

Abstract
Background:
The aim of this analysis was to evaluate the clinical factors influencing survival outcomes in patients with localized (clinical group I-III) FOXO1 fusion positive rhabdomyosarcoma (RMS).
Methods:
Patients with confirmed FOXO1 fusion positive RMS who were enrolled on three completed clinical trials for localized RMS were included in the analytic cohort. Outcomes were analyzed using the Kaplan-Meier method to estimate event-free survival (EFS) and overall survival (OS), and the curves were compared using the log-rank test. A Cox proportional hazards regression model was used to perform multivariate analysis of prognostic factors that were significant in the univariate analysis.
Results:
The estimated 4-year EFS and OS of 269 localized FOXO1 fusion positive RMS were 53% (95% CI: 47%-59%) and 69% (95% CI: 63%-74%), respectively. Univariate analysis revealed that a number of known favorable clinical characteristics including age at diagnosis between 1 and 9 years, complete surgical resection, tumor size ≤ 5 cm, favorable tumor site, absence of lymph node involvement, confinement to the anatomic site of origin, and PAX7-FOXO1 fusion were associated with improved outcomes. Multivariate analysis identified older age (≥10 years) and large tumor size (> 5 cm) as independent adverse prognostic factors for EFS within this population, and patients who had both adverse features experienced substantially inferior outcomes.
Conclusion:
Patients with localized FOXO1 fusion positive RMS can be further risk stratified based on clinical features at diagnosis and older patients with large primary tumors have the poorest prognosis.
Keywords: rhabdomyosarcoma, localized, translocation, risk stratification, prognosis, alveolar, PAX3, PAX7, FOXO1
Precis:
This study analyzes the effect of clinical features on outcomes for patients with localized FOXO1 fusion-positive rhabdomyosarcoma (RMS) treated on recent Children’s Oncology Group studies. Two clinical features, older age (≥10 years) and large tumor size (> 5 cm), were found to be independent adverse prognostic indicators for relapse in this patient group, and patients that had both features experienced outcomes similar to RMS patients with distant metastases.
Introduction
The classification of rhabdomyosarcoma (RMS) in children and adolescents has historically relied on histologic features, with embryonal (ERMS) and alveolar (ARMS) subtypes comprising the vast majority of cases.1 Recent developments in molecular testing have revealed that ARMS is correlated with the chromosomal translocations t(2;13) or t(1;13), which result in fusions of PAX3-FOXO1 or PAX7-FOXO1, respectively.1–3 A minority of tumors histologically classified as ARMS lack a PAX3/7-FOXO1 fusion and very rare cases of tumors bearing a PAX3/7-FOXO1 fusion are histologically classified as being ERMS or mixed histology. Nevertheless, the current classification of RMS remains a histologic definition.4
RMS treatment in North America is dictated by a risk stratification strategy that uses tumor histology, clinical stage including tumor site, nodal status, and presence or absence of distant metastases, and clinical group which is based on the extent of surgical resection prior to treatment and regional lymph node or distant spread.5 Although the use of clinical features for risk stratification has been in place for decades, a major challenge to this approach has been an evolving definition of what constitutes ARMS histology.6–11 The current criteria used by the Children’s Oncology Group (COG) to diagnose ARMS mandates that at least 50% of the sample have alveolar histology and emphasizes the presence of strong, diffuse myogenin staining to support the diagnosis.10
As molecular techniques have become more readily available, FOXO1 fusion testing has become integral to the diagnostic work-up for RMS, and has been recently incorporated into risk stratification by COG. Fusion positive (FP) ARMS (ARMSp) accounts for about 80% of ARMS, with PAX3 and PAX7 fusions representing 60% and 20% of all ARMS cases, respectively.11,12 Furthermore, there is a consensus that fusion negative (FN) ARMS (ARMSn) tumors are biologically and clinically distinct from ARMSp tumors,13–15 with inferior outcomes for ARMSp tumors16,17 compared to ARMSn, which have favorable outcomes similar to ERMS.10,12–14,18–22
Based on a recent analysis of patients treated on six prior COG studies, COG has proposed using a refined risk stratification using FOXO1 fusion status in addition to standard clinical characteristics in the next generation of clinical trials. In that analysis, FOXO1 status was the most significant factor impacting outcomes among patients with localized disease, with overall survival of 65% vs. 88% for FP and FN-RMS respectively.22 However, clinical factors influencing outcomes among a large group of patients with localized FOXO1 FP tumors have not been examined previously. The purpose of this study was to describe the outcomes for patients with localized (group I-III) FOXO1 FP-RMS treated contemporaneously on COG studies conducted between 1997 and 2013.
Methods
Patients with newly diagnosed localized FOXO1 FP-RMS enrolled on previously reported COG studies (D9602, D9803, and ARST0531) were included in this analysis.9,23,24 These trials were approved by institutional review boards (IRB) of each participating institution or the Pediatric Central IRB of the National Cancer Institute. Informed consent/assent from the patient and/or parent/guardian as appropriate was obtained prior to enrollment. Centralized FOXO1 fusion testing using RT-PCR or FISH was performed as previously described; if not available, institutional assessment was used.22
Outcomes were assessed by the event-free survival (EFS) and overall survival (OS), with an event defined by relapse/progression, second malignancy, or death of any cause, whichever occurred first. EFS and OS were assessed at 4 years, due to the limited follow-up of patients beyond that time point. Data were censored at the day of last contact for patients free of an event or who remained alive. Follow-up was current as of December 31, 2018. The Kaplan-Meier method was used to estimate the EFS and OS distributions and estimates with confidence interval calculated using the Peto-Peto method.25 Differences between survival curves were analyzed by the log-rank test. A Cox proportional hazards regression model was used to adjust comparisons of the EFS and OS by including prognostic factors that were significant in the univariate analysis.
Results
Characteristics for the 269 eligible patients with FP-RMS are shown in Table 1. Approximately half of the patients (n=141; 52%) were treated on ARST0531, and slightly fewer (n=115; 43%) were treated on D9803. A minority (n=13; 5%) were treated on D9602 prior to stage 1 and stage 2/3, group I/II ARMS patients being reclassified from low- to intermediate risk, and thus being made ineligible for that study. Histologically, the vast majority of tumors were classified as alveolar (n=257; 96%), although one tumor was classified as ERMS (0.4%), and few tumors were classified as mixed RMS (n=6; 2%), or RMS not otherwise specified (n=5; 2%). The specific FOXO1 fusion partner was available for most of the FP tumors (n=243; 90%), with PAX3 fusions representing the vast majority (n=192; 71%).
Table 1.
Characteristics of localized patients with FOXO1 fusion positive status (n=269).
| Characteristic | N (%) |
|---|---|
| Study | |
| D9602 | 13 (4.8) |
| D9803 | 115 (42.8) |
| ARST0531 | 141 (52.4) |
| Sex | |
| Male | 139 (51.7) |
| Female | 130 (48.3) |
| Age | |
| <1 | 17 (6.3) |
| 1-9 | 118 (43.9) |
| 10+ | 134 (49.8) |
| Primary tumor site | |
| Bladder/prostate | 3 (1.1) |
| Extremity | 81 (30.1) |
| Genitourinary, non-bladder/prostate* | 4 (1.5) |
| Head and neck* | 31 (11.5) |
| Intrathoracic | 1 (0.4) |
| Orbit* | 14 (5.2) |
| Parameningeal | 92 (34.2) |
| Perineum/anus | 13 (4.8) |
| Retroperitoneum | 8 (3.0) |
| Trunk | 19 (7.1) |
| Other | 3 (1.1) |
| Tumor size | |
| ≤5 cm | 158 (58.7) |
| >5 cm | 108 (40.1) |
| Unknown | 3 (1.1) |
| Nodal | |
| No, N0 | 181 (67.3) |
| Yes, N1 | 87 (32.3) |
| Unknown | 1 (0.4) |
| Invasiveness (T status) | |
| T1 | 146 (54.3) |
| T2 | 123 (45.7) |
| Group | |
| I | 17 (6.3) |
| II | 66 (24.5) |
| III | 186 (69.1) |
| Histology | |
| Alveolar rhabdomyosarcoma | 257 (95.5) |
| Embryonal rhabdomyosarcoma | 1 (0.4) |
| Not otherwise specified | 5 (1.9) |
| Mixed rhabdomyosarcoma | 6 (2.2) |
| FOXO1 fusion partner | |
| PAX3 | 192 (71.4) |
| PAX7 | 51 (19) |
| Unknown | 26 (9.7) |
Favorable site
Across all patients, the estimated 4-year EFS and OS rates were 53% (95% CI, 47%-59%) and 69% (95% CI, 63%-74%), respectively (Figure 1 and Supplemental Table 1). Among patient characteristics, age at diagnosis was the only prognostic factor correlated with both EFS (p=0.01) and OS (p=0.0001) based on univariate analysis. Patients between 1 and 9 years of age (4-year EFS 63%; 95% CI, 55%-72% and 4-year OS 80%; 95% CI, 73%-88%) had better EFS and OS, compared to patients < 1 year old (4-year EFS 35%; 95% CI, 13%-58% and 4-year OS 70%; 95% CI, 47%-92%) or patients ≥ 10 years (4-year EFS 46%; 95% CI, 38%-55% and 4-year OS 58%; 95% CI, 49%-67%) (Figure 2). There were no differences in EFS and OS based on sex (p=0.11 and p=0.15, respectively) or for patients treated on D9602/D9803, versus ARST0531 (p=0.57 and p=0.20, respectively) (Supplemental Figures S1 and S2).
Figure 1. EFS and OS of entire cohort.
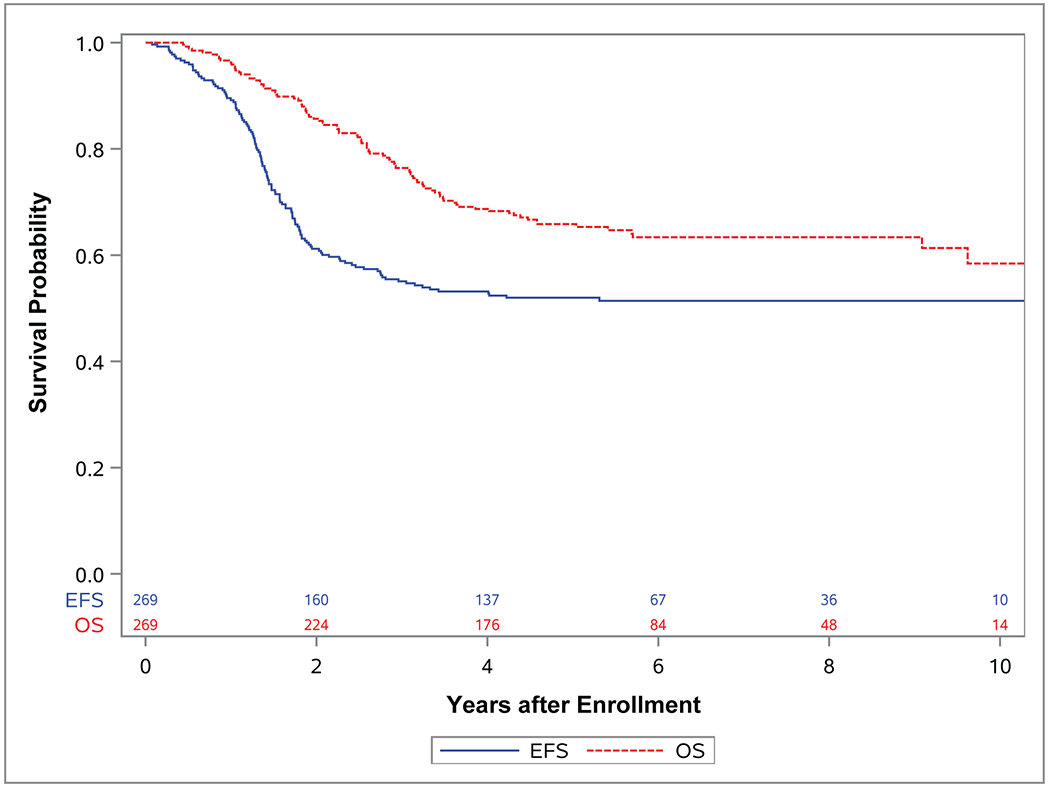
Kaplan-Meier curves representing EFS (blue) and OS (red) for for study population of localized (group I-III) FOXO1 fusion positive rhabdomyosarcoma (RMS) treated on either D9602, D9806, or ARST0531 between 1997 and 2013.
Figure 2. Event-free survival (EFS) and overall survival (OS) by age.


Kaplan-Meier curves representing (A) EFS and (B) OS for patients with localized (group I-III) FOXO1 fusion positive rhabdomyosarcoma (RMS) who, at diagnosis, were under 1 year of age (blue), between 1 and 9 years old (red) or 10 years or older (green).
Several tumor characteristics significantly correlated with OS in univariate analysis, including tumor site, regional lymph node involvement (N), tumor invasiveness (T), clinical group, and FOXO1 fusion partner. Patients with tumors located at favorable sites - orbit, head and neck (non-parameningeal), genitourinary (non-bladder/prostate) - had improved OS (p=0.005) compared to those with tumors at unfavorable sites (4-year OS 88%; 95% CI, 78%-97% vs. 64%; 95% CI, 58%-71%) (Supplemental Figure S3). Similarly, patients with no regional lymph nodes involved (N0) had superior OS (p=0.0014) compared to those with N1 (regional lymph nodes involved) disease (4-year OS 74%; 95% CI, 68%-81% vs. 56%; 95% CI, 46%-67%) (Supplemental Figure S4). Clinical group also affected OS, with group III patients (4-year OS 63%; 95% CI, 56%-70%) exhibiting inferior OS (p=0.01) compared to those classified as group I (4-year OS 82%; 95% CI, 64%-100%) or group II (4-year OS 81%; 95% CI, 71%-91%) (Supplemental Figure S5). Additionally, patients with tumors bearing PAX3-FOXO1 fusions (4-year OS 64%; 95% CI, 57%-71%) experienced inferior OS (p=0.0053) compared to those with PAX7-FOXO1 fusions (4-year OS 86%; 95% CI, 76%-96%) (Supplemental Figure S6).
While each of these factors (tumor site, nodal status, clinical group score and FOXO1 fusion partner) affected OS, differences in EFS for these factors did not reach statistical significance. Tumor size and invasiveness were the only tumor characteristics for which significant differences in both EFS and OS were observed. Patients who had tumors ≤ 5 cm experienced improved EFS (p=0.007) and OS (p<0.0001) compared to those with tumors > 5 cm (4-year EFS 60%; 95% CI, 52%-68% vs. 43%; 95% CI, 34%-53% 4-year OS 78%; 95% CI, 71%-84% vs. 55%; 95% CI, 46%-65%) (Figure 3). Similarly, patients with tumors confined to their anatomic site of origin (T1) experienced improved EFS (p=0.0125) and OS (p<0.0001) compared to those with tumors that had extended in to surrounding tissues (T2) (4-year EFS 59%; 95% CI, 51%-67% vs. 63%; 95% CI, 37%-55% 4-year OS 81%; 95% CI, 75%-88% vs. 54%; 95% CI, 45%-63%) (Figure 4).
Figure 3. Event-free survival (EFS) and overall survival (OS) by tumor size.
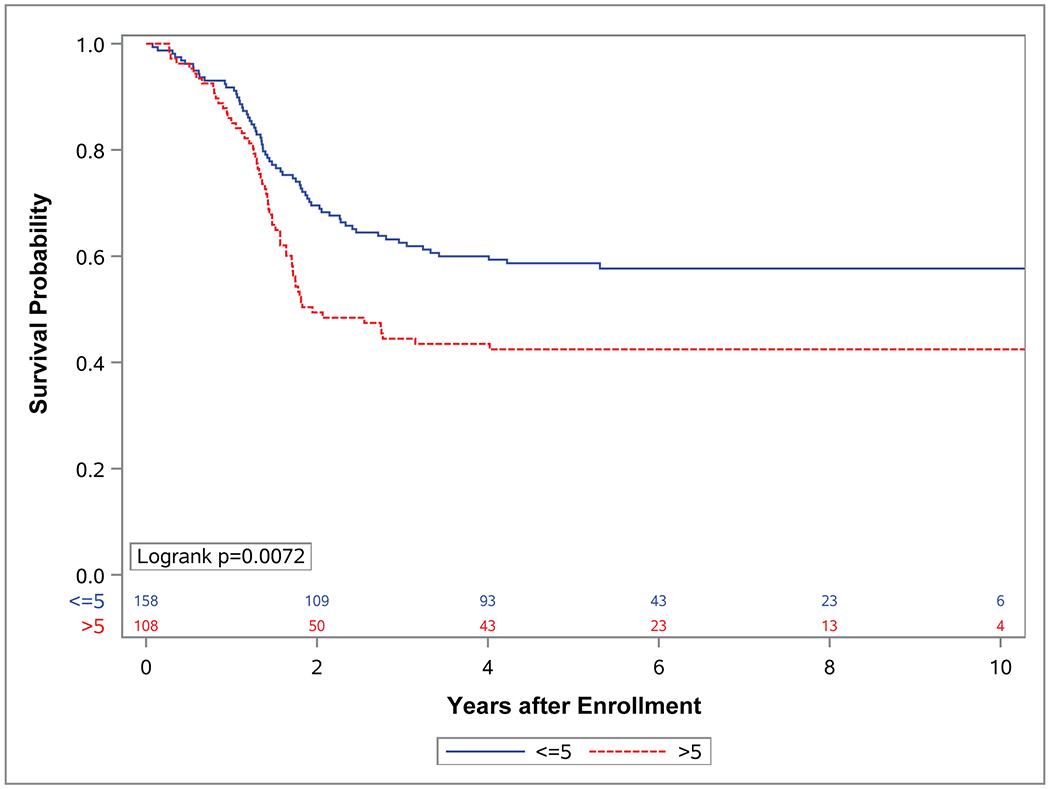
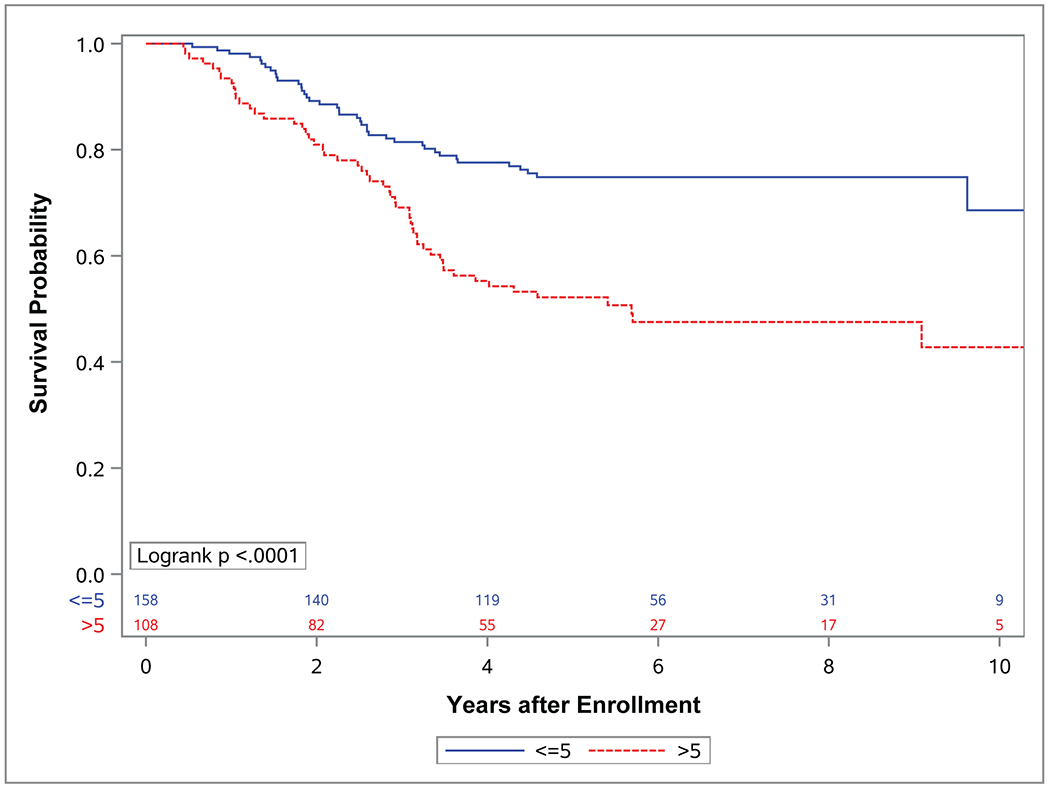
Kaplan-Meier curves representing (A) EFS and (B) OS for patients with localized (group I-III) FOXO1 fusion positive rhabdomyosarcoma (RMS) with tumors less than or equal to 5 cm (blue) or greater than 5 cm (red).
Figure 4. Event-free survival (EFS) and overall survival (OS) by tumor invasiveness.
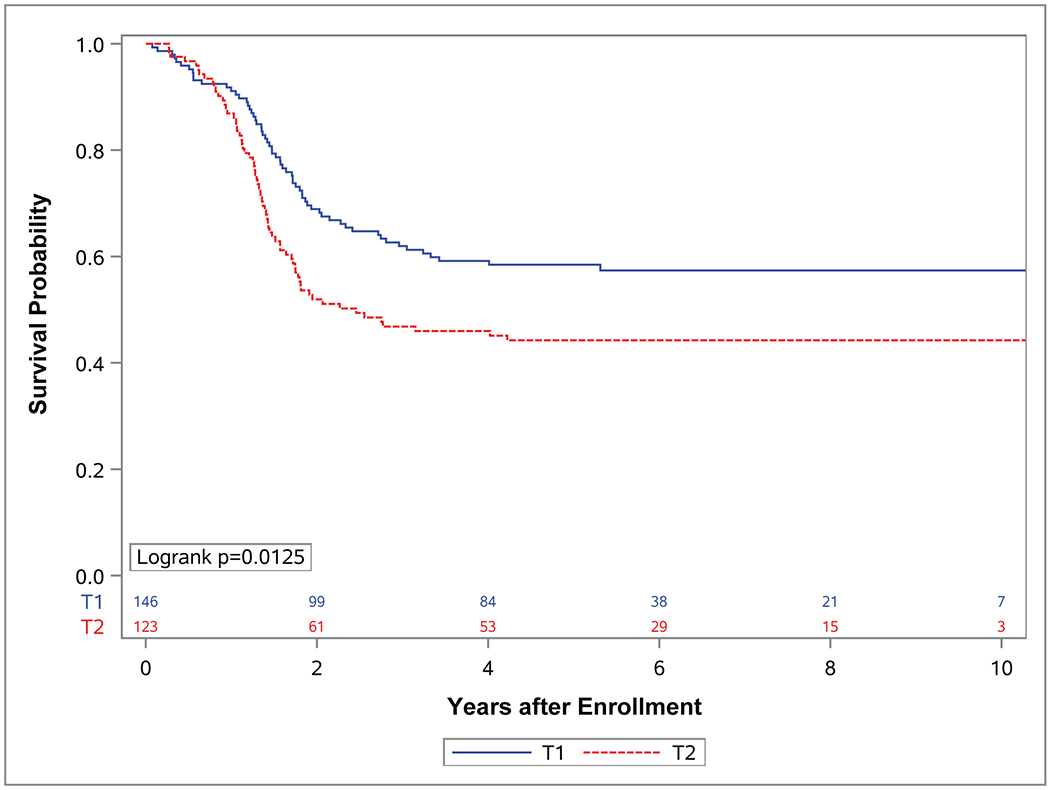

Kaplan-Meier curves representing (A) EFS and (B) OS for patients with localized (group I-III) FOXO1 fusion positive rhabdomyosarcoma (RMS) with tumors confined to the anatomic site (T1) (blue), or tumors that had extended into the surrounding tissues (T2) (red).
In multivariate analysis, only patient age and tumor size emerged as independent prognostic factors for EFS in this patient population (Table 2). Patients diagnosed at younger than 1 year or ≥ 10 years had significantly poorer EFS (p=0.026) compared to the group diagnosed between 1 and 9 years of age, as did those with large tumors (> 5 cm) (p=0.018). Strikingly, patients who were ≥ 10 years with primary tumors > 5 cm experienced significantly worse outcomes with 4-year EFS 31% (95% CI, 19%-43%) compared to patients with just one of these adverse features (Supplemental Figure S5 and Supplemental Table 1). For OS, multivariate analysis demonstrated that tumor size, tumor invasiveness, and FOXO1 fusion partner significantly correlated with OS after adjusting for other clinical factors (Table 2). Patients with large tumors (> 5 cm) experienced worse OS (p=0.017) compared to those with smaller tumors, as did patients with invasive tumors compared to non-invasive tumors (p=0.041). In addition, PAX3-FOXO1 fusion tumors were associated with inferior OS (p=0.044) compared to PAX7-FOXO1 fusion tumors. Patients with tumors harboring all three of these independent adverse factors (large tumor size, invasiveness, and PAX3 fusion) experienced the poorest OS (4-year OS 38% (95% CI, 24%-53%)) (Supplemental Table 1).
Table 2:
Cox proportional hazards model for multivariate analysis.
| HR | 95% CI | P | |
|---|---|---|---|
| Event-free survival | |||
| Age | 0.026 | ||
| 1-9 vs. <1 | 0.419 | 0.216-0.813 | |
| 1-9 vs. >=10 | 0.732 | 0.484-1.108 | |
| <1 vs. >=10 | 1.746 | 0.883-3.453 | |
| Tumor size | 0.018 | ||
| <=5 vs. >5 cm | 0.645 | 0.449-0.928 | |
| Invasiveness |
0.21 |
||
| T1 vs. T2 | 0.769 | 0.510-1.159 | |
|
| |||
| Overall survival | |||
| Age | 0.20 | ||
| 1-9 vs. <1 | 0.408 | 0.151-1.103 | |
| 1-9 vs. >=10 | 0.817 | 0.459-1.454 | |
| <1 vs. >=10 | 1.999 | 0.702-5.691 | |
| Primary site | 0.35 | ||
| favorable vs. unfavorable | 0.691 | 0.317-1.506 | |
| Tumor size | 0.017 | ||
| <=5 vs. >5 cm | 0.560 | 0.347-0.902 | |
| Nodal Status | 0.84 | ||
| N0 vs. N1 | 0.951 | 0.578-1.563 | |
| Group | 0.87 | ||
| I vs. II | 1.285 | 0.405-4.076 | |
| I vs. III | 1.103 | 0.372-3.273 | |
| II vs. III | 0.859 | 0.451-1.636 | |
| Invasiveness | 0.041 | ||
| T1 vs. T2 | 0.558 | 0.319-0.978 | |
| PAX | 0.044 | ||
| PAX3 vs. PAX 7 | 2.169 | 1.022-4.604 | |
Discussion
Our study describes the outcomes of the largest unbiased dataset of pediatric patients with localized FOXO1 FP-RMS to date, with several clinical features emerging as prognostic. Univariate analysis suggests that known clinical prognostic factors for localized RMS remain relevant in FOXO1 FP-RMS.26 Specifically, we observed significantly improved OS for patients between 1 and 9 years of age, clinical group of 1 or 2, and for those with tumors that were ≤ 5 cm, non-invasive (T1), arising in favorable sites, lacking regional lymph node involvement (N0), and bearing PAX7-FOXO1 fusions. Current risk stratification of RMS patients incorporates most of these factors, including clinical group, and tumor site, size, invasiveness, and nodal status, which comprise the established staging classification.5 Surprisingly, a statistically significant effect on EFS was only observed for age, tumor size, and tumor invasiveness. While the reasons for this are unclear, it is possible that, as other studies have suggested,27–31 some traditional unfavorable features may affect pattern of relapse, leading to a worse post-relapse prognosis.
There was no significant difference in outcomes between patients treated on different study regimens, suggesting that the lower cumulative dose of cyclophosphamide used in ARST0531 (cumulative dose 8.4-16.8 g/m2 compared to 28.6 g/m2 for D9602 and 25.1-30.8 g/m2 for D9803,)9 did not impact outcome in our study cohort. While earlier reports of outcomes for localized RMS have suggested that lower cyclophosphamide doses result in higher local failure rates, this has primarily been observed among specific subgroups of RMS patients, generally those with an ERMS predominance.32–36 Taken together, these data suggest that cyclophosphamide dose may play a lesser role in outcomes for FP-RMS than for FN-RMS.
Multiple studies have identified the presence of a FOXO1 fusion as an independent negative prognostic factor in localized RMS.12,13,16–22 Fewer studies have analyzed the impact of factors independent of fusion status. Our multivariate analysis identified two different sets of independent predictive factors for EFS and OS in this population. For EFS, older patient age at diagnosis (≥ 10 years) and tumor size (> 5 cm) were both independently associated with shorter EFS. For OS, tumor size (> 5 cm), tumor invasiveness (T2), and PAX3-FOXO1 fusion were independently associated with worse OS. As with the univariate analysis, the different factors identified for EFS and OS may suggest improved post-relapse outcomes for patients with non-invasive (T1) tumors and tumors bearing PAX7-FOXO1 fusions. It is thus plausible that patients with tumors identified with either of these features at diagnosis are more likely to experience local progression or isolated metastases, thus making them more amenable to definitive local control at the time of relapse, as has been suggested by others.29 However, definitive prospective data to support this are lacking, given that the only two prospective trials reported to date in patients with RMS at first relapse did not collect details on the local control approaches employed.37–39
Age has previously been identified as a prognostic factor independent of FOXO1 fusion status by Missiaglia et al, who reported that age between 1 and 9 years old was associated with improved outcomes.21 In that study however, age was an independent predictor of OS, whereas in our study, it was only significant for EFS. The reasons for this are not clear but may be related to the fact that age and fusion type were highly correlated in our study, with 82% of PAX7 fusion tumors found in patients aged between 1 and 9 years. We identified FOXO1 fusion type as an independent predictor of OS, with PAX3-FOXO1 fusion predicting a worse outcome. This is consistent with multiple prior studies reporting that PAX3-FOXO1 fusions are associated with less favorable clinical features and worse outcomes, compared to PAX7-FOXO1 fusions.8,12,16,18,21,40,41
We found tumor size to be the only independent indicator for both EFS and OS in FOXO1 FP-RMS in our study. While this has not been previously reported, two studies have identified higher IRS TNM stage as independently prognostic of poorer outcome with regard to FOXO1 fusion status and other clinical features.20,21 Neither of those studies analyzed tumor size separately from TNM stage; however, since tumor size is a component of the IRS TNM staging, those data may reflect the effect of tumor size. Indeed, in our analysis of EFS, tumor invasiveness was not an independent prognostic factor, suggesting that tumor size may better reflect likelihood of relapse rather than T status. Interestingly, we did find tumor invasiveness to be an independent prognostic factor for OS, along with tumor size and FOXO1 fusion partner. There is a single report of improved survival in patients with ARMS following first relapse if the relapse is circumscribed and adequate local therapy in the form of complete surgical resection and/or definitive radiation is delivered.31 Taken together, this may suggest that T2 tumors have a higher risk of metastatic relapse, making them less amenable to local control following relapse. These findings partially confirm results from a study conducted by the European Pediatric Soft-tissue sarcoma Study Group (EpSSG) that identified tumor invasiveness and fusion status as independent prognostic factors in a cohort that included only N1 ARMS patients.17 In that study however, tumor size was not found to be a significant predictor of outcome. This may be due to the inclusion of only N1 patients and the relatively few patients with ≤ 5 cm tumors in that cohort. EpSSG has now included N1 ARMS patients in the very high-risk group along with stage IV patients. Our results suggest that tumor size may contribute more than lymph node involvement in predicting the likelihood of relapse in patients with localized FP-RMS.
A key remaining question in the management of patients with localized FOXO1 FP-RMS is whether risk can be further stratified using additional clinical features to guide treatment. A newer risk classification system proposed by Missiaglia et al advocates simplifying the current system by using only fusion status, age, and stage to stratify all RMS patients.21 However, recent data from Hibbitts et al suggests that different clinical factors may be relevant depending on whether the tumor is FP or FN.22 Given this, risk stratification for RMS may require a different set of clinical criteria for FP and FN subtypes of RMS. Our analysis suggests that the combination of tumor size > 5 cm and age ≥ 10 years identifies the group at highest risk of relapse among those with localized FOXO1 FP tumors. EFS for this group was 31% (95% CI, 19%-43%), which is similar to that of patients with distant metastases (group IV) at diagnosis treated on COG high-risk studies during the same time period.22 Currently, this subgroup of patients is classified and treated as intermediate-risk. Our data suggest they should be classified and treated as high-risk in future COG studies, particularly given the extremely poor outcomes with salvage therapy post-relapse in this patient population.27,28,30,31
Interestingly, the presence of PAX3 or PAX7 fusion did not substantially change the outcomes of this highest risk group of patients ≥ 10 years with tumors > 5 cm. Those with PAX3-FOXO1 fusions had very similar EFS (36%; 95% CI, 22%-50%) to those unselected for fusion type, suggesting that the typically adverse prognostic effect of the PAX3-FOXO1 fusion may be less important when combined with the adverse clinical features of older age and large tumor size. This likely results from a high correlation between age and FOXO1 partner and the relatively low number of PAX7 fusions in this group; of the 63 patients in this study aged ≥ 10 years with tumors > 5 cm, only 4 had PAX7 fusions. Given the small number of patients in this group, we currently would not recommend further risk stratification using FOXO1 partner in the subgroup of older patients with large tumors. Additionally, missing fusion partner information on 10% of the patients may have attenuated the effect of FOXO1 fusion status on EFS in our analysis. The next generation of trials should aim to capture complete FOXO1 fusion partner data on all tumors to fully evaluate this group in future studies.
In conclusion, these analyses indicate that in patients with localized FOXO1 FP-RMS, age and tumor size at diagnosis will be useful to identify the group at highest risk for relapse and guide risk stratification for treatment and clinical investigation.
Supplementary Material
Figure 5. Event-free survival (EFS) and overall survival (OS) by age and tumor size.

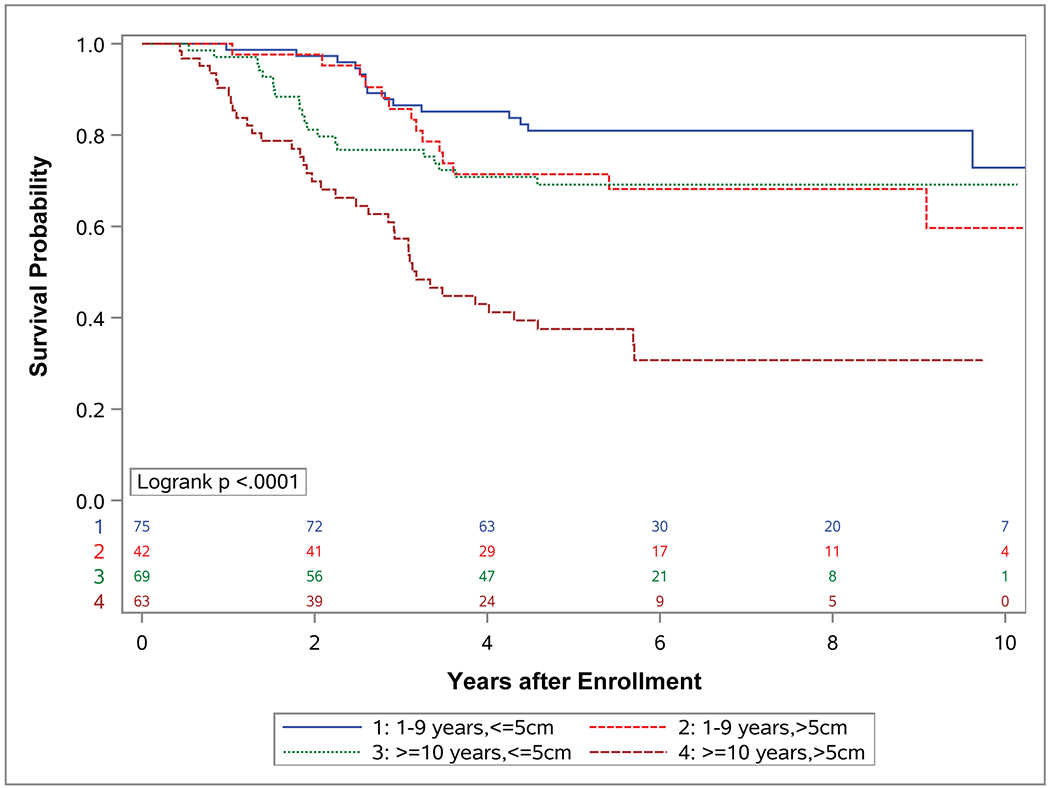
Kaplan-Meier curves representing (A) EFS and (B) OS for patients with localized (group I-III) FOXO1 fusion positive rhabdomyosarcoma (RMS) with age between 1 and 9 years and tumors less than or equal to 5 cm and (blue), age between 1 and 9 years and tumors greater than 5 cm (red), age 10 years or older and tumors less than or equal to 5 cm (green), or age 10 years or older and tumors greater than 5 cm (brown).
Funding:
Children’s Oncology Group [U10CA180886, U10CA180899, U10CA098543, U10CA098413]; St. Baldrick’s Foundation; Seattle Children’s Foundation, from Kat’s Crew Guild through the Sarcoma Research Fund.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013;20(6):387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galili N, Davis RJ, Fredericks WJ, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;5(3):230–235. [DOI] [PubMed] [Google Scholar]
- 3.Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54(11):2869–2872. [PubMed] [Google Scholar]
- 4.Arnold MA, Barr FG. Molecular diagnostics in the management of rhabdomyosarcoma. Expert Rev Mol Diagn. 2017;17(2):189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skapek SX, Ferrari A, Gupta AA, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newton WA Jr., Soule EH, Hamoudi AB, et al. Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: clinicopathologic correlation. J Clin Oncol. 1988;6(1):67–75. [DOI] [PubMed] [Google Scholar]
- 7.Newton WA Jr., Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer. 1995;76(6):1073–1085. [DOI] [PubMed] [Google Scholar]
- 8.Barr FG, Smith LM, Lynch JC, et al. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Intergroup Rhabdomyosarcoma Study-III trial: a report from the Children’s Oncology Group. J Mol Diagn. 2006;8(2):202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arndt CA, Stoner JA, Hawkins DS, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children’s oncology group study D9803. J Clin Oncol. 2009;27(31):5182–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudzinski ER. Histology and fusion status in rhabdomyosarcoma. Am Soc Clin Oncol Educ Book. 2013:425–428. [DOI] [PubMed] [Google Scholar]
- 11.Rudzinski ER, Teot LA, Anderson JR, et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Am J Clin Pathol. 2013;140(1):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnold MA, Anderson JR, Gastier-Foster JM, et al. Histology, Fusion Status, and Outcome in Alveolar Rhabdomyosarcoma With Low-Risk Clinical Features: A Report From the Children’s Oncology Group. Pediatr Blood Cancer. 2016;63(4):634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, Anderson MJ. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006;66(14):6936–6946. [DOI] [PubMed] [Google Scholar]
- 14.Davicioni E, Anderson MJ, Finckenstein FG, et al. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children’s Oncology Group. Am J Pathol. 2009;174(2):550–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun W, Chatterjee B, Shern JF, et al. Relationship of DNA methylation to mutational changes and transcriptional organization in fusion-positive and fusion-negative rhabdomyosarcoma. Int J Cancer. 2019;144(11):2707–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skapek SX, Anderson J, Barr FG, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatr Blood Cancer. 2013;60(9):1411–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallego S, Zanetti I, Orbach D, et al. Fusion status in patients with lymph node-positive (N1) alveolar rhabdomyosarcoma is a powerful predictor of prognosis: Experience of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer. 2018;124(15):3201–3209. [DOI] [PubMed] [Google Scholar]
- 18.Anderson J, Gordon T, McManus A, et al. Detection of the PAX3-FKHR fusion gene in paediatric rhabdomyosarcoma: a reproducible predictor of outcome? Br J Cancer. 2001;85(6):831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davicioni E, Anderson JR, Buckley JD, Meyer WH, Triche TJ. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the children’s oncology group. J Clin Oncol. 2010;28(7):1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williamson D, Missiaglia E, de Reynies A, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28(13):2151–2158. [DOI] [PubMed] [Google Scholar]
- 21.Missiaglia E, Williamson D, Chisholm J, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. 2012;30(14):1670–1677. [DOI] [PubMed] [Google Scholar]
- 22.Hibbitts E, Chi YY, Hawkins DS, et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children’s Oncology Group. Cancer Med. 2019;8(14):6437–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raney RB, Walterhouse DO, Meza JL, et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J Clin Oncol. 2011;29(10):1312–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawkins DS, Chi YY, Anderson JR, et al. Addition of Vincristine and Irinotecan to Vincristine, Dactinomycin, and Cyclophosphamide Does Not Improve Outcome for Intermediate-Risk Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J Clin Oncol. 2018;36(27):2770–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peto R, Peto J. Asymptotically efficient rank invariant test procedures. Journal of the Royal Statistical Society A. 1972;Series A 135:185–207. [Google Scholar]
- 26.Meza JL, Anderson J, Pappo AS, Meyer WH, Children’s Oncology G. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children’s Oncology Group. J Clin Oncol. 2006;24(24):3844–3851. [DOI] [PubMed] [Google Scholar]
- 27.Pappo AS, Anderson JR, Crist WM, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol. 1999;17(11):3487–3493. [DOI] [PubMed] [Google Scholar]
- 28.Mazzoleni S, Bisogno G, Garaventa A, et al. Outcomes and prognostic factors after recurrence in children and adolescents with nonmetastatic rhabdomyosarcoma. Cancer. 2005;104(1):183–190. [DOI] [PubMed] [Google Scholar]
- 29.Dantonello TM, Int-Veen C, Winkler P, et al. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J Clin Oncol. 2008;26(3):406–413. [DOI] [PubMed] [Google Scholar]
- 30.Chisholm JC, Marandet J, Rey A, et al. Prognostic factors after relapse in nonmetastatic rhabdomyosarcoma: a nomogram to better define patients who can be salvaged with further therapy. J Clin Oncol. 2011;29(10):1319–1325. [DOI] [PubMed] [Google Scholar]
- 31.Dantonello TM, Int-Veen C, Schuck A, et al. Survival following disease recurrence of primary localized alveolar rhabdomyosarcoma. Pediatr Blood Cancer. 2013;60(8):1267–1273. [DOI] [PubMed] [Google Scholar]
- 32.Baker KS, Anderson JR, Link MP, et al. Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: results from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2000;18(12):2427–2434. [DOI] [PubMed] [Google Scholar]
- 33.Walterhouse DO, Meza JL, Breneman JC, et al. Local control and outcome in children with localized vaginal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma committee of the Children’s Oncology Group. Pediatr Blood Cancer. 2011;57(1):76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walterhouse DO, Pappo AS, Meza JL, et al. Reduction of cyclophosphamide dose for patients with subset 2 low-risk rhabdomyosarcoma is associated with an increased risk of recurrence: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer. 2017;123(12):2368–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casey DL, Chi YY, Donaldson SS, et al. Increased local failure for patients with intermediate-risk rhabdomyosarcoma on ARST0531: A report from the Children’s Oncology Group. Cancer. 2019;125(18):3242–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casey DL, Wexler LH, Wolden SL. Worse Outcomes for Head and Neck Rhabdomyosarcoma Secondary to Reduced-Dose Cyclophosphamide. Int J Radiat Oncol Biol Phys. 2019;103(5):1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mascarenhas L, Lyden ER, Breitfeld PP, et al. Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2010;28(30):4658–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mascarenhas L, Chi YY, Hingorani P, et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination With Chemotherapy for First Relapse Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J Clin Oncol. 2019;37(31):2866–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mascarenhas L, Lyden ER, Breitfeld PP, et al. Risk-based treatment for patients with first relapse or progression of rhabdomyosarcoma: A report from the Children’s Oncology Group. Cancer. 2019;125(15):2602–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kazanowska B, Reich A, Stegmaier S, et al. Pax3-fkhr and pax7-fkhr fusion genes impact outcome of alveolar rhabdomyosarcoma in children. Fetal Pediatr Pathol. 2007;26(1):17–31. [DOI] [PubMed] [Google Scholar]
- 41.Duan F, Smith LM, Gustafson DM, et al. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes Chromosomes Cancer. 2012;51(7):662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.