

Abstract
Oligodendrocytes (OLs) produce myelin in the central nervous system (CNS), which accelerates the propagation of action potentials and supports axonal integrity. As a major component of CNS myelin, proteolipid protein 1 (Plp1) is indispensable for the axon-supportive function of myelin. Notably, this function requires the continuous high-level expression of Plp1 in OLs. Equally important is the controlled expression of Plp1, as illustrated by Pelizaeus–Merzbacher disease for which the most common cause is PLP1 overexpression. Despite a decade-long search, promoter–distal OL enhancers that govern Plp1 remain elusive. We have recently developed an innovative method that maps promoter–distal enhancers to genes in a principled manner. Here, we applied it to Plp1, uncovering two OL enhancers for it (termed Plp1-E1 and Plp1-E2). Remarkably, clustered regularly interspaced short palindromic repeats (CRISPR) interference epigenome editing showed that Plp1-E1 and Plp1-E2 do not regulate two genes in their vicinity, highlighting their exquisite specificity to Plp1. Assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) and chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) data show that Plp1-E1 and Plp1-E2 are OL-specific enhancers that are conserved among human, mouse and rat. Hi-C data reveal that the physical interactions between Plp1-E1/2 and PLP1 are among the strongest in OLs and specific to OLs. We also show that Myrf, a master regulator of OL development, acts on Plp1-E1 and Plp1-E2 to promote Plp1 expression.
Introduction
In the central nervous system (CNS), oligodendrocytes (OLs) extend and wrap their plasma membranes around axons, forming myelin sheaths (1). Myelin is essential for the development and function of the CNS. The classical function of myelin is to accelerate the propagation of action potentials along the axon by enabling saltatory conduction (2). We now know that myelin is more than a fatty insulation. For example, myelin provides axons with critical trophic and metabolic support (3–5), and adaptive myelination underlies learning and memory (6–10). Moreover, myelin promotes synaptogenesis and mediates the effect of social experience on animal behavior (11–13).
Proteolipid protein 1 (Plp1) is the most abundant membrane protein in CNS myelin (1), and PLP1 mutations are linked to neurological disorders such as Pelizaeus–Merzbacher disease (PMD) and spastic paraparesis (14). Gene knockout studies have shown that Plp1 is dispensable for myelin formation (15), a surprising finding in light of its super-abundance in myelin. However, Plp1 ablation in OL lineage cells led to axonal degeneration without overt demyelination (16,17), which is also observed in patients (18). These observations indicate that Plp1 is essential for the axon-supportive function of myelin. Fine regulation of PLP1 expression is physiologically important. More than half of PMD cases are caused by a duplication of the PLP1 locus that results in a 2-fold increase in PLP1 expression (19–21). Lowering PLP1 expression has been suggested as a curative approach for PMD (22,23). On the other hand, the axon-supportive function of myelin requires Plp1 to be expressed at a high level throughout life (24).
The OL-specific high-level expression of Plp1 has prompted the search for OL enhancers that regulate Plp1 (25–29). Enhancers are cis-regulatory DNA elements that direct cell type-specific gene expression by serving as transcription factor-binding platforms (30,31). A fascinating yet perplexing feature of enhancers is that they are often far away from target genes. This has made it difficult to find target genes of enhancers and vice versa. For this reason, the traditional approach to finding enhancers for a gene of interest is to find conserved sequence segments in its vicinity and to test whether they work as enhancers. If they do, the gene is assumed to be under their control. Following this spirit, wmN1, an OL enhancer in the first intron of Plp1, was discovered more than a decade ago (28). wmN1 has been assumed, but not proved, to govern Plp1 expression. It remains unknown whether there are any promoter–distal OL enhancers for Plp1. We have recently developed an innovative method that maps promoter–distal enhancers to genes in a principled manner (32). Its power was demonstrated for Myrf (32) and Rgcc (33). Encouraged by these successes, this study has applied it to Plp1, uncovering two promoter–distal OL enhancers for it.
Results
A principled method to find promoter–distal OL enhancers for Plp1
The key features of our new method are the rational search of enhancer candidates and CRISPRi (CRISPR interference) interrogation. It works in three steps (Fig. 1). First, chromatin conformation capture studies have shown that a gene and its enhancer tend to be found in the same topologically associating domain (TAD), a fundamental unit of genome organization and function (34,35). Thus, the Plp1 TAD is where we should look in search of Plp1 enhancers. In other words, the Plp1 TAD information allows us to rationally narrow down the search space for Plp1 enhancers. Of note, the internal detail of a TAD reflects cell type-specific gene-enhancer interactions, differing between cell types. In contrast, the boundary of a TAD tends to be conserved between cell types and species (34,36). This enabled us to identify the Myrf TAD by analyzing public chromatin interaction data for non-OL cell types (32). Now, OL chromatin interaction data are publicly available (37), greatly aiding TAD analysis for OL genes such as Plp1. Second, we systematically identify putative OL enhancers in the Plp1 TAD, which are qualified to be Plp1 enhancer candidates because they are in the same TAD as Plp1. Our previous study generated a genome-wide map of putative OL enhancers by integrating public OL ChIP-seq data (32). We compare this genome-wide map with the Plp1 TAD, uncovering all putative OL enhancers in the Plp1 TAD. Through the first and second steps (Fig. 1), we reduce the Plp1 enhancer search space from the entire genome to a few discrete loci in a principled manner. Third, we interrogate Plp1 enhancer candidates with CRISPRi, a cutting-edge epigenome editing technique (38–42), to determine whether they control Plp1 expression. CRISPRi potently silences promoters and enhancers in the genomic context, providing a revolutionary way to link enhancers to genes and vice versa.
Figure 1 .
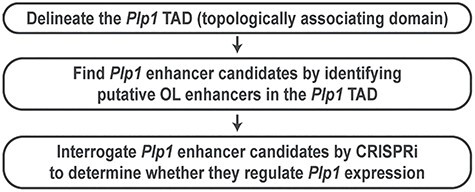
A principled method to find OL enhancers for Plp1.
TAD analysis for Plp1
To define the Plp1 TAD, we first examined public Hi-C data for five diverse human cell types (IMR90, K562, HMEC, HUVEC and NHEK) (35). PLP1 is on the plus strand of chromosome X. The location of the PLP1 promoter is indicated by thin crossing lines in Figure 2. In each panel of Figure 2, the diagonal represents the genome. Off the diagonal, the interaction strength between two loci is indicated by color. Red means the strongest interaction, and white no measurable interaction. The Hi-C data reveal that the PLP1 promoter is found in a densely self-associating region that spans 150 Kb (100 Kb upstream and 50 Kb downstream of PLP1). This TAD organization is conserved for the five cell types. Recently, a Hi-C dataset for human OLs was published (37), allowing us to test the validity of the PLP1 TAD that was inferred from non-OL Hi-C data. The OL Hi-C data indicate that the PLP1 promoter interacts with the same 150 Kb region in OLs (marked by a blue box in the bottom middle panel of Fig. 2). To check whether the PLP1 TAD is conserved in mouse, we examined the Hi-C data for CH12-LX (B cell lymphoma cells) (35). As in human, Plp1 is on the plus strand in the mouse genome. The CH12-LX Hi-C data reveals that the Plp1 promoter is found in a densely self-associating region of 95 Kb (75 Kb upstream and 20 Kb downstream of Plp1, marked in blue in the bottom right panel of Fig. 2). Given the smaller size of the mouse genome, this is an almost perfect conservation, highlighting the evolutionary conservation of the PLP1 TAD. Taken together, both OL and non-OL Hi-C data suggest that OL enhancers for PLP1 and Plp1 would be found in the 150 and 95 Kb regions, respectively.
Figure 2 .
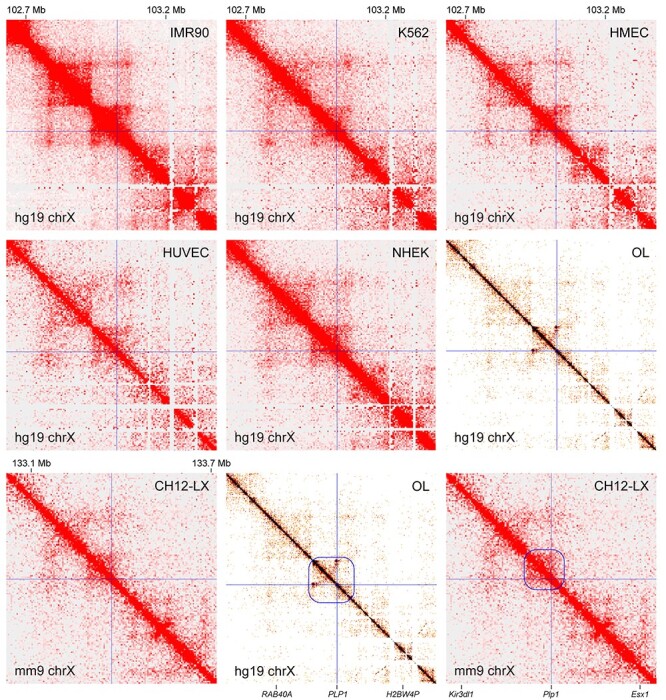
TAD analysis for Plp1. The public Hi-C data for seven diverse cell types from human and mouse. On the diagonal is the genome. Off the diagonal, the interaction strength between two loci is indicated by color. White means no interaction, and red the strongest interaction. The PLP1/Plp1 promoter locations are marked by thin crossing lines. The PLP1 TAD is marked by a blue box for OLs (bottom middle panel). The corresponding TAD for Plp1 is marked for CH12-LX (bottom right panel). IMR90, lung fibroblast; K562, chronic myelogenous leukemia cell; HMEC, human mammary epithelial cell; HUVEC, human umbilical vein endothelial cell; NHEK, normal human epidermal keratinocyte; CH12-LX, murine CH12 B cell lymphoma cell (please see Materials and Methods for the sources of these data). These figures were generated by Juicebox (85,86) and HiGlass (87).
Identification of two promoter–distal Plp1 enhancer candidates
We compared our genome-wide map of putative OL enhancers (32) with the Plp1 TAD, finding five putative OL enhancers in it. Since these five putative OL enhancers are in the same TAD as Plp1, they are qualified to be Plp1 enhancer candidates (EC1, 2, 3, 4 and 5 in Fig. 3A). The five ECs were ranked based on the strength of the underlying data: EC1 being the best enhancer candidate, EC2 the second best and so on. Of note, our criteria for calling putative OL enhancers were quite lenient. Thus, these five ECs may not work as OL enhancers, and even if they do, they may not regulate Plp1 expression. This is why we have to interrogate them with CRISPRi and other methods. The Wight and Peterson laboratories have extensively characterized conserved sequence segments in the vicinity of Plp1 in an effort to find OL enhancers for Plp1 (25–29). Their work led to the discovery of several enhancers around Plp1, including pk211, wmN1 and wmN2. Of these, wmN1 exhibited OL-specific enhancer activity (28,29). Due to its location in Plp1 (Fig. 3A), wmN1 has been assumed to regulate Plp1 expression in OLs. Of the five Plp1 ECs, EC4 corresponds to wmN1. EC3 is next to the 3′ untranslated region (UTR) of Plp1 and corresponds to pk211 (Fig. 3A) (28). EC5, which is in the Plp1 locus, maps to wmN2 (Fig. 3A) (28). Since our goal was to identify novel promoter–distal OL enhancers for Plp1, we focused on EC1 and 2, which happen to be the best two ECs. EC1 and EC2 are located at 66 and 73 Kb upstream of Plp1, respectively. Little is known about their role in Plp1 expression. As shown in Figure 3B, H3K27ac (histone 3 lysine 27 acetylation) peak-valley-peak patterns decorate EC1 and EC2 in differentiating OLs, whereas no H3K4me3 (histone 3 lysine 4 trimethylation) is observed for them. These are typical epigenetic features associated with active enhancers (43). Consistently, neither H3K27me3 (histone 3 lysine 27 trimethylation) nor H3K9me3 (histone 3 lysine 9 trimethylation) is observed for them, which are repressive histone marks. ChIP-seq data show that EC1 and EC2 are bound by Tcf7l2, Sox10, Olig2 and Myrf in OLs (Fig. 3B). Especially, they are bound by Sox10 in the rat spinal cord, indicative of their in vivo enhancer activity. The phastCons track (conservation in Fig. 3B) shows that EC1 and EC2 are evolutionarily conserved, suggesting that they may be important enhancers.
Figure 3 .
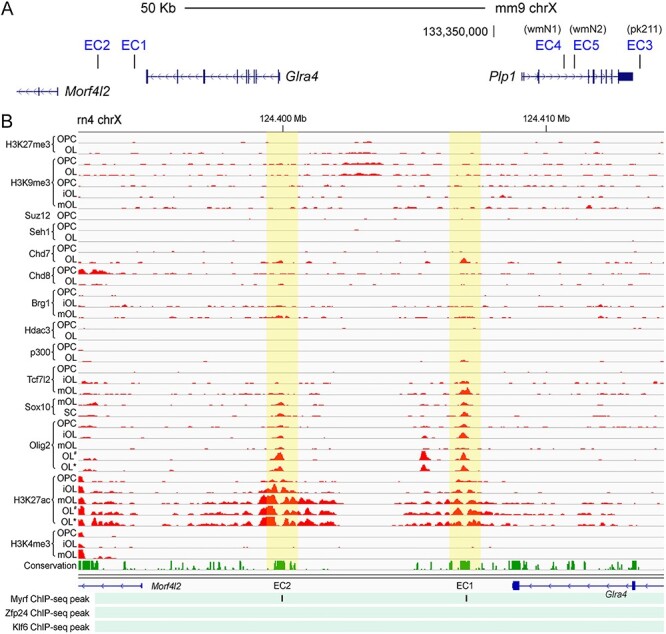
Five Plp1 ECs. (A) The locations of the five Plp1 ECs are shown. (B) Rat OL ChIP-seq data for EC1 and EC2. OPC, OL precursor cell; iOL, immature OL; mOL, mature OL; SC, spinal cord. For the Myrf ChIP-seq data, only peak locations are shown because the raw data is not available. The mouse Zfp24 and Klf6 ChIP-seq data were mapped to the rat genome by LiftOver. OL# and OL*: OLs treated with vehicle and lysophosphatidylcholine, respectively. Please see Materials and Methods for the sources of these data.
CRISPRi analysis of EC1 and EC2
CRISPRi is a state-of-the-art technique that can silence promoters and enhancers in the genomic context (38–42). In CRISPRi, dCas9–Krüppel-associated box (KRAB), a fusion protein between a nuclease-null Cas9 (dCas9) and a KRAB domain, is targeted to a specific locus by guide RNAs (gRNAs). When targeted to a promoter, dCas9–KRAB silences it by inducing H3K9me3 (40). When targeted to an enhancer, dCas9–KRAB silences it by the same mechanism, which in turn downregulates its target genes. This is how one can map promoter–distal enhancers to target genes by CRISPRi.
To silence EC1 and EC2, dCas9–KRAB was delivered to them by four independent gRNAs (G1–4) in Oli-neu cells, a widely used OL cell line (44). Specifically, gRNAs were cloned into an in-house piggyBac-based plasmid and inserted into the genome of an in-house Oli-neu cell line that expresses dCas9–KRAB in a doxycycline-dependent manner. In the resulting cell lines, gRNAs are expressed constitutively, whereas the expression of dCas9–KRAB is induced by doxycycline. As negative controls, two Oli-neu cell lines were generated where Scr1 and Scr2, two non-targeting scrambled gRNAs, were inserted into the genome. To determine whether silencing both EC1 and EC2 leads to a greater knockdown of Plp1, two Oli-neu cell lines were also generated where dCas9–KRAB is simultaneously targeted to EC1 and EC2 by two different sets of gRNAs. To interrogate EC1 and EC2 in a condition where endogenous Plp1 is expressed, the Oli-neu cell lines were cultured in the differentiation condition for 2 days in the presence of doxycycline. RNA was extracted from the 12 cell lines, and reverse transcription followed by quantitative polymerase chain reaction (RT-qPCR) performed to determine Plp1 expression. As expected, the expression level of Plp1 was comparable between Scr1 and Scr2 (Fig. 4A). In contrast, when dCas9–KRAB was delivered to EC1 or EC2 by any of the four gRNAs, Plp1 expression went down by 47–62% compared with Scr1 (Fig. 4A). Silencing both EC1 and EC2 by either EC1-G1 and EC2-G3 or EC1-G3 and EC2-G4 suppressed Plp1 expression to a greater extent (Fig. 4B). These results indicate that both EC1 and EC2 are required for Plp1 expression in Oli-neu cells.
Figure 4 .
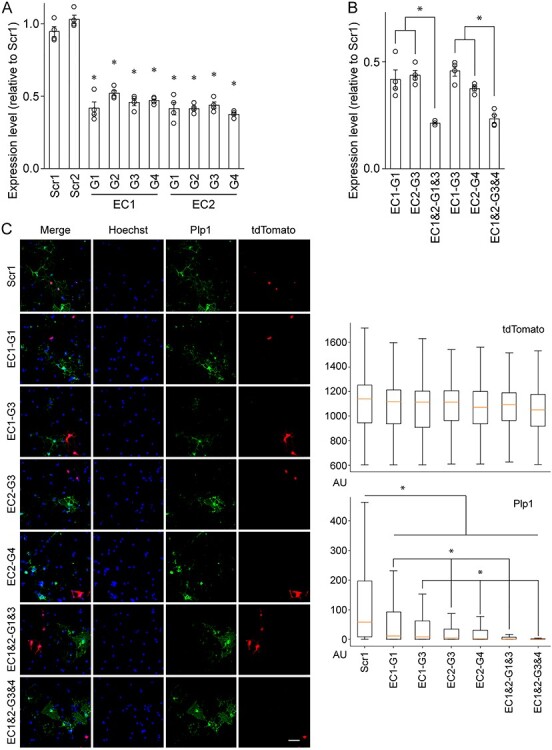
CRISPRi interrogation of Plp1 EC1 and EC2. (A) RT-qPCR analysis of Plp1 expression in Oli-neu cells after CRISPRi knockdown of EC1 and EC2. Shown are data points and their mean and standard error. *P <8.16 × 10−4 by Student’s t-test with Bonferroni correction. (B) RT-qPCR analysis of Plp1 expression in Oli-neu cells after CRISPRi knockdown of EC1 and EC2. Shown are data points and their mean and standard error. *P <1.73 × 10−2 by Student’s t-test with Bonferroni correction. (C) Quantitative immunofluorescence of Plp1 expression in mouse OLs after CRISPRi knockdown of EC1 and EC2. Left: Sample images. Scale bar, 50 μm. Right: The signal from each fluorescence channel was quantified for individual cells by CellProfiler. The number of cells analyzed is as follows: Scr1 (603), EC1-G1 (531), EC1-G3 (291), EC2-G3 (380), EC2-G4 (345), EC1 and 2-G1 and 3 (237) and EC1 and 2-G3 and 4 (194). *P <1.64 × 10−2 by Student’s t-test with Bonferroni correction. AU: arbitrary unit.
To check whether EC1 and EC2 also govern Plp1 expression in primary OLs, we repeated the CRISPRi experiment with mouse oligodendrocyte precursor cells (OPCs) purified by immunopanning (45,46). Transfection efficiency for mouse OPCs is neither high enough for RT-qPCR, nor is it possible to generate stable cell lines with them. Thus, we had to resort to quantitative immunofluorescence where individual cells are analyzed and thus high transfection efficiency is not needed. A plasmid expressing dCas9–KRAB and tdTomato was transfected into mouse OPCs, together with gRNA plasmids. Transfected OPCs were cultured in the differentiation condition for 2 days to induce differentiation. They were then stained for Plp1 and tdTomato (identifying transfected cells). As above, Scr1 was used for a control experiment. For an objective image analysis, the signal from each fluorescence channel (Hoechst, Plp1 and tdTomato) was quantified for individual OLs by CellProfiler (47). This quantitative single-cell image analysis revealed that while tdTomato signals were comparable across the seven samples, Plp1 signals were much lower when dCas9–KRAB was targeted to EC1 or EC2 (Fig. 4C). As for the Oli-neu RT-qPCR results, silencing both EC1 and EC2 had a greater effect on the Plp1 level compared with silencing EC1 or EC2 alone (Fig. 4C). Taken together, we conclude that EC1 and EC2 promote Plp1 expression in OLs.
EC1 and EC2 are dedicated to Plp1
It is not uncommon that a single enhancer controls multiple genes (41,42). Thus, EC1 and EC2 may also regulate other genes in the same TAD. Two genes are found in the EC1 and EC2 TAD, apart from Plp1: Morf4l2 and Glra4. Of these, Glra4 is not expressed in OLs according to the Brain RNA-seq database (Fig. 5A) (48). Apparently, EC1 and EC2 do not activate Glra4 expression in OLs. Morf4l2 is a ubiquitously expressed gene according to the Brain RNA-seq database (Fig. 5A) and the Genotype-tissue expression (GTEx) project (49). To determine whether EC1 and EC2 govern Morf4l2 expression in OL lineage cells, the Oli-neu RNA samples of Figure 4A were reanalyzed for Morf4l2 by RT-qPCR. CRISPRi silencing of EC1 and EC2 had no effect on the expression of Morf4l2 (Fig. 5B). Simultaneous knockdown of EC1 and EC2 did not affect it, either. These observations indicate that even though EC1 and EC2 are much closer to Glra4 and Morf4l2 than to Plp1 (Fig. 3A), they are specific to Plp1.
Figure 5 .
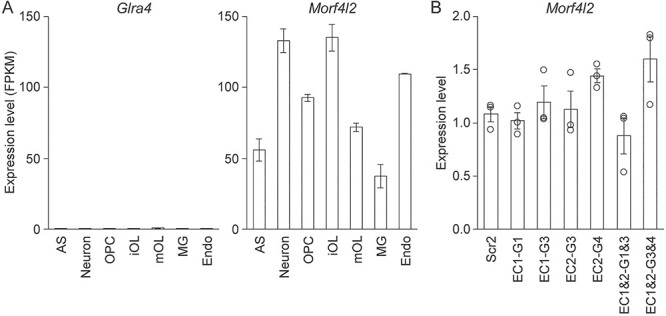
Two other genes in the EC1 and EC2 TAD. (A) Expression profiles of Glra4 and Morf4l2 in brain cell types. Data were taken from the Brain RNA-seq database. (B) RT-qPCR analysis of Morf4l2 expression in Oli-neu cells after CRISPRi knockdown of EC1 and EC2. Shown are data points and their mean and standard error. AS: astrocyte, iOL: immature OL, mOL: mature OL, MG: microglia, Endo: endothelial cell.
EC1 and EC2 are OL-specific enhancers
The epigenetic features (Fig. 3B) and the CRISPRi results (Fig. 4) strongly suggest that EC1 and EC2 work as enhancers in OL lineage cells. Furthermore, in light of their role in the activation of Plp1 expression in differentiating OLs, their enhancer activity may be stage specific. To address these, we performed a luciferase assay. EC1 and EC2 were cloned into pGL3-promoter and transfected into mouse OPCs. Rffl, an OL enhancer that is specifically active in differentiating OLs (50,51), was included as a control. pGL3-promoter (empty vector) was used to estimate baselines. Transfected OPCs were split into two. One set was cultured in the differentiation condition for 2 days to induce differentiation into OLs. The other set was kept in the proliferation condition for 2 days. As expected, the reporter activity of Rffl was significantly higher in the differentiation condition than in the proliferation condition (Fig. 6A). Post hoc analysis revealed that while there was no difference in the reporter activity between Rffl and pGL3-promoter in the proliferation condition, there was a significant difference in the differentiation condition (Fig. 6A). These results validate our culture conditions, allowing us to test the stage-specific enhancer activity of EC1 and EC2. EC1 exhibited strong enhancer activity regardless of the culture condition (Fig. 6A). In contrast, the enhancer activity of EC2 was significantly higher in the differentiation condition than in the proliferation condition (Fig. 6A). Indeed, post hoc analysis showed that the reporter activity of EC2 was not different from that of pGL3-promoter in the proliferation condition. Thus, the enhancer activity of EC2 was specific to the differentiation condition, mirroring the pattern observed for Rffl.
Figure 6 .
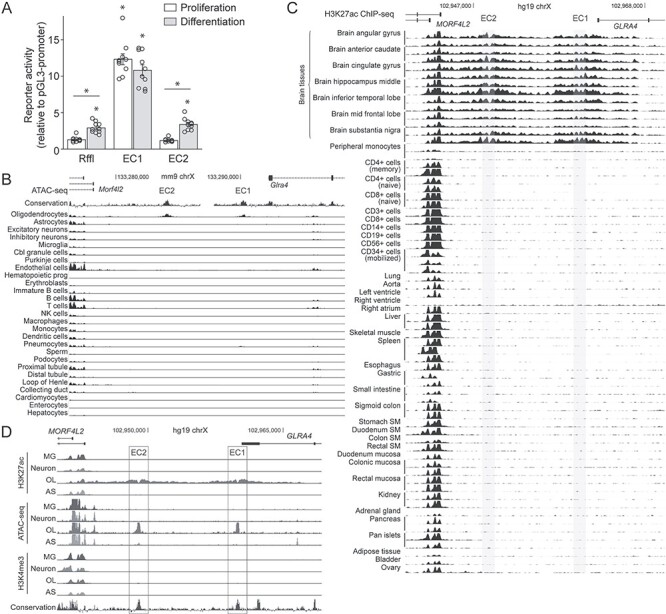
OL enhancer activity of EC1 and EC2. (A) Luciferase assay results for EC1 and EC2 that were transfected into mouse OPCs cultured in the proliferation and differentiation conditions. Shown are data points and their mean and standard error. *P <1.12 × 10−4 by Student’s t-test with Bonferroni correction. (B) Mouse single-cell ATAC-seq data for EC1 and EC2. (C) H3K27ac ChIP-seq data for EC1 and EC2 from the NIH Roadmap Epigenomics Project. The brain tissue results are clustered at the top. SM: smooth muscle. (D) Human brain cell type-specific ChIP-seq and ATAC-seq data for EC1 and EC2. MG, microglia; OL, oligodendrocyte; AS, astrocyte. Please see Materials and Methods for the sources of these data.
Having validated the OL enhancer activity of EC1 and EC2, we analyzed public datasets to further elucidate their properties. First, we examined the mouse single-cell ATAC-seq data from Shendure et al. (52). By using a single-cell ATAC-seq method, they determined chromatin accessibility for 13 different mouse tissues at the single-cell level. The resulting data were clustered into 27 broadly defined cell types. Strikingly, it reveals that EC1 and EC2 are accessible only in OLs (Fig. 6B), suggesting that EC1 and EC2 are OL-specific enhancers. Second, we looked up the H3K27ac ChIP-seq data from the NIH Roadmap Epigenomics Project (53). Consistent with the mouse single-cell ATAC-seq data, EC1 and EC2 are covered by broad H3K27ac peaks only in the brain tissues (Fig. 6C). Third, we examined the human brain cell type-specific ChIP-seq and ATAC-seq data from Glass et al. (54). EC1 and EC2 are marked by OL-specific ATAC-seq peaks that squarely overlap with OL-specific H3K27ac peak-valley-peaks (Fig. 6D). Consistent with their enhancer identity, no peak is observed for them in the H3K4me3 ChIP-seq data. Taken together, these data indicate that EC1 and EC2 are OL-specific enhancers that are conserved among human, mouse and rat. Since we have demonstrated that EC1 and EC2 are OL-specific enhancers that are dedicated to Plp1, they will henceforth be referred to as Plp1-E1 and Plp1-E2, respectively.
Plp1-E1 and Plp1-E2 physically interact with the PLP1 promoter
As mentioned above, the internal detail of a TAD reflects cell type-specific gene–enhancer interactions, differing between cell types. For this reason, non-OL Hi-C data cannot inform us about OL-specific gene–enhancer interactions. The OL Hi-C data fills this gap (37). Since Plp1-E1 and Plp1-E2 regulate Plp1 expression, we reasoned that the OL Hi-C data may disclose OL-specific physical interactions between Plp1-E1/2 and the PLP1 promoter. Indeed, it reveals strong physical interactions between them (Fig. 7A). To gauge the genome-wide significance of these physical interactions, all pairs of loci that are equidistant apart were examined. The interaction strength between Plp1-E1 and the PLP1 promoter, which are 72–Kb apart, is 0.0041 (see Materials and Methods). There are 1 011 512 pairs of loci that are equidistant apart in the OL Hi-C data. Of these, 1607 pairs physically interact, and five of them exhibit greater interaction strengths (Fig. 7B). So, the interaction of Plp1-E1 with PLP1 is the sixth strongest in OLs (P-value <5.94 × 10−6). The interaction between Plp1-E2 and the PLP1 promoter is more remarkable. It is the second strongest among the 1 011 428 pairs of loci that are 83 Kb apart (P-value <1.98 × 10−6). Clearly, the physical interactions of Plp1-E1/2 with PLP1 are highly significant from the genome-wide perspective.
Figure 7 .
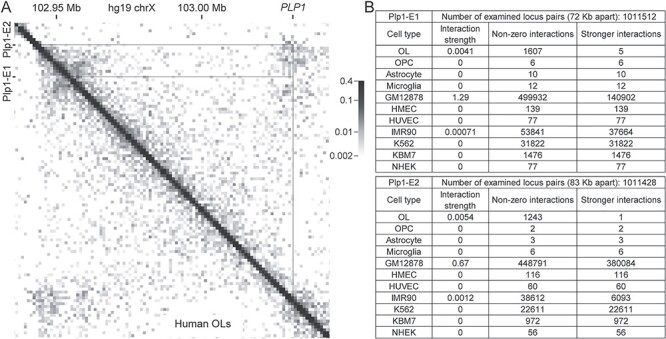
Physical interaction between Plp1-E1/2 and PLP1. (A) Hi-C data for human OLs zoomed in on PLP1 and Plp1-E1/2. (B) Comparative analysis of Hi-C data from diverse cell types. Please see Materials and Methods for the sources of these data.
To assess the cell type specificity of these interactions, we analyzed non-OL Hi-C data in the same manner. However, accurate statistical analysis was difficult because many Hi-C data are extremely sparse, as can be seen in Figure 7B. Nonetheless, the available data support that the interactions between Plp1-E1/2 and PLP1 are OL specific. For the six sparse datasets (OPC, astrocyte, microglia, HMEC, HUVEC and NHEK), Plp1-E1 and Plp1-E2 are not observed to contact the PLP1 promoter (Fig. 7B). This absence of interaction may well be because these datasets are sparse. So, we cannot draw any conclusions from the six datasets. For the K562 and KBM7 datasets, greater or comparable numbers of non-zero interactions are detected, yet Plp1-E1 and Plp1-E2 still do not interact with PLP1 (Fig. 7B), supporting the OL specificity of these interactions. For the two remaining sets (GM12878 and IMR90), Plp1-E1 and Plp1-E2 are observed to interact with PLP1 (Fig. 7B). Yet, these interactions are likely due to high background noises. For Plp1-E1, the fraction of stronger interactions in OLs is 0.0031 (5/1607). In GM12878 and IMR90, however, that is as high as 0.28 and 0.70, respectively. For Plp1-E2, the fraction of stronger interactions in OLs is 0.00081 (1/1243). In GM12878 and IMR90, that is 0.85 and 0.16, respectively. These suggest that the interactions between Plp1-E1/2 and PLP1 detected in GM12878 and IMR90 are likely spurious. Taken together, we conclude that the interactions of Plp1-E1/2 with the PLP1 promoter are OL specific, at least in comparison with GM12878, IMR90, K562 and KBM7.
Myrf regulates Plp1 expression via Plp1-E1 and Plp1-E2
Having shown that Plp1-E1 and Plp1-E2 govern Plp1, we wanted to identify transcription factors that act on them to regulate Plp1 expression. To this end, we surveyed public OL ChIP-seq data (Fig. 3B). Notably, we found that Myrf, a master regulator of OL development (50,55,56), binds to both Plp1-E1 and Plp1-E2 (Fig. 3B), leading us to hypothesize that Myrf promotes Plp1 expression by acting on them because Myrf mainly works as an activator (57). To test this hypothesis, we performed a luciferase assay in Oli-neu cells. Plp1-E1 and Plp1-E2 cloned in pGL3-promoter were transfected into Oli-neu cells, together with either pcDNA3 (empty vector) or Myrf cloned in pcDNA3. The reporter activity of Plp1-E1 and Plp1-E2 significantly went up in response to Myrf (Fig. 8A), supporting our hypothesis. We sought to map Myrf-binding sites in Plp1-E1 and Plp1-E2. Our previous study showed that Myrf binds to DNA as a homotrimer and that a 17 base pair long DNA motif termed the Myrf motif (Fig. 8B) mediates the sequence-specific DNA binding of Myrf (51). Motif incidence search with FIMO (58) revealed one good match in Plp1-E2 (Plp1-E2-WT in Fig. 8B). There was no match in Plp1-E1. To determine whether the motif incidence mediates the action of Myrf on Plp1-E2, we mutated it as shown in Figure 8B (Plp1-E2-MU), generating Plp1-E2-dMyrf, and repeated the luciferase assay. The mutation significantly dampened the response of Plp1-E2 to Myrf (Fig. 8A), indicating that Myrf acts on the motif incidence. However, post hoc analysis revealed that the reporter activity of Plp1-E2-dMyrf still went up significantly in response to Myrf (Fig. 8A). This suggests two possibilities. First, there is a cryptic binding site in Plp1-E2 that does not resemble the Myrf motif. Second, there is no additional Myrf binding site in Plp1-E2. In this case, Myrf may activate another activator or repress another repressor that acts on Plp1-E2, explaining why Plp1-E2-dMyrf still responds to Myrf despite the absence of an additional binding site.
Figure 8 .
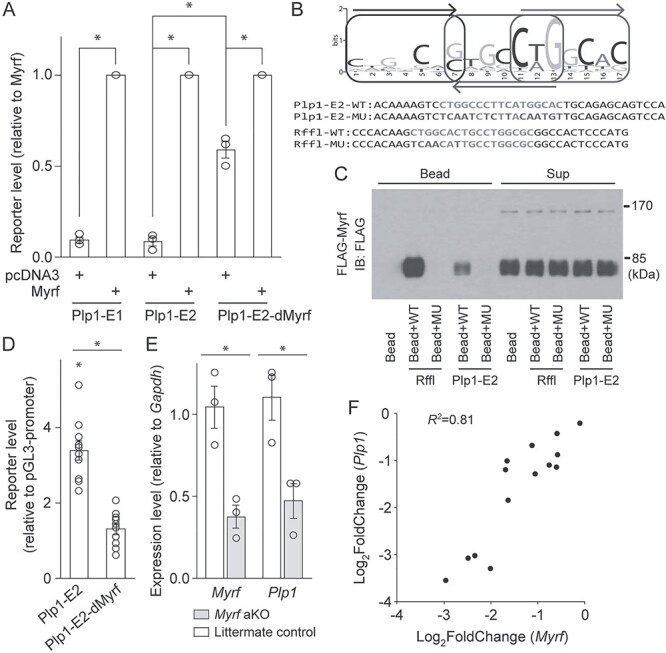
Myrf acts on Plp1-E1/2 to upregulate Plp1 expression. (A) Luciferase assay results showing the response of Plp1-E1/2 and Plp1-E2-dMyrf to Myrf in Oli-neu cells. Shown are data points and their mean and standard error. *P < 1.28 × 10−2 by Student’s t-test with Bonferroni correction. (B) The Myrf motif that mediates the sequence-specific DNA binding of Myrf. Also shown are the sequences of DNA oligos used for the DNA pulldown assay of (C). (C) DNA pulldown assay results. IB: immunoblotting. (D) Luciferase assay results for Plp1-E2 and Plp1-E2-dMyrf that were transfected into mouse OPCs cultured in the differentiation condition for 2 days. Shown are data points and their mean and standard error. *P <7.07 × 10−6 by Student’s t-test with Bonferroni correction. (E) RT-qPCR analysis of Myrf and Plp1 expression in mouse OLs upon acute Myrf knockout. Shown are data points and their mean and standard error. *P <4.52 × 10−2 by Student’s t-test with Bonferroni correction. aKO: acute knockout. (F) Correlation analysis of Myrf and Plp1 expression changes across 14 public OL gene expression datasets. Please see Materials and Methods for the sources of these data.
To test the physical interaction between Myrf and the motif incidence of Plp1-E2, we performed a DNA pulldown assay. FLAG-Myrf (Myrf with an N-terminal FLAG tag) was expressed in HEK293FT cells, and cell lysate mixed with either bare beads or beads coated with duplex DNA oligos. The duplex oligo contained either the motif incidence (Plp1-E2-WT in Fig. 8B) or the mutated one (Plp1-E2-MU in Fig. 8B). As in our previous studies (51,57,59), we used the motif incidence found in the Rffl locus and a mutant version of it (Rffl-WT and Rffl-MU in Fig. 8B) as the positive and negative controls. The mixture was separated into the sup and bead fractions by centrifuge, and both fractions were probed by FLAG antibodies. Immunoblotting showed that Myrf avidly bound beads coated with Rffl-WT, but neither bare beads nor beads coated with Rffl-MU (Fig. 8C). Under the same condition, Myrf-bound beads conjugated with Plp1-E2-WT, but not those coated with Plp1-E2-MU. The sup fraction results show that comparable amounts of proteins were used for the five binding reactions (Fig. 8C), ruling out the trivial possibility that the specific binding of Myrf to Rffl-WT and Plp1-E2-WT is due to unequal protein amounts used for the binding reactions.
To test the importance of the motif incidence of Plp1-E2 for the action of endogenous Myrf, we repeated the luciferase assay in primary OLs. Plp1-E2 and Plp1-E2-dMyrf were transfected into mouse OPCs. pGL3-promoter (empty vector) was used to estimate baselines. Transfected OPCs were cultured in the differentiation condition for 2 days to induce their differentiation into OLs. The reporter activity of Plp1-E2-dMyrf was significantly lower than that of Plp1-E2 (Fig. 8D). In fact, post hoc analysis showed that the reporter activity of Plp1-E2-dMyrf is not different from that of pGL3-promoter. These results demonstrate that the motif incidence is essential for the action of Myrf on Plp1-E2.
To corroborate our conclusion that Myrf promotes Plp1 expression via Plp1-E1 and Plp1-E2, we assessed the effect of acute Myrf knockout on Plp1 expression. To this end, we crossed mice harboring a floxed allele of Myrf (MyrfF/+ (55)) with CAG-CreER mice to generate MyrfF/F; CAG-CreER mice. OPCs were purified from them by immunopanning and cultured in the differentiation condition for 2 days to induce differentiation into OLs. For a control experiment, OPCs were purified from MyrfF/F littermates and cultured in the same condition in parallel. On the third day of differentiation, 4-hydroxytamoxifen was added for 8 h to acutely knockout Myrf in differentiating OLs. RNA was extracted and analyzed by RT-qPCR. This experimental strategy kept the initial differentiation process intact and allowed us to acutely delete Myrf in differentiating OLs, minimizing secondary effects associated with long-term perturbation. RT-qPCR analysis showed that Myrf was successfully knocked out (Fig. 8E), and this was accompanied by a significant drop in Plp1 expression (Fig. 8E). These observations further support that Plp1 is a target gene of Myrf. Finally, we examined 14 public OL gene expression datasets (see Materials and Methods) to assess the relationship between Myrf and Plp1 expression changes. If Plp1 is a target gene of Myrf, then changes in Myrf expression would be correlated with those in Plp1 expression. Indeed, there is a strong correlation between them (R2 = 0.81, Fig. 8F). When this correlation analysis was expanded to all of the 1807 transcription factors predicted in the mouse genome, we were surprised to find that Myrf is the one that exhibits the strongest correlation with Plp1.
Discussion
Plp1 is the most abundant membrane protein in CNS myelin (1). Even though it is dispensable for myelin formation (15), it is essential for the axon-supportive function of myelin (16–18). This function requires the continuous high-level expression of Plp1 (24). Equally important is the controlled expression of Plp1, as illustrated by PMD. More than half of PMD cases are caused by a duplication of the PLP1 locus that leads to a 2-fold increase in PLP1 expression (19–21). Lowering PLP1 expression may be a promising approach to curing PMD (22,23). A thorough understanding of how PLP1 expression is regulated in OLs would be essential for the development of effective PMD therapeutics.
To understand Plp1 expression in OLs, one has to identify OL enhancers that govern Plp1. Before the advent of CRISPRi, it was almost impossible to map promoter–distal enhancers to genes in a principled manner. For this reason, most studies focused on conserved sequence segments in the vicinity of the target gene. Following this spirit, the Wight and Peterson laboratories have extensively characterized conserved sequence segments in and around Plp1, identifying wmN1 (25–29). Since wmN1, located in the first intron of Plp1, showed an OL-specific enhancer activity, it has been assumed, but not proved, to control Plp1 expression. Apart from wmN1, it remained unknown whether there are any promoter–distal OL enhancers for Plp1. Since genes are often regulated by promoter–distal enhancers, we have recently developed an innovative method that rationally maps promoter–distal enhancers to genes (32). Its power was demonstrated for Myrf (32) and Rgcc (33). Encouraged by these successes, we have applied it to Plp1, uncovering Plp1-E1 and Plp1-E2. A notable aspect of Plp1-E1 and Plp1-E2 is that even though they are next to Glra4 and Morf4l2, they do not regulate their expression in Oli-neu cells, suggesting the exquisite specificity of Plp1-E1 and Plp1-E2 to Plp1. Importantly, public genomic data indicate that Plp1-E1 and Plp1-E2 are OL specific and conserved among human, mouse and rat. In the future, we plan to investigate the in vivo role and target specificity of Plp1-E1 and Plp1-E2 by mouse genetics. Another interesting topic is the epistatic relationship between Plp1-E1 and Plp1-E2, which may be context dependent. For example, would both Plp1-E1 and Plp1-E2 be required for Plp1 expression during developmental myelination, myelin maintenance and remyelination? Or would one be enough for a certain context?
Given the large body of circumstantial evidence implicating wmN1 in the regulation of Plp1 expression, we wanted to test it with CRISPRi and compare its potency with that of Plp1-E1 and Plp1-E2. CRISPRi silencing of wmN1 led to a significant drop in the expression of Plp1 (Supplementary Material, Fig. S1). However, it is likely a non-specific result because the same was also observed when dCas9–KRAB was targeted to wmN2 and two non-enhancer sites in the same intron. These complications precluded a meaningful CRISPRi analysis of wmN1 and its comparison with Plp1-E1 and Plp1-E2. It would be interesting to investigate whether Plp1-E1 and Plp1-E2 collaborate with wmN1 for Plp1 expression regulation by other means in the future.
The expression of a gene is regulated by positive and negative upstream regulators acting on the gene’s enhancers. Hence, one can find upstream regulators of a gene by identifying transcription factors that act on its enhancers. Following this logic, we identified Myrf as a positive upstream regulator of Plp1. The 14 public OL gene expression datasets reveal a striking correlation in the expression of Myrf and Plp1. Intrigued by this finding, we expanded the correlation analysis to the entire set of 1807 transcription factors predicted in the mouse genome. Remarkably, Myrf is the one that exhibits the strongest correlation with Plp1, reinforcing our conclusion that Plp1 is a target gene of Myrf. This correlation analysis also uncovered several transcription factors that are likely upstream regulators of Plp1. Among them are Tcf7l2 and Smad7. The ChIP-seq data (Fig. 3B) show that Tcf7l2 binds to Plp1-E1 in differentiating OLs (60), supporting that Tcf7l2 is an upstream regulator of Plp1. Consistently, the conditional knockout of Tcf7l2 was associated with a significant decrease in the expression of Plp1 (60,61). Regarding Smad7, a previous study indicated that it promotes OL differentiation by antagonizing the bone morphogenetic protein and Wnt/β-catenin signaling pathways (62). In light of the strong expression correlation between Smad7 and Plp1, Smad7 may also activate Plp1 expression for OL differentiation. To systematically find transcription factors that regulate Plp1 expression, we plan to perform a CRISPRi screen where each transcription factor is knocked down by CRISPRi and its effect on the enhancer activity of Plp1-E1 and Plp1-E2 is assessed by a luciferase assay. Since a fine balance between positive and negative upstream regulators would determine the final expression level of PLP1, identifying both types of regulators would be important and informative. Equally crucial would be the mapping of their binding sites in Plp1-E1 and Plp1-E2. For example, mutations of the binding site for a negative upstream regulator are likely to increase PLP1 expression, providing a rational explanation for those PMD cases where PLP1 expression goes up without a duplication of the PLP1 locus.
Materials and Methods
Constructs
A Myrf cDNA that encodes the 1139 amino acid long isoform was kindly provided by Dr Ben Emery. It was cloned into pcDNA3 with an N-terminal FLAG tag. To generate luciferase assay constructs, EC1 (mm10 chrX:136755392-136755991) and EC2 (mm10 chrX:136749215-136749814) were cloned into pGL3-promoter (Promega). Rffl was generated by cloning an Myrf ChIP-seq peak (rn4 chr10:70168592-70169175) into pGL3-promoter (50). For epigenome editing, dCas9–KRAB was amplified from pHAGE EF1α dCas9–KRAB (Addgene 50919) by PCR and inserted, together with an internal ribosome entry site-tdTomato cassette, into pCAG-Cre (Addgene 13775) after the Cre portion was removed. This construct, which is called ‘dCas9-KI’, was used for quantitative immunofluorescence. To generate an in-house Oli-neu cell line that expresses dCas9–KRAB in a doxycycline-dependent manner, pAAVS1-NDi-CRISPRi (Addgene 73497) was modified as follows. First, an RB (RFP and blasticidin resistance) cassette was fused to the rtTA via P2A. Second, the ITRs recognized by SB100X (Addgene 34879) were inserted. This construct and SB100X were transfected into Oli-neu cells (44). Cells where it was inserted into the genome were selected by blasticidin, and this selection process was followed by RFP. To generate gRNA constructs, the EF-1α promoter of pSBbi-RN (Addgene 60519) was replaced by the sgRNA scaffold taken from lentiCRISPR v2 (Addgene 52961), and gRNAs were cloned into it. These constructs were used for quantitative immunofluorescence. To generate gRNA constructs that can be inserted into the genome, the content of PB-CA (Addgene 20960) was replaced by the sgRNA scaffold, and a GP (GFP and puromycin resistance) cassette was inserted. This construct was called ‘PB-GP-U6’. gRNAs cloned into PB-GP-U6 and hypBase (63) were transfected into the Oli-neu cell line, and genomic insertion was selected by puromycin. Site-specific mutagenesis was performed by a PCR-based method, and sequence information for all constructs was verified by Sanger sequencing. The gRNA sequences are as follows.
Scr1: GCACTACCAGAGCTAACTCA
Scr2: TGCGAATACGCCCACGCGAT
EC1-G1: CTCATCGATTGGCACTACCT
EC1-G2: GTTACAGATCTTGTCCAGAT
EC1-G3: GCAATAAACCCTGTCCAACT
EC1-G4: CTGACACCCCAGCTCCATCA
EC2-G1: AAACAATAGAAGGTAGAATG
EC2-G2: AATTGATGTTTGCTCGAGAG
EC2-G3: CTCAGAGACTTACACCAGCC
EC2-G4: TTACAGCTGGGAGCCTGATG
Animal procedures, tissue harvest and cell culture
Animal husbandry was carried out in accordance with Institutional Animal Care and Use Committee-approved protocols. Mice with a floxed allele of Myrf (MyrfF/+ (55)) were purchased from Jax (010607). CAG-CreER mice were also purchased from Jax (004682). OPCs were purified from mouse pups of P7-9 by immunopanning that combines BSL1, O1 and O4 (45,46). Primary OPCs and Oli-neu cells were kept in a proliferative condition by supplementing the Sato media (46) with PDGF (10 μg/ml), NT3 (1 μg/ml) and CNTF (10 μg/ml). Primary OPCs and Oli-neu cells were maintained in a humidified 8% CO2 incubator at 37°C. HEK293FT cells were cultured in Dulbecco’s-modified Eagle’s medium supplemented with 10% fetal bovine serum and maintained in a humidified 5% CO2 incubator at 37°C. Transient transfection for OPCs, Oli-neu and HEK293FT cells was performed using Lipofectamine 2000 as per the manufacturer’s instructions.
RT-qPCR
Total RNA was purified by using Trizol (ThermoFisher 15596026), and cDNA synthesized by the SuperScript First-Strand kit (Invitrogen 11904-018). Quantitative PCR was performed on C1000 Touch thermal cycler with CFX384 optical reaction module (Bio-rad). Gapdh was used for a loading control. Each PCR reaction contained 2 μl of cDNA, 5 μl of the iTaq Universal SYBR Green Supermix (Bio-rad 1725124) and 500 nM of forward and reverse primers. The primer sequences are as follows.
Gapdh (forward): GGT GAA GGT CGG TGT GAA CGG
Gapdh (reverse): CTG GAA CAT GTA GAC CAT GTA GTT GAG G
Plp1 (forward): GCC AGA ATG TAT GGT GTT CTC CCA TG
Plp1 (reverse): GGT GGA AGG TCA TTT GGA ACT CAG C
Morf4l2 (forward): GGA ATC CTT GGA AGG GAA AGA AGG
Morf4l2 (reverse): GTT GTC CAC GAG TTT GAG AAG CC
Immunofluorescence
Cells were fixed with 4% formaldehyde and permeabilized with 0.1% Triton X-100. Upon blocking with 1% BSA, they were incubated with primary antibodies diluted in blocking buffer at 4°C overnight, followed by incubation with fluorochrome-conjugated secondary antibodies. Nuclei were stained with Hoechst 33342 (Invitrogen). Fluorescence was visualized with Leica DMi8 microscope with ORCA-Flash4.0 sCMOS camera. Reagents used for immunofluorescence are as follows: Plp1 (AA3), RFP (Rockland 600-401-379), donkey anti-rat, Alexa Fluor® 488 conjugate (ThermoFisher A-21208), and goat anti-rabbit, Alexa Fluor® 594 conjugate (ThermoFisher A-11037).
Luciferase assay
Luciferase assays were performed by using the Promega dual luciferase reporter assay kit as per the manufacturer’s instructions. pRL-TK was used as an internal control. The ratio between Firefly and Renilla luciferase activities was taken as the reporter activity.
DNA pulldown assay
HEK293FT cells were transfected with FLAG-Myrf. Cell lysate was cleared by centrifugation at 15 000 g for 20 min at 4°C. Biotinylated duplex oligonucleotides were conjugated to Dynabeads (Invitrogen) in buffer A (5 mM Tris pH 8.0, 0.5 mM EDTA, 1 M NaCl). Oligonucleotide-conjugated beads were washed twice with 500 μl of buffer A and three times with buffer C (20 mM Tris pH 8.0, 1 mM EDTA, 10% glycerol, 1 mM DTT, 50 mM NaCl). A total of 300 μg of cell lysate were incubated with oligonucleotide-conjugated beads in buffer C and sheared salmon sperm DNA (final concentration 0.2 mg/ml) for 20 min at room temperature with rotation. The mixture was spun down to separate into the bead and sup fractions. The bead fraction was washed five times with 500 μl buffer C, and both fractions were analyzed by immunoblotting with monoclonal ANTI-FLAG® M2 antibody (Sigma F1804, 1:1000).
OL ChIP-seq data
OL ChIP-seq data were downloaded from the Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra): GSE42454 (H3K9me3, Brg1, Olig2, H3K27ac, H3K4me3) (64), GSE72727 (Chd7, Sox10) (65), GSE119816 (Seh1) (66), GSE76411 (Hdac3, p300) (67), GSE82165 (Suz12) (68), GSE65119 (Tcf7l2) (60), GSE84011 (Olig2, H3K27ac) (69), GSE64703 (Sox10) (70), GSE107919 (Chd7, Chd8) (71), GSE101535 (Zfp24) (72), GSE79243 (Klf6) (73). The Myrf ChIP-seq data were downloaded from the journal website (50). H3K27me3 and H3K9me3 data were kindly provided by Dr Patrizia Casaccia (74). ChIP-seq reads were mapped to rn4 by Bowtie (75), and peaks called by MACS2 (76).
Public genomic data
Mouse single-cell ATAC-seq data (52) were downloaded from the Shendure laboratory website (https://atlas.brotmanbaty.org). Roadmap Epigenomics Project data (53) were visualized by the WASHU Epigenome Browser. Human brain cell type-specific ATAC-seq and ChIP-seq data (54) are available at https://genome.ucsc.edu/s/nottalexi/glassLab_BrainCellTypes_hg19. The following Hi-C data (35) were downloaded from 4DN Web Portal (https://4dnucleome.org): GM12878, HMEC, HUVEC, IMR90, K562, KBM7 and NHEK. The following Hi-C data (37) were downloaded from a public box directory at https://github.com/dixonlab/scm3C-seq: OL, OPC, astrocyte and microglia.
Hi-C data analysis
Cool files were analyzed by an in-house Python script that uses the cooler library (77). To compute the interaction strength between two loci, each locus was defined as a 6 Kb long segment. Then, the submatrix for the two loci was extracted, and its median value was taken as the interaction strength.
OL gene expression data
OL gene expression data were downloaded from SRA: GSE40510 (62), GSE19403 (78), GSE94067 (79), GSE130628 (80), GSE82210 (68), GSE74893 (81), GSE15303 (55), GSE135880 (82), GSE80439 (83), GSE124243 (84), GSE76410 (67), GSE42443 (64), GSE65118 (60) and GSE72726 (65). Each of these datasets has control and experimental groups, allowing us to compute the expression fold changes of Myrf and Plp1 between the two groups.
Conflict of Interest statement. Y.P. is currently applying for patents related to this paper.
Funding
The National Institutes of Health (R01NS094181, R21NS102558, R21NS112608, R21NS114476 to Y.P.).
Supplementary Material
Contributor Information
Dongkyeong Kim, Hunter James Kelly Research Institute, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14203, USA.
Hongjoo An, Hunter James Kelly Research Institute, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14203, USA.
Chuandong Fan, Hunter James Kelly Research Institute, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14203, USA.
Yungki Park, Hunter James Kelly Research Institute, Department of Biochemistry, Jacobs School of Medicine and Biomedical Sciences, State University of New York at Buffalo, Buffalo, NY 14203, USA.
References
- 1. Aggarwal, S., Yurlova, L. and Simons, M. (2011) Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol., 21, 585–593. [DOI] [PubMed] [Google Scholar]
- 2. Cohen, C.C.H., Popovic, M.A., Klooster, J., Weil, M.-T., Möbius, W., Nave, K.-A. and Kole, M.H.P. (2020) Saltatory conduction along myelinated axons involves a periaxonal nanocircuit. Cell, 180, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Funfschilling, U., Supplie, L.M., Mahad, D., Boretius, S., Saab, A.S., Edgar, J., Brinkmann, B.G., Kassmann, C.M., Tzvetanova, I.D., Mobius, W.et al. (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature, 485, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee, Y.J., Morrison, B.M., Li, Y., Lengacher, S., Farah, M.H., Hoffman, P.N., Liu, Y.T., Tsingalia, A., Jin, L., Zhang, P.W.et al. (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature, 487, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saab, A.S., Tzvetavona, I.D., Trevisiol, A., Baltan, S., Dibaj, P., Kusch, K., Möbius, W., Goetze, B., Jahn, H.M., Huang, W.et al. (2016) Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron, 91, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKenzie, I.A., Ohayon, D., Li, H., deFaria, J.P., Emery, B., Tohyama, K. and Richardson, W.D. (2014) Motor skill learning requires active central myelination. Science, 346, 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gibson, E.M., Purger, D., Mount, C.W., Goldstein, A.K., Lin, G.L., Wood, L.S., Inema, I., Miller, S.E., Bieri, G., Zuchero, J.B.et al. (2014) Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science, 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steadman, P.E., Xia, F., Ahmed, M., Mocle, A.J., Penning, A.R.A., Geraghty, A.C., Steenland, H.W., Monje, M., Josselyn, S.A. and Frankland, P.W. (2020) Disruption of oligodendrogenesis impairs memory consolidation in adult mice. Neuron, 105, 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan, S., Mayoral, S.R., Choi, H.S., Chan, J.R. and Kheirbek, M.A. (2020) Preservation of a remote fear memory requires new myelin formation. Nat. Neurosci., 23, 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang, F., Ren, S.-Y., Chen, J.-F., Liu, K., Li, R.-X., Li, Z.-F., Hu, B., Niu, J.-Q., Xiao, L., Chan, J.R.et al. (2020) Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat. Neurosci., 23, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang, F., Yang, Y.-J., Yang, N., Chen, X.-J., Huang, N.-X., Zhang, J., Wu, Y., Liu, Z., Gao, X., Li, T.et al. (2018) Enhancing oligodendrocyte myelination rescues synaptic loss and improves functional recovery after chronic hypoxia. Neuron, 99, 689–701.e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Makinodan, M., Rosen, K.M., Ito, S. and Corfas, G. (2012) A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science, 337, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu, J., Dupree, J.L., Gacias, M., Frawley, R., Sikder, T., Naik, P. and Casaccia, P. (2016) Clemastine enhances myelination in the prefrontal cortex and rescues behavioral changes in socially isolated mice. J. Neurosci., 36, 957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garbern, J.Y. (2007) Pelizaeus-Merzbacher disease: genetic and cellular pathogenesis. Cell. Mol. Life Sci., 64, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klugmann, M., Schwab, M.H., Pühlhofer, A., Schneider, A., Zimmermann, F., Griffiths, I.R. and Nave, K.-A. (1997) Assembly of CNS myelin in the absence of proteolipid protein. Neuron, 18, 59–70. [DOI] [PubMed] [Google Scholar]
- 16. Griffiths, I., Klugmann, M., Anderson, T., Yool, D., Thomson, C., Schwab, M.H., Schneider, A., Zimmermann, F., McCulloch, M., Nadon, N.et al. (1998) Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science, 280, 1610. [DOI] [PubMed] [Google Scholar]
- 17. Lüders, K.A., Patzig, J., Simons, M., Nave, K.A. and Werner, H.B. (2017) Genetic dissection of oligodendroglial and neuronal Plp1 function in a novel mouse model of spastic paraplegia type 2. Glia, 65, 1762–1776. [DOI] [PubMed] [Google Scholar]
- 18. Garbern, J.Y., Yool, D.A., Moore, G.J., Wilds, I.B., Faulk, M.W., Klugmann, M., Nave, K.A., Sistermans, E.A., van derKnaap, M.S., Bird, T.D.et al. (2002) Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain, 125, 551–561. [DOI] [PubMed] [Google Scholar]
- 19. Inoue, K., Osaka, H., Sugiyama, N., Kawanishi, C., Onishi, H., Nezu, A., Kimura, K., Yamada, Y. and Kosaka, K. (1996) A duplicated PLP gene causing Pelizaeus-Merzbacher disease detected by comparative multiplex PCR. Am. J. Hum. Genet., 59, 32–39. [PMC free article] [PubMed] [Google Scholar]
- 20. Wang, P.J., Hwu, W.L., Lee, W.T., Wang, T.R. and Shen, Y.Z. (1997) Duplication of proteolipid protein gene: a possible major cause of Pelizaeus-Merzbacher disease. Pediatr. Neurol., 17, 125–128. [DOI] [PubMed] [Google Scholar]
- 21. Sistermans, E.A., deCoo, R.F., De Wijs, I.J. and Van Oost, B.A. (1998) Duplication of the proteolipid protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology, 50, 1749–1754. [DOI] [PubMed] [Google Scholar]
- 22. Li, H., Okada, H., Suzuki, S., Sakai, K., Izumi, H., Matsushima, Y., Ichinohe, N., Goto, Y.I., Okada, T. and Inoue, K. (2019) Gene suppressing therapy for Pelizaeus-Merzbacher disease using artificial microRNA. JCI Insight, 4, e125052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elitt, M.S., Barbar, L., Shick, H.E., Powers, B.E., Maeno-Hikichi, Y., Madhavan, M., Allan, K.C., Nawash, B.S., Gevorgyan, A.S., Hung, S.et al. (2020) Suppression of proteolipid protein rescues Pelizaeus-Merzbacher disease. Nature, 585, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lüders, K.A., Nessler, S., Kusch, K., Patzig, J., Jung, R.B., Möbius, W., Nave, K.A. and Werner, H.B. (2019) Maintenance of high proteolipid protein level in adult central nervous system myelin is required to preserve the integrity of myelin and axons. Glia, 67, 634–649. [DOI] [PubMed] [Google Scholar]
- 25. Dobretsova, A. and Wight, P.A. (1999) Antisilencing: myelin proteolipid protein gene expression in oligodendrocytes is regulated via derepression. J. Neurochem., 72, 2227–2237. [DOI] [PubMed] [Google Scholar]
- 26. Meng, F., Zolova, O., Kokorina, N.A., Dobretsova, A. and Wight, P.A. (2005) Characterization of an intronic enhancer that regulates myelin proteolipid protein (Plp) gene expression in oligodendrocytes. J. Neurosci. Res., 82, 346–356. [DOI] [PubMed] [Google Scholar]
- 27. Pereira, G.B., Meng, F., Kockara, N.T., Yang, B. and Wight, P.A. (2013) Targeted deletion of the antisilencer/enhancer (ASE) element from intron 1 of the myelin proteolipid protein gene (Plp1) in mouse reveals that the element is dispensable for Plp1 expression in brain during development and remyelination. J. Neurochem., 124, 454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tuason, M.C., Rastikerdar, A., Kuhlmann, T., Goujet-Zalc, C., Zalc, B., Dib, S., Friedman, H. and Peterson, A. (2008) Separate proteolipid protein/DM20 enhancers serve different lineages and stages of development. J. Neurosci., 28, 6895–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamdan, H., Patyal, P., Kockara, N.T. and Wight, P.A. (2018) The wmN1 enhancer region in intron 1 is required for expression of human PLP1. Glia, 66, 1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buecker, C. and Wysocka, J. (2012) Enhancers as information integration hubs in development: lessons from genomics. Trends Genet., 28, 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shlyueva, D., Stampfel, G. and Stark, A. (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet., 15, 272–286. [DOI] [PubMed] [Google Scholar]
- 32. Kim, D., An, H., Shearer, R.S., Sharif, M., Fan, C., Choi, J.-O., Ryu, S. and Park, Y. (2019) A principled strategy for mapping enhancers to genes. Sci. Rep., 9, 11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim, D. and Park, Y. (2019) Molecular mechanism for the multiple sclerosis risk variant rs17594362. Hum. Mol. Genet., 28, 3600–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dixon, J.R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., Hu, M., Liu, J.S. and Ren, B. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rao, S.S., Huntley, M.H., Durand, N.C., Stamenova, E.K., Bochkov, I.D., Robinson, J.T., Sanborn, A.L., Machol, I., Omer, A.D., Lander, E.S.et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dixon, J.R., Jung, I., Selvaraj, S., Shen, Y., Antosiewicz-Bourget, J.E., Lee, A.Y., Ye, Z., Kim, A., Rajagopal, N., Xie, W.et al. (2015) Chromatin architecture reorganization during stem cell differentiation. Nature, 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee, D.-S., Luo, C., Zhou, J., Chandran, S., Rivkin, A., Bartlett, A., Nery, J.R., Fitzpatrick, C., O’Connor, C., Dixon, J.R.et al. (2019) Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat. Methods, 16, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gilbert, L.A., Horlbeck, M.A., Adamson, B., Villalta, J.E., Chen, Y., Whitehead, E.H., Guimaraes, C., Panning, B., Ploegh, H.L., Bassik, M.C.et al. (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell, 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kearns, N.A., Pham, H., Tabak, B., Genga, R.M., Silverstein, N.J., Garber, M. and Maehr, R. (2015) Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods, 12, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thakore, P.I., D'Ippolito, A.M., Song, L., Safi, A., Shivakumar, N.K., Kabadi, A.M., Reddy, T.E., Crawford, G.E. and Gersbach, C.A. (2015) Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods, 12, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fulco, C.P., Munschauer, M., Anyoha, R., Munson, G., Grossman, S.R., Perez, E.M., Kane, M., Cleary, B., Lander, E.S. and Engreitz, J.M. (2016) Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science, 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gasperini, M., Hill, A.J., McFaline-Figueroa, J.L., Martin, B., Kim, S., Zhang, M.D., Jackson, D., Leith, A., Schreiber, J., Noble, W.S.et al. (2019) A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell, 176, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Creyghton, M.P., Cheng, A.W., Welstead, G.G., Kooistra, T., Carey, B.W., Steine, E.J., Hanna, J., Lodato, M.A., Frampton, G.M., Sharp, P.A.et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. PNAS, 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jung, M., Krämer, E., Grzenkowski, M., Tang, K., Blakemore, W., Aguzzi, A., Khazaie, K., Chlichlia, K., vonBlankenfeld, G., Kettenmann, H.et al. (1995) Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci., 7, 1245–1265. [DOI] [PubMed] [Google Scholar]
- 45. Emery, B. and Dugas, J.C. (2013) Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb Protoc, 2013, 854–868. [DOI] [PubMed] [Google Scholar]
- 46. Dugas, J.C. and Emery, B. (2013) Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harb Protoc, 2013, 745–758. [DOI] [PubMed] [Google Scholar]
- 47. Carpenter, A.E., Jones, T.R., Lamprecht, M.R., Clarke, C., Kang, I.H., Friman, O., Guertin, D.A., Chang, J.H., Lindquist, R.A., Moffat, J.et al. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol., 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang, Y., Chen, K., Sloan, S.A., Bennett, M.L., Scholze, A.R., O'Keeffe, S., Phatnani, H.P., Guarnieri, P., Caneda, C., Ruderisch, N.et al. (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci., 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., Hasz, R., Walters, G., Garcia, F., Young, N.et al. (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bujalka, H., Koenning, M., Jackson, S., Perreau, V.M., Pope, B., Hay, C.M., Mitew, S., Hill, A.F., Lu, Q.R., Wegner, M.et al. (2013) MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS Biol., 11, e1001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim, D., Choi, J.-O., Fan, C., Shearer, R.S., Sharif, M., Busch, P. and Park, Y. (2017) Homo-trimerization is essential for the transcription factor function of Myrf for oligodendrocyte differentiation. Nucleic Acids Res., 45, 5112–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cusanovich, D.A., Hill, A.J., Aghamirzaie, D., Daza, R.M., Pliner, H.A., Berletch, J.B., Filippova, G.N., Huang, X., Christiansen, L., DeWitt, W.S.et al. (2018) A single-cell atlas of in vivo mammalian chromatin accessibility. Cell, 174, 1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. The Roadmap Epigenomics Consortium (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nott, A., Holtman, I.R., Coufal, N.G., Schlachetzki, J.C.M., Yu, M., Hu, R., Han, C.Z., Pena, M., Xiao, J., Wu, Y.et al. (2019) Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science, 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emery, B., Agalliu, D., Cahoy, J.D., Watkins, T.A., Dugas, J.C., Mulinyawe, S.B., Ibrahim, A., Ligon, K.L., Rowitch, D.H. and Barres, B.A. (2009) Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell, 138, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li, Z.H., Park, Y. and Marcotte, E.M. (2013) A bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLoS Biol., 11, e1001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Choi, J.-O., Fan, C., Kim, D., Sharif, M., An, H. and Park, Y. (2018) Elucidating the transactivation domain of the pleiotropic transcription factor Myrf. Sci. Rep., 8, 13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grant, C.E., Bailey, T.L. and Noble, W.S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics, 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fan, C., An, H., Sharif, M., Kim, D. and Park, Y. (2021) Functional mechanisms of MYRF DNA-binding domain mutations implicated in birth defects. J. Biol. Chem., 296, 100612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao, C., Deng, Y., Liu, L., Yu, K., Zhang, L., Wang, H., He, X., Wang, J., Lu, C., Wu, L.N.et al. (2016) Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nat. Commun., 7, 10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hammond, E., Lang, J., Maeda, Y., Pleasure, D., Angus-Hill, M., Xu, J., Horiuchi, M., Deng, W. and Guo, F. (2015) The Wnt effector transcription factor 7-like 2 positively regulates oligodendrocyte differentiation in a manner independent of Wnt/β-catenin signaling. J. Neurosci., 35, 5007–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weng, Q.J., Chen, Y., Wang, H.B., Xu, X.M., Yang, B., He, Q.J., Shou, W.N., Chen, Y., Higashi, Y., van denBerghe, V.et al. (2012) Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron, 76, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yusa, K., Zhou, L., Li, M.A., Bradley, A. and Craig, N.L. (2011) A hyperactive piggyBac transposase for mammalian applications. PNAS, 108, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu, Y., Chen, Y., Kim, B., Wang, H., Zhao, C., He, X., Liu, L., Liu, W., Wu, L.M.N., Mao, M.et al. (2013) Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell, 152, 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. He, D., Marie, C., Zhao, C., Kim, B., Wang, J., Deng, Y., Clavairoly, A., Frah, M., Wang, H., He, X.et al. (2016) Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci., 19, 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu, Z., Yan, M., Liang, Y., Liu, M., Zhang, K., Shao, D., Jiang, R., Li, L., Wang, C., Nussenzveig, D.R.et al. (2019) Nucleoporin Seh1 interacts with Olig2/Brd7 to promote oligodendrocyte differentiation and myelination. Neuron, 102, 587–601.e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang, L., He, X., Liu, L., Jiang, M., Zhao, C., Wang, H., He, D., Zheng, T., Zhou, X., Hassan, A.et al. (2016) Hdac3 interaction with p300 histone acetyltransferase regulates the oligodendrocyte and astrocyte lineage fate switch. Dev. Cell, 36, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. He, D., Wang, J., Lu, Y., Deng, Y., Zhao, C., Xu, L., Chen, Y., Hu, Y.C., Zhou, W. and Lu, Q.R. (2017) lncRNA functional networks in oligodendrocytes reveal stage-specific myelination control by an lncOL1/Suz12 complex in the CNS. Neuron, 93, 362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ou, Z., Sun, Y., Lin, L., You, N., Liu, X., Li, H., Ma, Y., Cao, L., Han, Y., Liu, M.et al. (2016) Olig2-targeted G-protein-coupled receptor Gpr17 regulates oligodendrocyte survival in response to lysolecithin-induced demyelination. J. Neurosci., 36, 10560–10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lopez-Anido, C., Sun, G., Koenning, M., Srinivasan, R., Hung, H.A., Emery, B., Keles, S. and Svaren, J. (2015) Differential Sox10 genomic occupancy in myelinating glia. Glia, 63, 1897–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhao, C., Dong, C., Frah, M., Deng, Y., Marie, C., Zhang, F., Xu, L., Ma, Z., Dong, X., Lin, Y.et al. (2018) Dual requirement of CHD8 for chromatin landscape establishment and histone methyltransferase recruitment to promote CNS myelination and repair. Dev. Cell, 45, 753–768.e758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Elbaz, B., Aaker, J.D., Isaac, S., Kolarzyk, A., Brugarolas, P., Eden, A. and Popko, B. (2018) Phosphorylation state of ZFP24 controls oligodendrocyte differentiation. Cell Rep., 23, 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Laitman, B.M., Asp, L., Mariani, J.N., Zhang, J., Liu, J., Sawai, S., Chapouly, C., Horng, S., Kramer, E.G., Mitiku, N.et al. (2016) The transcriptional activator Krüppel-like Factor-6 is required for CNS myelination. PLoS Biol., 14, e1002467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu, J., Magri, L., Zhang, F., Marsh, N.O., Albrecht, S., Huynh, J.L., Kaur, J., Kuhlmann, T., Zhang, W., Slesinger, P.A.et al. (2015) Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci., 35, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Langmead, B. and Salzberg, S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang, Y., Liu, T., Meyer, C.A., Eeckhoute, J., Johnson, D.S., Bernstein, B.E., Nusbaum, C., Myers, R.M., Brown, M., Li, W.et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abdennur, N. and Mirny, L.A. (2019) Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics, 36, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fancy, S.P.J., Baranzini, S.E., Zhao, C., Yuk, D.-I., Irvine, K.-A., Kaing, S., Sanai, N., Franklin, R.J.M. and Rowitch, D.H. (2009) Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev., 23, 1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Scaglione, A., Patzig, J., Liang, J., Frawley, R., Bok, J., Mela, A., Yattah, C., Zhang, J., Teo, S.X., Zhou, T.et al. (2018) PRMT5-mediated regulation of developmental myelination. Nat. Commun., 9, 2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang, W., Cho, H., Kim, D., Park, Y., Moon, J.H., Lim, S.J., Yoon, S.M., McCane, M., Aicher, S.A., Kim, S.et al. (2020) PRC2 acts as a critical timer that drives oligodendrocyte fate over astrocyte identity by repressing the Notch pathway. Cell Rep., 32, 108147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jiang, M., Liu, L., He, X., Wang, H., Lin, W., Wang, H., Yoon, S.O., Wood, T.L. and Lu, Q.R. (2016) Regulation of PERK-eIF2α signalling by tuberous sclerosis complex-1 controls homoeostasis and survival of myelinating oligodendrocytes. Nat. Commun., 7, 12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang, J., Yang, L., Dong, C., Wang, J., Xu, L., Qiu, Y., Weng, Q., Zhao, C., Xin, M. and Lu, Q.R. (2020) EED-mediated histone methylation is critical for CNS myelination and remyelination by inhibiting WNT, BMP, and senescence pathways. Sci. Adv., 6, eaaz6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang, H., Moyano, A.L., Ma, Z., Deng, Y., Lin, Y., Zhao, C., Zhang, L., Jiang, M., He, X., Ma, Z.et al. (2017) miR-219 cooperates with miR-338 in myelination and promotes myelin repair in the CNS. Dev. Cell, 40, 566–582.e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xu, H., Dzhashiashvili, Y., Shah, A., Kunjamma, R.B., Weng, Y.L., Elbaz, B., Fei, Q., Jones, J.S., Li, Y.I., Zhuang, X.et al. (2020) m(6)A mRNA methylation is essential for oligodendrocyte maturation and CNS myelination. Neuron, 105, 293–309.e295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Durand, N.C., Robinson, J.T., Shamim, M.S., Machol, I., Mesirov, J.P., Lander, E.S. and Aiden, E.L. (2016) Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst., 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Robinson, J.T., Turner, D., Durand, N.C., Thorvaldsdóttir, H., Mesirov, J.P. and Aiden, E.L. (2018) Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst., 6, 256–258.e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kerpedjiev, P., Abdennur, N., Lekschas, F., McCallum, C., Dinkla, K., Strobelt, H., Luber, J.M., Ouellette, S.B., Azhir, A., Kumar, N.et al. (2018) HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol., 19, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.