

Hao Wu, Li Wang, and Michael A. Crackower discuss recent advances in structure-based drug design to target NLRP3 and NLRP1.
Abstract
Inflammasome proteins play an important role in many diseases of high unmet need, making them attractive drug targets. However, drug discovery for inflammasome proteins has been challenging in part due to the difficulty in solving high-resolution structures using cryo-EM or crystallography. Recent advances in the structural biology of NLRP3 and NLRP1 have provided the first set of data that proves a promise for structure-based drug design for this important family of targets.
The innate immune system represents the first line of defense to invading foreign pathogens, being essential for efficient detection and clearance of the infection. It also plays a critical role in maintaining healthy tissue homeostasis, by detecting and removing damaged or dying cells, ensuring that organs maintain normal human physiological function throughout life. Following removal of pathogens or damaged cells, down-regulation of the innate immunity system is critical to ensure that unnecessary inflammation and tissue damage do not persist.
Many proteins and pathways of the innate immune system have been validated as compelling drug targets by virtue of their causal role in monogenic auto-inflammatory diseases (Harapas et al., 2018), their activation and up-regulation in many common inflammatory (Li and Wu, 2021) and neurodegenerative diseases (Heneka et al., 2018), and the extensive validation in preclinical models of diseases. Inflammasome proteins compose one key group of innate immune mediators of diseases (Lamkanfi and Dixit, 2012), which mediate the activation of IL-1β, IL-18, and Gasdermin D and the promotion of a pro-inflammatory response and disease. Thus, inflammasome proteins represent compelling drug targets.
Challenges of targeting inflammasome pathways
While inflammasomes represent highly compelling drug targets, the pursuit of drug discovery for many of these targets has been challenging due to the absence of high-resolution protein structures. The development of highly potent and selective small-molecule drugs requires the optimization of the precise molecular interactions between drug candidates and the intended protein targets. While optimization of small-molecule drugs can be pursued empirically, by engineering random modifications of small molecules in an effort to find those compounds that will have improved affinity and selectivity, it is far more powerful to design small molecules based on an intimate and precise understanding of how the molecules interact within a binding pocket on the protein. This understanding enables rational design of small molecules, which often leads to best-in-class designs and to expedited processes relative to using empirical approaches.
Recent advances in the structural biology of inflammasomes
Due to the heterogeneous, highly aggregated nature of inflammasome proteins, it has been difficult to generate high-resolution structures that can enable structure-based drug design (SBDD). In recent years, technological improvements in cryo-electron microscopy (cryo-EM) including direct electron detectors and computer programs are revolutionizing its utility for high-resolution structure determination. Together with x-ray crystallography, cryo-EM has generated excitement recently in structure analysis of inflammasome proteins and complexes.
NLRP3
Of particular interest are the recent advances in the structural biology of NLRP3, as it represents a most compelling drug target and has elicited significant drug discovery and development efforts in academia as well as pharmaceutical and biotechnology companies. NLRP3 can be activated by numerous self-derived damage-associated molecular patterns (Swanson et al., 2019) and is a critical regulator of the innate immune response in many diseases. Due to the difficulty in expressing the monomeric form of NLRP3 and the challenge of solving high-resolution protein structures, much of the drug discovery effort, however, has been based on empirical approaches resulting in limited structural diversity of the chemical matter (El-Sharkawy et al., 2020).
The first cryo-EM structure of NLRP3 was reported in 2019 as a complex with NEK7, the Ser/Thr kinase that acts as an essential scaffolding protein for NLRP3 activation (Sharif et al., 2019; see figure, panels A and B). This was accomplished with protein engineering that allowed for the expression of a stable form of monomeric NLRP3. While the resolution of the structure was not high enough to enable structure-based drug design, these data provided insights into the structural mechanism of NLRP3 activation. In an inactive state, NLRP3 adopts a closed conformation in its NACHT domain, preventing formation of an inflammasome assembly, and upon activation it may open up to allow for the formation of active NLRP3 inflammasome oligomers and the subsequent recruitment and activation of caspase-1. This cryo-EM study also suggested that not only the conformational change in NLRP3 is necessary, but also the leucine-rich repeat (LRR) domain-bound NEK7 bridges the gap between adjacent NLRP3 subunits to mediate the assembly of the higher order oligomeric complex.
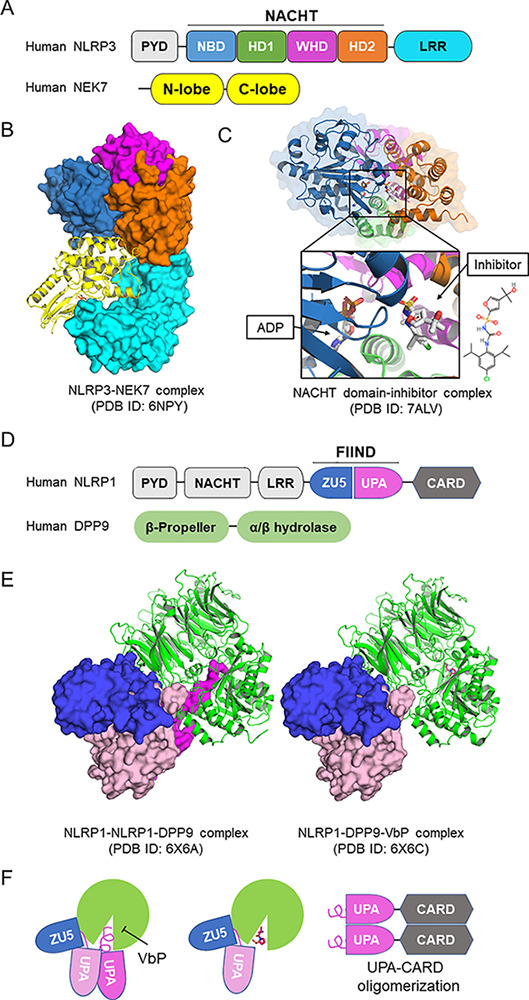
Structures of NLRP1 and NLRP3. (A) Domain architecture of NLRP3. NLRP3 is composed of an N-terminal pyrin domain (PYD); a central NACHT domain that is subdivided into NBD, helical domain 1 (HD1), winged helix domain (WHD), and helical domain 2 (HD2); and a C-terminal LRR domain. (B) Structure of monomeric NLRP3 in complex with NEK7 solved by cryo-EM (Protein Data Bank [PDB] ID: 6NPY). The structure is colored coded as in A by domains. (C) Crystal structure of NACHT domain of NLRP3 in complex with ADP and a small molecular inhibitor (PDB ID: 7ALV). The inhibitor locates at the multiple domain interface and locks protein in the inactive conformation. (D) Domain architecture of NLRP1 and DPP9. The autocatalytic FIIND is composed of ZU5 and UPA subdomains. (E) Cryo-EM structures of the ternary complex of NLRP1-NLRP1-DPP9 and the inhibition by VbP. In the ternary complex (left, PDB ID: 6X6A), two NLRP1 molecules bind to one DPP9 (green). The first NLRP1 has intact FIIND in which the ZU5 subdomain (blue) associates with the UPA subdomain (pink). The second NLRP1 has the UPA subdomain dissociated, and the N-terminal peptide of the UPA subdomain inserts into the DPP9 catalytic pocket (magenta). Vbp binding displaces the inserted N-terminal UPA peptide and reduces second UPA occupancy (PDB ID: 6X6C). (F) UPA-CARD released from DPP9 initiates inflammasome assembly.
Recently, cryo-EM structures of full-length wild-type human and mouse NLRP3 as inactive oligomers were reported in two preprints (Hochheiser et al., 2021 Preprint; Andreeva et al., 2021 Preprint). These structures exhibit decamer to hexadecamer double-ring cage-like arrangement, and the decameric human NLRP3 cage was solved in complex with the selective and potent NLRP3 inhibitor CRID3/MCC950 (Hochheiser et al., 2021 Preprint). In this 4-Å decameric structure, the mechanism of action of CRID3 was demonstrated for the first time. The data revealed that CRID3 binds in a pocket only present in the closed conformation created by multiple subdomains of the NACHT domain (see figure, panel A), thereby stabilizing this structure and preventing activation. They not only confirmed previous biochemical and modeling studies suggesting a similar mechanism of action (El-Sharkawy et al., 2020), but also refuted previous data suggesting that the mechanism of inhibition was via competition with nucleotide.
A crystal structure of the NACHT domain in complex with a CRID3-like small-molecule inhibitor at 2.8-Å resolution was also reported, which confirmed the binding of the inhibitor in a pocket formed by the four subdomains of the NACHT domain (Dekker et al., 2021; see figure, panel C). In an effort to help drive the discovery of differentiated chemical matter, we have recently successfully resolved a structure of monomeric NLRP3 complexed with a proprietary high affinity NLRP3 inhibitor (unpublished data). In this example we achieved a 2.6-Å resolution with a clear inhibitor bound pocket also formed by the subdomains of the NACHT domain. Thus, NLRP3 inhibitors, at least for the ones we have evidence for, act as an interdomain glue to lock the protein in an inactive conformation.
NLRP1
In addition to the recent advances on NLRP3, there have been significant advances in the structural biology for NLRP1. Like NLRP3, NLRP1 activation has been implicated in monogenic and complex pro-inflammatory diseases (Fenini et al., 2020). NLRP1 was the first inflammasome sensor to be identified, but only recently have we begun to elucidate the mechanism of activation of this protein. NLRP1 is unusual in that it contains a function-to-find domain (FIIND) that is auto-processed during protein synthesis to split into ZU5 and UPA subdomains, but the N- and C-terminal regions remain noncovalently bound (see figure, panel D). Through extensive recent studies, we now know that Anthrax lethal factor and viral proteases cleave an N-terminal region of NLRP1 to generate a neo-N-terminus, resulting in NLRP1 ubiquitination and N-terminal “functional” proteasomal degradation; this degradation leads to the release of the noncovalently associated C-terminal active fragment, which induce inflammasome assembly (Wang et al., 2021). Interestingly, the CARD8 protein, which also contains FIIND-CARD (caspase-1 recruitment domain) but lacks the nucleotide-binding domain (NBD)–LRR, was found to be able to form inflammasomes as well, and functional degradation can also explain the activation of the CARD8 inflammasome (Chui et al., 2020).
NLRP1 activation is further regulated by DPP8 and DPP9, which are related intracellular prolyl dipeptidases, and inhibition of DPP8/9 by the small molecule Val-boroPro (VbP) or genetic knockouts leads to NLRP1 inflammasome activation (Wang et al., 2021). Further evidence uncovered that DPP8/9 directly interacts with the FIIND and mediates inhibition of the NLRP1 inflammasome activity. Recently, cryo-EM structures of NLRP1-DPP9 complex alone and in complex with the DPP8/9 inhibitor VbP have been reported (Hollingsworth et al., 2021; Huang et al., 2021). Surprisingly, the NLRP1-DPP9 forms a ternary complex comprised of DPP9, one intact FIIND of an autoinhibited NLRP1, and one C-terminal fragment (UPA-CARD) freed by N-terminal degradation. The N-terminal residues of UPA-CARD insert into the DPP9 active site, and VbP competes for this interaction (see figure, panels E and F). The binding of UPA-CARD to DPP9 requires full-length NLRP1, and the ternary complex could prevent UPA-CARD from self-oligomerization during homeostatic protein turnover (see figure, panel F). This work provides important insights into how the unique domain architecture and negative regulatory protein DPP8/9 participate in NLRP1 activation and may pave the way for the discovery of novel inhibitors of NLRP1 and a starting point to enable SBDD.
Future prospects
These recent advances in our structural understanding of the mechanism of activation and inhibition of these key inflammasome proteins offer tremendous insight into inflammasome signaling. Equally important, they provide initial templates from which to conduct rational-based drug design for these highly compelling targets. In the future, we are certain to see the fruits of these seminal pieces of work by assisting the development of inflammasome inhibitors using rational drug design approaches. We envision that these efforts will generate best-in-class molecules for the treatment of numerous diseases of high unmet medical need where inflammasomes are key pathogenic drivers.
Acknowledgments
We apologize for incomplete citations due to space limitations.
This work was supported in part by National Institutes of Health (to H. Wu).
Disclosures: H. Wu is a co-founder of Ventus Therapeutics, and an institutional plan is in place to manage and monitor the conflicts of interest that arise from this relationship.
References
- Andreeva, L., et al. 2021. Biorxiv. 10.1101/2021.09.12.459968 (Preprint posted September 12, 2021) [DOI]
- Chui, A.J., et al. 2020. Cell Rep. 10.1016/j.celrep.2020.108264 [DOI] [Google Scholar]
- Dekker, C., et al. 2021. J. Mol. Biol. 10.1016/j.jmb.2021.167309 [DOI] [Google Scholar]
- El-Sharkawy, L.Y., et al. 2020. Molecules. 10.3390/molecules25235533 [DOI] [Google Scholar]
- Fenini, G., et al. 2020. Int. J. Mol. Sci. 10.3390/ijms21134788 [DOI] [Google Scholar]
- Harapas, C.R., et al. 2018. Curr. Rheumatol. Rep. 10.1007/s11926-018-0750-4 [DOI] [Google Scholar]
- Heneka, M.T., et al. 2018. Nat. Rev. Neurosci. 10.1038/s41583-018-0055-7 [DOI] [PubMed] [Google Scholar]
- Hochheiser, I.V., et al. 2021. Biorxiv. 10.1101/2021.07.22.453353 (Preprint posted July 22, 2021) [DOI]
- Hollingsworth, L.R., et al. 2021. Nature. 10.1038/s41586-021-03350-4 [DOI] [Google Scholar]
- Huang, M., et al. 2021. Nature. 10.1038/s41586-021-03320-w [DOI] [Google Scholar]
- Lamkanfi, M., and Dixit V.M.. 2012. Annu. Rev. Cell Dev. Biol. 10.1146/annurev-cellbio-101011-155745 [DOI] [PubMed] [Google Scholar]
- Li, D., and Wu M.. 2021. Signal Transduct. Target. Ther. 10.1038/s41392-021-00687-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif, H., et al. 2019. Nature. 10.1038/s41586-019-1295-z [DOI] [Google Scholar]
- Swanson, K.V., et al. 2019. Nat. Rev. Immunol. 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., et al. 2021. J. Allergy Clin. Immunol. 10.1016/j.jaci.2021.04.018 [DOI] [Google Scholar]