11, TAGLN and SMTN compared with non-transdifferentiated skin fibroblasts. As it was recently reported (32), smooth muscle myosin heavy chain, in combination with SMA is part of the cluster of markers, which allows discrimination between contractile and non-contractile cells, and calponin and smoothelin are auxiliary molecules of the contractile apparatus. Even though these markers are also presented in some myofibroblastic classes, the broad expression palette of VSMC markers in our cells indicates that they are close to VSMC. Based on previously published work by our groups, we chose SMA and SM22 as the two markers most technically reliable stable for both immunofluorescence and western blot to assess the transdifferentiation efficiency, to minimize any variability in experimental results that does not derive from the differences between control and patient cells (31).
Table 2.
Summary of findings
| Transdifferentiation potential | TGFβ responsiveness | Cytoskeleton function | |||||
|---|---|---|---|---|---|---|---|
| Immunofluorescence | Western blotting | pSMAD2/SMAD2 ratio (30 min) | Migration | Contractility | |||
| % SMA | SMA organization | SMA | SM22 | ||||
| Pathogenic Variant | Versus controls | Versus controls | Versus controls | Versus controls | Versus controls | Versus controls | Versus controls |
| ACTA2 #1 | ~ | ↓* | ↑ | ~ | ↓ | ↓ | ↓ |
| MYH11 #1 | ~ | ~ | ↑* | ↓* | ~ | ↓ | ~ |
| SMAD3 #1 | ↓ | ↓* | ↓ | ↓ | ~ | ~ | ~ |
| FBN1 #1 HI | ~ | ~ | ~ | ~ | ~ | ~ | ~ |
| FBN1 #2 HI | ~ | ~ | ~ | ~ | ~ | - | ~ |
| FBN1 #3 DN | ↓ | ~ | ↓ | ~ | ↓ | ~ | ~ |
~ = no difference; ↓ = trend of decrease; ↓* = significantly decreased; ↑ = trend of increase; ↑* = significantly increased.
ACTA2
ACTA2 #1 VSMC-like cells demonstrated similar transdifferentiation potential as controls. However, their new SMA fibers, encoded by ACTA2, appeared fragmented and disorganized. Guo et al. (4) previously showed lack of SMA fibers in VSMCs of patient with ACTA2 variants (p.R118Q and p.T353N). Our data showed that the p.R149C variant of ACTA2 #1 resembles other known ACTA2 pathogenic variants with its disorganized SMA fibers after transdifferentiation. Clinical data on ACTA2 p.R149C, p.R256H and p.T353N aortic sections confirmed that SMA protein is produced, but that VSMCs appear to be disorganized and lack structure (33). Complications with the assembly of mutant SMA (p.R256H) is further confirmed by analysis of mutant ACTA2 in yeast (33). Analysis of SMA fiber assembly by Malloy et al. (33) showed decreased SMA polymerization and elongated assembly times in ACTA2 p.R256H mutant yeast compared with wild-type yeast. Studies on ACTA2 pathogenic variants (p.R118Q, p.R256H and p.T353N) show overall disorganization of SMA fibers that is caused by dysfunctional assembly of SMA. Our results confirmed elucidated the disorganization of SMA for an additional ACTA2 pathogenic variant (p.R149C) in VSMC-like cells.
In addition, ACTA2 #1 VSMC-like cells showed decreased migration and contractility. During migration, the cytoskeleton is polymerized in the direction of movement, whereas depolymerization occurs at the rear end of cells. Variants in proteins that constitute the cytoskeleton, such as ACTA2 and MYH11, can be explanatory for the observed defective migration. Literature revealed that ACTA2 is induced during migration as well as during metastasis, suggesting that ACTA2 is essential for migration (34,35). Decreased contractility of multiple ACTA2 pathogenic variant VSMCs was previously found by Lu et al. (36) by performing a one-bead laser trap experiment to analyze force output. Our data on the reduced contractility of ACTA2 #1 VSMC-like cells is in line with this finding and highlights importance of ACTA2 for the contractility of VSMCs.
MYH11
MYH11 #1 VSMC-like cells showed decreased SM22 protein levels post transdifferentiation. Overexpression of MYH11 has been reported to induce degradation and turnover of thick filament-associated proteins and cytoskeleton proteins like SM22 but not SMA (37). Since MYH11 c.3879 + 2dup (MYH11 #1) is a relatively unknown variant, it is unclear whether this variant could lead to reduced levels or function of MYH11. Our finding that SMA is not degraded is in line with the results of Kwartler et al. (37) and suggests that this MYH11 pathogenic variant could indeed induce the described unfolded protein response that induces degradation of specific cytoskeleton proteins. Although the MYH11 #1 VSMC-like cells were able to transdifferentiate, migration velocity was decreased in MYH11 variant VSMC-like cells, similar to the ACTA2 #1 VSMC-like cells. MYH11 pathogenic variants have been predicted to compromise SMC migration, which is in line with our results (38,39).
SMAD3
SMAD3 #1 VSMC-like cells did not develop SMA fibers upon transdifferentiation, suggesting reduced transdifferentiation potential. This lack of SMA could be explained by disturbance in the TGFβ signaling pathway caused by the SMAD3 pathogenic variant. Induction of SMA by TGFβ was shown to occur via SMAD3 (40,41), suggesting that induction will not occur when SMAD3 is mutated. Like SMA, SM22 is induced via activation of the TGFβ pathway, specifically via SMAD3 (42). This also explains the reduced SM22 protein levels of SMAD3 pathogenic variant VSMC-like cells. The TGFβ signaling pathway is often considered physiologically active when SMAD2 is phosphorylated upon TGFβ stimulation, yet, for SMAD3 variants resulting in absent or defective SMAD3 proteins, this is not necessarily true (43). Although, the initial response to TGFβ stimulation is intact in SMAD3 knockout cells, downstream signaling is impaired due to absence of SMAD3 protein. This hampers induction of target genes, thereby leading to impaired induction of SMA and SM22 and thus diminished transdifferentiation.
FBN1
VSMC-like cells with a FBN1 DN pathogenic variant (FBN1 #3) express less SMA protein as wells as SMA fibers after transdifferentiation. This could be caused by defective ECM build-up resulting from the FBN1 variant, resulting in reduced binding sites for the cytoskeleton via integrins. The rigidity of the ECM is also important for the development of the cytoskeleton as, for example, a soft matrix results in a less well-defined cytoskeleton (44–46). The effect of a defective matrix on the cytoskeleton has recently also been shown in the Fibulin-4 VSMC-specific knockout mouse model in which absence of fibulin-4 led to absence of SMA protein and fibers (47).
Transdifferentiation of FBN1 DN and HI patient fibroblasts reveals differential results between two (predicted) types of FBN1 pathogenic variants. Although the DN FBN1 #3 VSMC-like cells show decreased induction of SMA and SM22, results from FBN1 #1 and #2 HI fibroblasts appear quite similar to controls. Clinical data have previously also suggested differences between the FBN1 DN and HI variant in their responsiveness to treatment with losartan (48,49). Patients with an FBN1 HI pathogenic variant that received treatment with losartan were responsive to the inhibition of aortic root dilation by losartan, whereas FBN1 DN patients did not respond to treatment (48). Furthermore, patients with an FBN1 HI variant showed improvement in biventricular end diastolic volume and stroke volume upon losartan treatment. This effect was not found in FBN1 DN patients that received losartan (49). This indicates that pathogenic variants in one gene can lead to different outcomes and could result in different clinical consequences for patients, which is in line with the nature of the FBN1 variants and their effects. HI variants result in half of the normal protein levels, whereas DN variants interfere with the normal protein and can result in non-functional protein levels. Our functional assays proved to be able to identify characteristics potentially distinguishing between FBN1 HI and DN pathogenic variants. Their application could potentially simplify the identification of HI and DN patients. Especially, the presence or absence of SMA, and potentially the pSMAD2/SMAD2 ratio, after TGFβ stimulation can distinguish FBN1 DN VSMC-like cells from FBN1 HI VSMC-like cells.
A possible limitation of our study is that only one cell line was used per pathogenic variant. We chose to analyze a set of pathogenic variants in the more frequently affected aneurysm genes and validate assays for assessing the roles of the pathogenic variant on RNA and protein expression and function. However, rather than drawing strong conclusions about each mutation, we wanted to demonstrate that these assays can be used to study the functional aspects of a wider range of mutations involved in AA pathogenesis. We plan to perform larger scale analysis in the future, with multiple cell lines of different pathogenicity, to use our assays to study the mechanistics of these mutations.
Conclusion
The assays which were tested showed promising applicability for distinguishing effects of pathogenic variants in the selected genes. We detected reduced transdifferentiation potential was noted in SMAD3 #1 and FBN1 #3 DN, defective migration in MYH11 #1 and ACTA2 #1 and defective contractility in ACTA2 #1. Further testing of pathogenic variants in these and other genes is needed to establish a panel of assays that can be used for testing effects of those variants. Our results suggest a potential application for these novel functional assays in assessing the functionality of a larger number of proteins encoded by the same genes. Hopefully, this will result in the ability to analyze the potential pathogenic effect of VUS in AA genes and thereby promote more accurate genetic diagnosis and family counseling for AA.
Materials and Methods
Cell lines and patient characteristics
Primary dermal fibroblasts of AA patients and controls were obtained from the biobanks of the departments of Clinical Genetics (Amsterdam University Medical Center—location VUmc and Erasmus University Medical Center, The Netherlands) for all experiments. Fibroblasts were cultured in Dulbecco’s-modified Eagle’s medium (DMEM, Lonza BioWhittaker) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (PS) at 37°C with 5% CO2. Fibroblasts of healthy controls and AA patients with heterozygous mutations in ACTA2 (cytoskeleton), MYH11 (cytoskeleton), SMAD3 (TGFβ signaling) and FBN1 (ECM) were used for all experiments (see Table 1 for genotypes of pathogenic variants). The pathogenic variants were evaluated by the Alamut Visual, a decision-support software for genome variant diagnostics, and were classified according to American College of Medical Genetics and Genomics (ACMG) as class 5.
Table 1.
Information on cell line numbers, genes, transcript numbers, pathogenic variants and expected effects on the affected protein in patient cell lines
| Gene | Cell line | Pathogenic variant | Expected effect on protein | Cellular process |
|---|---|---|---|---|
| ACTA2 (NM_001613.3) | ACTA2 #1 | c.445C > T, p.R149C | Missense mutation, presumably affecting the fiber formation due to the disappearance of a positive amino acid load | Cytoskeleton contractility |
| MYH11 (NM_002474.3) | MYH11 #1 | c.3879 + 2dup, p.? | Splice site mutation, premature stop codon. Possibly leading to happloinsufficiency and absence of functional protein | Cytoskeleton contractility |
| SMAD3 (NM_005902.3) | SMAD3 #1 | c.859C > T, p.R287W | Missense mutation, potential loss of H-bridges at interacting site that leads to reduced SMAD3/SMAD4 complex stability | TGFβ signaling |
| FBN1 (NM_000138.4) | FBN1 #1 HI | c.2369insC, p.C790Sfs*12 | Frameshift, premature stop codon resulting in haploinsufficiency | Extracellular matrix organization |
| FBN1 #2 HI | c.2851insG, p.L951Afs*2 | Frameshift, premature stop codon resulting in haploinsufficiency | Extracellular matrix organization | |
| FBN1 #3 DN | c.2132G > A, p.C711Y | Missense mutation in the TGFβ-binding protein-like domain that could affect binding to fibrillin-1 and results in a dominant negative effect on the protein | Extracellular matrix organization |
Transdifferentiation protocol
Fibroblasts were seeded at a density of 2.5 million cells/ml as previously described (31). Fibroblasts were seeded in DMEM with 10% FCS and 1% PS at a density of 2.5 million cells/ml on the corners of a 1 cm2 piece of Matriderm (MedSkin Solutions), meaning 250 000 cells per piece of Matriderm. Two days after seeding, the medium was changed to DMEM supplemented with 2% heat-inactivated FCS, 1% PS and 5 ng/ml human recombinant TGFβ1 (4342-5, BioVision). Heat inactivation was performed by placing the FCS in a 56°C water bath for 30 min and mixing regularly. Medium supplemented with TGFβ1 was changed every 4 days. After 14 days, the cells were enzymatically extracted from the Matriderm with the use of a collagenase solution in complete medium (2000 IU/ml, Worthington) at 37°C on a shaker for 3 h. Centrifugation was performed to remove collagenase and cells were transferred to new flasks and cultured for further experiments. Passage number of ‘VSMC-like’ cells after transdifferentiation was kept below five to prevent loss of transdifferentiation markers.
Immunofluorescent staining and image quantification
Dermal fibroblasts were seeded on 18 mm coverslips in 12-well plates with a density of 100 000 cells/well in DMEM with 10% FCS and 1%PS. Two days after seeding, the medium was changed to DMEM supplemented with 2% heat-inactivated FCS, 1% PS and 5 ng/ml human recombinant TGFβ1 (4342-5, BioVision). TGFβ supplemented medium was changed every 4 days, and after 14 days, cells were fixed with 2% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min. After fixation, cells were washed with PBS supplemented with 0.1% Triton X-100 and blocked with PBS+ (0.5% bovine serum albumine (BSA) and 0.15% glycine in PBS) for 30 min. Primary antibodies were incubated overnight at 4°C in PBS+; mouse monoclonal anti-SMA (1:1000, ab7817, Abcam) and rabbit polyclonal anti-SM22 (1:400, ab14106, Abcam). Cells were washed with PBS supplemented with Triton X-100 and incubated shortly with PBS+ prior to incubation with the secondary antibody in PBS+ (1:1000, anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 594, Molecular Probes) for 1 h at room temperature. Simultaneously, actin filaments were stained with SiR-Actin (1:1000, Cytoskeleton). After incubation, coverslips were mounted on glass slides with Vectashield supplemented with 4 ,6-diamidino-2-phenylindole (DAPI) (H-1200, Vector Laboratories) and sealed with nail polish. Images were recorded on a wide field epifluorescent microscope (Axio Imager D2, Zeiss).
,6-diamidino-2-phenylindole (DAPI) (H-1200, Vector Laboratories) and sealed with nail polish. Images were recorded on a wide field epifluorescent microscope (Axio Imager D2, Zeiss).
Quantification of the immunofluorescent signal was performed by calculating the corrected total cell fluorescence (CTCF) of SMA and actin. The CTCF of actin and SMA was determined by setting a color threshold to select the fibers in the image with Fiji image analyzing software (50) and determining the integrated density of this area (intensity of the fluorescence). This measurement was corrected for the background fluorescence and the total area of the fibers and results in the CTCF. The percentage of fluorescence was calculated by dividing SMA CTCF by total F-actin CTCF to determine what percentage of the actin cytoskeleton is positive for SMA compared with the total actin cytoskeleton [Eq. (1)].
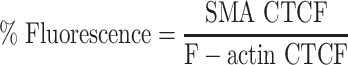 |
(1) |
Equation 1: Percentage of fluorescence.
Cytoskeletal fiber organization was assessed by measuring anisotropy on microscopic images using FibrilTool plug-in in Fiji (51). The anisotropy coefficient ranges from zero to one; parallel lines result in an anisotropy coefficient of one, whereas non-parallel structures have an anisotropy coefficient close to zero. The alignment of SMA fibers was assessed in confocal images of transdifferentiated cells of controls and patients with a pathogenic variant. According to the published protocol, X–Y fields of view were marked as regions of interest, and anisotropy was quantified within those regions.
Western blotting
VSMC-like cells were seeded at 100000 cells/six-well and allowed to attach for 48 h. Cells were scraped in PBS supplemented with protease inhibitor cocktail (1:100, 11836145001, Roche Applied Science) and phosphatase inhibitor cocktail (1:100, P0044, Sigma) and lysed in equal volumes of 2× Laemmli buffer (4% SDS, 20% glycerol, 120 mM Tris pH 6.8) supplemented with protease inhibitor cocktail and phosphatase inhibitor. Lysates were cleared of large DNA by passing through a 25G needle and then heated to 65°C for 10 min. Protein concentrations were measured with the Lowry protein assay (52). Equal amounts of protein were size separated by SDS-PAGE and separated proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane (1 h, 100 V, Immobilon). Membranes were blocked for 1 h at room temperature by either 5% BSA or 3% milk in PBS supplemented with 0.1% Tween-20. Membranes were incubated 45 min at room temperature with primary antibody. Membranes were washed five times with 0.1% Tween-20 in PBS and then incubated with horseradish peroxidase-conjugated secondary antibodies (1:2000, Jackson ImmunoResearch) for 1 h at room temperature. Bound secondary antibodies were detected with an Amersham Imager 600 (GE Healthcare Life Sciences) using chemiluminescence. Band intensity was quantified using Fiji image analyzing software (50).
Stimulation with TGFβ
To investigate potential differences in sensitivity to TGFβ stimulation between control and patient fibroblasts, cells were seeded in DMEM supplemented with 10% FCS and 1% PS in six-well plates to reach confluence and were allowed to attach for 24 h. The following day, medium was changed to DMEM supplemented with 1% PS and fibroblasts were serum deprived for 24 h prior to stimulation with human recombinant TGFβ. Protein samples were collected after 0, 15, 30 min, 1 and 4 h of stimulation with TGFβ1 in a concentration of 5 ng/ml (4342-5, BioVision). Lysis of cells and western blotting was performed as described above. Membranes were incubated overnight at 4°C with primary antibody. The TGFβ responsiveness of the control fibroblasts was used to establish baseline values to compare responsiveness values of patient cell lines: values outside the mean ± 2 × SD of the response in controls were considered abnormal.
Ring barrier migration assay
To measure the migration speed, a ring barrier migration assay was performed (53). In this assay, a barrier is placed in a cell culture chamber that prevents cells from entering the cell-free area. VSMC-like cells are seeded outside of this barrier and form a monolayer prior to the removal of the barrier. Upon removal of the barrier, VSMC-like cells can migrate into the cell-free area and migration can be monitored. Migration speed can be deducted. Migration velocity can be calculated based on total migration and the duration of the experiment. A total of 50 000 VSMC-like cells were seeded in the outer ring of the set-up and were allowed to attach for 24 h. After removing the ring barrier, cells were washed twice with DMEM supplemented with 1% PS (serum free) and were left in this medium during migration to prevent cell division. Multiple locations at the migration front were selected to monitor movement. Every 10 min, pictures were taken at the selected locations for 24 h. Data were analyzed with the AxioVision software (version 4.5.0.0) by measuring the total migration length of multiple cells and calculating the migration velocity. The migration velocity of the controls was used to establish baseline for the measurements in the patient cell lines: values outside the mean ± 2 × SD of the response were considered abnormal.
Measuring cell contractility
Cell contractility was measured and calculated according to previously published protocol using the ECIS (54). VSMC-like cells were seeded in duplicate in the array at a density of 30 000 cells/well in a sterile 96-well plate array well (96w10; Ibidi, Planegg, Germany). Cells were cultured for 48 h prior to stimulation. The impedance was recorded at a frequency of 4000 Hz.
VSMC-like cells were stimulated with 10 μg ionomycin/ml (Sigma Aldrich, Darmstadt, Germany). Contractile responses were measured in duplicate in each experiment. Contraction (C) of each well equals one minus the ratio between the resistance post- (PoS) and pre- (PrS) stimulation post empty well value subtraction, as depicted in Eq. (2):
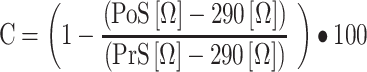 |
(2) |
Equation 2: Contractile response.
The maximum measured change in contraction was recorded and expressed as maximum contraction. To characterize the contractile output of patient fibroblasts, the mean response of the control group was used as a reference. Contraction of the patient VSMC-like cells was compared with a range between 2 × SD above and below the mean contractile response of the control group.
Statistics
Number of experiment replicates and independent samples are stated in the figure legends. Data were corrected for outliers with the Grubbs’ test for outliers. Statistical analysis was performed with a non-parametric Mann–Whitney test. Significance was tested two-tailed against the mean of the controls. Significance in gene expression analysis of stimulated controls was tested two-tailed against unstimulated controls. A P-value < 0.05 was considered to indicate a significant difference between groups. In the figures, P < 0.05 is shown with *P < 0.01 with **P < 0.001 with #. Results are expressed as mean ± SD, real-time PCR results are expressed as mean ± SD. All analyses were performed using Graphpad, version 7.03.
Supplementary Material
Acknowledgements
The authors would like to thank Hanny Odijk for expert technical assistance.
Conflict of Interest statement. None declared.
Contributor Information
Joyce Burger, Department of Molecular Genetics, Oncode Institute, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands; Department of Clinical Genetics, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Natalija Bogunovic, Department of Surgery, Institute for Cardiovascular Research, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands; Department of Physiology, Institute for Cardiovascular Research, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands; Department of Clinical Genetics, MOVE Institute, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands.
Nathalie P de Wagenaar, Department of Molecular Genetics, Oncode Institute, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands; Department of Cardiology, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Hui Liu, Department of Pathology, Erasmus University Medical Center, Rotterdam , The Netherlands.
Nicole van Vliet, Department of Molecular Genetics, Oncode Institute, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Arne IJpma, Department of Clinical Genetics, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands; Department of Pathology, Erasmus University Medical Center, Rotterdam , The Netherlands; Department of Bioinformatics, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Alessandra Maugeri, Department of Clinical Genetics, MOVE Institute, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands.
Dimitra Micha, Department of Clinical Genetics, MOVE Institute, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands.
Hence J M Verhagen, Department of Vascular Surgery, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Timo L M ten Hagen, Department of Pathology, Erasmus University Medical Center, Rotterdam , The Netherlands.
Danielle Majoor-Krakauer, Department of Clinical Genetics, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Ingrid van der Pluijm, Department of Molecular Genetics, Oncode Institute, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands; Department of Vascular Surgery, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Jeroen Essers, Department of Molecular Genetics, Oncode Institute, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands; Department of Vascular Surgery, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands; Department of Radiation Oncology, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Kak K Yeung, Department of Surgery, Institute for Cardiovascular Research, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands; Department of Physiology, Institute for Cardiovascular Research, Amsterdam University Medical Centers, location VU University Medical Center, Amsterdam 1081 HV, The Netherlands.
Funding
The authors would like to thank Stichting Lijf en Leven (Genexpressie analyse ter detective van de moleculaire mechanismen van aneurysmavorming—GAMMA), Erasmus MC Mrace grant and Amsterdam Cardiovascular Sciences Institute (ICaR AiO 2015 PhD grant) for supporting this research.
References
- 1. Kühnl, A., Erk, A., Trenner, M., Salvermoser, M., Schmid, V. and Eckstein, H.-H. (2017) Incidence, treatment and mortality in patients with abdominal aortic aneurysms: an analysis of hospital discharge data from 2005–2014. Dtsch. Arztebl. Int., 114, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ziganshin, B.A., Bailey, A.E., Coons, C., Dykas, D., Charilaou, P., Tanriverdi, L.H., Liu, L., Tranquilli, M., Bale, A.E. and Elefteriades, J.A. (2015) Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann. Thorac. Surg., 100, 1604–1611. [DOI] [PubMed] [Google Scholar]
- 3. van de Luijtgaarden, K.M., Heijsman, D., Maugeri, A., Weiss, M.M., Verhagen, H.J., IJpma, A., Bruggenwirth, H.T. and Majoor-Krakauer, D. (2015) First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum. Genet., 134, 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo, D.C., Pannu, H., Tran-Fadulu, V., Papke, C.L., Yu, R.K., Avidan, N., Bourgeois, S., Estrera, A.L., Safi, H.J., Sparks, E.et al. (2007) Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet., 39, 1488–1493. [DOI] [PubMed] [Google Scholar]
- 5. Zhu, L., Vranckx, R., Khau Van Kien, P., Lalande, A., Boisset, N., Mathieu, F., Wegman, M., Glancy, L., Gasc, J.M., Brunotte, F.et al. (2006) Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet., 38, 343–349. [DOI] [PubMed] [Google Scholar]
- 6. Wang, L., Guo, D.C., Cao, J., Gong, L., Kamm, K.E., Regalado, E., Li, L., Shete, S., He, W.Q., Zhu, M.S.et al. (2010) Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet., 87, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo, D.C., Regalado, E., Casteel, D.E., Santos-Cortez, R.L., Gong, L., Kim, J.J., Dyack, S., Horne, S.G., Chang, G., Jondeau, G.et al. (2013) Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet., 93, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loeys, B.L., Chen, J., Neptune, E.R., Judge, D.P., Podowski, M., Holm, T., Meyers, J., Leitch, C.C., Katsanis, N., Sharifi, N.et al. (2005) A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet., 37, 275–281. [DOI] [PubMed] [Google Scholar]
- 9. Loeys, B.L., Schwarze, U., Holm, T., Callewaert, B.L., Thomas, G.H., Pannu, H., De Backer, J.F., Oswald, G.L., Symoens, S., Manouvrier, S.et al. (2006) Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med., 355, 788–798. [DOI] [PubMed] [Google Scholar]
- 10. Mizuguchi, T., Collod-Beroud, G., Akiyama, T., Abifadel, M., Harada, N., Morisaki, T., Allard, D., Varret, M., Claustres, M., Morisaki, H.et al. (2004) Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet., 36, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lindsay, M.E., Schepers, D., Bolar, N.A., Doyle, J.J., Gallo, E., Fert-Bober, J., Kempers, M.J., Fishman, E.K., Chen, Y., Myers, L.et al. (2012) Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet., 44, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boileau, C., Guo, D.C., Hanna, N., Regalado, E.S., Detaint, D., Gong, L., Varret, M., Prakash, S.K., Li, A.H., d'Indy, H.et al. (2012) TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet., 44, 916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rienhoff, H.Y., Jr., Yeo, C.Y., Morissette, R., Khrebtukova, I., Melnick, J., Luo, S., Leng, N., Kim, Y.J., Schroth, G., Westwick, J.et al. (2013) A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys-Dietz syndrome. Am. J. Med. Genet. A, 161A, 2040–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matyas, G., Naef, P., Tollens, M. and Oexle, K. (2014) De novo mutation of the latency-associated peptide domain of TGFB3 in a patient with overgrowth and Loeys-Dietz syndrome features. Am. J. Med. Genet. A, 164A, 2141–2143. [DOI] [PubMed] [Google Scholar]
- 15. Bertoli-Avella, A.M., Gillis, E., Morisaki, H., Verhagen, J.M.A., deGraaf, B.M., van deBeek, G., Gallo, E., Kruithof, B.P.T., Venselaar, H., Myers, L.A.et al. (2015) Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol., 65, 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van de Laar, I.M., Oldenburg, R.A., Pals, G., Roos-Hesselink, J.W., deGraaf, B.M., Verhagen, J.M., Hoedemaekers, Y.M., Willemsen, R., Severijnen, L.A., Venselaar, H.et al. (2011) Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet., 43, 121–126. [DOI] [PubMed] [Google Scholar]
- 17. van de Laar, I.M., van derLinde, D., Oei, E.H., Bos, P.K., Bessems, J.H., Bierma-Zeinstra, S.M., vanMeer, B.L., Pals, G., Oldenburg, R.A., Bekkers, J.A.et al. (2012) Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet., 49, 47–57. [DOI] [PubMed] [Google Scholar]
- 18. Dietz, H.C., Pyeritz, R.E., Hall, B.D., Cadle, R.G., Hamosh, A., Schwartz, J., Meyers, D.A. and Francomano, C.A. (1991) The Marfan syndrome locus: confirmation of assignment to chromosome 15 and identification of tightly linked markers at 15q15-q21.3. Genomics, 9, 355–361. [DOI] [PubMed] [Google Scholar]
- 19. Milewicz, D.M. (1994) Identification of defects in the fibrillin gene and protein in individuals with the Marfan syndrome and related disorders. Tex. Heart Inst. J., 21, 22–29. [PMC free article] [PubMed] [Google Scholar]
- 20. Pepin, M., Schwarze, U., Superti-Furga, A. and Byers, P.H. (2000) Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med., 342, 673–680. [DOI] [PubMed] [Google Scholar]
- 21. Schwarze, U., Schievink, W.I., Petty, E., Jaff, M.R., Babovic-Vuksanovic, D., Cherry, K.J., Pepin, M. and Byers, P.H. (2001) Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet., 69, 989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Plancke, A., Holder-Espinasse, M., Rigau, V., Manouvrier, S., Claustres, M. and Khau Van Kien, P. (2009) Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur. J. Hum. Genet., 17, 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang, M.C., He, L., Giro, M., Yong, S.L., Tiller, G.E. and Davidson, J.M. (1999) Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN). J. Biol. Chem., 274, 981–986. [DOI] [PubMed] [Google Scholar]
- 24. Tassabehji, M., Metcalfe, K., Hurst, J., Ashcroft, G.S., Kielty, C., Wilmot, C., Donnai, D., Read, A.P. and Jones, C.J. (1998) An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa. Hum. Mol. Genet., 7, 1021–1028. [DOI] [PubMed] [Google Scholar]
- 25. Szabo, Z., Crepeau, M.W., Mitchell, A.L., Stephan, M.J., Puntel, R.A., Yin Loke, K., Kirk, R.C. and Urban, Z. (2006) Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J. Med. Genet., 43, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hucthagowder, V., Sausgruber, N., Kim, K.H., Angle, B., Marmorstein, L.Y. and Urban, Z. (2006) Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet., 78, 1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dasouki, M., Markova, D., Garola, R., Sasaki, T., Charbonneau, N.L., Sakai, L.Y. and Chu, M.L. (2007) Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am. J. Med. Genet. A, 143A, 2635–2641. [DOI] [PubMed] [Google Scholar]
- 28. Lee, V.S., Halabi, C.M., Hoffman, E.P., Carmichael, N., Leshchiner, I., Lian, C.G., Bierhals, A.J., Vuzman, D., Brigham Genomic, M., Mecham, R.P.et al. (2016) Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. U. S. A., 113, 8759–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo, D.C., Regalado, E.S., Gong, L., Duan, X., Santos-Cortez, R.L., Arnaud, P., Ren, Z., Cai, B., Hostetler, E.M., Moran, R.et al. (2016) LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ. Res., 118, 928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson, R.W., Liao, S. and Curci, J.A. (1997) Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron. Artery Dis., 8, 623–631. [DOI] [PubMed] [Google Scholar]
- 31. Yeung, K.K., Bogunovic, N., Keekstra, N., Beunders, A.A., Pals, J., van derKuij, K., Overwater, E., Wisselink, W., Blankensteijn, J.D., vanHinsbergh, V.W.et al. (2017) Transdifferentiation of human dermal fibroblasts to smooth muscle-like cells to study the effect of MYH11 and ACTA2 mutations in aortic aneurysms. Hum. Mutat., 38, 439–450. [DOI] [PubMed] [Google Scholar]
- 32. Bruijn, L.E., van denAkker, B.E.W.M., vanRhijn, C.M., Hamming, J.F. and Lindeman, J.H.N. (2020) Extreme diversity of the human vascular mesenchymal cell landscape. J. Am. Heart Assoc., 9, e017094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malloy, L.E., Wen, K.K., Pierick, A.R., Wedemeyer, E.W., Bergeron, S.E., Vanderpool, N.D., McKane, M., Rubenstein, P.A. and Bartlett, H.L. (2012) Thoracic aortic aneurysm (TAAD)-causing mutation in actin affects formin regulation of polymerization. J. Biol. Chem., 287, 28398–28408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rockey, D.C., Weymouth, N. and Shi, Z. (2013) Smooth muscle alpha actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS One, 8, e77166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee, H.W., Park, Y.M., Lee, S.J., Cho, H.J., Kim, D.H., Lee, J.I., Kang, M.S., Seol, H.J., Shim, Y.M., Nam, D.H.et al. (2013) Alpha-smooth muscle actin (ACTA2) is required for metastatic potential of human lung adenocarcinoma. Clin. Cancer Res., 19, 5879–5889. [DOI] [PubMed] [Google Scholar]
- 36. Lu, H., Fagnant, P.M., Bookwalter, C.S., Joel, P. and Trybus, K.M. (2015) Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc. Natl. Acad. Sci., 112, E4168―E4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kwartler, C.S., Chen, J., Thakur, D., Li, S., Baskin, K., Wang, S., Wang, Z.V., Walker, L., Hill, J.A., Epstein, H.F.et al. (2014) Overexpression of smooth muscle myosin heavy chain leads to activation of the unfolded protein response and autophagic turnover of thick filament-associated proteins in vascular smooth muscle cells. J. Biol. Chem., 289, 14075–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuang, S.Q., Kwartler, C.S., Byanova, K.L., Pham, J., Gong, L., Prakash, S.K., Huang, J., Kamm, K.E., Stull, J.T., Sweeney, H.L.et al. (2012) Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ. Res., 110, 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pannu, H., Tran-Fadulu, V., Papke, C.L., Scherer, S., Liu, Y., Presley, C., Guo, D., Estrera, A.L., Safi, H.J., Brasier, A.R.et al. (2007) MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet., 16, 2453–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu, B., Wu, Z. and Phan, S.H. (2003) Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am. J. Respir. Cell Mol. Biol., 29397–404. [DOI] [PubMed] [Google Scholar]
- 41. Vardouli, L., Vasilaki, E., Papadimitriou, E., Kardassis, D. and Stournaras, C. (2008) A novel mechanism of TGFbeta-induced actin reorganization mediated by Smad proteins and Rho GTPases. FEBS J., 275, 4074–4087. [DOI] [PubMed] [Google Scholar]
- 42. Qiu, P., Feng, X.H. and Li, L. (2003) Interaction of Smad3 and SRF-associated complex mediates TGF-beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J. Mol. Cell. Cardiol., 35, 1407–1420. [DOI] [PubMed] [Google Scholar]
- 43. van der Pluijm, I., vanVliet, N., von derThusen, J.H., Robertus, J.L., Ridwan, Y., vanHeijningen, P.M., vanThiel, B.S., Vermeij, M., Hoeks, S.E., Buijs-Offerman, R.et al. (2016) Defective connective tissue remodeling in Smad3 mice leads to accelerated aneurysmal growth through disturbed downstream TGF-beta signaling. EBioMedicine, 12, 280–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choquet, D., Felsenfeld, D.P. and Sheetz, M.P. (1997) Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell, 88, 39–48. [DOI] [PubMed] [Google Scholar]
- 45. Lo, C.M., Wang, H.B., Dembo, M. and Wang, Y.L. (2000) Cell movement is guided by the rigidity of the substrate. Biophys. J., 79, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Saez, A., Buguin, A., Silberzan, P. and Ladoux, B. (2005) Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys. J., 89, L52–L54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Burger, J., vanVliet, N., vanHeijningen, P., Kumra, H., Kremers, G.J., Alves, M., vanCappellen, G., Yanagisawa, H., Reinhardt, D.P., Kanaar, R.et al. (2019) Fibulin-4 deficiency differentially affects cytoskeleton structure and dynamics as well as TGFbeta signaling. Cell. Signal., 58, 65–78. [DOI] [PubMed] [Google Scholar]
- 48. Franken, R., denHartog, A.W., Radonic, T., Micha, D., Maugeri, A., vanDijk, F.S., Meijers-Heijboer, H.E., Timmermans, J., Scholte, A.J., van denBerg, M.P.et al. (2015) Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ. Cardiovasc. Genet., 8, 383–388. [DOI] [PubMed] [Google Scholar]
- 49. den Hartog, A.W., Franken, R., van denBerg, M.P., Zwinderman, A.H., Timmermans, J., Scholte, A.J., deWaard, V., Spijkerboer, A.M., Pals, G., Mulder, B.J.et al. (2016) The effect of losartan therapy on ventricular function in Marfan patients with haploinsufficient or dominant negative FBN1 mutations. Neth. Heart J., 24, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B.et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boudaoud, A., Burian, A., Borowska-Wykret, D., Uyttewaal, M., Wrzalik, R., Kwiatkowska, D. and Hamant, O. (2014) FibrilTool, an ImageJ plug-in to quantify fibrillar structures in raw microscopy images. Nat. Protoc., 9, 457–463. [DOI] [PubMed] [Google Scholar]
- 52. Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall, R.J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem., 193, 265–275. [PubMed] [Google Scholar]
- 53. Das, A.M., Eggermont, A.M. and tenHagen, T.L. (2015) A ring barrier-based migration assay to assess cell migration in vitro. Nat. Protoc., 10, 904–915. [DOI] [PubMed] [Google Scholar]
- 54. Bogunovic, N., Meekel, J.P., Micha, D., Blankensteijn, J.D., Hordijk, P.L. and Yeung, K.K. (2019) Impaired smooth muscle cell contractility as a novel concept of abdominal aortic aneurysm pathophysiology. Sci. Rep., 9, 6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.