

Abstract
Enveloped viruses have evolved diverse transmembrane proteins and protein complexes to enable host cell entry by regulating and activating membrane fusion in a target cell-specific manner. In general terms, the entry process requires a receptor binding step, an activation step and a membrane fusion step, which can be encoded within a single viral protein or distributed among multiple viral proteins. HIV and influenza virus, for example, encode all of these functions in a single trimeric glycoprotein, HIV env or influenza virus hemagglutinin (HA). In contrast, herpesviruses have the host receptor binding, activation and fusogenic roles distributed among multiple envelope glycoproteins (ranging from three to six), which must coordinate their functions at the site of fusion. Despite the apparent complexity in the number of viral entry proteins, herpesvirus entry is fundamentally built around two core glycoprotein entities: the gHgL complex, which appears to act as an ‘activator’ of entry, and the gB protein, which is thought to act as the membrane ‘fusogen’. Both are required for all herpesvirus fusion and entry. In many herpesviruses, gHgL either binds host receptors directly or assembles into larger complexes with additional viral proteins that bind host receptors, conferring specificity to the cells that are targeted for infection. These gHgL entry complexes (ECs) are centrally important to activating gB-mediated membrane fusion and establishing viral tropism, forming membrane bridging intermediates prior to gB triggering. Here we review recent structural and functional studies of Epstein-Barr virus (EBV) and Cytomegalovirus (CMV) gHgL complexes that provide a framework for understanding the role of gHgL in herpesvirus entry. Furthermore, a recently determined EM model of Herpes Simplex virus (HSV) gB embedded in exosomes highlights how gB conformational changes may promote viral and cellular membrane fusion.
Introduction
Herpesviruses are a large family of dsDNA viruses whose virions are encased within a lipid bilayer envelope. They have a broad host range, infecting almost all vertebrates and some invertebrates (bivalves). The family is divided into three subfamilies: alpha-, beta- and gammaherpesviruses. Nine members within these three sub-families infect humans (the human herpesviruses: HHVs) and constitute clinically important pathogens. Herpes Simplex viruses (HSV-1 and HSV-2), members of the alphaherpesvirus subfamily, cause infectious blindness, cold sores, genital herpes and encephalitis in rare cases [1]. Varicella zoster virus (VZV), another alphaherpesvirus, causes shingles and herpes zoster in the elderly [2]. Cytomegalovirus (CMV), a betaherpesvirus, can cause severe disease in transplant patients and is a leading cause of birth defects through congenital infection [3]. Epstein-Barr virus (EBV), which together with Kaposi’s sarcoma-associated herpesvirus (KSHV) form the human gammaherpesviruses, causes infectious mononucleosis in young adults and lymphoid or epithelial cell malignancies such as Burkitt and Hodgkin’s lymphoma, nasopharyngeal carcinoma, and gastric carcinomas in immune-system compromised patients [4,5]. KSHV causes Kaposi’s sarcoma of endothelial cells and B cell primary effusion lymphoma [6]. EBV and KSHV are both associated with HIV/AIDS related malignancies arising as a result of immunodeficiency.
Entry into target cells is the first step for virus infection, necessitating virus and host cell membrane fusion that requires overcoming a high energy barrier [7]. Fusion is catalyzed by a specialized membrane protein (the ‘fusogen’), which mitigates this energy barrier by executing a large conformational change from a meta-stable pre-fusion state to a stable post-fusion state. This conformational change brings opposing membranes together using energy from the protein refolding process. The core entry machinery for herpesviruses is formed by the gH, gL and gB proteins. The herpesvirus gB protein is a class III viral fusion protein that functions as a homotrimer [8,9]. The trigger for the gB conformational change is thought to be regulated by interactions with gHgL entry complexes (ECs), which are initiated after EC binding to host cell receptors [10,11]. Once gB is triggered, its fusion loops (FLs) insert into the target host cell lipid bilayer, followed by gB refolding to drive membrane merger and the onset of infection [8].
In addition to the core fusion proteins, many herpesviruses have also evolved additional, nonconserved proteins that establish virus tropism and engage cell-specific host receptors (Table 1). HSV-1/2 [12,13] and pseudorabies virus (PrV) express a gD protein that binds host cell receptors [14]. CMV expresses gO [15] and three UL proteins (UL128, 130 and 131) [16,17] to regulate tropism. HHV-6 expresses gO [18] and gQ1/gQ2 proteins [19], while EBV expresses gp42 [20,21].
Table 1.
Receptor binding gHgL entry complexes (EC) for Human Herpesvirus (HHV) that trigger entry
| Herpesvirus | Glycoprotein complex | Entry receptor | Cell type infected |
|---|---|---|---|
| Herpes Simplex Virus (HSV-1 and HSV-2) | gHgL-gD | Herpesvirus entry mediator (HVEM), Nectin-1, 3-O-sulphated heparan sulphate | Neurons, Fibroblasts, Epithelial cells |
| Varicella Zoster virus (VZV) | gHgL | Integrins | Respiratory mucosa, T-cells, Neurons |
| Cytomegalovirus (CMV) | gHgL/UL128/UL130/UL131 (Pentamer) | ErbB? | Epithelial, Endothelial and Myeloid cells |
| gHgL/gO (Trimer) | Platelet-derived growth factor receptor alpha (PDGFRα) | Fibroblasts, Epithelial, Endothelial and Myeloid cells | |
| Human Herpesvirus 6 (HHV-6A and HHV-6B) | gHgL/gQ1/gQ2 | CD46 (strain specific) | T-cells |
| gHgL/gO | ? | ? | |
| Epstein-Barr virus (EBV) | gHgL/gp42 | HLA-class II (bind gp42) | B cells |
| gHgL | αvβ3, αvβ6 integrins | Epithelial cells | |
| Kaposi’s sarcoma-associated herpesvirus (KSHV) | gHgL | EphA2 | Endothelial cells |
Many of these tropism-determining proteins assemble into complexes with gHgL to regulate herpesvirus entry. The CMV, HHV-6 and EBV gHgL complexes are particularly stable and of high affinity. CMV UL128–131 and gO assemble covalently into mutually exclusive cell entry complexes with gHgL in domain I or D-I region, forming a disulfide bond with gL Cys144 [17]. From earlier crystal structures of HSV-2, VZV and EBV gHgL, domain H1A/H1B of HSV-2 and VZV or domain I of EBV includes all gL residues and portion of gH forming the least conserved domain of gHgL among different sub-families and species of herpesvirus. EBV gp42 binds gHgL with nanomolar affinity through non-covalent interactions mediated by the gp42 N-terminal domain [22] and a peptide derived from this region of gp42 binds gHgL with similar affinity. Although the HSV gD protein is an exception to this “rule”, gD is thought to interact with gHgL, as well as with host receptors and gB, to coordinate HSV entry into cells.
A number of these virus-specific proteins that establish tropism have been shown to bind a cognate host receptor, thereby directing the infection of specific cells (Table 1). However, receptors for all of these have not been identified (Table 1). EBV gp42 binds to HLA class II expressed on B cells, targeting these cells for infection. The binding of gp42, or a even a 35 amino acid gp42-derived peptide, to EBV gHgL blocks virus entry into epithelial cells, thereby playing a central role in specifying viral tropism. HCMV gHgL/gO complexes bind to Platelet-Derived Growth Factor Receptor alpha (PDGFRα)[23]. This interaction is sufficient to specify HCMV entry into fibroblasts, but may also be required for entry into other cells. The CMV gHgL/UL128/UL130/UL131 complex (referred to as the ‘Pentamer’) is essential for the virus to enter epithelial, endothelial and myeloid cells, but a receptor for this complex has not been validated. Nonetheless, it is apparent that multiple gHgL entry complexes have evolved that dictate the breadth of cells infected by individual herpesviruses by targeting specific host receptors.
In addition to interactions mediated by these viral adaptor proteins, some of the herpesvirus gHgL proteins also exhibit the ability to bind directly with host receptors, most notably among the gammaherpesvirus sub-family. EBV gHgL has been reported to engage αvβ3/6 integrin receptors, triggering epithelial cell entry [24,25], and KSHV gHgL directly engages ephrin type A receptor A2 (EphA2) [6,26] (Table 1). Varicella zoster virus (VZV), from the alphaherpesvirus sub-family curiously lacks a gD homolog [2] and its gHgL could also engage cellular receptors directly. Recently, VZV has also been shown to require the αv integrin subunit for gB/gHgL mediated fusion [27].
Crystal structures of gHgL from different herpesviruses have been solved over the last decade, including gHgL from HSV-2 [28], VZV [29], PrV [30], and EBV [31] (Figure 1). gHgL crystal structures from the betaherpesvirus subfamily such as CMV have not yet been determined, but low resolution electron microscopy (EM) structures indicate its overall similarity to HSV-2 gHgL [17,32]. The gHgL heterodimer is a complex roughly 10 nm long, forming either a bent, boot-like shape in the case of HSV, VZV and CMV, or a more linear, rod-like shape in the case of EBV gHgL. In all of the gHgL structures, gL co-folds with the associated gH N-terminal end to form a membrane distal domain (D-I or H1A). Overall, the gHgL proteins are composed of four domains arranged linearly one after another (Figure 1a). gH is anchored to the membrane through a single pass transmembrane (TM) domain followed by a short but functionally important C-terminal domain [33,34].
Figure 1. Herpesvirus entry glycoproteins gHgL and gB crystal structures.
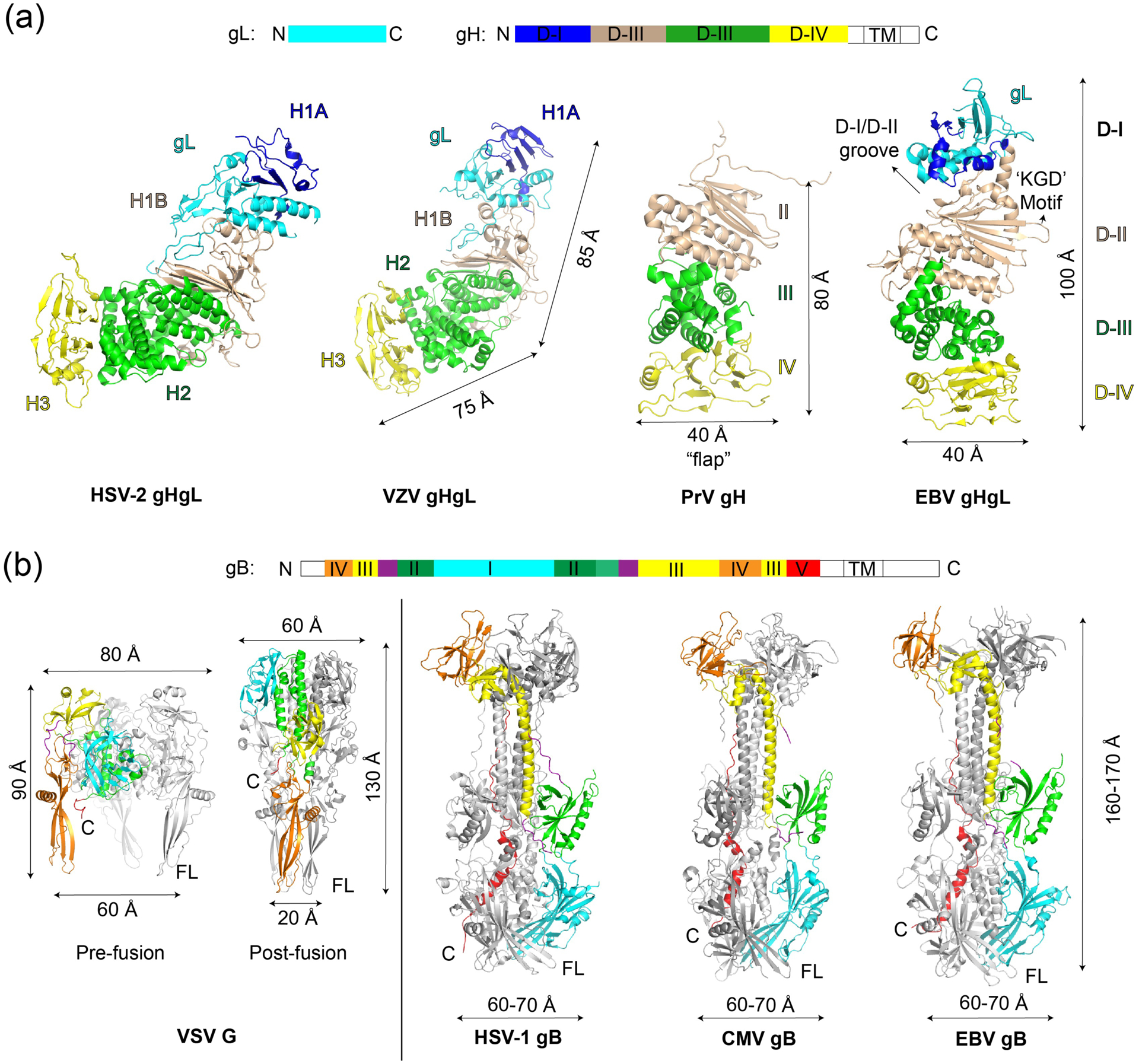
Known protein data bank (PDB) structures of (a) gHgL and (b) gB are summarized. Left of panel (b) also shows comparisons to the other class III fusion protein, VSV G, whose structures in both pre-fusion and post-fusion states are shown in the same color scheme for comparison. It must be noted that VSV G is much smaller than herpesvirus gB and does not have a gHgL equivalent. The individual glycoprotein structures provide a basis for understanding interactions in larger complexes as visualized by electron microscopy (EM).
Structures of postfusion gB have also been determined for HSV-1 [35], CMV [36,37], and EBV [38] (Figure 1b). Based on structural homology to VSV G fusion proteins in the prefusion [39] and postfusion state [40], gB domains have been anticipated to undergo large conformational changes to bring opposing membranes together. Despite important virus specific differences, the overall architecture of gHgL and gB across the herpesviruses are very similar consistent with a common fusion mechanism. Recent crystallographic and EM studies have revealed new information on the larger gHgL complexes and their interactions with host receptors [17,22,23,41], and provided the first low-resolution view of the full length gB structure that is not in the post-fusion conformation [42].
gHgL associated entry complexes
The larger gHgL associated ECs from CMV [17] and EBV [22] form highly stable complexes and this has facilitated their biochemical isolation and structural studies, as compared to HSV counterparts. Comparisons of these complexes indicate diversity in how gHgL ECs can be organized and confer important implications for the architecture that can be adopted in receptor bound state. EM data along with a recent crystal structure of the EBV gHgL/gp42 complex (Figure 2a) reveals the overall architecture of the complex and details of the high affinity gp42 interaction [22]. Flexible residues in the gp42 N-domain form an extended polypeptide structure that interacts with multiple gH domains (D-II to D-IV), making no interactions with gL. The structural data explain the ability of gp42-derived peptides to bind with nanomolar affinity to gHgL, through an extensive interface that involves interactions over 5 high affinity binding determinants (HABDs) [22]. The gp42 C-domain, which binds to the HLA receptor, is positioned in the middle of gHgL at the D-II–D-III junction (Figure 2a).
Figure 2. EM and crystal structures of EBV and CMV entry complexes.
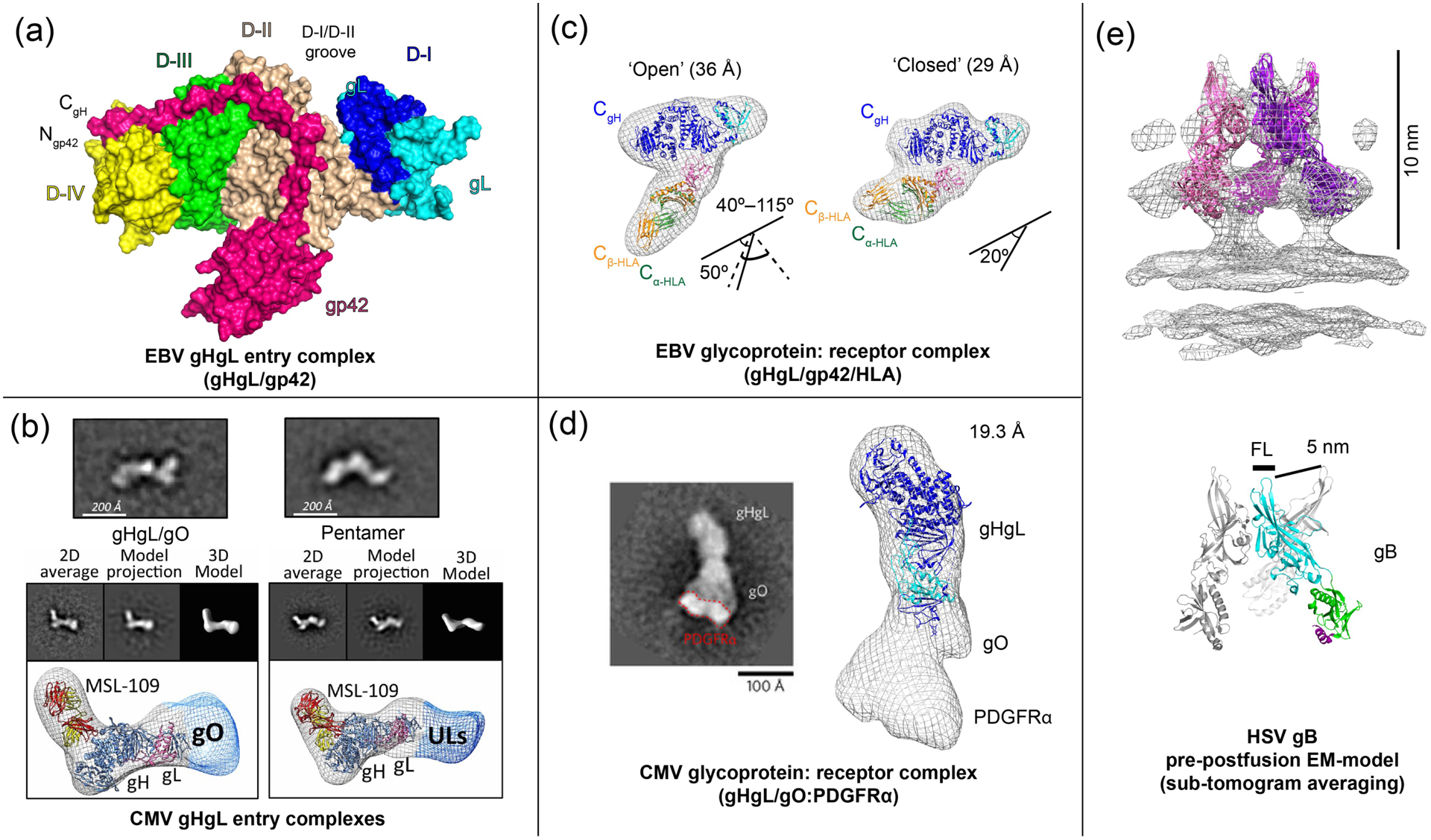
(a) The crystal structure of the EBV gHgL/gp42 complex (PDB ID: 5T1D) [22], showing interactions of the extended gp42 N-terminal domain and location of the HLA-binding gp42 C-terminal domain. (b) CMV gHgL forms gHgL/gO trimers and gHgL/UL128/UL130/UL131 Pentamers through its N-terminal domain, as visualized by negative stain EM [17]. (c) EM structure of EBV gHgL/gp42 in complex with HLA receptor [22,41]. (d) The EM structure of the CMV gHgL/gO trimer in complex with PDGFRα receptor model (EMD-3391) [23]. (e) A ‘pre-postfusion’ HSV-1 gB model derived from sub-tomogram averaging of intact gB on vesicles (EMD-3362) [42]. Figures adapted from original publications and public databanks as cited. Other structures were rendered in Chimera (UCSF) and MacPymol (Schrödinger LLC).
In contrast, EM studies of CMV gHgL ECs show a different overall architecture. CMV gHgL forms covalent, mutually exclusive gHgL/gO (trimer) or gHgL/UL128/UL130/UL131 (‘Pentamer’) complexes through disulfide bonds between gL-Cys144 and either gO-Cys351 or UL128-Cys162 [17]. Negative stain electron microscopy (EM) 3-D reconstructions of these complexes indicate that both gO and the UL128–131 complex assemble at the extreme N-terminus of gHgL in domain H1A, extending the elongated gHgL heterodimer (Figure 2b). Although the alphaherpesvirus gD does not form a high affinity interaction with gHgL, a PrV gD-gH chimera with gD replacing the N-terminal domain of gHgL is functional [14], indicating potential similarities with the CMV gHgL EC architectures. The EBV gHgL D-I domain has also been shown to affect entry into epithelial cells, as well as provide specificity for gB activation, consistent with a key and general functional role of D-I across the different herpesviruses [22]. Overall, the gHgL associated proteins in these ECs provide the potential for expanding the receptor and host cell repertoire in a highly modular fashion.
gHgL EC/receptor triggering complexes
gHgL EC complexes with host cell receptors have been visualized for the EBV B cell entry triggering complex (gHgL/gp42/HLA class II) [41] and the CMV gHgL/gO trimer bound to a fragment of PDGFRα [23]. The EBV B cell entry triggering complex (Figure 2c), shows a compact well defined ‘closed’ state and multiple variable ‘open’ states defined by the angle between gHgL and gp42/HLA arms. The ‘closed’ state brings the predicted C-terminal anchors of gH and HLA within 70 Å in the 170 Å long complex. The model also positions a functionally important hydrophobic pocket in the gp42 C-domain in contact with gH. Mutations in this pocket that disrupt fusion do not affect the overall formation of this triggering complex, but may decrease the stability of its closed conformation. These data suggest that the overall conformation of the complex between the viral and cell membranes is important in activating membrane fusion. The overall gHgL/gp42/HLA structure bears resemblance to V/Y shaped structures observed in cryoelectron tomography images of HSV virions [43] during entry, suggesting that similar membrane bridging structures form prior to fusion.
EM studies of the gHgL/gO/PDGFRα complex indicate that the receptor binds at the N-terminal end of the gHgL complex, consistent with the primary role of gO in mediating receptor interactions (Figure 2d). The receptor binding site location contrasts with the positioning of gp42/HLA near the midsection of the EBV gHgL structure. In this CMV EM study, the PDGFRα is truncated and it is not clear whether conformations analogous to the ‘closed’ and ‘open’ EBV structures might exist. Despite these differences, one common emerging theme is that gHgL ECs form membrane bridging structures with receptors to direct the cell specificity of entry prior to gB triggering. Membrane anchoring of the gHgL complexes and their orientation at the interface appears important to the efficiency of gB activation at least in some of the herpesviruses. Soluble EBV gHgL [44] does not activate fusion, although soluble HSV gHgL is functional in fusion [45]. Cell-cell fusion assays with the core fusion proteins transfected in trans have been shown to be active for some of the herpesviruses, but this orientation appears less efficient compared to proteins transfected in cis [46,47].
Structural model of “pre-postfusion” herpesvirus gB
Obtaining structural information on the pre-fusion state of the conserved fusogen gB is centrally important to understanding the mechanism of herpesvirus entry and potentially for the development of antiviral agents or vaccines. Recently, the full length HSV-1 gB in its native membrane anchored form was visualized by sub-tomogram cryo-ET averaging in membrane vesicles isolated as exosomes [42]. Extended and compact structures were visible in these exosomes with the extended form matching the postfusion gB crystal structures. The compact form of gB could be resolved to 23 Å resolution and yielded a partial model of three of five gB domains. Individual domains from the post-fusion HSV-1 gB crystal structure [35] were fitted in the EM envelope using hierarchical constrained density-fitting (Figure 2e). This model led to the surprising possibility that prior to membrane fusion, the gB fusion loops are exposed at the membrane distal end of gB and spread 5 nm apart in the three subunits of the trimer [42] (Figure 2e). Based on this model, a collapse of the fusion loop-containing domains towards each other and their projection along the trimer axis towards the target cell membrane would initiate the process of membrane fusion. Further high resolution studies are needed to validate this gB model and to gain insight into the mechanism of gB activation by gHgL ECs.
Conclusion
Although herpesviruses require multiple envelope glycoproteins to mediate host cell recognition and entry, the overall process can be simplified into two key functional steps analogous to those in other viruses: a receptor binding step followed by membrane fusogen activation. Accumulating data indicate that herpesvirus gHgL has evolved as a platform for the assembly of adaptor proteins that form larger entry complexes to modulate the specificity of host cell receptor binding, establishing key elements of virus-specific tropism (Figure 3). These gHgL complexes with host cell receptors lead to gB activation in a process that is still poorly understood. Structural studies of EBV and HCMV gHgL ECs indicate diversity in the overall architecture of these complexes, yet studies with the EBV B cell entry complex suggest that the conformation of the receptor-bound EC may be important to gB activation and membrane fusion. Further studies of gHgL EC interactions with receptors are needed to better understand whether they share common features that are important to virus entry and to clarify how receptor binding leads to gB activation. While the mechanism of gB triggering is assumed to be similar among the herpesviruses, visualizing details of the gB-gHgL interaction has been elusive due its transient and dynamic nature. Obtaining a high resolution structure of prefusion gB would represent a significant step forward in understanding not only the membrane fusion step of herpesvirus entry but also for gaining insight into its activation by gHgL and gHgL ECs.
Figure 3. Herpesvirus gHgL is a versatile entry glycoprotein.
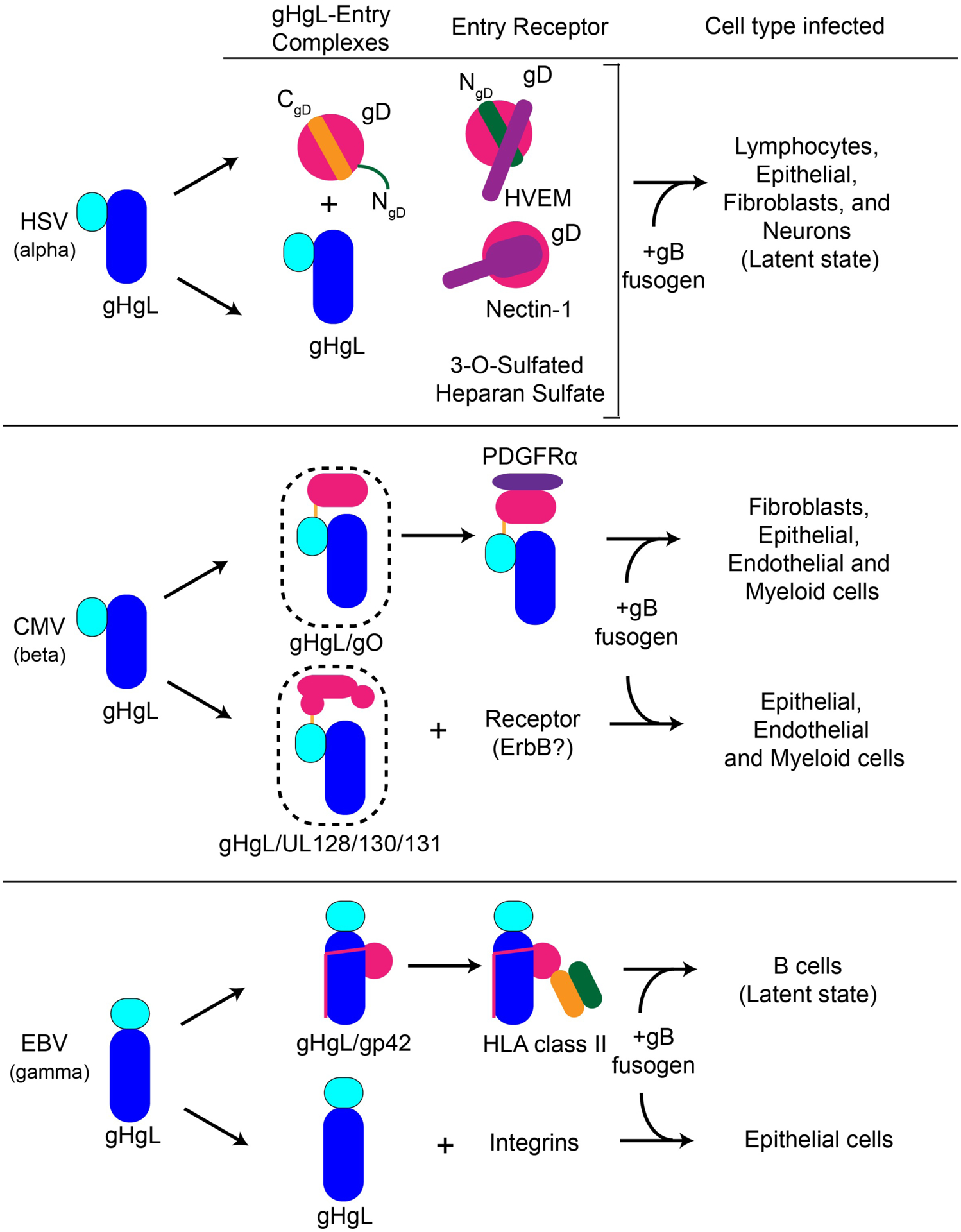
Entry mediated by various gHgL entry complexes and the corresponding cell types infected are schematically shown for HSV, CMV and EBV.
Acknowledgements.
We thank the members of the Jardetzky and Longnecker laboratories for assistance. This research was supported by AI119480 and AI076183 (R.L. and T.J.) from the National Institute of Allergy and Infectious Diseases, and by CA117794 (R.L. and T.J.) from the National Cancer Institute.
Footnotes
Conflicts of Interest. The authors declare no competing interests.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH: Herpes virus fusion and entry: a story with many characters. Viruses 2012, 4:800–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliver SL, Yang E, Arvin AM: Varicella-Zoster Virus Glycoproteins: Entry, Replication, and Pathogenesis. Curr. Clin. Microbiol. Reports 2016, 3:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanarsdall AL, Johnson DC: Human cytomegalovirus entry into cells. Curr. Opin. Virol 2012, 2:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chesnokova LS, Jiang R, Hutt-Fletcher LM: Viral Entry. Curr. Top. Microbiol. Immunol 2015, 391:221–35. [DOI] [PubMed] [Google Scholar]
- 5.Möhl BS, Chen J, Sathiyamoorthy K, Jardetzky TS, Longnecker R: Structural and Mechanistic Insights into the Tropism of Epstein-Barr Virus. Mol. Cells 2016, 39:286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar B, Chandran B: KSHV Entry and Trafficking in Target Cells—Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses 2016, 8:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison SC: Viral membrane fusion. Virology 2015, 479:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R: Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol 2011, 9:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baquero E, Albertini AA, Gaudin Y: Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr. Opin. Struct. Biol 2015, 33:52–60. [DOI] [PubMed] [Google Scholar]
- 10.Stampfer SD, Heldwein EE: Stuck in the middle: structural insights into the role of the gH/gL heterodimer in herpesvirus entry. Curr. Opin. Virol 2013, 3:13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heldwein EE: gH/gL supercomplexes at early stages of herpesvirus entry. Curr. Opin. Virol 2016, 18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Q, Longnecker R, Connolly SA: Substitution of herpes simplex virus 1 entry glycoproteins with those of saimiriine herpesvirus 1 reveals a gD-gH/gL functional interaction and a region within the gD profusion domain that is critical for fusion. J. Virol 2014, 88:6470–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan Q, Longnecker R, Connolly SA: A Functional Interaction between Herpes Simplex Virus 1 Glycoprotein gH/gL Domains I and II and gD Is Defined by Using Alphaherpesvirus gH and gL Chimeras. J. Virol 2015, 89:7159–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klupp BG, Mettenleiter TC: Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J. Virol 1999, 73:3014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC: Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol 2006, 80:710–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanarsdall AL, Chase MC, Johnson DC: Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol 2011, 85:11638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciferri C, Chandramouli S, Donnarumma D, Nikitin PA, Cianfrocco MA, Gerrein R, Feire AL, Barnett SW, Lilja AE, Rappuoli R, et al. : Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. U. S. A 2015, doi: 10.1073/pnas.1424818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mori Y, Akkapaiboon P, Yonemoto S, Koike M, Takemoto M, Sadaoka T, Sasamoto Y, Konishi S, Uchiyama Y, Yamanishi K: Discovery of a second form of tripartite complex containing gH-gL of human herpesvirus 6 and observations on CD46. J. Virol 2004, 78:4609–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jasirwan C, Furusawa Y, Tang H, Maeki T, Mori Y: Human herpesvirus-6A gQ1 and gQ2 are critical for human CD46 usage. Microbiol. Immunol 2014, 58:22–30. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM: Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol 1997, 71:4657–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS: Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol 2006, 80:9444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sathiyamoorthy K, Hu YX, Möhl BS, Chen J, Longnecker R, Jardetzky TS: Structural basis for Epstein–Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat. Commun 2016, 7:13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabanova A, Marcandalli J, Zhou T, Bianchi S, Baxa U, Tsybovsky Y, Lilleri D, Silacci-Fregni C, Foglierini M, Fernandez-Rodriguez BM, et al. : Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol 2016, 1:16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM: Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins αvβ6 or αvβ8. Proc. Natl. Acad. Sci. U. S. A 2009, 106:20464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutt-Fletcher L: The long and complicated relationship between Epstein-Barr virus and epithelial cells. J. Virol 2016, doi: 10.1128/JVI.01677-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, König S, et al. : The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma–associated herpesvirus. Nat. Med 2012, 18:961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang E, Arvin AM, Oliver SL: A role for the αV integrin subunit in Varicella Zoster Virus-mediated fusion and infection. J. Virol 2016, doi: 10.1128/JVI.00792-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE: Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol 2010, 17:882–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, et al. : A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc. Natl. Acad. Sci. U. S. A 2015, 112:6056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney M-C, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA: Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. U. S. A 2010, 107:22635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS: Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A 2010, 107:22641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciferri C, Chandramouli S, Leitner A, Donnarumma D, Cianfrocco MA, Gerrein R, Friedrich K, Aggarwal Y, Palladino G, Aebersold R, et al. : Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015, 11:e1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silverman JL, Heldwein EE: Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J. Virol 2013, 87:10139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Jardetzky TS, Longnecker R: The Cytoplasmic Tail Domain of Epstein-Barr Virus gH Regulates Membrane Fusion Activity through Altering gH Binding to gp42 and Epithelial Cell Attachment. MBio 2016, 7:e01871–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC: Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313:217–20. [DOI] [PubMed] [Google Scholar]
- 36.Chandramouli S, Ciferri C, Nikitin PA, Caló S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, et al. : Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat. Commun 2015, 6:8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burke HG, Heldwein EE: Crystal Structure of the Human Cytomegalovirus Glycoprotein B. PLoS Pathog. 2015, 11:e1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Backovic M, Longnecker R, Jardetzky TS: Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A 2009, 106:2880–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roche S, Rey FA, Gaudin Y, Bressanelli S: Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315:843–8. [DOI] [PubMed] [Google Scholar]
- 40.Roche S, Bressanelli S, Rey FA, Gaudin Y: Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313:187–91. [DOI] [PubMed] [Google Scholar]
- 41.Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, Jiang W, Mellins ED, Longnecker R, Zhou ZH, et al. : Assembly and Architecture of the EBV B Cell Entry Triggering Complex. PLoS Pathog. 2014, 10:e1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeev-Ben-Mordehai T, Vasishtan D, Hernández Durán A, Vollmer B, White P, Prasad Pandurangan A, Siebert CA, Topf M, Grünewald K: Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc. Natl. Acad. Sci 2016, 113:4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maurer UE, Sodeik B, Grünewald K: Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A 2008, 105:10559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowe CL, Connolly SA, Chen J, Jardetzky TS, Longnecker R: A soluble form of Epstein-Barr virus gH/gL inhibits EBV-induced membrane fusion and does not function in fusion. Virology 2013, 436:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ: Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 2010, 84:12292–12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanarsdall AL, Ryckman BJ, Chase MC, Johnson DC: Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J Virol 2008, 82:11837–11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chesnokova LS, Ahuja MK, Hutt-Fletcher LM: Epstein-Barr Virus Glycoproteins gB and gHgL Can Mediate Fusion and Entry in Trans; Heat Can Act as a Partial Surrogate for gHgL and Trigger a Conformational Change in gB. J. Virol 2014, doi: 10.1128/JVI.01597-14. [DOI] [PMC free article] [PubMed] [Google Scholar]