

SUMMARY
Impaired DNA crosslink repair leads to Fanconi anemia (FA), characterized by a unique manifestation of bone marrow failure and pancytopenia among diseases caused by DNA damage response defects. As a germline disorder, why the hematopoietic hierarchy is specifically affected is not fully understood. We find that reprogramming transcription during hematopoietic differentiation results in an overload of genotoxic stress, which causes aborted differentiation and depletion of FA mutant progenitor cells. DNA damage onset most likely arises from formaldehyde, an obligate by-product of oxidative protein demethylation during transcription regulation. Our results demonstrate that rapid and extensive transcription reprogramming associated with hematopoietic differentiation poses a major threat to genome stability and cell viability in the absence of the FA pathway. The connection between differentiation and DNA damage accumulation reveals a novel mechanism of genome scarring and is critical to exploring therapies to counteract the aplastic anemia for the treatment of FA patients.
Graphical Abstract
INTRODUCTION
Defects in various DNA repair mechanisms give rise to more than 13 cancer-prone genetic syndromes (Rass et al., 2007; Weinberg, 2014). While a number of these disorders are accompanied by aberrant lymphocyte profiles owing to the impaired V(D)J recombination (Rooney et al., 2004), Fanconi anemia (FA) patients present a distinct and severe manifestation of bone marrow failure and aplastic anemia, presented by a general and progressive deficit in blood and bone marrow cellularity (D’Andrea, 2010).
The FA pathway is primarily responsible for the processing of strong replication-stalling lesions such as interstrand crosslinks (ICLs) and DNA-protein crosslinks (DPCs) (Ceccaldi et al., 2016; Duxin and Walter, 2015). Among the 24 identified FA genes, nine act upstream in response to replication stress to activate the FA pathway via monoubiquitination of FANCD2 and promote FANCD2 recruitment at the sites of DNA lesions or stalled forks (Garcia-Higuera et al., 2001; Meetei et al., 2003; Shen et al., 2009). The FA core complex consisting of FANCA, B, C, E, F, G, and L monoubiquitinates the FANCD2 and FANCI complex, which in turn orchestrates the recruitment of FA pathway nuclease scaffold and nucleases for the removal of crosslinking lesions (Huang et al., 2014; Rajendra et al., 2014; Smogorzewska et al., 2007). A group of recombination factors (FANCD1/Brca2, FANCN/PALB2, FANCO/RAD51C), whose biallelic mutations leads to FA, are involved in the downstream processing of DNA double strand breaks (DSBs), as a potential intermediate of ICL/DPC repair or resulting from replication fork collapse (Petermann et al., 2010).
In additional to DNA crosslinking damage repair, there is growing evidence that the FA pathway is important for stabilization/fork protection during replication stalling. Defects in FA genes lead to accelerated erosion of daughter strands when replication is interrupted by dNTP depletion or polymerase inhibition (Schlacher et al., 2012). The replication fork erosion phenotype can be mitigated by suppression of exonucleases such as ExoI (Karanja et al., 2012). A commonly perceived model based on these observations is that the FA pathway is required to prevent erroneous and excessive processing of replication forks when fork progression is stalled (Karanja et al., 2014; Tian et al., 2017).
FA pathway functions in DNA crosslinking damage repair and replication stress response are essential across all cell types. Why the hematopoietic system is particularly affected remains to be fully defined. In humans, bi-allelic mutations in any of the 24 FA genes are singly causative for FA disease manifestations. In mice, however, onset of the hematopoietic phenotype requires co-inactivation of the aldehyde dehydrogenase genes ALDH2 or ADH5, plus the exogenous challenge of alcohol or methanol (Langevin et al., 2011; Rosado et al., 2011). These observations revealed a genetic interaction between the FA pathway and aldehyde metabolism and a potential role for aldehyde-mediated DNA damage as a cause of genomic instability (Garaycoechea et al., 2018). However, the endogenous source of aldehydes and the triggering event for the distinctive blood and bone marrow cell loss remains unclear.
In this study, we tested the idea that cellular differentiation is a potential source of endogenous genotoxic stress in hematopoietic cells. Blood progenitor and stem cell models deficient in the FA mechanism showed severe deficiency in their differentiation capacity and a strong accumulation of endogenous DNA damage as a result of differentiation induction. The endogenous DNA damage appears to originate from formaldehyde, which accumulates as a byproduct of protein demethylation during rapid and extensive epigenetic reprograming of blood progenitor cells. Impaired repair of formaldehyde-induced DNA crosslinking damage in FA mutant cells attenuated lineage progression and caused cell death, which likely leads to the attrition of blood and bone marrow cells. These findings establish a more precise cause of the hematopoietic manifestation in Fanconi anemia patients.
RESULTS
Deletion of FANCL attenuates hematopoietic differentiation
Biallelic mutation of FA genes is singly causative for the degenerative blood phenotypes in human. To determine whether a defective FA pathway is sufficient to impair the viability of blood cells, we disrupted the FANCL E3 ligase gene, which is essential for the FA pathway activation, in two distinct types of progenitor cells (Fig. S1A–C). The HL60 promyelocyte is a bipotent progenitor capable of differentiating into monocytes or neutrophils. The K562 myelogenic cells are a pluripotent precursor capable of transitioning into four myeloid lineages. In the FANCL−/− HL60 and K562 knockout mutants, ablation of the FA mechanism was verified by the FANCL-null status, elimination of damage-induced FANCD2 monoubiquitination, and hypersensitivity to mitomycin C (Fig. 1A, S1G, H). The fact that knockout mutants were obtained with typical frequencies suggests that deletion of FANCL does not render a significant proliferation disadvantage to these progenitor cells. Indeed, cell proliferation of the FANCL mutants appears indistinguishable from their isogenic parental controls (Fig. 1B). These results suggest that the FA mechanism is not essential for blood progenitor survival and proliferation.
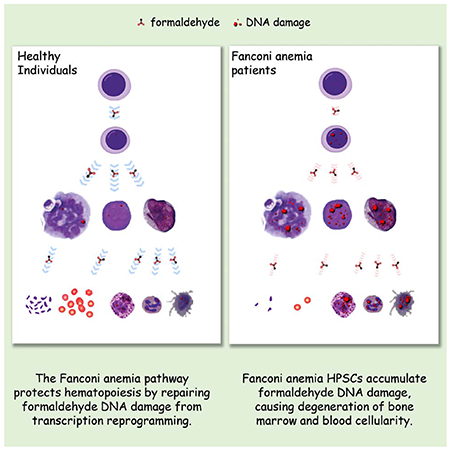
Fig. 1. Disruption of the FA pathway attenuates hematopoietic cellular differentiation and causes cell death.
(A) FANCL−/− cells generated via CRISPR/CAS9 in HL60 and K562 cells. Parental FANCL+/+ and FANCL−/− mutant cells were exposed to mitomycin C (MMC; 200 nM, 12 hours) prior to harvesting, and cell extracts were immunoblotted with FANCL (top panels) and FANCD2 (middle panels). *Corresponds to a non-specific band detected by the FANCL antibody in K562 cells.
(B) Proliferation of FANCL+/+ and FANCL−/− HL60 (left) and K562 (right) cells. Cell growth analyses were performed within log phase range.
(C) Differentiation of HL60 FANCL+/+ and FANCL−/− cells into monocytes was induced by V-D3 treatment or mock induction. The extent of differentiation was determined by CD14 positivity via flow cytometry.
(D) Differentiation of K562 FANCL+/+, FANCL−/− knockout mutants (FANCL−/−-1 and FANCL−/−-2), and the FANCL−/−-1 mutant complemented with wild-type FANCL cDNA stable expression (FANCL−/−-1C) was induced by PMA. The extent of differentiation into megakaryocytes was determined by CD41 positivity via flow cytometry.
(E) Yields of adherant monocytes from FANCL+/+, ADH5−/−, and FANCL−/− HL60 cells induced by V-D3. Adherent cells were visualized by crystal violet staining 7 days after V-D3 treatment.
(F) Quantification of experiments as shown in (E).
(G) Yields of adherant megakaryocytes from FANCL+/+, FANCL−/−-1 and FANCL−/−-2 K562 cells induced by PMA. Adherent cells were visualized the crystal violet staining 5 days after PMA treatment.
(H) Quantification of experiments as shown in (G).
(I) Apoptosis of FANCL+/+ and FANCL−/− HL60 cells induced to differentiate into monocytes by V-D3 treatment (5 days).
(J) Apoptosis of FANCL+/+ and FANCL−/− K562 cells induced to differentiate into megakaryocytes by PMA treatment (3 days).
(K) Apoptosis of FANCL+/+ and FANCL−/− K562 cells induced to differentiate into erythroids by hemin treatment (5 days).
(L) CD71- and GFP-positive cell population as measured by bivariate flow cytometry. CD34+ human cord blood cells were co-transfected with the LentiCRISPRv2GFP and gRNA vectors expressing FANCL gRNA (left) or LacZ gRNA as a control (right). Erythroid induction was initiated by SCF/IL-3/EPO incubation for 5 days before cell harvesting for FACS analyses.
(M) Quantification of (L) from three biological repeats.
P values were generated from unpaired t-tests. Error bars represent standard deviations from three or more biological repeats with duplicates or triplicates.
We next tested the differentiation competence of the FANCL−/− mutants into their designated lineages. When wild-type HL60 cells were differentiated to monocytes by treatment with vitamin D3 (V-D3), more than 67.6 ± 1.8% of the cells expressed the monocytic marker CD14, reflecting a normal progression of differentiation. V-D3-induced differentiation of an HL60 ADH5−/− mutant was similar to that of the FANCL+/+ cells (Fig. S2A), indicating that neither loss of ADH5 nor the CRISPR knockout manipulation affected HL60 differentiation into monocytes. By contrast, loss of FANCL in HL60 cells led to a significantly reduced CD14-positive cell population (34 ± 2.93%) (Fig. 1C). Similarly, loss of FANCL in K562 cells substantially hampered their differentiation into megakaryocytes induced by PMA (phorbol myristate acetate) (Fig. 1D).
Cellular adhesion is an acquired functional characteristic of monocytes and megakaryocytes. As a result of FANCL deletion, FANCL−/− HL60 and K562 mutants exhibited markedly reduced yields of adherent monocytes and megakaryocytes, respectively, compared to that of the FANCL+/+ cells and the ADH5−/− mutant (Fig. 1E–H). These results indicate an aborted lineage progression in the absence of an intact FA pathway. Moreover, V-D3-induced differentiation resulted in a pronounced increase in apoptosis of the FANCL−/− mutant cells, compared to the parental HL60 cells or the ADH5−/− mutant (Fig. 1I, S2B–D). In the K562 FANCL−/− mutants, differentiation into either megakaryocytes or erythroids led to significant increases in apoptosis (Fig. 1J, K, S2E–F). In contrast, disruption of FANCL in HeLa cells had no effect on apoptosis, with or without V-D3 exposure (Fig. S2G–H). Moreover, K562 knockout mutants of the XPA and MSH2 genes are able to differentiate normally as the wild-type cells (Fig. S2I–M), suggesting that attenuated differentiation is specifically attributed to the loss of the FA pathway, rather than a general mutational stress from unrepaired nucleotide excision repair lesions or mismatches.
To extend the results generated from immortalized progenitor models, we tested primary human hematopoietic stem cells (CD34+ HSCs) isolated from healthy bone marrow donors. HSCs were subjected to gRNA-mediated FANCL depletion to disable the Fanconi pathway. A lentiviral vector expressing both CAS9 and GFP was co-transfected with gRNA expression vectors producing either FANCL gRNA or LacZ gRNA as a negative control. Ablation of the of Fanconi mechanism was confirmed by the abolished FANCD2 monoubiquitination (Fig. S2N). The resulting HSCs were induced with SCF/IL-3/EPO for five days to drive their differentiation toward the erythroid lineage. Flow cytometry was performed to determine the proportion of cells positive for both GFP and CD71, a marker for erythroid lineage. As shown in Fig. 1L–M, 18.28% HSCs treated with a control gRNA (anti-LacZ) exhibited GFP-CD71 positivity, whereas loss of FANCL substantially decreased the GFP-CD71-positive cells to 8.12%, indicating that the FA pathway is critical for the differentiation of primary HSCs.
Taken together, these results indicate that, in the absence of the FA pathway, the onset of cellular differentiation is detrimental to the survival and lineage progression of hematopoietic progenitor and stem cells.
The FA mechanism is essential for the protection of transcription re-programming during hematopoietic differentiation.
Tissue morphogenesis and cellular differentiation requires transcription reprogramming of a defined set of genes to attain the tightly orchestrated lineage transition. In particular, hematopoietic differentiation involves rapid and extensive reprogramming to accomplish the drastic changes in both cellular functionality and morphology (Ramirez et al., 2017). Such drastic transcription adjustments are likely associated with acute elevation of genotoxic stress including accumulation of the R-loop structure (Schwab et al., 2015) and increased nuclear production of formaldehyde as a result of histone demethylation (Kooistra and Helin, 2012).
To determine whether FANCL-deficient blood cells exhibit disrupted differentiation reprogramming, we analyzed transcription response upon differentiation induction. FANCL+/+, ADH5−/−, and FANCL−/− HL60 cells were subjected to V-D3 induction and their transcription activities were determined by transient tritium uridine labeling. As shown in Fig. 2A, FANCL+/+ and ADH5−/− HL60 cells exhibited profound increase (7.6 ± 0.6-fold and 6.9 ± 0.7-fold, respectively) in their transcription activities upon V-D3 induction. Loss of FANCL, in contrast, severely hindered transcription upregulation and presumably interrupted lineage progression at various stages. Consistent with this result, inactivation of FANCL in K562 cells substantially reduced the transcription activities when driven to differentiate into megakaryocytes (Fig. 2B), compared to wild-type K562 cells. These results suggest that a defective FA pathway is likely sufficient and causative for the disruption of lineage progression of blood progenitors.
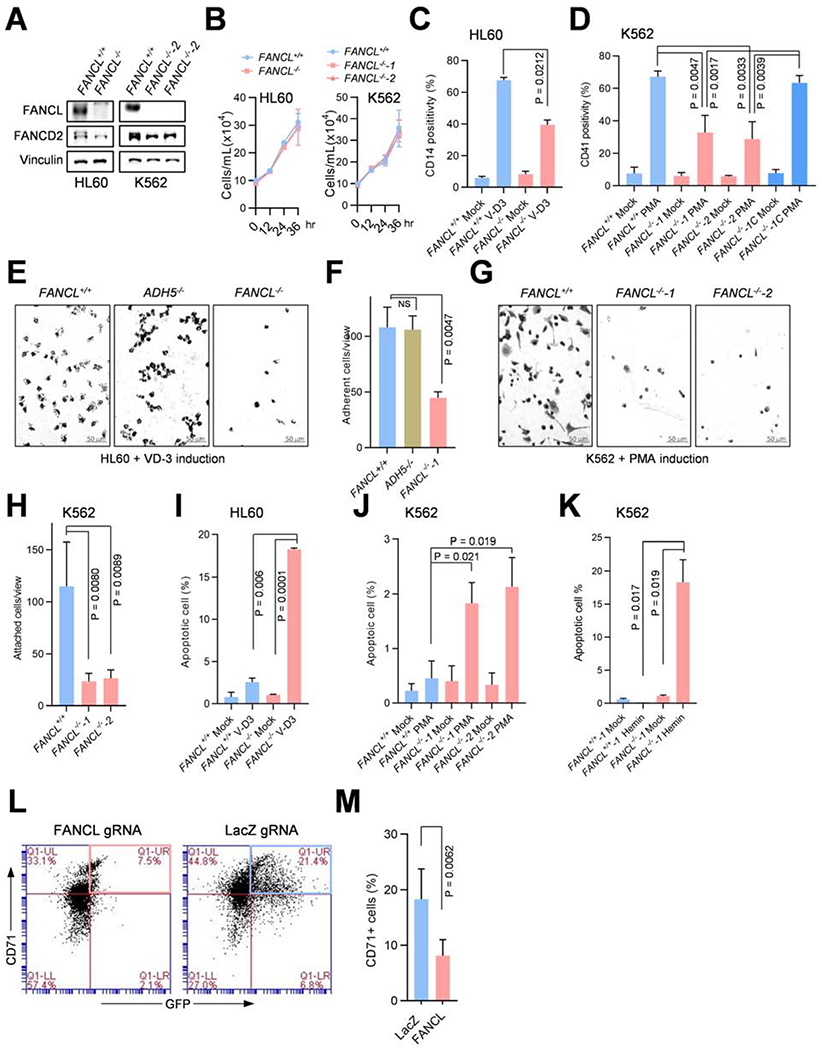
Fig. 2. FA pathway function is required to protect transcription reprograming of progenitor cell differentiation.
(A) Transcription activities in FANCL+/+, ADH5−/−, and FANCL−/− HL60 cells at the indicated times after V-D3 treatment or mock-treatment, as measured by transient [3H]-uridine incorporation into cellular RNA.
(B) Transcription activities in FANCL+/+, FANCL−/−-1, and the complemented FANCL−/− mutant (FANCL−/−-1C) K562 cells at the indicated times after PMA treatment or mock treatment, measured as in (A).
(C) Transcription activities of MSCs differentiating to adipocytes at the indicated times after induction, measured as in (A). The inset figure shows representative adipocytes differentiated from MSCs, visualized by oil red-O staining of fatty acids. The adipogenic differentiation was induced by the combination of dexamethasone/indomethacin/IMBX.
(D) Transcription activities of MSC cells differentiating to neuron-like cells at the indicated times, measured as in (A). The inset figure shows representative neuron-like cells differentiated from MSCs, visualized by immunofluorescent staining of the mature neuron marker NeuN. The neuronal differentiation was induced by sequential treatment with bFGF and β-mercaptoethanol.
Error bars representing standard deviations were derived from three or more biological repeats.
To assess the extent of transcription activity changes among distinct types of cellular differentiation, human bone marrow-derived mesenchymal stem cells (MSCs) were induced to differentiate into adipocytes, and transcription activities during the course of differentiation were measured (Fig. 2C). We found no significant changes in transcription levels despite the complete transition of cell type, suggesting that differentiation per se is not necessarily accompanied by a strong burst of transcription regulation. Given that the MSC to adipocyte differentiation spans a course of 18 days, transcription reprogramming is likely distributed along the duration of differentiation without an acute increase of transcription. Consistently, SaSO2 cells differentiating into osteocytes also showed no detectable variations in transcription activities (Fig. S3A, B). To determine whether more rapid differentiation leads to a dramatic change in transcription, MSCs were driven to neuron-like cells, which occurs within 24 hours. Analysis of transcription activities found a moderate increase of 0.82 ± 0.3-fold between 0 to 4 hours after induction (Fig. 2D), compared to the 7.6-fold increase in differentiating wild-type HL60 cells. These observations suggest that hematopoietic progenitors undergo an extensive transcription upregulation during their differentiation than other cell types and that the regulated transcription is disrupted by the impaired FA pathway.
Differentiation causes strong endogenous DNA damage in FA mutant cells
The disadvantageous outcomes of the differentiating FANCL−/− mutant progenitors suggest an elevated presence of endogenous genotoxic stress during the course of their differentiation. To determine whether genotoxic stress occurs during differentiation and whether it elicits a response from the Fanconi anemia pathway, we examined differentiating blood progenitor cells for their FANCD2 focus formation, which reflects the activation of the FA mechanism. In HL60 cells induced to differentiate by V-D3 for 8 hrs (Fig. 3A, B), the amount of FANCD2 foci exhibited a significant increase compared to the mock-induced control. The amplitude of FANCD2 foci in differentiating HL60 cells is comparable to that of an 8-hr exposure to 500 μM exogenous formaldehyde. In differentiating K562 cells induced by PMA, a similar increase in FANCD2 foci was observed compared to uninduced cells (Fig. 3C, D). Thus, differentiation of hematopoietic progenitors appears to generate endogenous DNA damage or replication stress, triggering the response from the FA pathway.
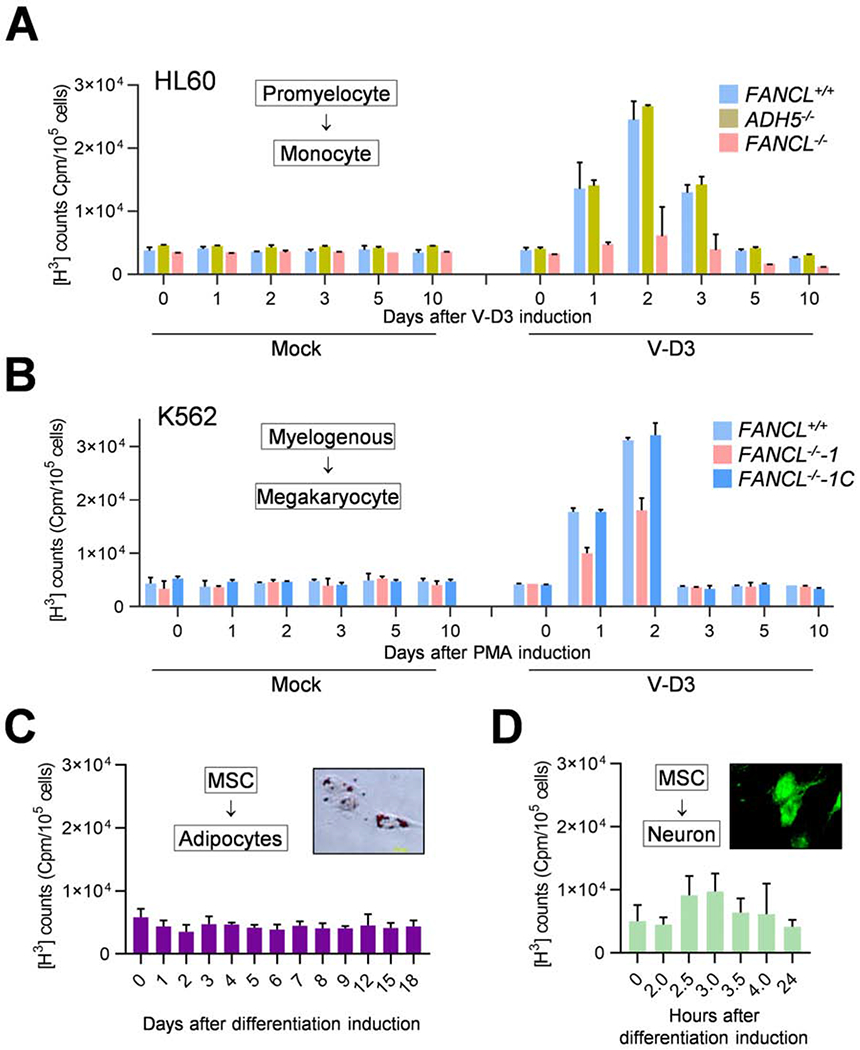
Fig. 3. Loss of FANCL elevates DNA damage in differentiating progenitor cells.
(A) FANCD2 foci in HL60 cells treated with V-D3 to initiate monocyte differentiation or formaldehyde (CH2O, 500 μM) for 8 hrs. Mock treated cells were used as a negative control. Bar = 10 μm.
(B) Quantified results as in (A) from three independent experiments.
(C) FANCD2 focus in K562 cells treated with PMA to initiate megakaryocyte differentiation or formaldehyde (CH2O, 500 μM) for 8 hrs. Mock treated cells were used as a negative control.
(D) Quantified results as in (C) from three independent experiments.
(E) γH2AX foci in HL60 FANCL+/+ and FANCL−/− cells treated with V-D3 for 3 days.
(F) γH2AX foci in K562 FANCL+/+ and FANCL−/− cells treated with PMA for 3 days.
(G) γH2AX foci in HL60 ADH5+/+ and ADH5−/− cells treated with V-D3 for 3 days.
(H) 53BP1 focus formation in HL60 FANCL+/+ and FANCL−/− cells treated with V-D3 to initiate monocyte differentiation for 3 days.
(I) CSB foci in HL60 FANCL+/+ and FANCL−/− cells treated with V-D3 to initiate monocyte differentiation for 7 days.
(J) Quantification of γH2AX nuclear fluorescence intensity in experiment shown in (E).
K) Quantification of γH2AX nuclear fluorescence intensity in experiment shown in (F).
(L) Quantification of γH2AX nuclear fluorescence intensity in experiment shown in (G). P values were generated from unpaired t-tests with three or more biological repeats. Error bars represent standard deviations.
(M) Immunoblotting of γH2AX in mutant cells with indicated genotypes. Top: HL60 FANCL+/+ and FANCL−/− cells upon 3-day treatment with V-D3. Bottom: K562 FANCL+/+ and FANCL−/− cells upon 3-day treatment with PMA.
(N) γH2AX ChIP-Seq analysis of K562 FANCL+/+ and FANCL−/− cells upon 3-day treatment with PMA. The SUM of peak sequences mapped to coding regions are shown for each sample.
(O) γH2AX ChIP-Seq analysis of K562 FANCL+/+ and FANCL−/− cells upon 3-day treatment with PMA. The SUM of peak sequences mapped to promoter regions (±2 kb of transcription start sites) are shown for each sample. Error bars were generated from duplicated samples and P values were generated by one-way ANOVA analyses.
Consistent with differentiation-mediated FANCD2 activation, analysis of the DNA damage surrogate marker γH2AX revealed a drastic increase in γH2AX foci formation and γH2AX level in FANCL−/− HL60 cells induced by V-D3 (Fig. 3E, J, M, S4A, C). Differentiation of FANCL−/− K562 cells into megakaryocytes also resulted in markedly increased γH2AX (Fig. 3F, K, M, S5A, C). Loss of ADH5 produced no detectable effect on γH2AX levels during differentiation (Fig. 3G, L, S6C). These results suggest that inactivation of FANCL strongly elevated the amount of DNA damage during blood progenitor differentiation.
Congruent with these findings, 53BP1 staining showed a substantial increase of foci formation in FANCL−/−, compared to FANCL+/+ cells treated with V-D3, reflecting the presence of DNA double strand breaks arising from differentiation of the FANCL−/− mutant cells (Fig. 3H). Similarly, K562 FANCL−/− mutant driven to megakaryocytes displayed significant increase in 53BP1 foci compared to wild-type cells (Fig. S5B, D).
In contrast to the discrete and uniformly dispersed γH2AX foci induced by ionizing radiation or DNA-damaging agents, differentiating FANCL−/− mutant cells exhibited a patched/clustered pattern of γH2AX staining (Fig. 3A, B), raising the possibility that the DNA damage may occur at localized nuclear compartments such as active clusters of transcription or replication. To this end, we examined focus formation of CSB, a key factor in transcription-coupled DNA repair, which is recruited specifically to the site of DNA lesions stalling transcription elongation (Fousteri and Mullenders, 2008; Sarker et al.). As shown in Fig. 3I, we found that CSB staining in the FANCL−/− cells exhibited a remarkable increase upon V-D3 induction, suggesting that differentiation-induced DNA damage most likely resides at areas of active transcription.
To further establish that differentiation-induced DNA damage is linked to transcription re-programming, we performed ChIP-Seq analyses to identify the genomic regions enriched by γH2AX pulldown. FANCL+/+ and FANCL−/− K562 cells were subjected to γH2AX ChIP with and without PMA induction. We used HOMER software to identify γH2AX binding regions which were significantly increased in IP pulldown samples over the control input sample. Shown in Fig. 3N, S6D, PMA-treatment led to a significantly increased reads of coding sequences in FANCL−/− than in the FANCL+/+ cells, suggesting that loss of FANCL is responsible for the increased DNA damage at transcribed regions during differentiation. When comparing HOMER-identified regions with increased γH2AX signals, we observed a marked increase of chromosomal regions mapped to promotors (defined as ± 2kb of TSS) ( Fig. 3O) in differentiating FANCL−/− cells compared to FANCL+/+ cells identically induced. Also, differentiating FANCL−/− mutant cells exhibited a PMA-dependent enrichment of promotor sequences when compared to the untreated FANCL−/− cells. This finding is further exemplified by comparing normalized γH2AX reads in the 4kb TSS region for top 632 genes in PMA-treated K562 cells exhibiting at least 2-fold more sum reads than the untreated cells. As shown in the heat map (Fig. S6E), PMA-induced differentiating FANCL+/+ K562 cells displayed a visibly increased DNA damage in the TSS regions compared to mock-induced controls. In the FANCL−/− mutants, induction of differentiation led to more extensive damage in the TSS regions compared to the mock-induced mutants.
These results suggest that, in the absence of FA pathway function, hematopoietic differentiation inflicts DNA damage preferentially in the coding and transcription regulatory regions of the genome, most likely causing severe disruption of transcription reprogramming during differentiation.
Given that differentiation-mediated DNA damage appears to localize at transcribed regions of the genome, we asked whether the drastic elevation of transcription during reprogramming prompted R-loop accumulation and gave rise to secondary damage that disrupts differentiation and causes cell death. We performed immunohistochemistry analyses with the S9.6 antibody in wild-type and FANCL−/− mutants (HL60 and K562) with and without differentiation induction and quantified the formation of nuclear R-loops. In wild-type HL60 cells, V-D3 treatment caused a modest increase of R-loop formation, likely reflecting the augmented transcription activities. The FANCL−/− mutant exhibited a higher level of V-D3-induced R-loop formation, albeit slightly latent to the onset of γH2AX foci formation (Fig. S4B, D), suggesting that loss of the FA pathway results in both DNA damage and the R-loop accumulation in differentiating HL60 cells. In the K562 FANCL−/− mutant cells, the onset of γH2AX foci and R-Loop signal appears to be nearly concurrent (Fig. S5A, C; Fig. S6 A, B), suggesting that R-Loop accumulation might occur independent of DNA damage during K562 differentiation. Together, these results indicate that differentiating FA mutant cells encounter a significantly elevated level of genotoxic stress.
Differentiation of blood cells leads to accumulation of nuclear formaldehyde
A broad array of posttranslational modifications, such as histone ubiquitination, acetylation, and methylation, are reversed in order to switch on and switch off the genes specific to the committed lineage during hematopoietic differentiation. Among them, oxidative protein demethylation, via either LSD1 or the JMJ family of protein demethylases, yields the highly reactive formaldehyde as an obligatory final product (Shi et al., 2004; Tsukada et al., 2005). Therefore, a differentiation-dependent increase in nuclear formaldehyde would be expected if rapid and intense transcription reprogramming takes place during the blood cell differentiation. Indeed, using an enzyme-coupled fluorometric assay, we detected significantly increased cellular levels of formaldehyde in VD-3–induced wild-type and FANCL−/− HL60 cells (Fig. 4A). A significant increase of cellular formaldehyde also occurred in differentiating wild-type and FANCL−/− K562 cells (Fig. 4B). These results suggest that the observed rise in cellular formaldehyde levels is a function of blood cell differentiation rather than defects in the Fanconi anemia pathway.
Fig. 4. Differentiation-dependent formaldehyde release in hematopoietic progenitor cells.
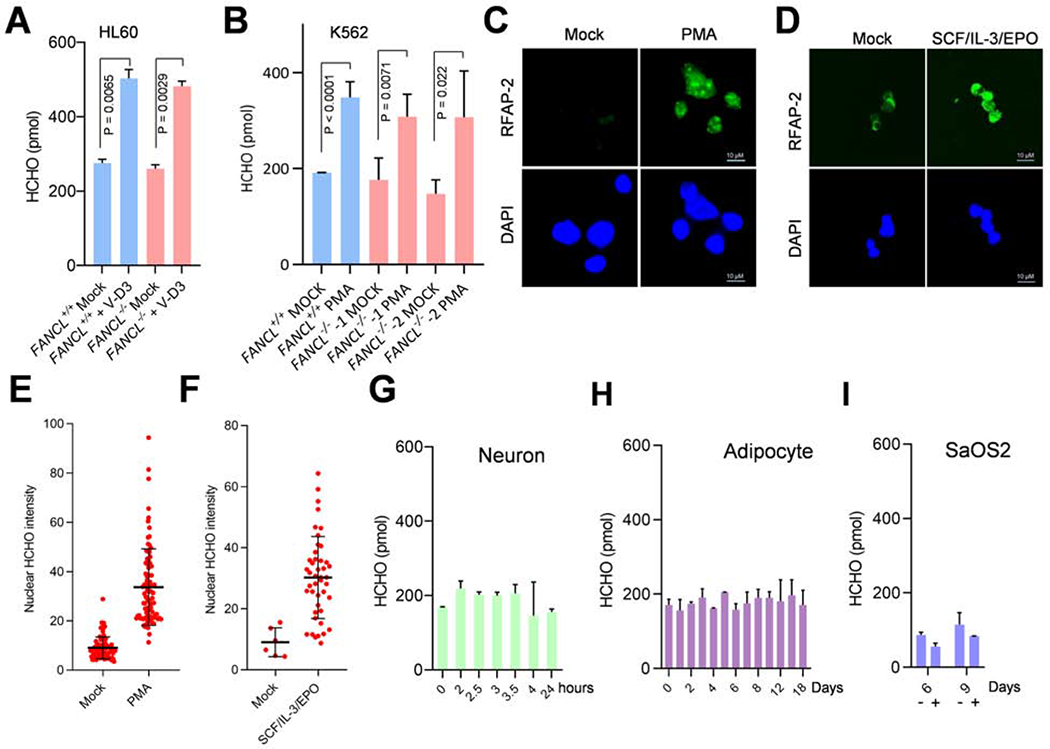
(A) Intracellular formaldehyde concentration in HL60 FANCL+/+ and FANCL−/− cells 3 days after V-D3 treatment. The formaldehyde concentrations are obtained by using a fluorometric assay on cell lysate.
(B) Intracellular formaldehyde concentration in K562 FANCL+/+ and FANCL−/− cells induced to differentiate into megakaryocytes by PMA treatment. Cells were harvested after 3 days and measured as in (A).
(C) Detection of nuclear formaldehyde in PMA-treated (48 hrs) K562 cells by staining with the formaldehyde fluorescent probe RFAP-2. Confocal images were acquired from permeabilized nuclei preparations.
(D) Detection of nuclear formaldehyde in SCF/IL-3/EPO-treated (72 hrs) CD34+ primary HSCs cells. Confocal images were acquired as in (C).
(E) Quantification of nuclear fluorescence signal as shown in (C).
(F) Quantification of nuclear fluorescence signal as shown in (D).
(G) Intracellular formaldehyde concentration of MSC cells driven to neuron-like cells. Samples were collected at indicated times (days) upon differentiation induction by the bFGF/β-mercaptoethanol combination and measured as in (A).
(H) Intracellular formaldehyde concentration of MSC cells driven to adipocytes. Samples were collected at indicated times (days) upon differentiation induction by the dexamethasone/indomethacin/IMBX combination and measured as in (A).
(I) Intracellular formaldehyde concentration of SaOS2 osteoblasts driven to osteocytes. Samples were collected at indicated times (days) upon differentiation induction by the ascorbic acid/β-glycerophosphate combination (+) or control (−) and measured as in (A).
P values were generated from unpaired t-tests with three or more biological repeats. Error bars represent standard deviations.
To further test this premise, HeLa wild-type and FANCL−/− mutant were treated with V-D3 and no altered formaldehyde levels were detected (Fig. S7A). When a FANCD2−/− mutant derived from the terminally differentiated TK6 human lymphoblastoid was treated with V-D3, the same level of intracellular formaldehyde was maintained (Fig. S7B). Thus, generation of formaldehyde appears to depend on blood cell differentiation potency and that deficiency in FA pathway per se is not a cause of the rising formaldehyde levels.
To visualize accumulation of formaldehyde in differentiating blood cells, we used a formaldehyde-responsive fluorescent probe RFAP-2 (Brewer et al., 2017). As shown (Fig. 4C, E), PMA-induced K562 cells displayed a strong nuclear formaldehyde signal as a result of differentiation. To confirm this result, we tested primary human hematopoietic stem cells (HSCs, CD34+). HSCs were driven toward erythroids by SCF/IL-3/EPO (Fig. S7C) and we observed a strong increase of nuclear formaldehyde marked by the RFAP-2 probe (Fig. 4D, F), further confirming a differentiation-induced accumulation of nuclear formaldehyde in blood progenitor/stem cells.
To examine the connection between formaldehyde generation and the extent of transcription reprogramming, we analyzed formaldehyde production during the MSC differentiation to neuron-like cells and found no significant increases during the course of transcription upregulation (Fig. 4G), presumably because the extent of transcription reprogramming is not sufficiently large. Similarly, analysis of formaldehyde levels in MSC cells driven to adipocytes showed nearly unaltered levels during the course of differentiation (Fig. 4H). In the case of human SaOS2 osteoblasts transitioning to osteocytes (Fig. S3A), slight decreases of cellular formaldehyde levels were observed (Fig. 4I). Furthermore, FANCL−/− MSC cells can differentiate into neuron-like cells with the same efficiency as wild-type cells, indicating that Fanconi pathway deficiency does not broadly impair cellular differentiation (Fig. S7D, E). Together these data suggest that differentiation-mediated increase in nuclear formaldehyde likely arises from extensive and rapid transcription reprogramming of blood progenitor/stem cells during the lineage transition.
Increased accumulation of DNA-protein crosslinking damage in FANCL mutant cells
Release of formaldehyde within the proximity of transcribed regions is expected to inflict DNA crosslinking damage to genomic DNA. The affected deoxyguanine residuals in genomic DNA can be converted via the NaCNBrH3-mediated reductive reaction into N2-methyl-dG (NmdG), a chemical marker of formaldehyde-modified DNA that is detectable by mass spectrometry (Yu et al., 2015). We prepared genomic DNA from FANCL+/+ and FANCL−/− cells with or without a 5-day V-D3 induction and treated the DNA with NaCNBrH3 to convert formaldehyde-prevalent DNA lesions to NmdG. Using heavy isotope–labeled NmdG as a quantitative standard, we analyzed the formation of formaldehyde-inflicted DNA lesions (Fig. 5A, S8A–D). We found that differentiation of wild-type cells resulted in a moderate increase in NmdG, consistent with the accumulation of formaldehyde during normal progenitor/stem cells differentiation. In the FANCL−/− mutant, the NmdG level showed a modest increase compared to FANCL+/+ cells, likely reflecting a higher basal level of formaldehyde-induced DNA lesions in the absence of repair by the FA pathway. However, a significantly elevated NmdG level is detected in FANCL−/− cells treated with V-D3, demonstrating that a proficient FA pathway is important to protect differentiating cells from formaldehyde-inflicted DNA damage.
Fig. 5. DPC damage accumulation in FANCL mutant cells during hematopoietic differentiation.
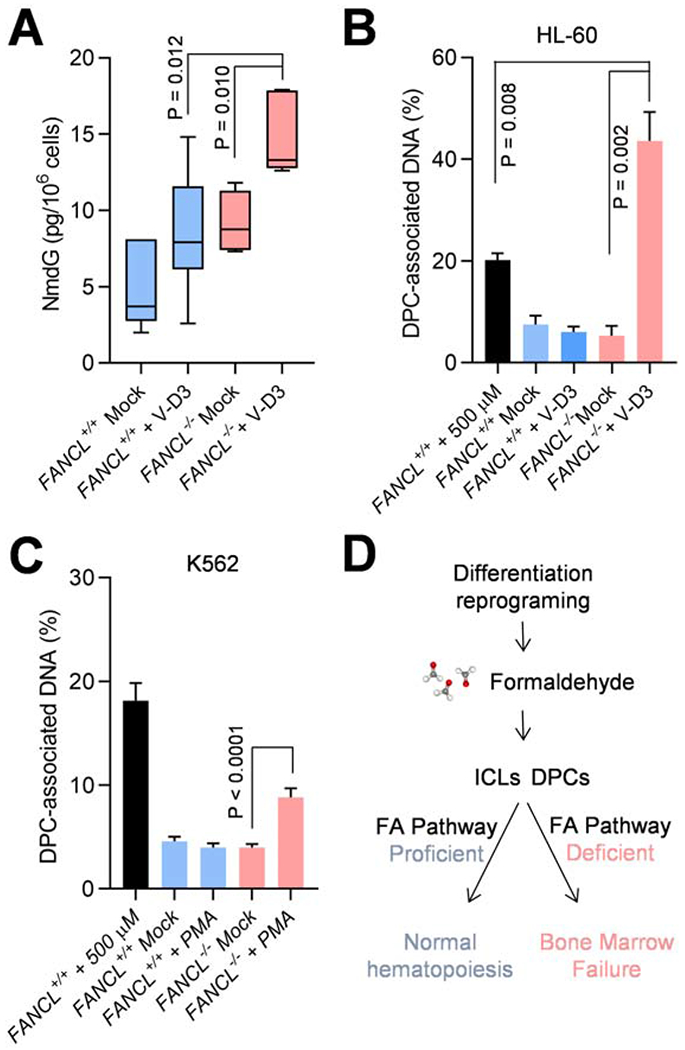
(A) Amount of NmdG in HL60 FANCL+/+ and FANCL−/− cells 5 days after V-D3 treatment, as measured by mass spectrometry using 13C-NmdG as standard. P values are derived from two-tailed Student’s t-test.
(B) Analyses of DPCs in HL60 FANCL+/+ and FANCL−/− cells 5 days after V-D3 induction. The first sample represents FANCL+/+ cells exposed to extracellular formaldehyde (500 μM, 4 hr). DPCs in the genomic DNA were measured by a modified KCl/SDS precipitation assay.
(C) Analyses of DPCs in K562 FANCL+/+ and FANCL−/− cells 3 days after PMA induction. The first sample represents FANCL+/+ cells exposed to extracellular formaldehyde (500 μM, 4 hr). DPCs in the genomic DNA were measured by a modified KCl/SDS precipitation assay.
(D) A model depicting the role of differentiation reprogramming in the pathogenesis of Fanconi anemia.
P values were generated from unpaired t--tests with three or more biological repeats. Error bars represent standard deviations.
To validate this premise further, we analyzed levels of DNA-protein crosslinks (DPCs) using a modified indirect assay (Hu et al., 2020). Wild-type and FANCL−/− cells were induced by V-D3 and protein-conjugated DNA was isolated by chaotropic denaturation and K-SDS precipitation. We found that differentiation of FANCL−/− cells resulted in a profound increase in DPCs compared to the untreated FANCL−/− cells (Fig. 5B). The amount of DPCs in the differentiating HL60 FANCL−/− cells also significantly exceeded that of the FANCL+/+ cells exposed to 500 μM of exogenous formaldehyde, a lethal dose to most cultured cells. These results illustrate extensive formation of endogenous DPCs in differentiating FANCL−/− HL60 cells. Supporting this notion, K562 FANCL−/− exhibited a significant increase of DPCs when driven to megakaryocytes by PMA induction (Fig. 5C). On the other hand, HeLa wild-type and FANCL−/− mutant cells treated with V-D3 did not show any increase in DPCs, suggesting that in non-differentiating cells, FA pathway defect does not give rise to measurable DPC accumulation (Fig. S8E). Furthermore, K562 knockout mutants deficient in the mismatch repair and nucleotide excision repair mechanisms did not show any increase in DPC formation as a result of PMA-mediated differentiation (Fig. S8F). Collectively, these results suggest that the FA pathway is critical in minimizing endogenous formaldehyde-induced DNA crosslinking damage and protecting hematopoietic differentiation.
Discussion
Reprogramming gene transcription is essential to any cellular differentiation and inevitably causes formaldehyde release at transcribed regions. However, the magnitude of formaldehyde release likely differs vastly depending on the scale and the dynamics of the orchestrated gene expression and epigenetic regulations. Results from this study demonstrate that the Fanconi anemia pathway plays a critical role in preventing the accumulation of endogenous DNA damage primarily formed during blood progenitor cell or blood stem cell differentiation.
Hematopoietic progenitors and stem cells continuously undergo rapid and nonreplicative differentiation to sustain the fast replenishing rate of terminally differentiated blood cells (Grinenko et al., 2018). The rapid and extensive transcription reprogramming during hematopoietic differentiation creates a temporal and spatial burst of nuclear formaldehyde, which potentially generates DNA crosslinks in genes participating in the reprogramming process and likely gene cascades involved in the ensuing stress responses. This is reflected by the increased DNA damage susceptibility of TSS regions during differentiation (Fig. 3N–O). Conversely, for cellular differentiation with less extensive transcription reprogramming and/or spanning a prolonged duration, formation of formaldehyde-induced DNA damage is expected to be significantly lower and remain at a sustainable level. Supporting this notion, we observed differentiation-mediated accumulation of nuclear formaldehyde only in blood progenitor/stem cells, but not in other types of cellular differentiation including adipocytes, neurons, and osteocytes (Fig. 4). This is further substantiated by the result that FANCL knockout MSC cells can differentiate normally into neuron-like cells as wild-type cells. These results are consistent with the fact many FA patients undergo normal development into adulthood despite their germline mutations.
Repair mechanism of DNA crosslink damage, include ICLs and DPCs, are likely to be complex. Give the excessive bulkiness of the protein moiety in a DPC lesion, proteolytic size reduction seems to be a necessary to reduce the steric hindrance. Biochemical studies in xenopus egg extracts using a defined DPC lesion suggests that the DNA-dependent proteinase SPRTN and proteasome are involved in processing DPCs and the DPC repair relies on replication but not on FANCI/D2 (Duxin et al., 2014; Duxin and Walter, 2015; Larsen et al., 2019). Our results showed significant accumulation of global DPCs in differentiating FANCL−/− cells (Fig. 5B, C), suggesting that the FA mechanism is involved in DPC removal during differentiation. As formaldehyde is cable of generating a large variety of DPCs, it is plausible that a subset of DPCs is repaired in a replication dependent manner, while the others reply the FA mechanism to introduce lesion-removing incisions, especially in differentiating progenitor/precursor cells which generally cease to replicate DNA.
Our study shows that loss of the FA pathway during differentiation leads to acute accumulation of endogenous DNA damage in two different types of myeloid progenitors and primary HSCs. Interestingly, reprogramming FA cells during iPSC generation was found to cause cell death (Raya et al., 2009), which could be mediated, at least partially, by an acute onset of endogenous DNA damage from extensive transcription reprogramming. The rapid increase of formaldehyde-induced DNA crosslinks creates a strong deleterious impact and causes aborted differentiation or DNA damage-induced apoptosis. The elevated accumulation of R-Loops in FA-deficient cells may also cause additional disruption of differentiation (Schwab et al., 2015). Deriving this mechanism generally in hematopoiesis, abrogated differentiation and cell death upon differentiation are expected to gradually deplete stem cell and progenitor pools from the hematopoiesis hierarchy (Fig. 5D). Such a mechanism may delineate, at least in part, the progressive and general attrition of blood/bone marrow cellularity in FA patients. It is also plausible that formaldehyde-mediated damage during differentiation constitutes a frequent source of DNA strand breaks to promote translocations in both lymphoid and myeloid cells, elevating predisposition to hematopoietic malignancies observed in FA patients (Nalepa and Clapp, 2018). Additionally, our findings shed light on the observation that aldehyde dehydrogenases are a frequent biomarker of stem cells and particularly hematopoietic progenitors (Armstrong et al., 2004; Dalerba et al., 2007; Hess et al., 2006; Moreb, 2008). The finding that differentiation reprogramming is the pathogenic trigger of Fanconi anemia may offer a new avenue of therapeutic exploration for the treatment of aplastic anemia and bone marrow failure in FA patients.
Limitations
The main conclusion of this work depends partly on the notion that transcription reprogramming during hematopoietic differentiation is more extensive and rapid compared to solid tissue differentiation, which would likely result in HSPC differentiation causing an acute accumulation of nuclear formaldehyde from histone demethylation.
It would have been ideal to include data comparing the extents of transcription reprogramming of HSPCs and other cell types. However, selection of cell types and differentiation routes/destinations can be arbitrary if only a limited number of cell types are profiled. Moreover, the transcription cascades, of HSPCs or other tissue types, are unlikely to spread out uniformly over the course of differentiation, rendering the sampling time difficult to justify. Therefore, a large number of HSPC and other types of cellular differentiations would have to be analyzed to test the notion that the extent of reprogramming in HSPCs explains our observations.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Li L. leili852002@yahoo.com
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The data for γH2AX ChIP-Seq analysis of K562 FANCL+/+ and FANCL−/− cells upon 3-day treatment with PMA reported in this paper was deposited in the Gene Expression Omnibus (GEO) database (accession number: GSE161891). Original data for images in the paper is available at Mendeley Data https://data.mendeley.com/sckwd9jgnc
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and Reagents
HL-60 cells (acquired from ATCC, CCL-240) were maintained in RPMI 1640 medium supplemented with 50 units/ml penicillin, 50 μg/ml streptomycin, and 20% heat-inactivated fetal bovine serum (FBS). TK-6 cells were grown in RPMI 1640 medium supplemented with 10% FBS and antibiotics. The hTERT immortalized adipose derived mesenchymal stem cells (ATCC, SCRC. 4000) were maintained in Complete Growth Medium (Mesenchymal Stem Cell Basal Medium + Mesenchymal Stem Cell Growth Kit and 0.2 mg/mL G418). HeLa cells and SaOS-2 cells were obtained from ATCC maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS and 50 units/ml penicillin-50 μg/ml streptomycin. Human cord blood CD34+ cells were purchased from STEMCELL Technologies (Cat 70008.3) and maintained in StemSpan™ SFEM II medium (Cat 09655). All cells were cultured at 37 °C in 5% CO2 incubators. 1a,25-Dihydroxyvitamin D 3 (V-D3) (Cat. D1530), Phorbol 12-myristate 13-acetate (PMA) (Cat. P1585), Hemin (Cat H9039-1G), Mitomycin C (Cat. M0503), Oil Red O solution (Cat. O-1391), β-glycerophosphate (Cat. G9422), and ascorbic acid (A4403) were purchased from Sigma Life Sciences. Anti-ADH5 antibody (ab91385), and anti-NeuN antibody (ab177487) were purchased from Abcam. Anti-FANCL antibody was a gift from Dr. Weidong Wang (National Institute on Ageing, NIH). FITC anti-human CD14 (Cat 325603), PE anti-human CD41 (Cat 303705) were purchased from Biolegend. StemSpan™ Erythroid Expansion Supplement (100X) (Cat 02692) and StemSpan™ SFEM II medium were purchased from STEMCELL Technologies.
METHODS DETAILS
Construction of CRISPR-CAS9 mutants
Construction of the knockout mutants followed the principal procedure described by the Church group (Mali et al., 2013). Ten to 15 independent PCR product clones were sequenced to genotype each knockout mutant candidate, along with antibody-based validations.
For HL-60 cell lines, electroporation was carried out by the Neon transfection system (Invitrogen) following the protocol from the manufacture. gRNA and CAS9 expression vectors were co-transfected with pEGFP and subsequently sorted into 96-well plates using a BD FACSJazz sorter.
The gRNAs used in this study are: FANCL: 5’ GGGAAGAGACTTCCACCTT 3’. ADH5: 5’ GTCACGGATTCTGGTCGGCG 3’; XPA: 5’ GGCGGCTTTAGAGCAACCCG 3’; MSH2: 5’ GGGTCTTGAACACCTCCCGGG 3’.
All knockout mutants generated for this study were authenticated by the Characterized Cell Line Core Facility at MD Anderson using no less than 13 STR markers to confirm isogenicity with the parental cell lines.
Apoptosis analysis
The apoptosis of cells was assessed by FITC Annexin V Apoptosis Detection Kit I (BD Biosciences). Briefly, HL60 cells were treated or mock-treated with 100nM vitamin D3 to induce differentiation for 5 days. K562 cells were either treated or mock-treated with 5nM of PMA to induce differentiation. Upon harvesting, the cells were washed with PBS and resuspended at 1 × 106 cells/ml in 1X binding buffer. 100 μl of cell suspension with 1x105 cells were incubated with 5 μL of Annexin V-FITC and 5 μL of 0.5 mg/mL propidium iodide for 15 min at RT (25°C) in the dark. An additional 400 μl of 1X Binding Buffer was added to each sample. Bivariate flow cytometric analysis was carried out on a BD Accuri™ C6 Flow Cytometer (BD Biosciences).
Cellular differentiation
HL60 monocytic differentiation: cells were first seeded at 1.25x105 cells/ml and then treated with 100nM 1a,2 5-Dihydroxyvitamin D3 (VD3) for 2 days. The monocytic marker CD14 was used to assess the differentiation by FACS analysis. After 7 days of VD3 treatment the adherent HL-60 cells were fixed by crystal violet staining and the numbers of attached cells were scored as field/per view from multiple random fields. To induce K562 megakaryocytic differentiation, cells were seeded at 2 x 105 cells/ml and then treated with 5nM of Phorbol 12-myristate 13-acetate (PMA). The megakaryocytic lineage marker CD41 was use to evaluate the cell differentiation. After 5 days of PMA induction the adherent megakaryocytes were similarly assessed as for HL-60 cells. To induce K562 erythroid differentiation, cells were treated with 50μM Hemin for 6 days. For MSC to neuron induction, 10 ng/mL basic fibroblastic growth factor (bFGF) were added to the medium for 24hrs. The cells then were washed with PBS and transferred to fresh complete growth medium with 1 mM β-mercaptoethanol (βME) for 24 hours. Neuronal differentiation was confirmed by Neuron N (NeuN) immunostaining and morphology changes 24 hours after induction. For MSC to adipocyte differentiation, cells were cultured in complete growth medium with 0.1 μM of dexamethasone, 60 μM of indomethacin, and 0.5 mM of IMBX. Medium was replaced every 3 days. Oil Red O (ORO) staining was applied to visualize the presence of lipids.
To induce osteocyte differentiation from SaOS-2 cells, 50 μg/ml ascorbic acid (A4403, Sigma) and 7.5 mM β-glycerophosphate (G9422, Sigma) were added into the culture. Alkaline phosphatase (ALP) activity was measured to determine the differentiation progression. Briefly, 1.25x104 cells were lysed in 50μL 0.5% Triton X-100 in PBS with 3 freeze/thaw cycles. Then, 100 μl of pNPP substrate solution (5mg pNPP tablet in 5mL diethanolamine buffer (1.02 M diethanolamine, 0.5mM MgCl2, pH 9.8)) was added to 50μL of the test sample and incubated at 37°C for 30min. The absorbance at 405 nm was measured by Synergy 4 microplate reader (Bioteck).
Primary human cord CD34+ cell to erythroid differentiation: StemSpan™ Erythroid Expansion Supplement (100X) (Cat# 02692), consisting of recombinant human stem cell factor (SCF), recombinant human interleukin 3 (IL-3), recombinant human erythropoietin (EPO), was added into the StemSpan™ SFEM II medium (Cat# 09655) to induce the production of erythroid cell. Medium was changed every 3 days. Erythroid differentiation was assessed by the CD71 marker via FACS analysis and visual inspection of cell color. LentiCRISPR-EmGFP-sgFANCL or LentiCRISPR-EmGFP-sgLacZ were used for knockdown of FANCL and control, respectively. Transfection of HSCs was carried out by the Neon™ Transfection System (Thermofisher), using buffer T following manufacturer protocol. 50% transfection efficiency was observed as assessed by GFP expression.
RNA synthesis assay with metabolic incorporation of tritium-labeled uridine
RNA synthesis was measured by incorporation of tritiated uridine. 105 cells were incubated with 50 μCi of H3-Uridine for 60 min. After labeling, cells were washed with PBS and then lysed with lysis buffer (0.1 ml of 10 mM TRIS buffer (pH 7.4) with 0.5 % SDS, 0.2 M NaCl, 10 mM EDTA, and 5 mM vanadyl ribonucleoside complex). Cell lysates were precipitated by 10% ice cold TCA and then followed by 5 % TCA wash, 95 % ethanol wash. The incorporation of tritium uridine was measured by TriCard 3100 Scintillation Counter (Perkin Elmer).
Formaldehyde fluorometric analysis
To quantitatively analyze formaldehyde concentration in cell extracts, 105 cells were analyzed by the formaldehyde assay kit (Abcam) for detection. In brief, cells were collected at various time points and washed with cold PBS. After re-suspension in 100 μl of cold formaldehyde assay buffer, insoluble material was removed by centrifugation. Deproteinization was achieved by the PCA-KOH method. Supernatant was incubated with Formaldehyde reaction mix (assay buffer, formaldehyde enzyme mix, formaldehyde developer and PicoProbe) at 25 °C for 20 minutes protected from light. The fluorescence at 535/587 nm was recorded by Synergy 4 microplate reader (Bioteck).
Formaldehyde in situ detection with the RFAP2 probe
For in vivo nuclear FA detection, two days after K562 cells were either mock treated or induced by PMA, the nuclei were isolated and permeabilized using procedures from the Cold Spring Harbor Protocol (doi:10.1101/pdb.top074583) and re-suspended in PBS. The formaldehyde florescent probe RFAP2 (exciting wavelength 488 nm, emission 550nm) and DAPI were then added immediately to the buffer at a final concentration of 5 μM and gently shaken for 30 min at room temperature. The images from multiple random fields were taken by Zeiss LSM 880 laser scan microscope and then analyzed using the ZEN (blue edition) software. Average intensities along laser tracks were measured and ratios of line intensities over selected region areas were calculated and compared.
Cell proliferation assay
HL-60 and K562 cells were seeded at 1 x 105/ml per well in triplicates in 96-well plates. The proliferation of cells was measured by calculating cell densities every 12 hr for a total of 36 hr. The results were plotted against time with an interval of 12 hr.
Immunohistochemistry
For CSB, gammaH2AX, 53BP1, and S9.6 staining, HL60 cells were fixed in 3 % paraformaldehyde for 10 min at room temperature, and then permeabilized for 10 min with 0.5 % Triton-X-100. For R-loop staining, cells were fixed in ice cold methanol for 5 min and washed once with phosphate buffered saline (PBS). Then the cells were stained with antibodies overnight at 4°C. After primary antibody incubation, the cells were washed three times with PBS and incubated with Alexa Fluor 488-labeled anti-rabbit IgG antibody and Alexa Fluor 561 labeled anti-mouse IgG antibody (Invitrogen) for 1 hr at room temperature. Finally, cells were mounted in mounting solution ProLong Gold with DAPI (Invitrogen).
Image acquisition of multiple random fields was performed with a Zeiss LSM 880 laser scan microscope. Microscopy image analyses were performed using the ZEN (blue edition) software. Nuclei were manually segmented using the DAPI channel and the resulting regions of interest transferred to the fluorescence channel of interest (488 for CSB, gammaH2AX and 53BP1, 561 for S9.6). For intensity quantifications of 488/561 laser lines, average intensities along laser tracks were measured and ratios of line intensities over selected region areas were calculated and compared. Prism 6 (GraphPad Software) was used to calculate P values based on T-test analyses. Data were considered statistically significant for P values < 0.05.
ChIP and ChIP-Seq analysis
ChIP analysis was performed as described previously (Li et al., 2014) with minor modifications. In brief, cells were cross-linked with 1% formaldehyde for 10 min. The crosslinking reaction was stopped by adding glycine to 125 mM and incubating for 5 min. Fixed cells were scraped from dishes and washed 2x with PBS. Cell pellets were resuspended in cell lysis buffer (5 mM PIEPES pH 8.0, 85 mM KCl, 1% NP-40) containing the complete protease inhibitor cocktail (Roche) and 1 mM PMSF. After 20 min rotation in 4 °C, cell nuclei were harvested by centrifugation and resuspended in nuclei lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS) containing protease inhibitors prior to sonication (30 sec on 30 sec off for 60 min/round, 3 rounds) with a Bioruptor Sonicator (Diagenode) to a length of approximately 100-400 bp. Sonicated chromatin samples containing approximately 60 μg of DNA were diluted 4-fold with dilution buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.01% SDS) and were immunoprecipitated overnight at 4 °C with 2 μg of γH2AX antibody. Protein A/G beads (Millipore) were added and samples were incubated for another 2 hr. The immunoprecipitates were washed 2x with low-salt buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2mM EDTA, 1% Triton X-100, 0.01% SDS), 2x with high-salt buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2mM EDTA, 1% Triton X-100, 0.01% SDS), and 1x with LiCl buffer (20 mM Tris-HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 1% NP40, 1% Na-Deoxycholate). DNA was eluted in elution buffer (50 mM NaHCO3, 1% SDS) by vigorously shaking for 15 min. Crosslinks were reversed in the presence of 0.3 M NaCl at 67 °C for 12 hr. DNA was purified using a PCR purification kit (Qiagen) and was sequenced using the Illumina Solexa HiSeq 2000.
Sequencing reads were mapped to human genome (version hg19) using the BWA software. Top γH2AX binding regions were identified using the HOMER software (peak finding option “histone”, fold change 2, p<0.0001) by comparing γH2AX pulldown samples with a control input sample without IP (wild type K562 untreated). Peaks are mapped to coding sequences or to promoters (within ±2kbp of TSS). To generate a heatmap for read counts near TSS, we focused on top 632 genes in any samples having at least 2-fold more sum reads than control in the 4kb genomic region encompassing TSS. ChIP-seq raw counts were slightly normalized for sequencing depth by scaling to allow equal background noise read levels. Sum normalized reads for 4kbp TSS region from γH2AX pulldown samples were logged after subtracting reads from the control input sample and used for generating a heatmap.
Mass spectrometry
Deoxy Glucose, sodium cyanoborohydride (NaCNBH3), DNase I, alkaline phosphatase, phosphodiesterase were purchased from Sigma. Formaldehyde (13C, D2) was purchased from Cambridge Isotope Labs. 2’-Deoxyguanosine was generated by Berry and Associates.
Preparation of internal standard 13CD2-N2-mehtyl-deoxyguanosine (*N2-mDG): The 13CD2 heavy labelled methyl-deoxyguanosine (*N2-mDG) was synthesized based on previously published method (Lu et al., 2012) with modifications. Briefly, 10 mM deoxy glucose solution was treated with 100 mM 13CD2 formalin in 100 mM phosphate buffer (pH 7.2) for overnight at 37 °C. The reaction product, 13CD2-hydroxymethyl-deoxyguanosine (*N2-OHMeDG) was eluted at 19.5 min from HPLC using 4.6 mm X 250 mm C18 analytical column. The gradient was continuous 4 to 25% methanol within 35 min at the flow rate of 500 microliter per min using 0.1% formic acid and 100% methanol with 0.1% formic acid as mobile phase. The fractions containing *N2-OHMeDG were pooled, vacuum dried and resuspended with 200 microliters of 50 mM NaCNBH3. The mixture was incubated at 37 °C for overnight, followed by further separated by HPLC using previous condition. The *N2-mDG was eluted at 20.9 min. The purity and concentration of purified *N2-mDG was determined by Fusion™ Tribrid™ Mass Spectrometer by monitoring m/z 285.2 peak with corresponding unlabeled N2-mDG (m/z 282.1).
DNA digestion: Fifty micrograms of extracted genomic DNA was reduced by incubation with 50 mM NaCNBH3 in 100mM sodium phosphate buffer (pH 7.2) for 6 hr at 37°C. Reduced DNA was digested with 200 units of Dnase I, 5 units of alkaline phosphatase and 0.05 units of phosphodiesterase for 1 hr at 37°C along with 3 pico gram of internal standard (*N2-mDG). Undigested DNA and enzymes were removed by a Pall Nano Sep 3 kDa filter at 10 kg spin for 50 min. The resulting filtrate was dried under vacuum and resuspended with 0.1% formic acid for further in housed C18 tip column purification (Jung et al., 2017). A micro-pipette tip C18 column was made from a 200 micro liter pipette tip by layering 2 mg of C18 matrix (Reprosil-Pur Basic C18, 3 mm, Dr. Maisch GmbH, Germany) on top of the C18 disk (3M, EmporeTM C18) plug. Resuspended samples were loaded on the C18 tip equilibrated with 0.1% FA. Bounded DNA was eluted with 200 micro liters of 50% MeOH with 0.1% FA. The eluent was vacuum dried, resuspended with 0.1% FA and subject to nanoHPLC-MS/MS analysis.
Mass spectrometry analysis: Orbitrap Fusion™ Tribrid™ (Fusion) mass spectrometer (Thermo Scientific) with NSI source been used to measure N2-mDG. The samples were loaded onto an in-house Reprosil-Pur Basic C18 (3 μm, Dr.Maisch GmbH, Germany) trap column which was 2 cm × 100 μm size. Then the trap column was washed with loading solution and switched in-line with an in-house 5 cm × 150 μm column packed with Reprosil-Pur Basic C18 (2 μm) equilibrated in 0.1% formic acid/water. The DNA adducts were separated with a 25 min linear gradient of 0 to 25 % acetonitrile/0.1% formic acid at a flow rate of 800 nl/min. Fusion mass spectrometer was operated in targeted mass analyzing mode. Full MS spectrum was scanned at 150- 400 m/z in 120,000 resolutions, 5×105 AGC target (50 ms maximum injection time) by Orbitrap. Two targeted mass, m/z 282.1 and m/z 285.1 was continuously isolated using quadrupole and fragmented by HCD with 30 normalized collision energies, and detected by Orbitrap with 30,000 resolutions, 50-300 m/z scan range, 5x103 AGC target, and 100 ms of maximum injection time. The area-under-curve (AUC) of transitions of mz 282.1 → 166.0723 from endogenous N4-mDG, and m/z 285.1 → 169.0883 from internal standard *N4-mDG was manually calculated using Thermo Xcalibur Qual Browser (Ver. 2.2) to calculate absolute amount.
Isolation of DPC-associated DNA
The ARK protocol (Hu et al., 2020) for measuring DPC coefficient was modified from the indirect DPC assay (Costa et al., 1996). HL60 or K562 cells (2.5 × 106) were lysed in 950 μl of M buffer (5.6 M GTC, 10 mM Tris–HCl (pH 6.8), 20 mM EDTA, 4% Triton X-100, 1% Sarkosyl and 1% dithiothreitol) and mildly sheared with a 1-mL pipet tip (passing 6 times). DNA/DPC was recovered by precipitation with an equal volume of pre-chilled ethanol (−20°C) and as a pellet after a full speed centrifugation (micro-centrifuge) at 4°C for 20 min. The pellet was washed at 4°C one time in the buffer composed of 20 mM Tris–HCl pH 6.5, 150 mM NaCl and 50% ethanol. To dissolve the DNA/DPC pellet 0.5 ml of pre-warmed 1% SDS, 20 mM Tris-HCl (pH 7.5) was added and incubated at 42°C for 6 min. The samples were briefly spun (3500 x g, 30 seconds, on micro-centrifuge) and shorn by passing through a 25-gauge needle. 0.5 ml of 200 mM KCl, 20 mM Tris-HCl (pH 7.5) buffer was added to precipitate SDS-bound DPCs. The samples were incubated on ice for 6 min to complete the DPC precipitation. The precipitate was pelleted after a full speed microcentrifugation for 5 min at room temperature. The supernatant was collected and set aside on ice for DNA measurement. The DPC pellet was washed in 1.5 ml of 100 mM KCl, 20 mM Tris-HCl (pH 7.5) by incubating at 55°C for 10 min, on ice for 6 min, followed by centrifugation at 20,000 × g speed in a micro-centrifuge at 4°C for 5 min. The supernatant was collected and combined with the previously collected supernatant for total DNA measurement. The wash procedure was repeated one more time before dissolving the DPC pellet in 1 ml of the proteinase K buffer (100 mM KCl, 20 mM Tris-HCl, pH 7.5, and 10 mM EDTA). DNA-crosslinked proteins were digested by adding proteinase K to a final concentration of 0.2 mg/ml and incubating at 55°C for 45 min. The digestion was chilled on ice for 6 min and centrifuged at 20,000 × g for 10 min at 4 °C to remove any debris. The 1 ml supernatant in this tube contains the DPC-associated DNA.
To determine the DPC coefficient, 10 μl from the 4 ml of the recovered free DNA supernatant, and 62.5 μl from the 1 ml supernatant from the DPC resuspension were used for DNA quantification by the PicoGreen assay kit. The DPC coefficient is expressed as the percentage of DNA from DPCs over the total DNA in each sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
P values were generated from unpaired t-tests or one-way ANOVA as indicated in the figure legends. Error bars represent standard deviations from three or more biological repeats with duplicates or triplicates.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-ADH5 Antibody | Abcam | Cat#ab91385, RRID:AB_2049142 |
| Recombinant Anti-NeuN antibody [EPR12763] - Neuronal Marker | Abcam | Cat# ab177487, RRID:AB_2532109 |
| FANCL | Dr. Weidong Wang | N/A |
| FANCL Polyclonal antibody | Invitrogen | Cat#PA5-57768, RRID:AB_2641499 |
| FITC anti-human CD14 | Biolegend | Cat#325603, RRID:AB_830676 |
| PE anti-human CD41 | Biolegend | Cat#303705, RRID:AB_314375 |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 for IHC | EMD Millipore | Cat#05-636, RRID:AB_309864 |
| Anti-CSB antibody | Abcam | Cat#ab96089, RRID:AB_10679889 |
| Anti-53BP1 antibody | Abcam | Cat#ab21083, RRID:AB_722496 |
| Alexa Fluor 488 Anti-Rabbit | Invitrogen | Cat#A21206, RRID:AB_141708 |
| Alexa Fluor 568 Anti-Mouse | Invitrogen | Cat#A10037, RRID:AB_2534013 |
| DNA-RNA Hybrid, clone S9.6 | EMD Millipore | Cat#MABE1095 |
| gamma H2A.X (phospho S139) [EP854(2)Y] - ChIP Grade | Abcam | Cat#ab81299 , RRID:AB_1640564 |
| RFAP2 | Dr. Christopher Chang | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1α,25-Dihydroxyvitamin D3 | Sigma-Aldrich | Cat#D1530 |
| Mitomycin C | Sigma-Aldrich | Cat#M0503 |
| Oil Red O solution | Sigma-Aldrich | Cat#O1391 |
| β-Glycerophosphate | Sigma-Aldrich | Cat#G9422 |
| L-Ascorbic Acid | Sigma-Aldrich | Cat#A4403 |
| Dexamethasone | Sigma-Aldrich | Cat#D4902 |
| Indomethacin crystalline | Sigma-Aldrich | Cat#I7378 |
| 3-isobutyl-1-methylxanthine | Sigma-Aldrich | Cat#I7018 |
| Fibroblast Growth Factor- Basic Human | Sigma-Aldrich | Cat#F0291 |
| pNPP | Thermo Fisher | Cat#34047 |
| Uridine [5, 6-3H]- | Perkin Elmer | Cat#NET367 |
| Deoxy Glucose | Sigma | Cat#D8375 |
| Sodium cyanoborohydride | Sigma-Aldrich | Cat#156159 |
| Phosphodiesterase I | Sigma-Aldrich | Cat#P3243 |
| Formaldehyde (13C, D2) | Cambridge Isotope Labs | Cat#CDLM-4599-1 |
| Deoxyguanosine | Berry and Associates | N/A |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat#P1585 |
| Hemin | Sigma-Aldrich | Cat#H9039 |
| Deoxyribonuclease I | Sigma-Aldrich | Cat#D4263 |
| StemSpan SFEM II medium | STEMCELL Technologies | Cat#09655 |
| StemSpan Erythroid Expansion Supplement | STEMCELL Technologies | Cat#02692 |
| Critical Commercial Assays | ||
| FITC Annexin V Apoptosis Detection Kit I | BD Biosciences | Cat#556547 |
| Formaldehyde Assay Kit (Fluorometric) | Abcam | Cat#ab196997 |
| Experimental Models: Cell Lines | ||
| HL-60 | ATCC | ATCC CCL-240; RRID:CVCL_0002 |
| TK6 | Dr. Shunichi Takada | N/A |
| ASC52telo, hTERT immortalized adipose derived Mesenchymal stem cells | ATCC | ATCC SCRC-4000; RRID:CVCL_U602 |
| HeLa | ATCC | ATCC CCL-2; RRID:CVCL_0030 |
| K562 | Dr. Michael Andreeff | N/A |
| Saos-2 | ATCC | ATCC HTB-85; RRID:CVCL_0548 |
| Human cord blood CD34+ | STEMCELL Technologies | Cat#70008.3 |
| Deposited Data | ||
| γH2AX ChIP-Seq analysis of K562 FANCL+/+ and FANCL−/− cells upon 3-day treatment with PMA | This paper | GEO: GSE161891 |
| Unprocessed images | This paper | https://data.mendeley.com/drafts/sckwd9jgnc |
| Oligonucleotides | ||
| FANCL gRNA | This paper | GGAAGAGACTTCCACCTTA |
| ADH5 gRNA | This paper | GTCACGGATTCTGGTCGGCG |
| XPA gRNA | This paper | GGCGGCTTTAGAGCAACCCG |
| MSH2 gRNA | This paper | GGGTCTTGAACACCTCCCGGG |
| Software and Algorithms | ||
| Zen 2.6 (blue edition) | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| GraphPad Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| HOMER (Hypergeometric Optimization of Motif EnRichment) v4.11 | UCSD | http://homer.ucsd.edu/homer/ngs/ |
ACKNOWLEDGMENTS
We thank Weidong Wang (NIA, NIH) for providing FANCL antibodies, Lee Zou (MGH, Harvard Medical School) for providing the S9.6 antibody and technical advice, and Margaret (Peggy) Goodell (Baylor College of Medicine) for technical discussions on HSCs. Founding source include CA190635 and CA193124-Project 3 (to L. L.), ES 4705 and ES 28096 (to C.J.C.), CA204020 (to X.S.), and CA157448 (to J. C.). Cancer Prevention and Research Institute of Texas Grant RP150538 (to J. C. and L. L.). C.J.C. is an Investigator with the Howard Hughes Medical Institute and T.F.B. was partially supported by a Chemical Biology Training Grant from the NIH (T32 GM066698). This research was partly supported by the Fundamental Research Funds for the Central Universities. The M. D. Anderson Flow Cytometry, DNA Sequencing, and Characterized Cell Line core facilities are supported by the National Cancer Institute Cancer Center Support Grant CA016672.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF Interests: The authors declare no conflict of interests.
REFERENCES
- Armstrong L, Stojkovic M, Dimmick I, Ahmad S, Stojkovic P, Hole N, and Lako M (2004). Phenotypic characterization of murine primitive hematopoietic progenitor cells isolated on basis of aldehyde dehydrogenase activity. Stem Cells 22, 1142–1151. [DOI] [PubMed] [Google Scholar]
- Brewer TF, Burgos-Barragan G, Wit N, Patel KJ, and Chang CJ (2017). A 2-aza-Cope reactivity-based platform for ratiometric fluorescence imaging of formaldehyde in living cells. Chem Sci 8, 4073–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nature Reviews Molecular Cell Biology 17, 337. [DOI] [PubMed] [Google Scholar]
- Costa M, Zhitkovich A, Gargas M, Paustenbach D, Finley B, Kuykendall J, Billings R, Carlson TJ, Wetterhahn K, Xu J, et al. (1996). Interlaboratory validation of a new assay for DNA-protein crosslinks. Mutat Res 369, 13–21. [DOI] [PubMed] [Google Scholar]
- D’Andrea AD (2010). Susceptibility pathways in Fanconi’s anemia and breast cancer. The New England journal of medicine 362, 1909–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalerba P, Dylla SJ, Park I-K, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. (2007). Phenotypic characterization of human colorectal cancer stem cells. Proceedings of the National Academy of Sciences 104, 10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014). Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, and Walter JC (2015). What is the DNA repair defect underlying Fanconi anemia? Current opinion in cell biology 37, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fousteri M, and Mullenders LHF (2008). Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Research 18, 73. [DOI] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, Guilbaud G, Park N, Roerink S, Nik-Zainal S, et al. (2018). Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, and D’Andrea AD (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7, 249–262. [DOI] [PubMed] [Google Scholar]
- Grinenko T, Eugster A, Thielecke L, Ramasz B, Krüger A, Dietz S, Glauche I, Gerbaulet A, von Bonin M, Basak O, et al. (2018). Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nature Communications 9, 1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DA, Wirthlin L, Craft TP, Herrbrich PE, Hohm SA, Lahey R, Eades WC, Creer MH, and Nolta JA (2006). Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood 107, 2162–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Klages-Mundt N, Wang R, Lynn E, Kuma Saha L, Zhang H, Srivastava M, Shen X, Tian Y, Kim H, et al. (2020). The ARK Assay Is a Sensitive and Versatile Method for the Global Detection of DNA-Protein Crosslinks. Cell reports 30, 1235–1245 e1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, and Li L (2014). Modularized functions of the Fanconi anemia core complex. Cell reports 7, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SY, Choi JM, Rousseaux MWC, Malovannaya A, Kim JJ, Kutzera J, Wang Y, Huang Y, Zhu WM, Maity S, et al. (2017). An Anatomically Resolved Mouse Brain Proteome Reveals Parkinson Disease-relevant Pathways. Mol Cell Proteomics 16, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanja KK, Cox SW, Duxin JP, Stewart SA, and Campbell JL (2012). DNA2 and EXO1 in replication-coupled, homology-directed repair and in the interplay between HDR and the FA/BRCA network. Cell Cycle 11, 3983–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanja KK, Lee EH, Hendrickson EA, and Campbell JL (2014). Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 13, 1540–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra SM, and Helin K (2012). Molecular mechanisms and potential functions of histone demethylases. Nature Reviews Molecular Cell Biology 13, 297. [DOI] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, and Patel KJ (2011). Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Raschle M, Walter JC, and Duxin JP (2019). Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol Cell 73, 574–588 e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Wen H, Xi YX, Tanaka K, Wang HB, Peng DN, Ren YF, Jin QH, Dent SYR, Li W, et al. (2014). AF9 YeAtS Domain Links Histone Acetylation to DOT 1 L-Mediated H3k79 Methylation. Cell 159, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Craft S, Nakamura J, Moeller BC, and Swenberg JA (2012). Use of LC-MS/MS and Stable Isotopes to Differentiate Hydroxymethyl and Methyl DNA Adducts from Formaldehyde and Nitrosodimethylamine. Chem Res Toxicol 25, 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. (2003). A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35, 165–170. [DOI] [PubMed] [Google Scholar]
- Moreb JS (2008). Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther 3, 237–246. [DOI] [PubMed] [Google Scholar]
- Nalepa G, and Clapp DW (2018). Fanconi anaemia and cancer: an intricate relationship. Nature reviews. Cancer 18, 168–185. [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, and Helleday T (2010). Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, and Passmore LA (2014). The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell 54, 858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez RN, El-Ali NC, Mager MA, Wyman D, Conesa A, and Mortazavi A (2017). Dynamic Gene Regulatory Networks of Human Myeloid Differentiation. Cell Syst 4, 416–429 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, Ahel I, and West SC (2007). Defective DNA Repair and Neurodegenerative Disease. Cell 130, 991–1004. [DOI] [PubMed] [Google Scholar]
- Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, Barrero MJ, Consiglio A, Castella M, Rio P, Sleep E, et al. (2009). Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 460, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney S, Chaudhuri J, and Alt FW (2004). The role of the non-homologous end-joining pathway in lymphocyte development. Immunological Reviews 200, 115–131. [DOI] [PubMed] [Google Scholar]
- Rosado IV, Langevin F, Crossan GP, Takata M, and Patel KJ (2011). Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nature Structural &Amp; Molecular Biology 18, 1432. [DOI] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, and Cooper PK Recognition of RNA Polymerase II and Transcription Bubbles by XPG, CSB, and TFIIH: Insights for Transcription-Coupled Repair and Cockayne Syndrome. Molecular Cell 20, 187–198. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, and Niedzwiedz W (2015). The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell 60, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Do H, Li Y, Chung WH, Tomasz M, de Winter JP, Xia B, Elledge SJ, Wang W, and Li L (2009). Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell 35, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DnA repair. Cell 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, and Jentsch S (2015). DNA-protein crosslink repair. Nat Rev Mol Cell Biol 16, 455–460. [DOI] [PubMed] [Google Scholar]
- Tian Y, Shen X, Wang R, Klages-Mundt NL, Lynn EJ, Martin SK, Ye Y, Gao M, Chen J, Schlacher K, et al. (2017). Constitutive role of the Fanconi anemia D2 gene in the replication stress response. J Biol Chem 292, 20184–20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y.-i., Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, and Zhang Y (2005). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811. [DOI] [PubMed] [Google Scholar]
- Weinberg RA (2014). The biology of cancer, Second edition. edn (New York: Garland Science, Taylor & Francis Group; ). [Google Scholar]
- Yu R, Lai Y, Hartwell HJ, Moeller BC, Doyle-Eisele M, Kracko D, Bodnar WM, Starr TB, and Swenberg JA (2015). Formation, Accumulation, and Hydrolysis of Endogenous and Exogenous Formaldehyde-Induced dNa Damage. Toxicol Sci 146, 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A, and Costa M (1992). A simple, sensitive assay to detect DNA-protein crosslinks in intact cells and in vivo. Carcinogenesis 13, 1485–1489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data for γH2AX ChIP-Seq analysis of K562 FANCL+/+ and FANCL−/− cells upon 3-day treatment with PMA reported in this paper was deposited in the Gene Expression Omnibus (GEO) database (accession number: GSE161891). Original data for images in the paper is available at Mendeley Data https://data.mendeley.com/sckwd9jgnc