

Abstract
Tumor necrosis factor-α (TNFα) activates NADPH Oxidase 1 (Nox1) in vascular smooth muscle cells (VSMCs), producing superoxide (O2•-) required for subsequent signaling. LRRC8 family proteins A-E comprise volume-regulated anion channels (VRACs). The required subunit LRRC8A physically associates with Nox1, and VRAC activity is required for Nox activity and the inflammatory response to TNFα. VRAC currents are modulated by oxidants, suggesting that channel oxidant sensitivity and proximity to Nox1 may play a physiologically relevant role. In VSMCs, LRRC8C knockdown (siRNA) recapitulated the effects of siLRRC8A, inhibiting TNFα-induced extracellular and endosomal O2•- production, receptor endocytosis, NF-κB activation, and proliferation. In contrast, siLRRC8D potentiated NF-κB activation. Nox1 co-immunoprecipitated with 8C and 8D, and colocalized with 8D at the plasma membrane and in vesicles. We compared VRAC currents mediated by homomeric and heteromeric LRRC8C and LRRC8D channels expressed in HEK293 cells. The oxidant chloramine T (ChlorT, 1 mM) weakly inhibited 8C, but potently inhibited 8D currents. ChlorT exposure also impaired subsequent current block by the VRAC blocker DCPIB, implicating external sites of oxidation. Substitution of the 8D extracellular loop domains (EL1, EL2) onto 8C conferred significantly stronger ChlorT-mediated inhibition of 8C currents. Our results suggest that LRRC8A/C channel activity can be effectively maintained in the oxidized microenvironment expected to result from Nox1 activation at the plasma membrane. Increased ratios of 8D:8C expression may potentially depress inflammatory responses to TNFα. LRRC8A/C channel downregulation represents a novel strategy to reduce TNFα-induced inflammation.
Keywords: Volume Regulated Anion Channel, Leucine Rich Repeat Containing 8, Tumor Necrosis Factor-α, Superoxide, NADPH Oxidase 1, Chloramine T, Oxidation
INTRODUCTION
TNFα stimulates the production of extracellular superoxide (O2•-) by activating NADPH Oxidase 1 (Nox1) at the plasma membrane (Choi et al., 2016). O2•- production continues within “signaling endosomes” formed by TNFα receptor internalization (Miller et al., 2007). Inhibition of either Nox1 activity or the scavenging of reactive oxygen species (ROS) blocks TNFα inflammatory signaling in VSMCs (Miller et al., 2007; Gimenez et al., 2016). Nox proteins transfer two electrons from cytoplasmic NADPH to molecular oxygen on the opposite side of an insulating membrane. Sustained Nox activity therefore requires ongoing charge compensation (DeCoursey et al., 2003). HV1 proton channels serve this role by operating in parallel to Nox2 in plasmalemmal and phagosomal membranes of white blood cells (DeCoursey, 2016). It has been speculated that another channel(s) provides charge compensation for Nox1 in other cell types, but this has not been directly established (Miller et al., 2007; Lamb et al., 2009; Choi et al., 2016).
We recently demonstrated that Leucine Rich Repeat Containing 8A (LRRC8A), an essential component of Volume-Regulated Anion Channels (VRACs), is part of the multi-protein Nox1 signaling complex in VSMCs. LRRC8A physically associates with Nox1, and VRAC activity is required for full Nox1 activation in response to TNFα. Furthermore, targeted knockdown of LRRC8A disrupts multiple aspects of the downstream inflammatory response to TNFα (Choi et al., 2016). These findings are consistent with prior observations that VRAC inhibitors and siLRRC8A modulate VSMC proliferation (Qian et al., 2009; Liang et al., 2014; Lu et al., 2019), and suggest that LRRC8 channels play a critical role in TNFα signaling. The nature of the functional dependence of Nox1 on VRAC remains unclear, but the voltage-dependence and rectification properties of VRACs are appropriate to support a role in charge compensation.
VRACs are hexameric proteins (Deneka et al., 2018), requiring LRRC8A in combination with at least one additional LRRC8 subunit (B,C,D,E) (Qiu et al., 2014; Voss et al., 2014; Pedersen et al., 2015) to produce active plasma membrane channels. Different subunit combinations yield currents with distinct biophysical properties in terms of single channel conductance, open probability and ion selectivity (Voss et al., 2014; Syeda et al., 2016). Heterologous co-expression of LRRC8A/C, A/D, and A/E isoforms in Xenopus oocytes resulted in currents with differential sensitivity to the oxidant chloramine T (ChlorT), which activated A/E channel currents, but inhibited A/C and A/D channel currents (Gradogna et al., 2017). The physical association of Nox1 with redox-sensitive channels capable of regulating Nox1 activity and ROS production raises the possibility of reciprocal functional interdependence between these two proteins. Redox-dependent modification of VRACs could potentially provide highly localized feedback regulation of Nox1 activity.
We sought to determine which of the non-A LRRC8 channel subtypes associate with Nox1, and how they support oxidase activity and TNFα signaling in VSMCs. siRNA-mediated knockdown of LRRC8C had an anti-inflammatory impact on TNFα signaling similar to that previously identified for siLRRC8A (Choi et al., 2016), while siLRRC8D knockdown had significant but opposing effects. siLRRC8C also reduced TNFα-induced extracellular O2•- production, demonstrating an impact on Nox1 activity, while siLRRC8D did not. Neither siLRRC8B nor siLRRC8E significantly altered TNFα signaling. VRAC currents derived from LRRC8C expression were relatively resistant to ChlorT-induced inhibition compared to LRRC8D channel currents, which were potently inhibited. LRRC8A/C VRACs thus appear selectively suited to support sustained Nox1 activity and pro-inflammatory signaling. Changing patterns of LRRC8 isoform expression and differential regulation by oxidation may represent important mechanisms for modulation of the inflammatory response to TNFα, as well as other pro-inflammatory signaling molecules that activate Nox1.
MATERIALS AND METHODS
Reagents
All chemicals and enzymes unless otherwise noted were obtained from Sigma-Aldrich (St. Louis, MO).
Cell culture
Primary murine aortic VSMC were isolated from C57/BL6 mice as described previously (Choi et al., 2015). The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 1X minimum essential medium non-essential amino acids, 1X vitamin, and 20 mM HEPES. Human aortic VSMCs were purchased from Cell Applications (San Diego, CA) and maintained in human SMC growth medium (#311–500, Cell Applications).
HEK293T cells were obtained from the American Tissue Culture Collection. HEK293 cells lacking LRRC8A were the generous gift of Dr. Rajan Sah (Washington University, St. Louis MO). These cells were maintained at 37° C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS, Hyclone) and 0.1% Pen/Strep (25U/ml, Life Technologies).
Polymerase Chain Reaction
Qualitative comparisons of the expression of LRRC8 isoforms were made by reverse transcription-PCR analysis of mRNA isolated from VSMCs and HEK293 cells. The primers employed are provided in Table 1.
Table 1.
Primers used for RT-PCR
| LRRC8A-fwd | GCATCCCTCACTCCATCTTTAG | TTGAACAGGTCCTCCTCCAC |
| LRRC8A-rev | TTGTTGCGGTTCAGGTAGAG | CCACCCAGCTCTTCTACTGC |
| LRRC8B-fwd | CCTGAAGCTCTGGCACAATAA | CCTTGCAGCTTTTCCTATGC |
| LRRC8B-rev | CAGATTGGTCAGGTACTGGATTT | TCAGACAGTTCCGTTTTAGGG |
| LRRC8C-fwd | CACACCATGGCTCATTTGTTC | TCTGCCTTCCGAAAAGAGTG |
| LRRC8C-rev | AGAGAGGGTCATACTGGTCTATC | CTTCCCCAGAATGGAGATGA |
| LRRC8D-fwd | GGCATCTTCAGCCTCTCTAATC | GGTGCCCAGACTCTACCAAA |
| LRRC8D-rev | AAACACTGCCGTCGGTAAA | TCAAAGCTGATTGCGTTGAC |
| LRRC8E-fwd | TGACAGGGATGGGTACTTAGA | GCCGAGTTCAAGCAGTTCAC |
| LRRC8E-rev | CTGGCCTCGAACTCAGAAAT | TGACCACGAGGTAAGGGAAG |
Plasmid Modification and Expression
Wild-type LRRC8A (RC508632), 8C (RC203642), and 8D (RC222603) plasmids were obtained from Origene Technologies or from ViGene Biosciences (LRRC8E; CH871303). Chimeric constructs in which the intracellular loop of LRRC8A was inserted into LRRC8C, D or E were kindly provided by Dr. Kevin Strange (Novo Biosciences and Vanderbilt University). Modifications to these plasmids were made using the QuikChange Lightning site-directed mutagenesis kit (Agilent). The C-termini were tagged with mGFP or mCherry to facilitate protein localization. Substitutions of the two extracellular loops were made as detailed in the Results. Plasmids were transfected using Lipofectamine 2000 (DMEM + 1.5–2.5 μg/μl cDNA, 2.5% lipofectamine 2000, +10% FBS). For co-transfections of LRRC8A and a second isoform, a cDNA ratio of 3:1 to 4:1 (nonA:A) was used.
siRNA transfection
siRNA (negative control, LRRC8A, B, C, D, E) were purchased from Dharmacon (Lafayette, CO). siRNA (100 nM) was incubated with Lipofectamine 2000 (Life Technologies) in serum-free medium for 15 min. The resultant complex of siRNA-Lipofectamine 2000 was added to cells in DMEM containing 5% FBS, and maintained for 3 days before performing experiments.
NF-κB activity
VSMC were infected following 2 days of siRNA transfection with replication-deficient adenovirus expressing a luciferase reporter driven by NF-κB transcriptional activation for 24 hours in DMEM containing 5% FBS followed by exposure to TNFα (10 ng/mL) in serum-free DMEM for 6 hours. Luciferase activity (relative light units) was quantified according to the manufacturer’s protocol (Promega, Madison, WI) and normalized to protein concentration (BCA protein assay).
Immunoprecipitation (IP)
Human aoritic VSMCs were stimulated with TNFα (10 ng/mL, 10 min) then lysed (50mM Tris base, 150mM NaCl, 1mM EDTA, 10% glycerol, 1mM DTT, 1% Triton X-100, 0.1% Na-DOC, 0.1% SDS, 10mM β-glycerophosphate, 20mM para-nitrophenyl phosphate, 2mM sodium pyrophosphate, 1mM Na3VO4, 5mM NaF, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride (PMSF) at pH 7.4) for 1 hour with nutation at 4°C, and centrifuged for 30min at 20,000g. Supernatants were pre-cleared with protein-G sepharose beads for 1h at 4°C and cleared-supernatants were incubated with antibody (2 μg) for 1.5h, then incubated with protein-G sepharose for 1h. Beads were washed with lysis buffer, resuspended in SDS sample buffer, boiled and the associated proteins were then analyzed by western blot. Antibodies for IP were used as follows; Nox1 (sc-5821, Santa Cruz), LRRC8C (HPA029347, Sigma-Aldrich), LRRC8D (HPA014745, Sigma-Aldrich).
Immunofluorescence
HEK293T and human aortic VSMCs were grown on coverslips and fixed in 4% paraformaldehyde for 15min at room temperature. For HEK293T cells the coverslips were coated with 20 μg/ml polyethyleneimine (50–100,000 MW) to facilitate adherence. Cells were permeabilized with 0.5% Triton-X 100 and blocked with 1% BSAprior to incubation with primary and secondary antibody (1:100) (Nox1 (Santa Cruz, sc-5821), (LRRC8D (Santa Cruz, sc-515070)) at 37°C for 1h in the dark. They were then mounted in ProLong Gold (ThermoFisher). Images were obtained on a Leica SPE confocal microscope.
Western blot analysis
Cells were stimulated with TNFα (10ng/mL). Protein extracts (40 μg) were separated by electrophoresis on a polyacrylamide gel (10%) and transferred to nitrocellulose membranes. Nonspecific binding was blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) for 1 hour at room-temperature. Membranes were incubated with primary antibodies overnight at 4°C. Antibodies included: Tubulin (Vanderbilt Antibody Core), LRRC8A (#A304–175A, Bethyl Laboratories, Montgomery, TX), LRRC8C (HPA029347, Sigma-Aldrich), LRRC8D (HPA014745, Sigma-Aldrich), PCNA (05–347, Millipore). Signals were developed using fluorescent secondary antibodies with the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE) and quantified densitometrically.
Detection of TNFα receptor endocytosis
Cells were grown on coverslips in six well plates and incubated with siRNA for 3 days. Human TNFα conjugated with biotin (R&D Systems) was incubated with FITC-labeled avidin (Life Technologies) for 1 hr at 4°C, and then exposed to cells for 2 hrs at 4°C. After washing with cold media, cells were warmed to 37°C for 15 min. Cells were then fixed in 3.7% formaldehyde and nuclear counterstaining was performed with TO-PRO-3 Iodide (Life Technologies) for 5 min. Cover-slides were mounted with ProLong Gold anti-fade reagent (Life Technologies). Cells were imaged by Leica SPE confocal laser microscope and the intracellular punctate FITC signal quantified using ImageJ software.
Superoxide detection
Extracellular and endosomal O2•- was quantified using the membrane-impermeable electron spin resonance (ESR) probe 1-Hydroxy-2,2,6,6-tetramethylpiperidin-4-yl-trimethylammonium chloride (CAT1H; Enzo Life Sciences). Cells were incubated with Krebs/HEPES containing CAT1H (0.5 mmol/L) and TNFα (10 ng/mL) for 20 minutes then the incubation buffer was collected and the cells were washed, scraped and snap frozen and placed in an ESR finger Dewar under liquid nitrogen. ESR spectra were recorded from the cell samples (endosomal) or from the Krebs buffer (extracellular) using the following settings: field sweep, 80 G; microwave frequency, 9.39 GHz; microwave power, 2 mW; modulation amplitude, 5 G; conversion time, 327.68 ms; time constant, 5242.88 ms; 512 points resolution; and receiver gain, 1×104. The ESR signal was normalized to protein concentration.
Gene Expression in Human Disease
We utilized the GTExPortal (gtexportal.org) to compare LRRC8 isoform mRNA expression among 54 different tissues in samples from hundreds of individuals. GEO2R was used to analyze microarray data from the NIH Gene Expression Omnibus (GEO datasets, www.ncbi.nlm.nih.gov) and assess LRRC8 isoform expression in human biopsy samples from control patients and individuals with inflammatory aortic disease (GSE57691) or psoriasis (GSE13355).
Electrophysiology
HEK 293T cells were seeded into 6- or 12-well plates for a minimum of 2 hrs, and transfected overnight with plasmid. Whole-cell patch clamp recordings were made at room temperature (21–22 °C) from freshly trypsinized and dissociated cells collected on the 1st day (18–24 hrs) or 2nd day (42–48 hrs) post transfection. LRRC8 protein expression level and distribution was documented in all recorded cells by visualizing mGFP- and/or mCherry fluorescence on an Olympus IX71 microscope (60X objective), equipped with a Hamamatsu C10600 digital camera and Till Photonics Oligochrome and image acquisition/analysis hardware and software (Hunt Optics & Imaging, Inc, Pittsburgh, PA). Isotonic external recording saline (330 mosm/L, pH 7.4) contained (in mM): 130 NaCl, 1.8 MgCl2, 1.8 CaCl2, 10 HEPES, ~70 Mannitol, 1–2 NaOH. Pipette saline (315 mosm/L, pH 7.2) contained (in mM): 120mM CsCl, 4 TEACl, 2 MgCl2, 5 Na2ATP, 10 HEPES, ~35 Mannitol, 1–2 CsOH, 1.186 CaCl2, 5 EGTA (estimated free Ca2+ of 61 nM using WEBMAXC). Final solution osmolality was adjusted by addition of 1M mannitol, measured with a Precision Systems mOsmette osmometer (Natick, MA). Mannitol was excluded from the hypotonic external saline (250–255 mosm/L). DCPIB (30 μM; Tocris Bioscience, Bristol UK) was added daily from freshly thawed 30 mM (DMSO) stock aliquots. ChlorT (0.25–1.0 mM) and TCEP were added daily from 0.5M (dH2O) stock solutions that were freshly remade every ~4 wks. DTT (10 mM) was directly added on the day of use. The recording chamber volume was maintained at ~500 μl and superfused constantly at ~1.2 ml/min. Solution changes were effected using an Automate ValveBankII system (Berkeley, CA) with a multiport manifold that delivered new solution to the recording chamber with a latency of 8–10s.
Amplified whole-cell currents were low-pass filtered at 2 kHz and sampled at 10 kHz using a Molecular Devices Axopatch 200B amplifier driven with a pClamp 10 interface (Molecular Devices, Sunnyvale, CA). Pipette resistances were typically 2.0–3.0 MΩ. Fast pipet capacitance was compensated following gigaseal formation. Cell capacitance (Cm; 20 ± 1 pF) and series resistance (RS; 6.6 ± 0.2 MΩ) were measured using the Clampex 10 membrane test utility while applying a +20 mV test pulse from a holding potential (VH) of −40 mV. 65%−70% RS compensation (75% prediction) was applied. Current-voltage (I-V) relationships were recorded for 100 ms test pulses (−100 to +120 mV, in 20 mV increments) applied from a prepulse level of 0 mV. Current amplitude was measured 2 ms after initiation of the voltage pulse. Oxidant effects were assessed while recording current responses to 250 ms voltage ramps (−100 to +120 mV) applied at 30 s intervals. Ramp current values at +120 mV applied potential were quantified and reported. Current density is expressed as pA/pF, and current-voltage (I-V) plots are corrected for a 5 mV liquid junction potential measured between the pipette and isotonic bath saline. Zero-current level is indicated in displayed traces by a dashed line.
Statistical analysis
Statistical calculations and comparisons were made with Excel (2013) and Graphpad Prism 5 analysis software (GraphPad Software, San Diego, CA). Values are reported as Mean ± Standard Deviation (S.D.), except where indicated in Figure legends. Current-voltage (I-V) plots show Mean ± S.E.M. current density values at each voltage. N represents the number of independently performed experiments in cultured cells, or the number of recorded cells in electrophysiological experiments. Plots were generated using Graph Pad Prism. For quantification of biochemical results, statistical differences were assessed by unpaired t-test, one sample t-test (control hypothetical value is 1) or one-way ANOVA. Post hoc comparisons were performed using Newman-Keuls analysis to compare all groups. Reported significances for GEO gene expression data represent corrected P values for comparisons between groups, as calculated by the GEO2R software based on the log2 transformed data. Comparison of ratios of LRRC8C to 8D expression in psoriasis were made after 2X retransformation of the data, followed by one-way ANOVA and either Dunnett’s or Tukey’s multiple comparison analysis, as indicated. Ratio T-tests were applied to log-transformed VRAC current amplitude data to assess the significance of changes in paired measurements in control vs. oxidant conditions. The Mann-Whitney nonparametric test was used for other two-way comparisons. A P value less than 0.05 was considered to be statistically significant.
RESULTS
LRRC8C isoform supports TNFα –dependent inflammatory response in VSMCs
Messenger RNA for all five members of the LRRC8 family (isoforms 8A through 8E) was detected by RT-PCR in murine VSMCs (Fig. 1A). Qualitatively, 8C and 8D mRNA appeared the most abundant while 8E mRNA was the least. Efficacy of siRNA was verified by RT-PCR, which confirmed pronounced knockdown of all isoforms (Fig. 1B). To explore isoform-dependent roles in inflammatory signaling, we first assessed the effect of siRNA knockdown of individual LRRC8 isoforms on TNFα-mediated NF-κB activation. Resting NF-κB level was not significantly altered by any of the siRNAs. In control cells, 6 hours treatment with TNFα (10ng/ml) increased NF-κB level by ~10-fold (Fig. 1C). Consistent with our previous report (Choi et al., 2016), siLRRC8A reduced the TNFα-mediated increase in NF-κB activity by 32% compared to Control + TNFα (P < 0.05). NF-κB activation was also decreased by siLRRC8C (38%, P < 0.05 vs. Control + TNFα, N.S. vs. siLRRC8A + TNFα). By contrast, NF-κB activation was significantly potentiated by siLRRC8D (26%, P < 0.05). Knockdown of LRRC8B and D had no significant effect on TNFα-mediated NF-κB activation (Fig. 1C). We therefore focused our subsequent work on the 8C and 8D isoforms.
Figure 1.
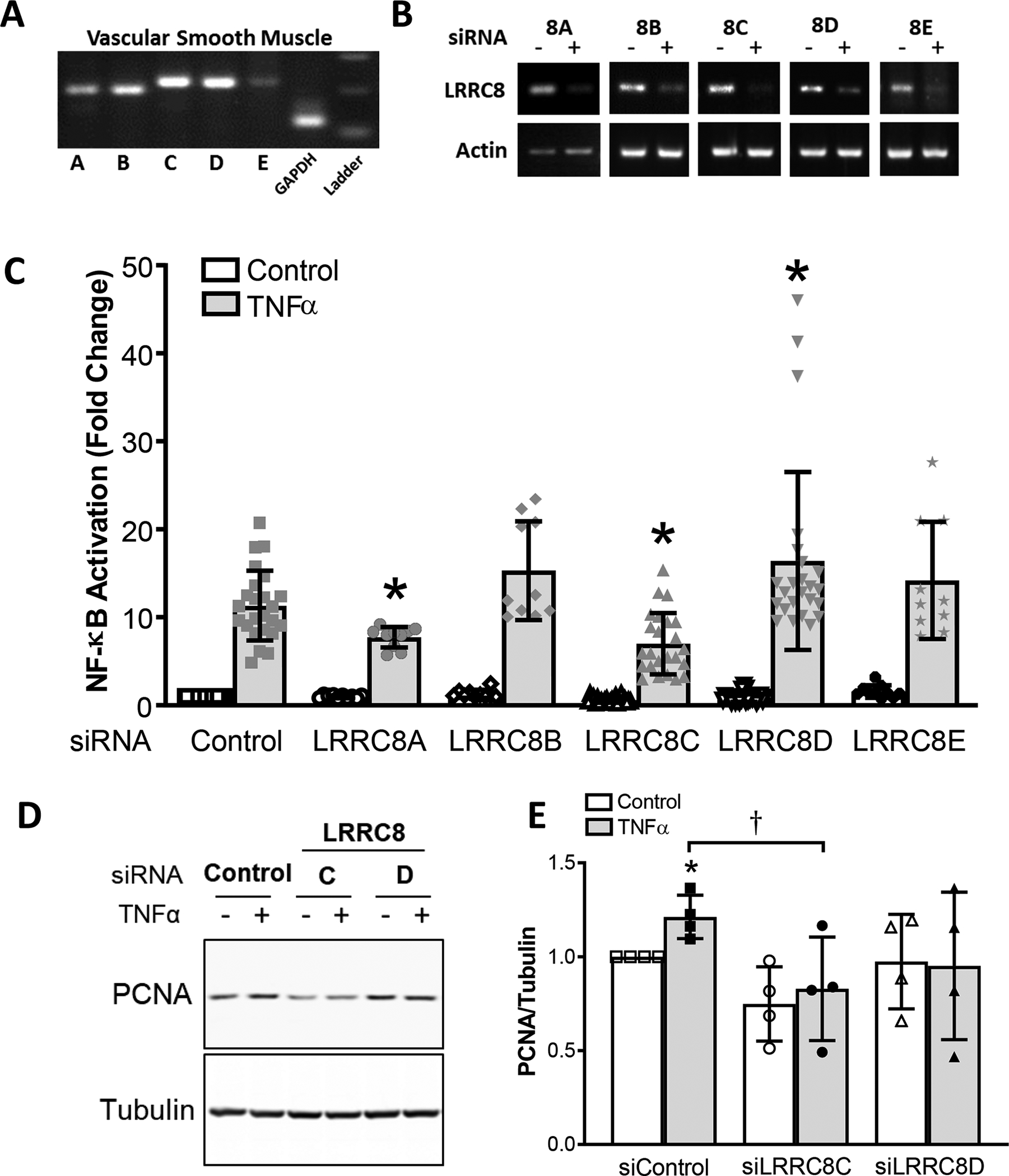
A, All LRRC8 isoforms (8A-8E) are expressed in cultured murine aortic VSMCs. RT-PCR was performed with cDNA derived from mRNA obtained from cultured VSMCs. B, siRNA knockdown of mRNA for LRRC8 isoforms was detected by RT-PCR. C, NF-κB activation 6 hrs after exposure to TNFα (10ng/ml) in siRNA-treated cells. Data are expressed as fold change (Mean ± S.D.) relative to cells treated with scrambled siRNA (Control bars) without TNFα exposure (first bar). N = 21 for siControl, siLRRC8C and siLRRC8D; N =10 for siLRRC8A, siLRRC8B, and siLRRC8E. Asterisks indicate statistical significance compared to Control + TNFα exposure (*P = 0.0009, siLRRC8C; P = 0.0339, siLRRC8D; P = 0.003, siLRRC8E; unpaired t test). D and E, Activation of proliferation in control VSMCs by TNFα is reflected by an increase in PCNA expression (D, lane 2; E, siControl bars). E, The TNFα-dependent increase in siControl cells (*P = 0.0351 vs. control; one sample t-test) was prevented in cells treated with siLRRC8C or siLRRC8D. siLRRC8C-treated cells had significantly reduced PCNA levels after TNFα exposure compared to siControl (†P = 0.0427, unpaired t test; n = 4).
The nuclear protein PCNA (Proliferating Cell Nuclear Antigen) is involved in DNA replication and is an effective marker of proliferation. TNFα caused an increase in PCNA expression in murine VSMCs after 24 hrs of siRNA treatment (21 ± 5.7% over control, n=4; P < 0.05) (Fig. 1D, E). LRRC8C siRNA did not significantly reduce PCNA level under control conditions, but reduced PCNA after TNFα exposure (P < 0.05 vs. siControl + TNFα; n=4 (Fig. 1E). In contrast, siLRRC8D had no significant effect on either resting or stimulated PCNA levels.
We previously demonstrated that TNFα-induced NF-κB activation in VSMC is Nox1-dependent, and that LRRC8A physically associates with Nox1 based upon immunoprecipitation and immunohistochemistry (Choi et al., 2016). Immunoprecipitation of Nox1 and western blotting for LRRC8C and 8D revealed that both proteins associate with Nox1 (Fig. 2A). Neither association was significantly altered by TNFα treatment (Fig. 2A). The antibodies used for western blot analysis were validated in HEK293 cells heterologously expressing Flag-tagged LRRC8C or 8D (Fig. 2C). For both proteins, the respective anti-LRRC8 antibodies identified the same bands as an anti-Flag antibody. Immunostaining of LRRC8D and Nox1 was performed in human aortic VSMCs, which provided superior discrimination compared to murine VSMCs. LRRC8D is co-localized with Nox1 in numerous punctate regions at both the plasma membrane and in intracellular vesicles (Fig. 2B). Effective immunostaining was confirmed in HEK293 cells expressing fluorescently tagged Nox1 and LRRC8D. The pattern of tag fluorescence and anti-LRRC8D was indistinguishable (Fig. 2D). Because the anti-LRRC8C and anti-Nox1 antibodies that demonstrated effective immunostaining were both raised in goat, we were unable to compare the localization of LRRC8C to Nox1.
Figure 2.
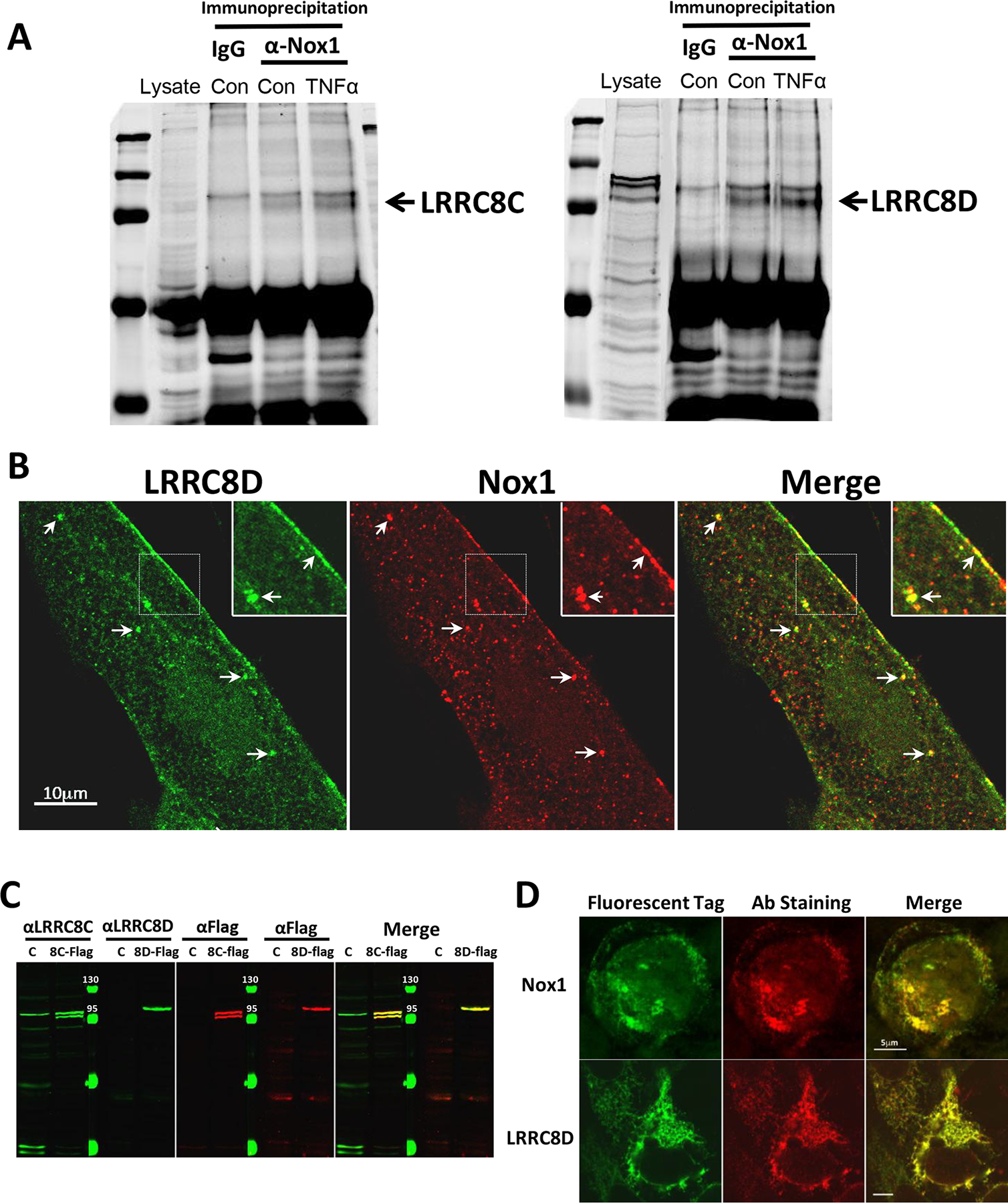
A, Control IgG or anti-Nox1 Ab were used to immunoprecipitate proteins from cultured human VSMC cell lysates under control conditions or following a 10 min exposure to TNFα. Western blotting for LRRC8C (left) or LRRC8D (right) proteins was performed on whole cell lysates (Lysate) or the immunoprecipitated proteins. Anti-Nox1 pulled down both LRRC8 proteins but this association was not enhanced by exposure to TNFα (n = 3). B, Immunostaining of cultured human VSMCs with anti-LRRC8D and anti-Nox1 reveals co-localization both at the plasma membrane and in intracellular vesicles (arrows; insets reflect 2X magnification). C, Western blots of protein obtained from Control HEK293 cells (C), or HEK cells expressing flag-tagged LRRC8C (8C-Flag, left) or LRRC8D (8D-Flag, right). Blots were probed using either αLRRC8C or αLRRC8D and αFlag and imaged using the Odyssey Imaging System. The protein-specific (green) and αFlag antibodies (red) identified the same bands in expressing cells (yellow signal in Merged image). αLRRC8C identified a band in the Control lysate of the same size as expressed LRRC8C but LRRC8D was not identified in the HEK293 cell lysate. D, HEK293 cells heterologously expressing GFP-tagged Nox1 (top) or LRRC8D (bottom) were immunostained with anti-LRRC8D or anti-Nox1 (red, middle panels) to validate antibody specificity. GFP and antibody labeling were strongly colocalized for both proteins (yellow signal, Merge panels).
TNFα-induced O2•- production by Nox1 occurs at both the plasma membrane and within endosomes (Miller et al., 2007). Extracellular O2•- production is primarily dependent on Nox1, as reflected by the markedly reduced O2•- signal detected by the membrane-impermeable CAT1H spin trap in Nox1 null cells (Choi et al., 2019). We previously showed that siLRRC8A significantly reduced the quantity of extracellular O2•- produced by TNFα-stimulated VSMCs (Choi et al., 2016). Here we quantified both extracellular and endosomal O2•- production using CAT1H in control and siRNA-treated murine VSMCs cells. The abundance of O2•- increased in both compartments following exposure to TNFα. Knockdown of LRRC8C significantly reduced TNFα-induced O2•- production at both sites, while siLRRC8D had no effect (Fig. 3A, B).
Figure 3.
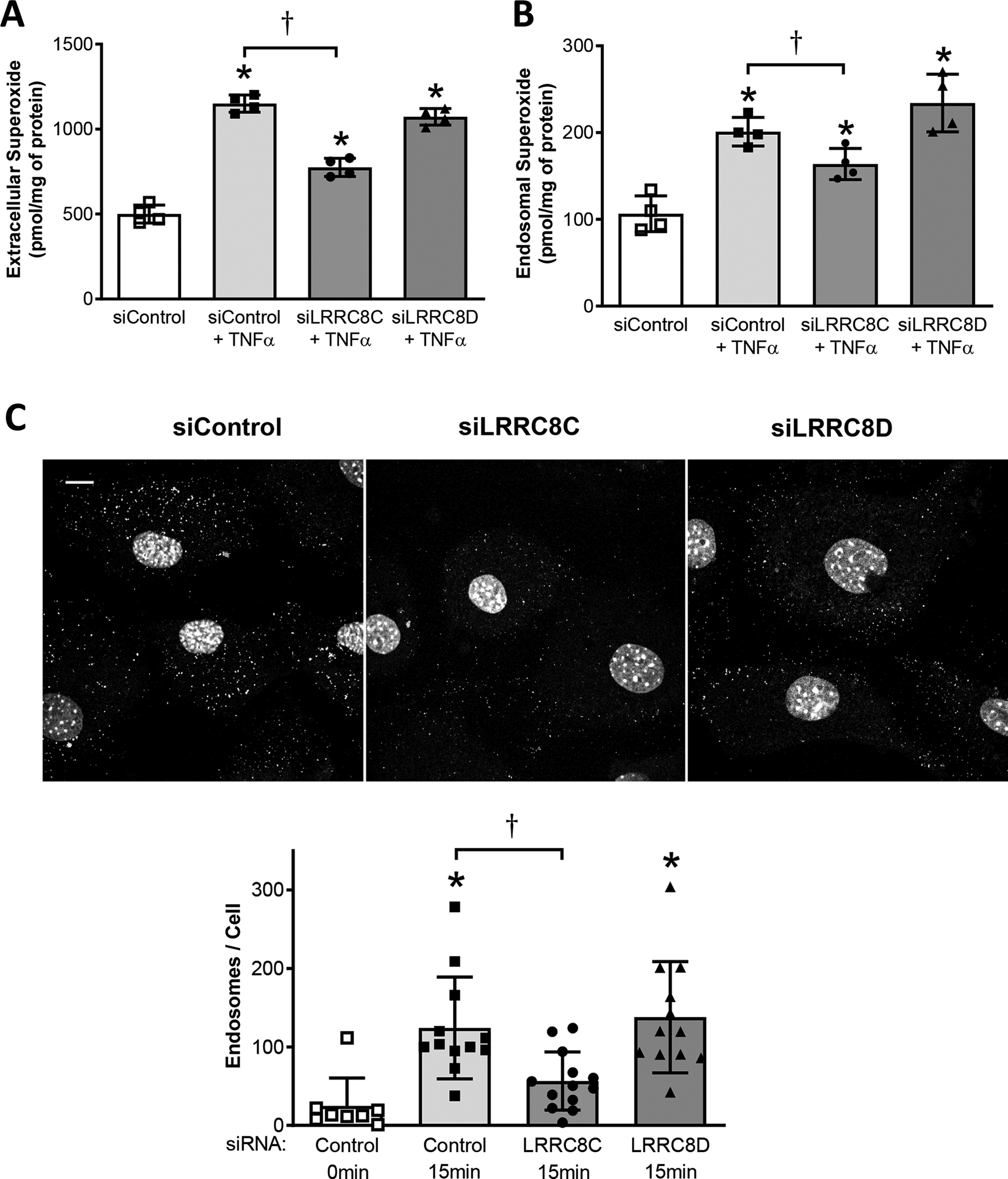
A, Extracellular superoxide production is increased in response to TNFα (CAT1H signal in media). This effect is inhibited by siLRRC8C but not by siLRRC8D. B, Endosomal superoxide production is also increased in response to TNFα (CAT1H signal in cell fraction); this effect also was only significantly inhibited by siLRRC8C. A, B: *P < 0.05 vs. siControl cells not exposed to TNFα, †P < 0.05 vs. TNFα –treated siControl (One-way ANOVA, Newman Keuls paired test; N = 4). C, Endocytosis of FITC-biotin-labelled TNFα is inhibited by siLRRC8C but not by siLRRC8D. Top: Typical images of fluorescent intracellular puncta in VSMCs treated for 15 mins with TNFα. Nuclei are stained with To-Pro-3. Scale (upper left): 10 μm. Bottom: Quantitation of puncta, expressed as endosomes/cell. *P < 0.005 compared to siControl cells not exposed to TNFα; †P = 0.0037, siLRRC8C vs. Control, 15 min., unpaired t-test (n = 3). Results are representative of three independent experiments.
TNFα receptor endocytosis is important for NF-κB activation (Choi et al., 2015). Impaired endosomal deposition of O2•- could reflect reduced endosomal oxidase activity, as previously observed in cells lacking a ClC-3 transporter-mediated anion conductance (ClC-3) (Miller et al., 2007). However, a reduced CAT1H endosomal O2•- signal could also result from impairment of the endocytic process itself. TNFα receptor endocytosis in VSMCs is O2•--dependent, and was thus impaired in cells lacking Nox1 or LRRC8A (Choi et al., 2016). We assessed endocytosis of fluorescently labelled TNFα as previously (Choi et al., 2015) using biotin-labeled TNFα and FITC-avidin. The ability of murine VSMCs to form TNFα-containing endosomes was significantly reduced by siLRRC8C, while siLRRC8D had no effect (Fig. 3C)
LRRC8 Expression in Human Tissues
Our siRNA data demonstrate that induced changes in LRRC8 isoform expression impact inflammatory cell signaling. However, the pathophysiological significance of these signaling changes depends on whether isoform expression is significant in tissues of interest, and if expression is altered by inflammatory disease states in vivo. Among 54 different tissues catalogued by the GTExPortal, LRRC8A expression is remarkably high in tissues containing an abundance of smooth muscle; the aorta (1st), tibial artery (2nd), coronary artery (6th), vagina (9th), and uterus (10th) rank in the top ten highest-expressing tissues (Fig. 4A). In the three blood vessels tested, the other LRRC8 isoforms, including 8C and 8D, are expressed at much lower copy number (<10% of LRRC8A), and their relative expression ranks much lower compared to LRRC8A (Fig. 4A).
Figure 4.
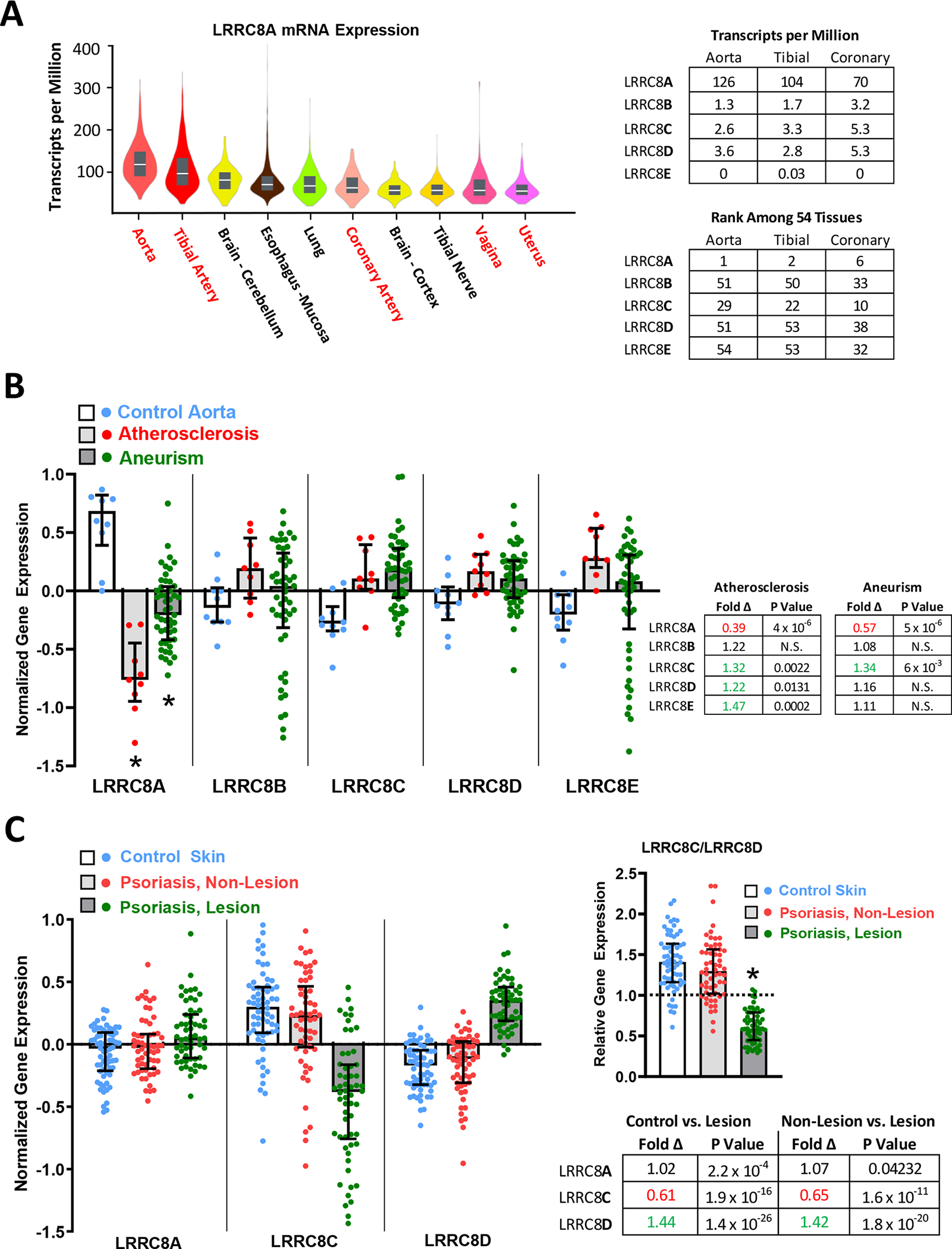
LRRC8 gene expression in health and disease. A, left, Violin plots of LRRC8A expression (transcripts/million) in the 10 highest expressing among 54 human tissues assessed, including median values (white lines) and interquartile range (grey bars). LRRC8A is highly expressed in blood vessels and in other organ tissues with abundant smooth muscle (shown in red). The accompanying tables (right) quantify the relative expression of all LRRC8 isoforms (8A-8E) in the three blood vessels tested (top), and rank the relative expression of each LRRC8 isoform in these vessels against expression in all 54 tissues (bottom). N values vary between 142 (uterus) and 663 (tibial artery). B, Comparison of LRRC8 isoform expression in abdominal aortic biopsies from organ donors (Control, blue), and patients with either aortic atherosclerosis (red) or abdominal aneurisms (green) (LRRC8A: (ILMN_1739840), 8B: (ILMN_1712128), 8C: (ILMN2094396), 8D: (ILMN_1763409), 8E: (ILMN_1669052)). Normalized values are defined as the ratio of specific gene expression to the average expression levels for all gene products in the tissue, plotted as log2. An average expression level (1.0) for the gene of interest is thus plotted at 0 (log21 = 0); an expression ratio of 2 is plotted at 1.0 (log22 = 1); a 50% decrease in expression ratio is plotted at (−1.0). LRRC8A expression is strongly reduced in both disease states. Fold differences and statistical significance of all changes are provided in the accompanying table (corrected P values obtained by GEO2R analysis). Fold change is determined from the difference between normalized expression values under two different conditions and raising 2 to that power (2X), thus a change in expression level from 1 to −1 reflects a fold change of 22 = 4. C, Expression of LRRC8A (23347_at_s), 8C (228314_at) and LRRC8D (218684_at) was compared in skin biopsies from healthy patients (Control, N = 64), and from uninvolved (Non-lesion) and involved (Lesion) skin from psoriasis patients (N = 58). Psoriatic lesions demonstrate a small (7%) increase in LRRC8A expression, but remarkable changes in LRRC8C (decreased) and LRRC8D (increased). The shift in the ratio of LRRC8C to 8D was compared after 2X transformation of the data (right). * P < 0.05, ANOVA and Tukey’s multiple comparison test.
We next compared LRRC8 isoform expression in biopsy samples from patients with inflammatory aortic disease or psoriasis. Both disease conditions are associated with significant TNFα-mediated inflammation (Chima & Lebwohl, 2018; Lamb et al., 2020), and psoriasis is widely treated with selective anti-TNFα biological agents (e.g., Etanercept) as first line therapy (Chima & Lebwohl, 2018). Geo series GSE57691 compares genome-wide expression in abdominal aortic biopsies from 10 healthy control patients (organ donors), 9 patients with aortic atherosclerosis, and 49 patients with abdominal aortic aneurisms (Biros et al., 2015). Normalized transcript expression data for all LRRC8 isoforms is summarized in Fig. 4B. The most striking disease-related change is a marked reduction in LRRC8A expression in both atherosclerotic (61% decrease) and aneurismal vessels (43% decrease). By contrast, modest but statistically significant increases in expression occurred for LRRC8C, D and E in atherosclerosis, and for 8C in aneurismal disease. The relative LRR8C:8D expression ratio was not remarkably altered under disease conditions, as both proteins increased to comparable degrees.
LRRC8A, 8C, and 8D gene expression was explored in GEO series GSE13355, which compares skin biopsies from healthy volunteers (control, n = 64) to both healthy (non-lesion) and inflamed (psoriatic lesion) skin from 58 patients with psoriasis (Nair et al., 2009). In psoriatic lesions, LRRC8A expression is statistically but not meaningfully affected (increased 1.02-fold), but 8C expression is dramatically decreased while 8D expression is markedly increased (Fig 4C). The LRR8C:8D expression ratio in psoriatic lesions is consequently reversed to a median of 0.59 (interquartile range 0.448 – 0.788) from the positive ratio demonstrated in the control 1.41 (1.16 – 1.63) and non-lesion groups 1.3 (1.02 – 1.56).
Chloramine-T inhibits native VRAC current and disrupts current block by DCPIB
The essential secondary structure of an LRRC8A monomer includes cytoplasmic N- and C-termini, four highly conserved transmembrane domains, two extracellular loop domains (EL1, EL2), and a single intracellular loop (IL) (Fig. 5A). Seventeen leucine-rich repeat elements are clustered at the C-terminus. The primary amino acid sequence of the extracellular loops of LRRC8A, C and D is compared in Fig. 5B, including potential sites of redox modification (cysteine and methionine residues), and the site of DCPIB interaction with EL1 (Kern et al., 2019). A color-coded overview of the essential elements of the chimeric plasmids utilized in these experiments is provided in Fig. 5C.
Figure 5.
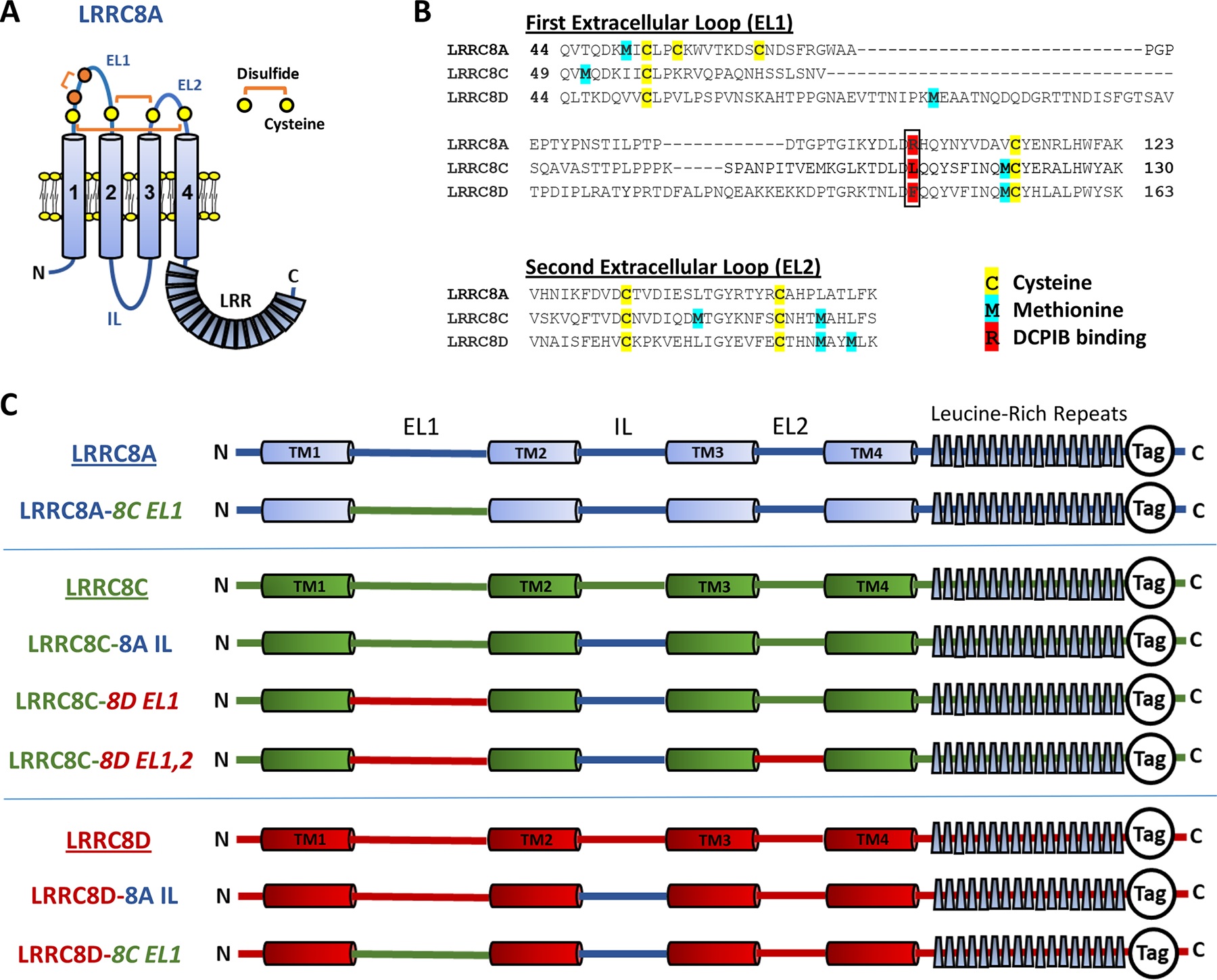
A, Schematized topography of an LRRC8A monomer. The first extracellular loop (EL1) contains two conserved (C1, C4; yellow) and two non-conserved cysteines (C2, C3; orange). C1 and C4 form disulfide bonds with conserved cysteines in EL2, as shown. C2 and C3 form a third disulfide within EL1. The intracellular loop (IL, blue) sequence (P147-R262) of LRRC8A was substituted for the homologous regions of LRRC8C and 8D for the experiments shown in Figs. 8, 10, and 11. B, The EL1 and EL2 amino acid sequences for LRRC8A, C, and D, with potential redox-responsive cysteines (yellow) and methionines (blue) highlighted. R108 (red, boxed) of LRRC8A is implicated in DCPIB binding and block of LRRC8A homohexameric channels (Kern et al., 2019). LRRC8C and 8D have lysine and phenylalanine, respectively, in the analogous position. C, Structure and nomenclature of the C-terminal tagged LRRC8 “wild-type” and chimeric constructs utilized in this study. LRRC8A sequence in shown in blue, LRRC8C in green, and LRRC8D in red. All chimeras included the 8A IL substitution. EL1 and EL2 substitutions are indicated in italics. Tag refers to either monomeric GFP or mCherry fluorescent proteins.
RT-PCR detected all native LRRC8 isoforms (8A-8E) in wild-type (WT) HEK cells (Fig. 6A). In accord with a previous quantitative assessment of LRRC8 transcript levels (Friard et al., 2017), LRRC8D appeared to be the most abundantly expressed. Expression of LRRC8B-8E isoforms was quantitatively similar in LRRC8A(−/−) mutant HEK cells (Fig. 6A). In whole-cell recordings, application of hypotonic external saline (330 reduced to 255 mosm/L) to WT cells activated robust native VRAC currents that were nearly completely (>95%) blocked by 30 μM DCPIB (Fig. 6B, E). Comparable hypotonic current was absent in LRRC8A(−/−) mutant cells (Fig. 6C, F), consistent with recent reports (Friard et al, 2017; Friard et al, 2019). Expression of LRRC8A-mCh in 8A(−/−) mutant cells reconstituted DCPIB-sensitive hypotonic currents with largely WT behavior (Fig. 6D, F). Under isotonic recording conditions, the majority of WT cells (14/20 cells) displayed insignificant background currents (<6 pA/pF at +120 mV) (Fig. 6B,a). Despite the use of a pipette solution osmolality 5% lower (315 mosm/L) than the isotonic bath solution, a subset (6/20) of WT cells displayed significantly larger initial currents (22 ± 6 pA/pF; P = 0.0006 vs. majority group) that increased in amplitude with recording time (3–10 min) and resembled VRAC currents (Fig. 6G, H). These isotonic currents were effectively blocked by DCPIB (Fig 6G,c and H), confirming that VRACs can be activated to varying degrees even under isotonic recording conditions. This finding is supported by prior observations that inclusion of 10–20 μM external DCPIB reliably prevents VRAC activation when recording other classes of anion current (J. Rohrbough, unpublished data). The addition of C-terminal fluorescent tags to LRRC8 expression constructs has been shown to constitutively activate current under isotonic conditions (Gaitan-Penas et al., 2016; Yamada & Strange, 2018). Accordingly, in 8A(−/−) mutant cells expressing 8A-mCh, we recorded significantly larger isotonic currents (50 ± 20 pA/pF at +120 mV, n = 3; Fig. 6D, F; P = 0.016 vs. WT). These modest current densities nevertheless indicate that minimal activation occurs for heteromeric channels containing both tagged and untagged (endogenous) subunits, and that hypotonic conditions are required for robust current activation.
Figure 6.
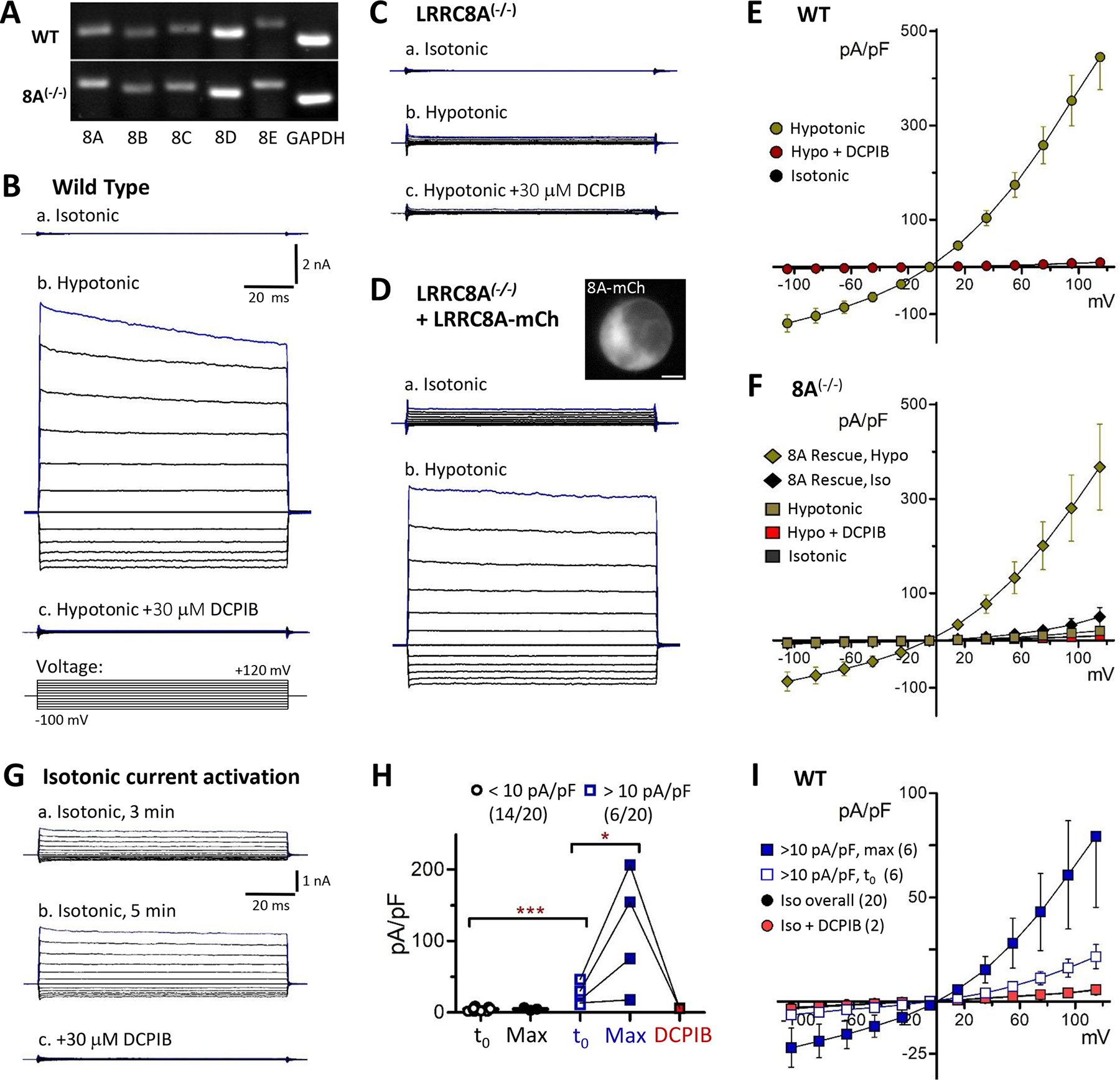
LRRC8A is required for endogenous VRAC current activation by hypotonic conditions. A, Qualitative PCR assessment of LRRC8 isoform expression in wild-type (WT) and LRRC8A−/− HEK293 cells. B-D, Whole-cell currents recorded in HEK cells in response to 100 ms voltage test pulses (−100 to +120 mV). B, Endogenous currents in a wild-type (WT) cell in isotonic (a, 330 msm/L) and hypotonic conditions (b, 255 mosm/L). DCPIB (c, 30 μM) potently blocks hypotonic current. The voltage pulse paradigm is depicted in c (bottom). Test pulses were preceded by a 10 ms prepulse to 0 mV from a holding potential of −40 mV. Scale in B-D: 2 nA, 20 ms. C, In LRRC8A(−/−) mutant cells, hypotonic currents are virtually absent (b). D, Expression of 8A-mCh in 8A(−/−) mutant cells results in small currents under isotonic conditions (a), and restores a robust response to hypotonic conditions (b). Inset image (a) shows 8A-mCh distribution in the recorded cell. E, Current-voltage (I-V) relationships for wild-type cells. Mean (S.E.M) current densities are plotted at each voltage under the indicated conditions (N ≥ 14). DCPIB blocks >95% of hypotonic-activated currents. F, I-V relationships for LRRC8A(−/−) mutant cells (square symbols; N = 3–12) and 8A(−/−) cells expressing 8A-mCh (Rescue; diamonds; N = 4–6). G, Example of time-dependent activation of endogenous VRAC in isotonic saline in a wild-type cell. Currents increased over the course of 5 mins after establishing whole cell configuration (a, b). Addition of 30 μM DCPIB (c) completely blocked current. Qualitatively similar currents were observed under isotonic conditions in 6 of 20 WT recordings. H, Quantified time-dependent changes in isotonic current amplitude (+120 mV) in wild-type cells. The majority (14/20; black symbols) exhibited small initial currents (<10 pA/pF) and showed no significant amplitude increase over 4–10 minutes (“Max”: maximal time-dependent current). The remainder (6/20 cells; blue symbols) exhibited significantly larger initial currents (*** P = 0.0006 vs. majority group) with amplitudes that increased substantially over several minutes (* P = 0.0302, Max vs. t0). These currents were blocked by DCPIB (red symbols; N = 2; connecting lines represent same-cell recordings). I, I-V relationships in wild-type cells displaying VRAC currents (>10 pA/pF) under isotonic conditions. Open squares: Initial (t0; 1–2 min) current density. Blue squares: Maximal current density (6–10 min). Red: Isotonic + DCPIB. Black circles: Overall I-V for wild-type cells in isotonic saline.
PCR gel 50% orig
IV traces: 75% orig width
Iso/DCPIB/8A(−/−) 10 nA scale. Scaled to 15nA x2/3 orig height
Rescue (15nA scale): 75% orig. width Cell image: 5 um scale
IV plots: 80% (D, G), 85% (H)
Oxidant sensitivity of VRAC currents was examined in WT cells and in LRRC8A(−/−) mutant cells expressing various LRRC8 constructs. Externally applied chloramine-T (ChlorT) was reported to similarly inhibit currents mediated by heteromeric LRRC8A/C and A/D channels expressed in Xenopus oocytes (Gradogna et al., 2017). In HEK cells, externally applied ChlorT likewise inhibited endogenous VRAC in WT cells (Fig. 7A), and reconstituted current in 8A(−/−) mutant cells expressing 8A-mCh (Fig. 7B). The inhibitory response to ChlorT was rapid, observable within 10–20 sec after initial exposure (see Methods), and largely complete within 1.5–2 mins. 1 mM ChlorT inhibited current amplitudes by 50–60% (+120 mV; Fig. 7C). Surprisingly, following ChlorT exposure DCPIB was far less effective in blocking residual current (“post-ox. DCPIB”; Fig. 7A–C) compared to pre-oxidation control conditions. Structural analysis has indicated that DCPIB binds and blocks the outer channel pore of LRRC8A hexahomomers via a “cork-in-bottle” mechanism, interacting with Arg103 (Kern et al., 2019) located in extracellular loop 1 (EL1; Fig. 5A, B). Our results therefore suggest that oxidation results in altered VRAC extracellular structure.
Figure 7.
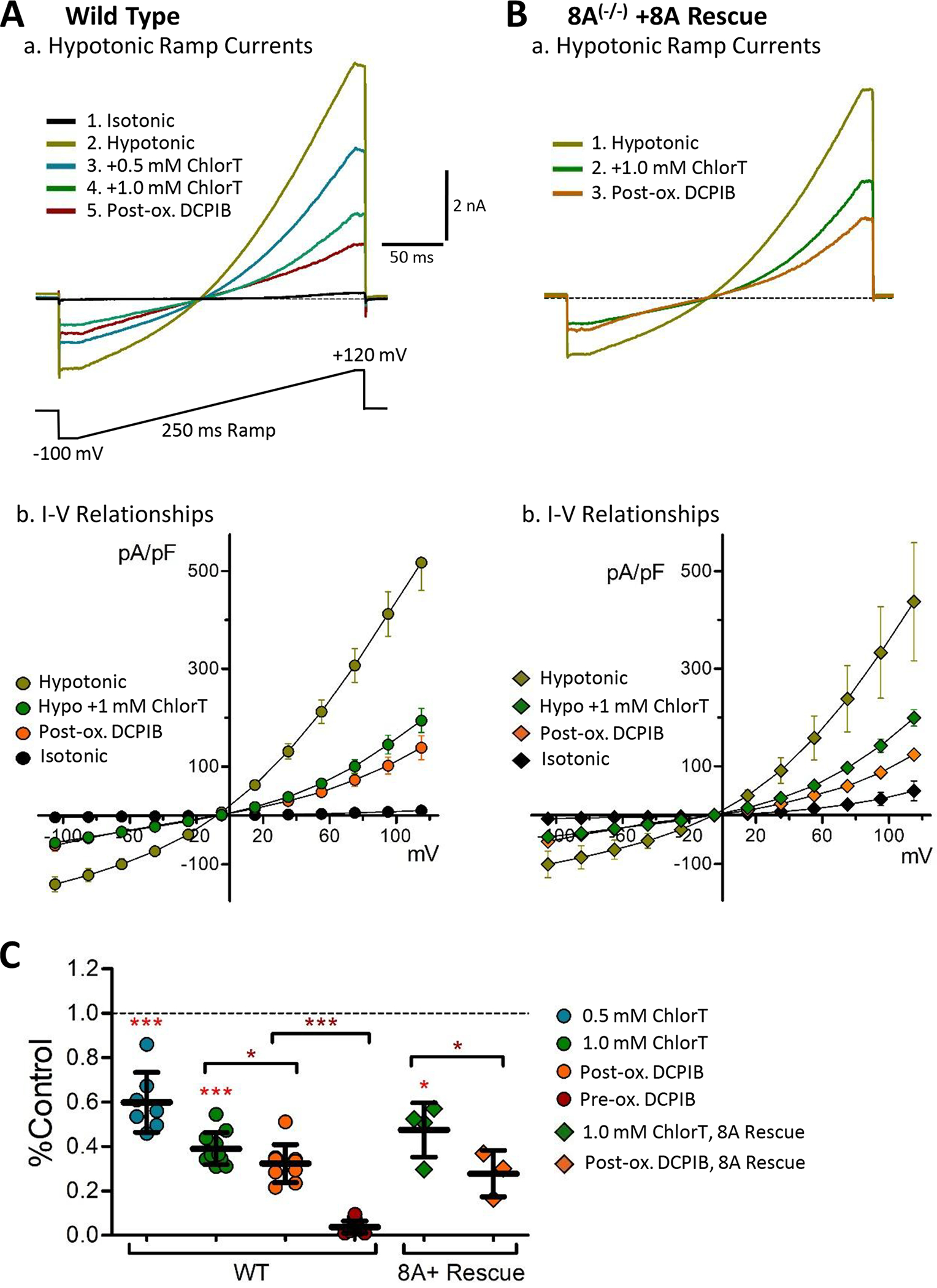
Inhibition of endogenous VRAC currents by the oxidant chloramine-T. A, Wild-type (WT) HEK293 cells. a, Currents elicited by 250 ms voltage ramps (−100 mV to +120 mV; voltage protocol is depicted below) under the indicated sequential conditions (isotonic, hypotonic, hypotonic + chloramineT (ChlorT), hypotonic + 30 μM DCPIB). Current is progressively inhibited by 0.5 mM and 1 mM ChlorT. Following ChlorT exposure, DCPIB shows greatly reduced block efficacy (post-ox. DCPIB; red). b, I-V relationships recorded in wild-type cells under the indicated conditions (n ≥ 8). B, LRRC8A(−/−) mutant cell expressing 8A-mCh (8A Rescue). a, Ramp current responses recorded in the indicated sequence (hypotonic, hypotonic + 1 mM ChlorT, hypotonic + 30 μM DCPIB). Current is inhibited by ChlorT, and post-oxidant DCPIB block (post-ox DCPIB; orange) is greatly reduced. b, I-V relationships for 8A Rescue (n ≥ 3) are similarly altered by oxidant. C, Normalized inhibitory effects of ChlorT and DCPIB on hypotonic ramp current amplitude (+120 mV) for wild-type and 8A Rescue conditions. 1 mM ChlorT reduces wild-type peak current by 61% (P < 0.0001 vs. control, n = 7) and 8A Rescue current by 53% (P = 0.0136 vs. control; n = 4). Post-oxidant DCPIB produces only partial block of residual current (P = 0.0108, WT; P = 0.0355, 8A Rescue) compared to pre-oxidant DCPIB block (***P < 0.0001, pre-ox vs. post ox, WT; Mann-Whitney test. Brackets compare the indicated pair of bars. N ≥ 7 for wild type; n = 3–4 for 8A Rescue.
LRRC8C and 8D channel currents display differential oxidant sensitivity
We hypothesized that oxidants produced by Nox1 can regulate activity of closely associated VRAC channels. Having established the association of Nox1 with LRRC8C and 8D, we examined the ChlorT oxidant sensitivities of 8C vs. 8D channels, which has not previously been compared in mammalian cells. The trafficking of assembled VRAC channels to the plasma membrane is regulated by the LRRC8A intracellular loop (IL) sequence (P147-R262) (Yamada & Strange, 2018). 8C, D, and E chimeric proteins with the 8A IL sequence were demonstrated to create functional homomeric channels in LRRC8 global knockout cells (Yamada & Strange, 2018). We expressed 8C-A IL and 8D-A IL chimeras with C-terminal-tagged mGFP or mCh in 8A(−/−) mutant cells. Under these conditions, VRAC current is expected to be mediated predominantly, if not exclusively, by homomeric 8C or 8D channels. In both 8C- and 8D-expressing cells, VRAC currents were substantially active under isotonic conditions, and further activated (~2.5-fold) by hypotonic saline (Fig. 8A–B). LRRC8C currents were on average weakly inhibited by 1 mM ChlorT (23 ± 4% reduction; n = 7; Fig. 8D, G). In contrast, 8D currents were potently inhibited by 0.5–1.0 mM ChlorT (Fig. 8Bc), displaying over 85% inhibition at 1 mM (Fig. 8H; P < 0.0005 vs. 8C, n = 6). We also assayed a tagged LRRC8A protein substituted with the LRRC8C EL1 sequence (Yamada & Strange, 2018). 8A-C EL1-expressing cells likewise displayed robust isotonic and hypotonic currents (Fig. 8C) with variable responses to 0.5 mM – 1.0 mM ChlorT, but no average current amplitude change (96 ± 10% of control at 1 mM; n = 14; Fig. 8C, F, I). For each construct, 30 μM DCPIB blocked currents present under either isotonic or hypotonic conditions by ≥ 90% (Fig. 8G–I). This indicates that effective channel block by DCPIB does not strictly require the Arg103 found in LRRC8A, since both 8C (lysine) and 8D (phenylalanine) have alternative residues at that position (Fig. 5B). As observed for endogenous currents, DCPIB was ineffective in blocking residual 8C or 8D currents following ChlorT exposure (post-ox. DCPIB; Fig. 8G, H). In 8A-8C EL1-expressing cells, which were insensitive to ChlorT, post-ox DCPIB resulted in partial (~40%) current block (Fig 8F, I). Oxidant-mediated structural changes to the EL1 domain therefore appear to similarly disrupt DCPIB access or binding in both native VRACs and expressed channel constructs.
Figure 8.
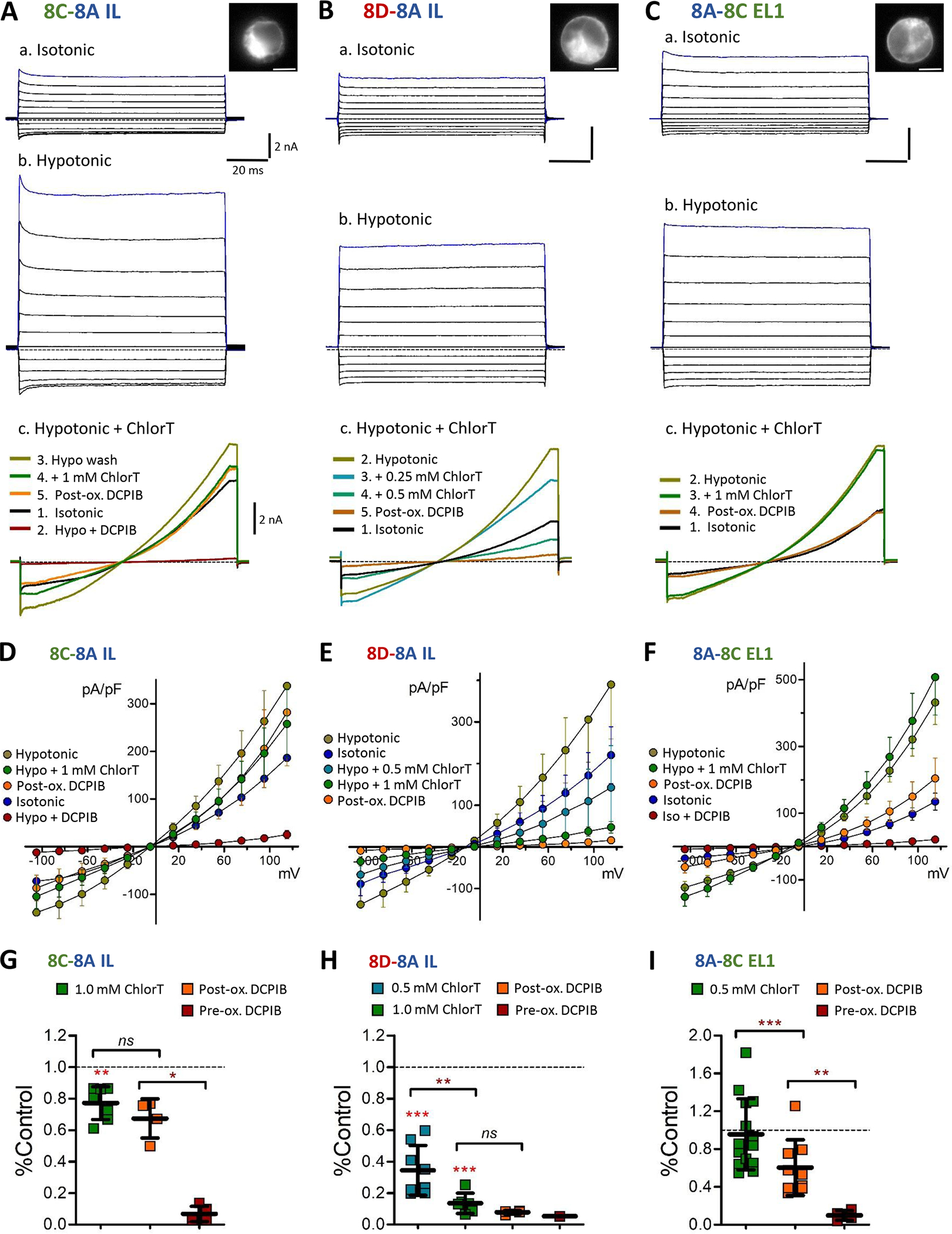
Differential oxidant sensitivity of LRRC8C, 8D and 8A currents. The 8A intracellular loop (IL) was substituted onto the 8C (A, 8C-8A-IL) and 8D (B, 8D-8A-IL) constructs tagged with mGFP or mCh and expressed in LRRC8A(−/−) cells. Inset images (a) show construct fluorescence in the recorded cell. Panels a–b: Representative currents evoked by 100 ms voltage pulses (−100 to +120 mV) under isotonic (a) and hypotonic conditions (b). Scale: 2 nA, 20 ms (A-C). Panel c: Current responses in individual cells elicited by 250 ms voltage ramps (−100 to +120 mV) under the indicated series of conditions (isotonic, hypotonic, hypotonic + ChlorT, DCPIB). 8C-8A IL current is weakly inhibited by ChlorT (Ac), while 8D-8A IL current (Bc) is strongly inhibited in a concentration-dependent manner by 0.25 mM and 0.5 mM ChlorT. C, The first extracellular loop (EL1) of 8C was substituted onto LRRC8A-mGFP (8A-8C EL1). Expressing cells display currents in both isotonic and hypotonic conditions (a,b). 8A-8C EL1 current is unaffected by 1 mM ChlorT (c). D–F, I-V relationships for the three expression conditions, showing mean current densities (S.E.M.) at each voltage level under the indicated conditions. G-I, Quantified inhibitory effects of ChlorT (0.5 – 1.0 mM) and DCPIB (pre- and post-oxidant exposure). Horizontal brackets compare the indicated pair of bars (Paired ratio t-test or Mann-Whitney test). 1 mM ChlorT weakly inhibits 8C-8A IL currents (G)) by 23% (**P = 0.0024 vs. control; n = 7), but potently inhibits 8D-8A IL currents (H) by >85% (***P = 0.0001 vs. control; P = 0.0012 vs. 8C-8A IL). ChlorT exerts no overall effect on 8A-8C EL1 current amplitudes (I) (P = 0.2837 vs. control; n = 14). Post-ox. DCPIB exposure results in insignificant additional current block for 8C (P = 0.1949; n = 5) and 8D (P = 0.067; n = 4), and 40% additional block for 8A-8C EL1 (P = 0.0028; n = 9).
Cell images 30um×30um 20% orig cropped size 10um scale bar
IV traces 75% orig
Ramp traces 75%
IV plots 77%
Endogenous VRAC currents are thought to be carried by heterohexamers composed of LRRC8A and one or more non-A subunits. We therefore assessed oxidant responses in 8A(−/−) mutant cells co-expressing 8A-mCh with either 8C-mGFP (A/C) or 8D-mGFP (A/D) (Fig. 9). Both A/C and A/D co-expression resulted in currents under isotonic conditions that were augmented by hypotonicity. 1 mM ChlorT moderately inhibited current in A/C-expressing cells (40 ± 6% reduction, n = 8; Fig. 9Ab, C, E), but exerted significantly stronger current inhibition in A/D-expressing cells (71 ± 4% reduction, n = 6; P < 0.005 vs A/C) (Fig. 9Bb, D, E). When co-expressed with 8C or 8D, 8A-mCh is likely to also form some channels with endogenous non-A protein(s) that may contribute to hypotonically-activated VRAC current. However, the differential current inhibition by ChlorT in cells co-expressing A/C vs. A/D paralleled the behavior of the 8C-A IL and 8D-A IL constructs. Currents in A/C and A/D-expressing cells likewise displayed insignificant post-oxidant current block by DCPIB (Fig. 9C, D, E).
Figure 9.
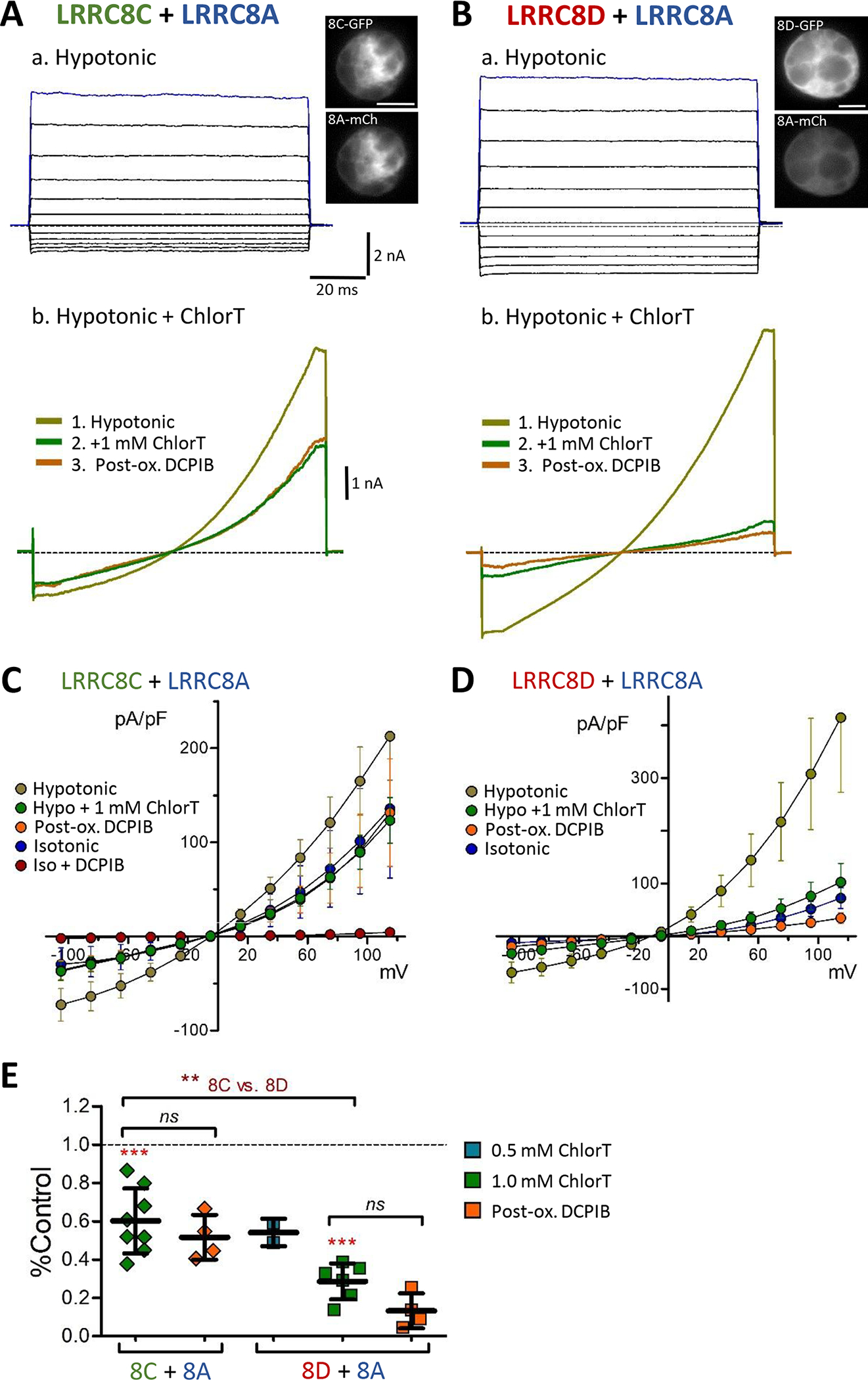
Differential oxidant sensitivity of 8C-8A and 8D-8A heteromeric channel currents. A, B, Hypotonic current responses to 100 ms voltage pulses (−100 to +120 mV; a), and 250 ms voltage ramps (−100 to +120 mV; b) in cells co-expressing 8C + 8A (A) or 8D + 8A (B). The C-terminals of all isoforms were tagged with eGFP or mCh (see Fig. 5C); inset images at top show co-expressed fluorescence in the recorded cells. 1 mM ChlorT moderately inhibits current in 8C/A-expressing cells (Ab), and more strongly inhibits current in 8D/A-expressing cells (Bb). Post-oxidant DCPIB is ineffective or only weakly blocks residual currents. C, D. I-V relationships showing Mean (± S.E.M) current densities at each voltage under the indicated conditions. 8C/A co-expression (C) resulted in substantial activation of DCPIB-sensitive currents in isotonic saline. E, Summary of current responses to ChlorT (0.5 – 1.0 mM) and pre/post DCPIB block. Brackets compare the indicated pairs of bars. 1 mM ChlorT inhibits 8C/8A currents by 39.9% (P = 0.001 vs. control) and 8D/8A currents by 71.4% (P = 0.0004 vs. control; paired ratio t-tests). ChlorT inhibition is significantly greater in cells expressing 8D + 8A (P = 0.0013 vs 8C+8A, Mann-Whitney test). Following ChlorT exposure, DCPIB exerts insignificant block on remaining current (Post-ox. DCPIB; P = 0.7814, 8C + 8A; P = 0.0890, 8D + 8A; Paired ratio t-tests). N = 6–8, 1 mM ChlorT; n = 4–6, post-ox. DCPIB.
ChlorT plot: 90% orig.
Reducing agents exert minimal effects on ChlorT sensitive currents
The membrane-permeable reducing agent dithiothreitol (DTT) activated heteromeric LRRC8A/C and 8A/D channel currents in oocytes, suggesting a baseline level of oxidation-dependent inhibition of these channels, although DTT did not reverse ChlorT-mediated current inhibition (Gradogna et al., 2017). In WT HEK293 cells, DTT application (10 mM, 4–5 min) exerted no consistent effect on endogenous current amplitudes under hypotonic conditions (111% (66%) of control amplitude, P = 0.9549, n = 5; Fig. 10A), and had no measurable effect on the efficacy of current block by 30 μM DCPIB (95% (4%) current block, n = 5; Fig. 10A). The membrane-impermeant reducing agent TCEP (tris (2-carboxyethyl) phosphine; 0.5 mM) likewise did not significantly alter WT hypotonic current amplitude (85% (39%) of control; P = 0.80, n =2; data not shown), and was ineffective in reversing current inhibition by 1 mM ChlorT (n = 2; Fig. 10B). However, TCEP application following ChlorT exposure appeared to partially restore sensitivity to DCPIB, which blocked ~50% of the remaining current (P = 0.038 vs. ChlorT current; paired ratio t-test n=2; Fig. 10B).
Figure 10.

Wild-type and 8D channel currents are insensitive to reducing agents. A, The membrane-permeant reductant dithiothretiol (DTT, 10 mM) does not significantly alter the amplitude of endogenous wild-type currents activated by hypotonic conditions (+120 mV; n = 5). Following DTT exposure, DCPIB (30 μM) addition results in nearly complete block of current (N = 3). B, The membrane-impermeant reductant TCEP (0.5 mM) is ineffective in reversing ChlorT-mediated (1 mM) inhibition of wild-type hypotonic currents. Subsequent addition of DCPIB further inhibits current (post-red DCPIB; *P = 0.038 vs. TCEP current level; ratio paired t-test, n = 2). C-E, Effects of TCEP application and ChlorT washout on isotonic currents in LRRC8A(−/−) mutant cells expressing 8D-8A IL. C, TCEP (0.5–1.0 mM) exerted no effect on isotonic current amplitudes (P = 0.4063; n = 5). Following TCEP exposure, addition of 1 mM ChlorT produced strong inhibition (post-red. ChlorT; N = 2), while DCPIB resulted in current block (post-red. DCPIB; N = 3). D, Current amplitude changes recorded in two cells (+120 mV). Addition of 1 mM ChlorT at t0 (2–3 min exposure) resulted in rapid inhibition by 60–80%. In cell 1 (black symbols), subsequent TCEP application (0.5 mM; 6 min) results in partial amplitude recovery, and addition of DCPIB results in >80% block of remaining current. In cell 2 (red symbols) washout of ChlorT results in a similar current amplitude recovery (red arrow, 6 min wash with control isotonic saline). Subsequent TCEP application (1 mM, 3 min) results in only minimal increase, while DCPIB blocks ~70% of remaining current. E, Summary of current changes during TCEP and wash experiments depicted in D. Symbols indicate individual recordings; values are plotted relative to pre-ChlorT control amplitude. +ChlorT (green symbols): amplitude following exposure to 1 mM ChlorT (n = 13). Post-ox TCEP (blue symbols): amplitude following exposure to 0.5 – 1.0 mM TCEP after ChlorT (no intervening wash); P < 0.0001 vs. ChlorT, paired ratio t-test (n = 13). Red symbols: Amplitude recovery following ChlorT washout only (post-ox. wash); this value was not distinguishable from the effects of TCEP (n = 4; P = 0.9399 vs TCEP; Mann-Whitney test). Post-reductant DCPIB current (orange symbols) was assessed following sequential exposure to ChlorT and TCEP, with or without intervening wash (n = 9; ††P = 0.0074 vs. ChlorT current level).
We next examined the effects of sequential ChlorT and TCEP exposure under isotonic conditions in 8A(−/−) cells expressing 8D-8A IL, which exhibited potent oxidant sensitivity (Fig. 10C–E). Initial exposure to TCEP (0.5 – 1.0 mM, 2–5 min) did not significantly alter current amplitudes (92% (26%) of control, P = 0.4063, n = 5; Fig. 10C), while subsequent DCPIB application blocked current by >90% (post-red. DCPIB, n = 3; Fig. 10C). ChlorT applied immediately following TCEP strongly inhibited current amplitudes (post-red. ChlorT, n = 2; Fig. 10C), indicating that ChlorT inhibitory action was not impaired by a reduced environment. We then tested whether TCEP was able to reverse ChlorT-mediated current inhibition. ChlorT was first applied to cells (1 mM, 1.5 – 3 min exposure) to induce current inhibition, followed by TCEP exposure (2.5 – 6 min) to assess reversibility (Fig. 10D). Current amplitudes demonstrated partial recovery in TCEP, ranging from fold increases of 20%−30% to >2-fold over the minimal ChlorT current level (66% (12%, SEM) increase, n = 10; Fig. 10D, E). To test whether current recovery was attributable to ChlorT washout, we performed additional recordings that incorporated a wash period following ChlorT (Fig. 10D). We observed a comparable 50% (27%, SEM) current amplitude increase associated with washout alone (n = 4; Fig. 10D, E) that did not differ statistically from the TCEP-associated increase (P = 0.9399 vs. TCEP, Mann-Whitney test; Fig. 10E). Addition of TCEP following a wash period (Fig. 10D) did not further significantly increase current amplitude (P = 0.2306 vs. wash alone; paired ratio t-test, n = 4). We conclude that ChlorT-mediated changes underlying current inhibition are not readily reversible by a reducing agent under these experimental conditions.
LRRC8D extracellular domains confer the inhibitory response to oxidation
The EL1 and EL2 amino acid sequences of LRRC8A, 8C and 8D and the general structural organization of LRRC8 proteins reveal multiple potential targets for redox active agents to mediate external structural changes to VRACs (Fig. 5A, B). In crystalized LRRC8A homohexamers, EL1 and EL2 are stabilized by three disulfide bonds (Deneka et al., 2018; Kasuya et al., 2018; Kern et al., 2019). In contrast, the EL1 and EL2 domains of LRRC8C and 8D contain each contain 2 conserved cysteines (Fig. 5B). The LRRC8C and 8D EL1 sequences differ significantly from each other and from 8A, with 8D EL1 containing 34 more residues compared to 8C. Methionine can also be oxidized by ChlorT, representing another potential target for regulation (Drazic & Winter, 2014) and methionine redox-dependent modulation can regulate protein function (Drazic & Winter, 2014). LRRC8C and 8D share a conserved methionine in each EL, while other methionine locations differ significantly between 8C, 8D, and 8A (Fig. 5B). Changes in the redox state of any of these sites could potentially regulate external channel structure, including the architecture of the DCPIB binding site in EL1 (Kern et al., 2019).
To directly test the involvement of LRRC8D extracellular structure in ChlorT-mediated current inhibition, we created LRRC8D and 8C EL chimeras. Substitution of EL1 of 8D onto the LRRC8C-8A IL construct (8C-8D EL1; Fig. 5C) resulted in currents that did not respond significantly to 1 mM ChlorT (88.6% (27.8%) of control, n = 4; P = 0.4308; Fig. 11C). By contrast, substitution of both EL1 and EL2 of 8D onto 8C (8C-8D EL1,2; Fig. 11A, B) greatly enhanced current inhibition (68.8% (6.8%) inhibition, n = 7; P = 0.0006 vs. 8C-8A IL; Fig. 11C). Conversely, substitution of EL1 of 8C onto 8D-8A IL abolished inhibition and resulted in mild current augmentation in response to 1 mM ChlorT (34% (20%) increase; n = 5; P= 0.0109 vs. control; Fig. 11C). Post-ox. DCPIB block was absent in each EL1 and EL1/EL2 variant (Fig. 11B, and data not shown). These results demonstrate that extracellular structural elements of both EL1 and EL1 of LRRC8D are required for the strong current inhibition by ChlorT.
Figure 11.
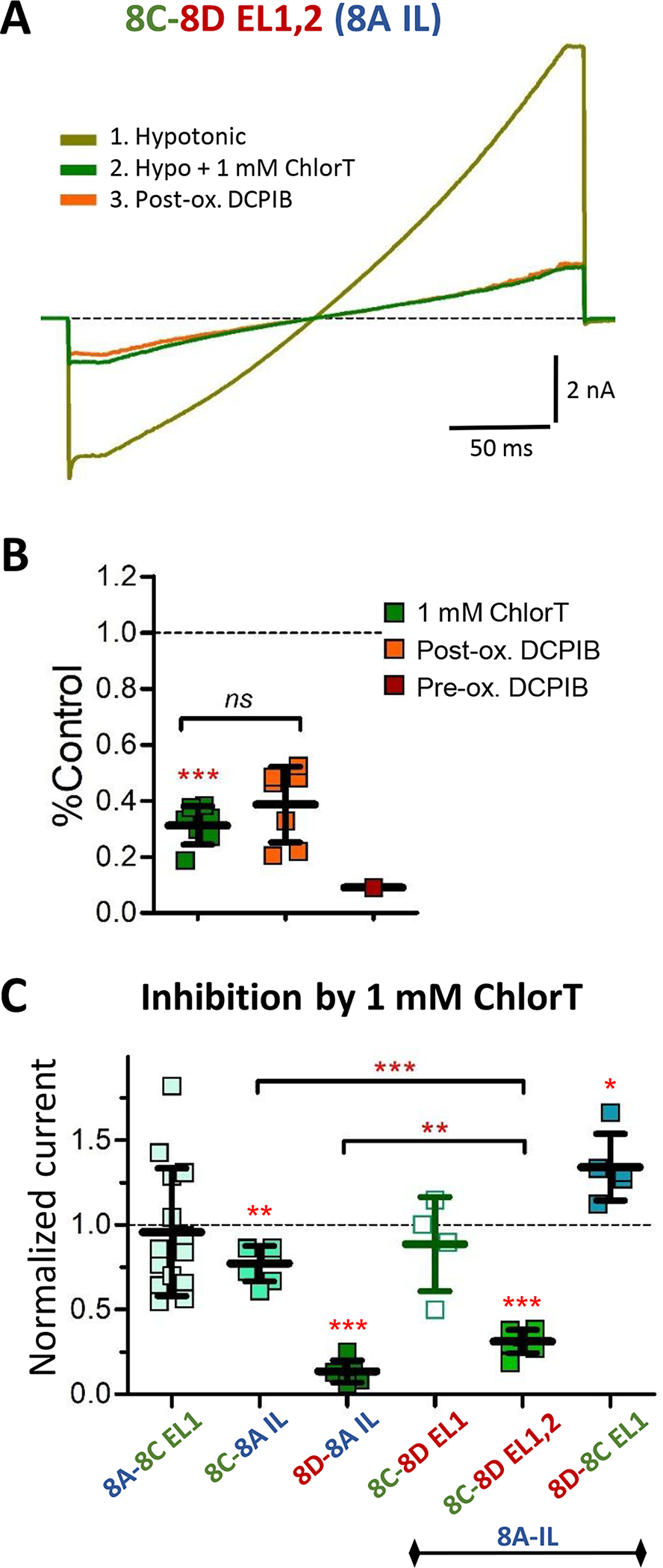
The extracellular loop domains of LRRC8D confer enhanced inhibitory ChlorT response to LRRC8C. A, Currents evoked by 250 ms voltage ramps (−100 to +120 mV) in hypotonic saline, recorded from a cell expressing the LRR8C-8A IL protein substituted with both 8D extracellular loops (8C-8D EL1,2). Current is strongly inhibited by 1 mM ChlorT (green). DCPIB is ineffective in blocking residual current following ChlorT exposure (post-ox. DCPIB; orange). B, Summary of 8C-8D EL1,2 current inhibition by ChlorT and DCPIB. 1 mM ChlorT inhibits current by 69% (***P < 0.0001 vs hypotonic control current; paired ratio t-test, n = 7). Post-ox. DCPIB produces no further current block (P = 0.1431, n = 7). C, Comparison of current sensitivity to 1 mM ChlorT in LRRC8A(−/−) mutant cells expressing various LRRC8A, 8C, and 8D chimeric constructs. Symbols represent individual recordings; values are normalized to current amplitude at +120 mV in control saline. LRRC8A (8A-8C EL1; n = 14) displays variable individual responses, but no overall oxidant sensitivity (P = 0.2837 vs. Control; same data as Fig. 8I). Homomeric LRRC8C-8A IL shows weak inhibition (Fig. 8G), while LRRC8D-8A IL shows strong inhibition (Fig. 8H, 1 mM ChlorT). Substitution of EL1 of LRRC8D onto 8C (8C-8D EL1) does not alter the response to ChlorT (P = 0.4308 vs 8C-8A IL; n = 4). Substitution of both EL1 and EL2 (8C-8D EL1,2) confers a strong inhibitory oxidant response (n = 7; as in B. ***P = 0.0006 vs. 8C-8A IL, **P = 0.0023 vs. 8D-8A IL; Mann-Whitney tests). Substitution of EL1 of LRRC8C onto 8D abolishes inhibition and results in mild augmentation of current amplitude (*P = 0.0109 vs. Control; n = 5).
DISCUSSION
Differential roles for LRRC8C and 8D in TNFα –dependent inflammatory signaling
We previously established that the requisite VRAC subunit LRRC8A associates with Nox1 and is required for TNFα signaling in VSMCs (Choi et al., 2016). We now show differential roles for the LRRC8A/C and LRRC8A/D channel subtypes. Nox1 physically associates with both 8C and 8D, but A/C channels most efficiently support Nox1 oxidase activity. LRRC8C knockdown depresses TNFα-dependent extracellular O2•- production, TNFα receptor endocytosis, NF-κB activation, and cell proliferation as effectively as siLRRC8A. We conclude that TNFα signaling critically depends on LRRC8A/C channel activity. In contrast, LRRC8D knockdown does not significantly alter O2−• production and receptor endocytosis, and enhances NF-κB activation. The direct proximity of Nox1 and LRRC8 likely exposes the channels to an oxidized microenvironment. The relative oxidant insensitivity of A/C channel currents may promote sustained O2•- production by Nox1, facilitating pro-inflammatory signaling. Inhibitory oxidant sensitivity is conferred predominantly by the LRRC8D extracellular loop domains.
TNFα-dependent inflammation is implicated in atherosclerosis (Kosmas et al., 2019), and in aortic (Rabkin, 2015) and cerebral aneurysms (Signorelli et al., 2018). Our analysis of gene expression data shows a pronounced downregulation of LRRC8A in vascular diseases. Gene groupings with strongly downregulated expression in atherosclerotic tissue included cytokine-cytokine receptor interaction (hsa04060; including TNFα and IL-1β and receptors), and chemokine signaling (hsa04062) (Biros et al., 2015). A straightforward interpretation is that downregulated tissue expression of these genes reflects a compensatory response to a poorly controlled inflammatory disease state. LRRC8A downregulation reduces VSMC Nox1-and Angiotensin II-dependent signaling (Choi et al., 2016; Lu et al., 2019), and is protective against ischemic stroke (Zhou et al., 2020). The VRAC inhibitor DCPIB is likewise anti-inflammatory in multiple contexts in vivo (Zhang et al., 2008; Alibrahim et al., 2013; Han et al., 2014). The vascular diseases analyzed here are associated with moderately increased expression of LRRC8C and LRRC8D, but no change in 8C:8D expression ratio. However, a different pattern of regulation of 8C vs. 8D is seen in psoriasis. Affected skin is associated with a >50% reduction in 8C:8D expression. Psoriasis is commonly treated by targeting TNFα signaling (Chima & Lebwohl, 2018), and Nox1 inhibition reduced inflammation and improved human keratinocyte survival in an experimental model of psoriasis (Emmert et al., 2020). The expression changes favoring 8D over 8C in psoriatic skin again suggest a compensatory response to downregulate pro-inflammatory Nox-dependent signaling. Relatively little is currently known regarding how VRAC subtype composition and trafficking are regulated. Regulation of Nox1 association with 8A/D vs. 8A/C channel subtypes might also contribute to a local compensatory response to increased cytokine activity. These results suggest an important link between LRRC8 isoform expression and human disease, and highlight the potential therapeutic utility of isoform-selective pharmacologic agents.
Nox-dependent ROS production is induced by cell swelling, and ROS can be required for subsequent VRAC activation (Lambert, 2003; Holm et al., 2013). VRACs can also be activated under isotonic conditions by agents known to activate Nox1, including epidermal growth factor, Angiotensin II and TNFα (Browe & Baumgarten, 2004; Varela et al., 2004; Matsuda et al., 2010; Crutzen et al., 2012). The LRRC8 channel subtype(s) underlying these currents is unknown. In oocyte expression studies, currents mediated by heteromeric LRRC8A/E channels were activated by oxidants (Gradogna et al., 2017), a potential role for A/E channels. However, it is also conceivable that ROS indirectly activates A/C or A/D channels by cytoplasmic mechanisms that are independent of the direct extracellular effects of oxidation. Importantly, Nox oxidases can deposit O2•- only into the extracellular space, or within the lumen of intracellular vesicles (reviewed in (Lamb et al., 2020)). It has been speculated that O2•- may cross membranes via anion channels (reviewed in (Lamb et al., 2020)). H2O2 clearly does cross membranes via aquaporin channels (Bienert et al., 2007), and therefore may act on either side of the bilayer. Defining the specific site where oxidants acts on transmembrane proteins remains critically important.
Extracellular loop domains confer differential oxidant sensitivities for LRRC8C and 8D channel currents
The differential roles for LRRC8C and LRRC8D in TNFα signaling suggested that 8C and 8D channel currents might be differentially sensitive to oxidation. We showed that both native VRAC currents and reconstituted currents in LRRC8A(−/−) HEK293 cells are inhibited 50%−60% by 1 mM ChlorT. LRRC8C and 8D proteins substituted with the 8A-IL sequence were previously demonstrated to form functional homomeric VRAC channels in LRRC8 knockout HCT116 cells (Yamada & Strange, 2018). We utilized these chimeric proteins in LRRC8A(−/−) HEK cells to assess 8C- and 8D-specific differences in oxidant sensitivity in the absence of contributions from other subunits. 8D-8A IL currents displayed ~4-fold greater inhibition to 1 mM ChlorT than 8C-8A IL currents, while 8A-8C EL1 currents were unresponsive. A reasonable interpretation is that these currents are predominantly mediated by homomeric channels formed by the expressed chimeric proteins. This conclusion is supported by the substantial currents present under isotonic conditions, attributable to the activating effect of the C-terminal fluorescent tag on all subunits (Gaitan-Penas et al., 2016), although hypotonicity is required for complete current activation (Gradogna et al., 2017; Yamada & Strange, 2018). We cannot exclude heteromeric association of the expressed chimeras with endogenous non-A LRRC8 proteins. However, such channels containing untagged native proteins would be minimally activated under isotonic conditions (Gaitan-Penas et al., 2016). The observed (2- to 3-fold) augmentation of isotonic currents by hypotonic conditions is consistent with the recorded currents being mediated predominantly by homomeric channels of the expressed chimeric proteins. In isomer co-expression experiments (Fig. 9), the combination 8A-mCh with native non-A proteins likely contributed to recorded hypotonic currents. 8A/D co-expression nevertheless resulted in currents displaying ~2-fold greater inhibition to ChlorT than did 8A/C co-expression. The results indicate that enhanced oxidant inhibition is not unique to homomeric 8D-8A IL channels, but is primarily conferred by LRRC8D. The oxidant sensitivity of native VRAC current suggests a predominant expression of LRRC8A/D-type channels, consistent with our qualitative PCR assay as well as prior quantitative RT-PCR analysis (Friard et al., 2017).
Our results indicate that oxidant-mediated current inhibition is related to structural modification of the extracellular loop (EL1, EL2) domains. The crystal structure of homomeric LRRC8A channels reveals two disulfide bonds between conserved pairs of cysteines linking EL1 with EL2, and a third disulfide within EL1 (Deneka et al., 2018; Kern et al., 2019). LRRC8C and 8D subunits can potentially form at most two disulfide bonds between EL1 and EL2. The status and importance of these bonds in functional channels under physiological conditions is unknown. Both EL1 and EL2 of LRRC8D are required for current inhibition, or to confer enhanced inhibition to LRRC8C. This suggests that ChlorT must oxidize targets in both loops to effect inhibition, perhaps by inducing inter-loop disulfide formation. Disulfide bond formation could also potentially occur between channel subunits. ChlorT readily oxidizes glutathione (GSH) to a disulfide (GSSG), but cannot further oxidize cysteines to sulfinic or sulfonic acids (Shechter et al., 1975). ChlorT is also commonly used to target methionine residues, several of which are present in the EL loops of LRRC8C and 8D, and redox-dependent methionine modulation can regulate protein function (Drazic & Winter, 2014). Currents mediated by heteromeric 8A/D channels expressed in oocytes were inhibited by MTSET, a membrane-impermeant modifier of extracellular cysteines (Gradogna et al., 2017), consistent with an alteration of extracellular loop structure.
Membrane permeant (DTT) and impermeant (TCEP) reducing agents are theoretically capable of reversing these changes. However, these reagents did not significantly alter LRRC8D currents in HEK293 cells, suggesting that either the channel extracellular structure is largely reduced, or that the redox active sites are inaccessible to TCEP (Metcalfe et al., 2011). Extracellular redox potential (Cysteine/Cystine) in vivo is subject to change, and becomes progressively oxidized in cardiovascular disease and with aging, smoking, and obesity (Go & Jones, 2011). Nox1 activation will create an even more oxidized local environment. Somewhat unexpectedly, TCEP was unable to reverse ChlorT-mediated LRRC8D current inhibition. A potential explanation is that key structural aspects of the oxidized extracellular loops cannot be restored or “reset” by simple reduction. Disulfides can act as “switches”, sensing extracellular redox potential and modulating protein function (Yi & Khosla, 2016). Redox-reactive proteins such as thioredoxin or protein disulfide isomerases (PDI) exist in cytoplasmic and secreted extracellular forms and interact with the local redox environment to regulate these disulfides. PDI is known to modulate Nox1 intracellularly via disulfide formation with p47phox, a cytoplasmic subunit required for oxidase activity (Janiszewski et al., 2005; Gimenez et al., 2019).
ChlorT exposure impaired subsequent DCPIB block of endogenous VRAC currents, and currents resulting from expressed LRRC8 constructs, further suggesting that oxidation modifies extracellular loop structure. DCPIB binding and block of LRRC8A homomers is localized to a ring within the channel pore, formed by the association of R108 in EL1 of all six subunits (Kern et al., 2019), and is thought to form the channel ion selectivity filter (Denecka et al., 2018). Non-arginine residues in the same positions are apparently capable of subserving the same function in LRRC8C and LRRC8D channels. Residual post-oxidant DCPIB block efficacy was most evident for 8A-8C EL1 currents, which lacked ChlorT sensitivity, and most difficult to quantify for LRRC8D currents (8D-8A IL and 8A/D co-expression) because ChlorT alone exerts a profound inhibitory effect. Further investigation will be necessary to distinguish the relationship between oxidant-dependent channel modification, and channel binding and block by DCPIB. Post-oxidant DCPIB sensitivity in 8D appeared to be partially restored by TCEP exposure, even though TCEP did not significantly current inhibition by ChlorT. The baseline potency of DCPIB block of both native VRACs and currents resulting from expressed LRRC8 constructs again suggests that under control conditions, channels are mostly in a reduced state.
LRRC8A/D channel inhibition by extracellular oxidants may be relevant to pathophysiologic conditions associated with oxidative stress. VRACs have been broadly implicated in cancer cell proliferation, migration and invasion, and consequently have been proposed as targets for cancer therapy (reviewed in (Xu et al., 2020)). A/D channels are permeable to uncharged osmolytes such as taurine, as well as to platinum-based anticancer therapeutics (Planells-Cases et al., 2015). An unbiased genome-wide screen identified LRRC8A and 8D knockdown as inducers of resistance to these drugs. Furthermore, low LRRC8D expression was correlated with decreased survival in ovarian cancer patients, suggesting that reduced drug uptake via A/D channels may reduce treatment effectiveness (Planells-Cases et al., 2015). Given the near ubiquity of increased ROS production by cancer cells (Gorrini et al., 2013), endogenous LRRC8A/D channel oxidation could reduce the uptake of chemotherapeutics, a limitation that might be amenable to anti-oxidant therapy (Ghoneum et al., 2020).
In summary, LRRC8A/C VRACs are the primary LRRC8 isoform responsible for support of Nox1 activity and TNFα signaling, which may be facilitated by the relative oxidant insensitivity of LRRC8A/C currents. The relative abundance of the LRRC8C vs. LRRC8D significantly influences the inflammatory response of VSMCs to TNFα. These observations may have clinical relevance, as LRRC8 isoform expression is altered in people with atherosclerosis and psoriasis, suggesting compensation for an inflamed state. Thus, LRRC8 VRACs are exciting potential targets for anti-inflammatory therapeutic agents.
Key Points.
LRRC8A-containing anion channels associate with Nox1 and regulate superoxide production and TNFα signaling. Here we show that LRRC8C and 8D also co-immunoprecipitate with Nox1 in vascular smooth muscle cells.
LRRC8C knockdown inhibited TNFα-induced O2•- production, receptor endocytosis, NF-κB activation, and proliferation while LRRC8D knockdown enhanced NF-κB activation. Significant changes in LRRC8 isoform expression in human atherosclerosis and psoriasis suggest compensation for increased inflammation.
The oxidant chloramine-T (ChlorT, 1 mM) weakly (~25%) inhibited LRRC8C currents but potently (~80%) inhibited LRRC8D currents. Substitution of the extracellular loop (EL1, EL2) domains of 8D onto 8C conferred significantly stronger (69%) ChlorT-dependent inhibition.
ChlorT exposure impaired subsequent current block by DCPIB, which occurs through interaction with EL1, further implicating external oxidation sites.
LRRC8A/C channels most effectively sustain Nox1 activity at the plasma membrane. This may result from their ability to remain active in an oxidized microenvironment.
ACKNOWLEDGMENTS
This project was supported by R01 HL128386 (F. Lamb) and by a Career Development Award from the American Heart Association 16SDG30610002 (H. Choi). We wish to thank Dr. Kevin Strange (Novo Biosciences and Vanderbilt University) for providing chimeric LRRC8 plasmids and Dr. Rajan Sah (Washington University) for the LRRC8A null HEK293 cells.
ABBREVIATIONS
- Ad
adenovirus
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- CAT1H
1-Hydroxy-2,2,6,6-tetramethylpiperidin-4-yl-trimethylammonium chloride
- DCPIB
4-(2-Butyl-6,7-dichloro-2-cyclopentyl-indan-1-on-5-yl) oxobutyric acid
- DMEM
Dulbecco’s modified Eagle’s medium
- eGFP
enhanced green fluorescent protein
- ESR
electron spin resonance
- FBS
fetal bovine serum
- FITC
Fluorescein isothiocyanate
- LRRC8
Leucine Rich Repeat Containing 8
- NADPH
reduced nicotinamide-adenine dinucleotide phosphate
- NF-κB
nuclear factor-kappa B
- Nox
NADPH oxidase
- ROS
reactive oxygen species
- SEM
standard error of the mean
- siRNA
small interfering ribonucleic acid
- TNFα
tumor necrosis factor-α
- TNFR
tumor necrosis factor-α receptor
- VRAC
volume-regulated anion channel
- VSMC
vascular smooth muscle cell
Footnotes
CONFLICT OF INTEREST
None declared.
REFERENCES
- Alibrahim A, Zhao LY, Bae CY, Barszczyk A, Sun CL, Wang GL & Sun HS. (2013). Neuroprotective effects of volume-regulated anion channel blocker DCPIB on neonatal hypoxic-ischemic injury. Acta Pharmacol Sin 34, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK & Jahn TP. (2007). Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem 282, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Biros E, Gabel G, Moran CS, Schreurs C, Lindeman JH, Walker PJ, Nataatmadja M, West M, Holdt LM, Hinterseher I, Pilarsky C & Golledge J. (2015). Differential gene expression in human abdominal aortic aneurysm and aortic occlusive disease. Oncotarget 6, 12984–12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browe DM & Baumgarten CM. (2004). Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl- current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol 124, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chima M & Lebwohl M. (2018). TNF inhibitors for psoriasis. Semin Cutan Med Surg 37, 134–142. [DOI] [PubMed] [Google Scholar]
- Choi H, Dikalova A, Stark RJ & Lamb FS. (2015). c-Jun N-terminal kinase attenuates TNFalpha signaling by reducing Nox1-dependent endosomal ROS production in vascular smooth muscle cells. Free Radic Biol Med 86, 219–227. [DOI] [PubMed] [Google Scholar]
- Choi H, Ettinger N, Rohrbough J, Dikalova A, Nguyen HN & Lamb FS. (2016). LRRC8A channels support TNFalpha-induced superoxide production by Nox1 which is required for receptor endocytosis. Free Radic Biol Med 101, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Stark RJ, Raja BS, Dikalova A & Lamb FS. (2019). Apoptosis signal-regulating kinase 1 activation by Nox1-derived oxidants is required for TNFalpha receptor endocytosis. Am J Physiol Heart Circ Physiol 316, H1528–H1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutzen R, Shlyonsky V, Louchami K, Virreira M, Hupkens E, Boom A, Sener A, Malaisse WJ & Beauwens R. (2012). Does NAD(P)H oxidase-derived H2O2 participate in hypotonicity-induced insulin release by activating VRAC in beta-cells? Pflugers Arch 463, 377–390. [DOI] [PubMed] [Google Scholar]
- DeCoursey TE. (2016). The intimate and controversial relationship between voltage-gated proton channels and the phagocyte NADPH oxidase. Immunol Rev 273, 194–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Morgan D & Cherny VV. (2003). The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 422, 531–534. [DOI] [PubMed] [Google Scholar]
- Deneka D, Sawicka M, Lam AKM, Paulino C & Dutzler R. (2018). Structure of a volume-regulated anion channel of the LRRC8 family. Nature. [DOI] [PubMed] [Google Scholar]
- Drazic A & Winter J. (2014). The physiological role of reversible methionine oxidation. Biochim Biophys Acta 1844, 1367–1382. [DOI] [PubMed] [Google Scholar]
- Emmert H, Fonfara M, Rodriguez E & Weidinger S. (2020). NADPH oxidase inhibition rescues keratinocytes from elevated oxidative stress in a 2D atopic dermatitis and psoriasis model. Exp Dermatol 29, 749–758. [DOI] [PubMed] [Google Scholar]
- Gaitan-Penas H, Gradogna A, Laparra-Cuervo L, Solsona C, Fernandez-Duenas V, Barrallo-Gimeno A, Ciruela F, Lakadamyali M, Pusch M & Estevez R. (2016). Investigation of LRRC8-Mediated Volume-Regulated Anion Currents in Xenopus Oocytes. Biophys J 111, 1429–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneum A, Abdulfattah AY, Warren BO, Shu J & Said N. (2020). Redox Homeostasis and Metabolism in Cancer: A Complex Mechanism and Potential Targeted Therapeutics. International journal of molecular sciences 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez M, Schickling BM, Lopes LR & Miller FJ Jr. (2016). Nox1 in cardiovascular diseases: regulation and pathophysiology. Clin Sci (Lond) 130, 151–165. [DOI] [PubMed] [Google Scholar]
- Gimenez M, Verissimo-Filho S, Wittig I, Schickling BM, Hahner F, Schurmann C, Netto LES, Rosa JC, Brandes RP, Sartoretto S, De Lucca Camargo L, Abdulkader F, Miller FJ Jr. & Lopes LR. (2019). Redox Activation of Nox1 (NADPH Oxidase 1) Involves an Intermolecular Disulfide Bond Between Protein Disulfide Isomerase and p47(phox) in Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 39, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM & Jones DP. (2011). Cysteine/cystine redox signaling in cardiovascular disease. Free Radic Biol Med 50, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS & Mak TW. (2013). Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12, 931–947. [DOI] [PubMed] [Google Scholar]
- Gradogna A, Gavazzo P, Boccaccio A & Pusch M. (2017). Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J Physiol 595, 6719–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Liu S, Li Z, Hu F, Zhang Q, Zhou M, Chen J, Lei T & Zhang H. (2014). DCPIB, a potent volume-regulated anion channel antagonist, attenuates microglia-mediated inflammatory response and neuronal injury following focal cerebral ischemia. Brain Res 1542, 176–185. [DOI] [PubMed] [Google Scholar]
- Holm JB, Grygorczyk R & Lambert IH. (2013). Volume-sensitive release of organic osmolytes in the human lung epithelial cell line A549: role of the 5-lipoxygenase. Am J Physiol Cell Physiol 305, C48–60. [DOI] [PubMed] [Google Scholar]
- Janiszewski M, Lopes LR, Carmo AO, Pedro MA, Brandes RP, Santos CXC & Laurindo FRM. (2005). Regulation of NAD(P)H Oxidase by Associated Protein Disulfide Isomerase in Vascular Smooth Muscle Cells. J Biol Chem 280, 40813–40819. [DOI] [PubMed] [Google Scholar]
- Kasuya G, Nakane T, Yokoyama T, Jia Y, Inoue M, Watanabe K, Nakamura R, Nishizawa T, Kusakizako T, Tsutsumi A, Yanagisawa H, Dohmae N, Hattori M, Ichijo H, Yan Z, Kikkawa M, Shirouzu M, Ishitani R & Nureki O. (2018). Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat Struct Mol Biol 25, 797–804. [DOI] [PubMed] [Google Scholar]
- Kern DM, Oh S, Hite RK & Brohawn SG. (2019). Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig B & Stauber T. (2019). Biophysics and Structure-Function Relationships of LRRC8-Formed Volume-Regulated Anion Channels. Biophys J 116, 1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmas CE, Silverio D, Sourlas A, Montan PD, Guzman E & Garcia MJ. (2019). Anti-inflammatory therapy for cardiovascular disease. Ann Transl Med 7, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb FS, Choi H, Miller MR & Stark RJ. (2020). TNFalpha and Reactive Oxygen Signaling in Vascular Smooth Muscle Cells in Hypertension and Atherosclerosis. American journal of hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb FS, Moreland JG & Miller FJ Jr. (2009). Electrophysiology of reactive oxygen production in signaling endosomes. Antioxid Redox Signal 11, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert IH. (2003). Reactive oxygen species regulate swelling-induced taurine efflux in NIH3T3 mouse fibroblasts. J Membr Biol 192, 19–32. [DOI] [PubMed] [Google Scholar]
- Liang W, Huang L, Zhao D, He JZ, Sharma P, Liu J, Gramolini AO, Ward ME, Cho HC & Backx PH. (2014). Swelling-activated Cl- currents and intracellular CLC-3 are involved in proliferation of human pulmonary artery smooth muscle cells. Journal of hypertension 32, 318–330. [DOI] [PubMed] [Google Scholar]
- Lu J, Xu F, Zhang J. (2019). Inhibition of angiotensin II-induced cerebrovascular smooth muscle cell proliferation by LRRC8A downregulation through suppressing PI3K/AKT activation. Human Cell 32, 316–325. [DOI] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Moreland JG, Miller FJ & Lamb FS. (2010). Activation of swelling-activated chloride current by tumor necrosis factor-alpha requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem 285, 22864–22873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe C, Cresswell P, Ciaccia L, Thomas B & Barclay AN. (2011). Labile disulfide bonds are common at the leucocyte cell surface. Open Biol 1, 110010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FJ Jr., Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ & Lamb FS. (2007). Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res 101, 663–671. [DOI] [PubMed] [Google Scholar]
- Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, Ruether A, Schreiber S, Weichenthal M, Gladman D, Rahman P, Schrodi SJ, Prahalad S, Guthery SL, Fischer J, Liao W, Kwok PY, Menter A, Lathrop GM, Wise CA, Begovich AB, Voorhees JJ, Elder JT, Krueger GG, Bowcock AM, Abecasis GR & Collaborative Association Study of P. (2009). Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, Klausen TK & Nilius B. (2015). The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol (Oxf) 213, 868–881. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, Kucukosmanoglu A, Xu G, Voss FK, Reincke SM, Stauber T, Blomen VA, Vis DJ, Wessels LF, Brummelkamp TR, Borst P, Rottenberg S & Jentsch TJ. (2015). Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J 34, 2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian JS, Pang RP, Zhu KS, Liu DY, Li ZR, Deng CY & Wang SM. (2009). Static pressure promotes rat aortic smooth muscle cell proliferation via upregulation of volume-regulated chloride channel. Cell Physiol Biochem 24, 461–470. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP & Patapoutian A. (2014). SWELL1, a Plasma Membrane Protein, Is an Essential Component of Volume-Regulated Anion Channel. Cell 157, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabkin SW. (2015). Accentuating and Opposing Factors Leading to Development of Thoracic Aortic Aneurysms Not Due to Genetic or Inherited Conditions. Front Cardiovasc Med 2, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter Y, Burstein Y & Patchornik A. (1975). Selective oxidation of methionine residues in proteins. Biochemistry 14, 4497–4503. [DOI] [PubMed] [Google Scholar]
- Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, Mathur J, Cahalan SM, Peters EC, Montal M & Patapoutian A. (2016). LRRC8 Proteins Form Volume-Regulated Anion Channels that Sense Ionic Strength. Cell 164, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela D, Simon F, Riveros A, Jorgensen F & Stutzin A. (2004). NAD(P)H oxidase-derived H(2)O(2) signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem 279, 13301–13304. [DOI] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T & Jentsch TJ. (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638. [DOI] [PubMed] [Google Scholar]
- Xia Y, Liu Y, Xia T, Li X, Huo C, Jia X, Wang L, Xu R, Wang N, Zhang M, Li H & Wang X. (2016). Activation of volume-sensitive Cl- channel mediates autophagy-related cell death in myocardial ischaemia/reperfusion injury. Oncotarget 7, 39345–39362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Wang X & Shi C. (2020). Volume-regulated anion channel as a novel cancer therapeutic target. Int J Biol Macromol 159, 570–576. [DOI] [PubMed] [Google Scholar]
- Yamada T & Strange K. (2018). Intracellular and extracellular loops of LRRC8 are essential for volume-regulated anion channel function. J Gen Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, Kleinridders A, Wunderlich T, Kashkar H, Utermohlen O, Bruning JC, Schutze S & Kronke M. (2009). Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460, 1159–1163. [DOI] [PubMed] [Google Scholar]
- Yi MC & Khosla C. (2016). Thiol-Disulfide Exchange Reactions in the Mammalian Extracellular Environment. Annu Rev Chem Biomol Eng 7, 197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ & Kimelberg HK. (2008). DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol 210, 514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JJ, Luo Y, Chen SR, Shao JY, Sah R & Pan HL. (2020). LRRC8A-dependent volume-regulated anion channels contribute to ischemia-induced brain injury and glutamatergic input to hippocampal neurons. Exp Neurol 332, 113391. [DOI] [PMC free article] [PubMed] [Google Scholar]