

Abstract
CD3-bispecific antibodies represent an important therapeutic strategy in oncology. These molecules work by redirecting cytotoxic T cells to antigen-bearing tumor cells. Although CD3-bispecific antibodies have been developed for several clinical indications, cases of cancer-derived resistance are an emerging limitation to the more generalized application of these molecules. Here, we devised whole-genome CRISPR screens to identify cancer resistance mechanisms to CD3-bispecific antibodies across multiple targets and cancer types. By validating the screen hits, we found that deficiency in IFNγ signaling has a prominent role in cancer resistance. IFNγ functioned by stimulating the expression of T-cell killing–related molecules in a cell type–specific manner. By assessing resistance to the clinical CD3-bispecific antibody flotetuzumab, we identified core fucosylation as a critical pathway to regulate flotetuzumab binding to the CD123 antigen. Disruption of this pathway resulted in significant resistance to flotetuzumab treatment. Proper fucosylation of CD123 was required for its normal biological functions. In order to treat the resistance associated with fucosylation loss, flotetuzumab in combination with an alternative targeting CD3-bispecific antibody demonstrated superior efficacy. Together, our study reveals multiple mechanisms that can be targeted to enhance the clinical potential of current and future T-cell–engaging CD3-bispecific antibody therapies.
Introduction
CD3-bispecific antibodies are an important emerging strategy for the treatment of both hematologic and solid tumors (1). Although present in various formats, a typical CD3-bispecific antibody is capable of binding the CD3 subunit of the T-cell receptor complex on cytotoxic T cells and a tumor-associated antigen on tumor cells simultaneously, resulting in CD3-bispecific antibodies recruiting T cells to engage and destroy target cancer cells via redirected T-cell cytotoxicity (RTCC; ref. 2). To date, this type of therapy has achieved remarkable clinical success, primarily in hematologic indications. Blinatumomab is approved for the treatment of B-cell acute lymphoblastic leukemia (ALL), and it has been demonstrated to extend patient survival compared with conventional chemotherapy in pivotal clinical trials (3). Other CD3-bispecific antibodies, such as flotetuzumab (CD123-DART), have also shown notable therapeutic potential in patients with relapsed/refractory acute myeloid leukemia (AML; ref. 4).
Although efficacious, resistance to CD3-bispecific antibody therapy has been reported (5), rendering retreatment with the same agent ineffective. A variety of mechanisms can contribute to such resistance or nonresponsiveness. One well-acknowledged example is evasion through loss of the target antigen. In relapsed blinatumomab [CD19-bispecific T-cell engager (BiTE)]-treated patients, approximately 20% were found to have no or lowered expression of the target antigen CD19 (5, 6). Interestingly, in the context of CD19-BiTE resistance, CD19 loss was likely associated with disrupted CD19 membrane trafficking, suggesting a different mechanism compared with that in CAR T-cell therapy (6). Besides antigen loss, tumor-derived immunosuppression is also shown to undermine the potency of CD3-bispecific antibodies (7). A deeper understanding of resistance mechanisms to CD3-bispecific antibody therapy could provide multiple rationales for patient selection or combination strategies.
To discover novel resistance mechanisms to CD3 bispecifics, we devised a genome-wide CRISPR screen to interrogate the impact of more than 19,000 genes in cancer cells on sensitivity or resistance to T-cell killing directed by CD3 bispecifics. In this study, we performed the CRISPR screen in three cell lines of different indications using two clinically relevant CD3-bispecific molecules. From the screens, we identified multiple pathways involved in the resistance phenotype and uncovered biological insights in the resistance mechanism.
Materials and Methods
Cell lines
All cell lines (MOLM13, SEM, HCC827, HCC1954, and MM1S) involved in this study were acquired from Novartis Cell Line Ordering repository (CLEO). The CLEO cell line repository was derived from early passages of the Cancer Cell Line Encyclopedia (8) in 2012. The cells have passed mycoplasma testing on a regular basis, and their identity is regularly confirmed by SNP testing. For cell killing assays, MOLM13, SEM, and HCC827 were engineered to constitutively express firefly luciferase using EF1a-Luciferase lentivirus (GeneTarget Inc.; Catalog #LVP435). All cell lines are cultured in RPMI1640 (Invitrogen) with 10% FBS (Lonza). For TF-1 cells, GM-CSF (5 ng/mL; Peprotech) was added to support cell growth.
Animals and CD3-bispecific antibody in vivo efficacy study
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (8 weeks old) were purchased from The Jackson Laboratory and maintained in a specific pathogen-free facility. Animals were maintained and handled in accordance with Novartis Biomedical Research Institutes Animal Care and Use Committee protocols and regulations. All studies were conducted under an approved Institutional Animal Care and Use Committee protocol. For the in vivo study, 5 × 106 MOLM13 cells of wild-type (Scramble), FUT8−/−, and IL3RA−/− were implanted at day 0 by s.c. injection into the right flank of each mouse. On day 1, 20 × 106 freshly thawed human peripheral blood mononuclear cells (PBMC; Lonza; Catalog #CC-2702) were infused into each animal by i.v. injection. On day 6, mice were randomized into experimental groups based on average tumor volume.
The CD123-DART was formulated by dialyzing the CD123-DART protein in in 20 mmol/L citrate, 100 mmol/L sodium chloride, 50 mmol/L sucrose, and 0.02% Tween 80 at pH 5.5. Starting from day 6 after PBMC infusion, mice were dosed with 0.03 mg/kg CD3-bispecific antibodies for 6 consecutive days by i.p. injection. Two times a week, tumor volume of each mouse was monitored by a caliper, and body weight was measured by a scale. Mice were sacrificed once the body weight dropped by 20% from the initial weight, the tumor volume was over 2,000 mm3, or until graft-versus-host disease (GVHD) onset. The GVHD onset was determined by weight loss of over 20% and reduced mobility as reported in the literature (9).
Lentiviral transduction
Lentivirus was packaged by transfecting Lenti-X 293T cells (Clontech; Catalog #632180) with the lentivirus plasmid and helper plasmids (Cellecta; Catalog #CPCP-K2A) using the lipofectamine 2000 reagent (Invitrogen; Catalog #11668027). The viral supernatant was collected 72 hours after transfection, followed by virus concentration (Clontech; Catalog #631231). For lentiviral transduction, the concentrated lentivirus was added to the target cells in the presence of 10 μg/mL polybrene (Sigma; Catalog #TR-1003-G) and centrifuged at 32°C for 90 minutes at 800 × g. The viral supernatant was then removed and switched to fresh DMEM with 10% FBS (Invitrogen; Catalog #11965084).
CRISPR knockout
To engineer CRISPR knockout cell lines, the MOM13, SEM, HCC827, MM1S, and TF-1 cells were transduced with Cas9-overexpressing lentivirus with blasticidin resistance marker. The cells were selected in 10 μg/mL blasticidin (Sigma; Catalog #CAS589205) for at least 14 days. This step generated Cas9-engineered cell lines. Next, the Cas9-engineered cell lines were transduced with lentivirus containing the single-guide RNA (sgRNA) sequence, followed by selection by 2 μg/mL puromycin (Invitrogen; Catalog #A1113803) for at least 7 days. For knockout validation, the knockout efficiency was assessed by flow cytometry, Western blot, or Sanger sequencing of the targeted genome. The sgRNA sequences for each targeted genes are provided in Supplementary Table S1.
qPCR quantification of sgRNA-transduced cells
The qPCR for sgRNA was designed through IDT PrimeTime portal (https://www.idtdna.com/pages/products/qpcr-and-pcr/gene-expression/primetime-primer-only-assays). Briefly, the forward primer and reverse primer covered the flanking sequence, and a probe with FAM dye and ZEN quencher was complementary to the sgRNA sequence. For the qPCR, the genomic DNA of the cells was extracted using the QIAcube HT automation system (Qiagen; Catalog #51331). The PrimeTime qPCR reaction was performed on the Vii7 Real-Time PCR System (Applied Biosystems) using the IDT-recommended PrimeTime qPCR protocol. For each reaction, 1 μL genomic DNA (~10 ng) was used in a total reaction volume of 20 μL. The relative abundance of the target sgRNA over control sgRNA was given by . The sequences for PrimeTime primers are fwd: 5′-CTTGGCTTTATATATCTTGTGGAAAGG-3′ and rev: 5′-TTAAACTTGCTATGCTGTTTCCAG-3′. The probe sequences are sgScramble: 5′-TGAAGAAGATGGTGCGCTCC-3′, sgCD123: 5′-AGGTCGTACTGGACGTCCGG-3′, and sgCDH3: 5′-ACCCACCCTGAGAGCAACCAG-3′.
RTCC assay and cytokine ELISA
CD3-bispecific antibodies involved in this study are generated from published sequences of CD123-DART (4), PCAD-DART (10), CD33-BiTE (11), and CD19-BiTE (12). The antibodies were dissolved in PBS and frozen in −80°C in small aliquots for long-term storage.
Firefly luciferase-expressing MOLM13, SEM, and HCC827 were used as target cells. The cells were seeded in a 384-well plate at 2,000 to 3,000 cells/well in complete RPMI. After overnight culture in 37°C, T cells and CD3-bispecific antibodies were added using the Tecan Freedom EVO platform (TECAN). The CD3-bispecific antibodies were diluted in a 10-point dilution series by a factor of 3 in complete RPMI; complete RPMI was used as the diluent. In the experiment in which L-fucose was used, L-fucose (Sigma; Catalog #F2252) of 100 mmol/L or indicated concentrations was added to the coculture. The target cell abundance was quantified by the Bright-Glo luciferase assay (Promega; Catalog #E2650) at designated timepoints. A killing curve was derived from normalizing the luminescence at each concentration point to the untreated.
IFNγ concentrations from the RTCC assay were quantified using the MSD system (MSD; Catalog #K151TTK-2). Briefly, the 96-well MSD plates were blocked for 1 hour at room temperature by the kit-provided blocking solution. Supernatants were diluted 1:3 using kit-provided diluent and applied to the plates, together with serially diluted IFNγ standards (ranging from 6 pg/mL to 100 ng/mL) provided by the MSD kit. Detection antibodies were added and incubated at room temperature for 1 hour. Plate washing in between incubations was performed using the Biotek EL406. The plate wash buffer used was PBS with 0.05% Tween-20. The data were collected and analyzed using the Meso Sector 600 instrument (MSD).
CD3-bispecific antibody CRISPR screen
Human T cells were prepared from purified human PBMCs (Lonza; Catalog #CC-2702) using the Pan T-cell Isolation Kit (Miltenyi; Catalog #130–096–535), then stimulated with anti-CD3/CD28 activator beads (Invitrogen; Catalog #11131D) at bead:T-cell ratio of 3:1, and cultured for 10 days in complete RPMI. The complete RPMI was formulated with RPMI1640 (Invitrogen), 10% FBS (Lonza), nonessential amino acid (Invitrogen), L-glutamine (Invitrogen), penicillin–streptomycin (Sigma), and β-mercaptoethanol (Sigma). To expand T cells, human IL2 (10 ng/mL; Peprotech; Catalog #200–02) was added.
For CRISPR screen, lentivirus was generated from the sgRNA plasmid library as described in an earlier study (13). The library contains approximately 180,000 sgRNA sequences, with 10 guides targeting each gene in the genome. Note that 180 × 106 target cells were transduced with the pooled lentivirus to ensure an infection rate of approximately 30%. After 3 days of culture, the cells were selected with 2 μg/mL puromycin for 4 additional days. At the end of the culture, at least 1,000 cells/guide were achieved to ensure adequate representation of each knockout species. Note that 180 × 106 target cells were mixed with T cells at indicated target cell:T-cell ratio and CD3-bispecific antibody at optimized concentrations, which was 1 pmol/L CD123-DART for MOLM13 and SEM screen, and 10 pmol/L PCAD-DART for the HCC827 screen. Trace amounts of the cells were collected during the coculture for flow cytometric analysis. For the MOLM13 and SEM screens, the cells were collected 14 days after coculture, and for the HCC827 screen, cells were collected 7 days after coculture. The genomic DNA from each group was purified by the QIAamp DNA Blood Maxi Kit (Qiagen; Catalog #51194). Targeted sequencing of the incorporated sgRNA region was performed on the HiSeq 2500 system (Illumina). Data normalization and analysis are delineated in Fig. 1C.
Figure 1.

Genome-wide CRISPR screens reveal resistance mechanisms to CD3-bispecific antibodies. A, Schematic of the genome-wide CRISPR screen. The cancer cells were engineered to stably express CAS9 and then infected with a sgRNA lentivirus library. NGS, next-generation sequencing. B, Time-course analysis of T cells and MOLM13 cells in the screen coculture. The T cells were CD3+, and the sgRNA-transduced MOLM13 cells were RFP+. Top row shows isotype antibody treatment, and bottom row shows CD123-DART antibody treatment. Data represent three independent experiments. C, Flowchart of the screen data analysis. Guide calls were quantified from targeted RNA-seq of the guide sequences and subsequently normalized to internal nontargeting controls in the guide library. The data were then normalized to the initial pool sample, and statistical test was performed between the DART group and isotype group. D, Data presentation for the three screens. Each point represents one gene, and the axes show enrichment scores from CD3-bispecific treatment group (DART) or isotype antibody treatment group (Isotype) on the log2 scale. Genes with high DART enrichment and low isotype enrichment scores were considered as resistance hits and vice versa for sensitizing hits. E and F, Venn diagram of the shared resistance (E) or sensitizing (F) hits from all three screens. Hits were determined by running two-tailed t test between the DART group and isotype group for each cell line, with a cutoff of −log10 P value >2 (Supplementary Table S3).
Size-exclusion chromatography–small-angle X-ray scattering study of core fucosylation–deficient CD123
CD123His-Scramble and CD123His-FUT8KO proteins were concentrated to 7 mg/mL and submitted to Argonne National Laboratory for the size-exclusion chromatography (SEC)–SAXS (small-angle X-ray scattering) study. SEC-SAXS experiments were performed at BioCAT (beamline 18-ID; Advanced Photon Source, Argonne National Laboratory). The setup included a focused 12-keV (1.03 ) X-ray beam, a coflow sample cell (14), a sample-to-detector distance of approximately 3.5 m, and a Pilatus3 1M detector. The momentum transfer (scattering vector) q was sampled from approximately 0.0043 to 0.3576−1. In order to ensure sample monodispersity, we used an in-line SEC setup, which included an Äkta pure fast-performance liquid-chromatography unit and a Superdex200 10/300 GL column (GE Healthcare Life Sciences). The column outlet was directly connected to the SAXS sample cell. Note that 0.5-second exposures were collected every 2 seconds during the gel filtration chromatography run. Exposures preceding those corresponding to the sample elution were averaged to generate the buffer I(q) versus q curve, which was then subtracted from the protein + buffer exposures. All the data reduction and processing were performed using RAW (https://sourceforge.net/projects/bioxtasraw/; ref. 15). Each peak fraction frame was analyzed using the PRIMUS, GNOM, CRYSOL, SASREF, and SUPCOMB programs of the ATSAS package (16, 17). Chains A and G, representing the monomeric CD123 form, the crystal structure PDBID—5UV8, were separated and allowed to rotate and translate with respect to each other within SASREF, with side-chain and subunit orientation constraints derived from the crystal structure to obtain configurations that best fit the corresponding experimental scattering data.
Flow cytometry and Western blot
To detect surface markers by flow cytometry, 5 × 105 MOLM13, SEM, and HCC827 cells were resuspended in 100 μL autoMACS running buffer (Miltenyi; Catalog #130–091–221). Prior to staining, cells were blocked with Fc-blocking antibody (5 μg/mL; Invitrogen; Catalog # 14–9161–73) for 5 minutes at room temperature. Fluorescently labeled antibodies were used to stain the cells on ice for 30 minutes. To detect intracellular proteins, including phosphorylated proteins, the cells were processed with the BD Transcription Factor Phospho Buffer Set (BD Biosciences; Catalog #565575). Flow cytometry was performed on a BD LSR FORTESSA instrument (BD Biosciences). Flow cytometry data were analyzed by FlowJo v10 (BD Biosciences). Antibodies used for flow cytometry are listed in Supplementary Table S2.
For lens culinaris agglutinin (LCA) staining, 0.5 × 106 to 2 × 106 MOLM13 or SEM cells were harvested from cell culture, washed once with 1 mL autoMACS running buffer, and then resuspended in 100 μL autoMACS running buffer. LCA-FITC (Vector Laboratories; Catalog #FL-1041) was stocked at 5 mg/mL and was added to stain 5 × 105 cells at a 1:500 dilution (final concentration = 10 μg/mL) on ice for 30 minutes before flow cytometry analysis.
For Western blot, protein concentration was quantified with BCA Protein Assay Kit (Invitrogen; Catalog #23225). A typical Western blot workflow was followed and included cell lysis in RIPA buffer (Invitrogen; Catalog #89900), denaturing in SDS buffer (Sigma; Catalog #T7777), 4% to 20% gel electrophoresis (Bio-Rad), and transfer using the Trans-Blot Turbo Transfer System (Bio-Rad). For each lane, 15 to 20 μg protein was loaded. The membranes were blocked using SuperBlock Blocking Buffer (Thermo Scientific; Catalog # 37537) and blotted with primary and secondary antibodies. The membranes were developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific; Catalog # 34577) and documented using the ImageQuant LAS 4000 system (GE Life Sciences). Antibodies used for Western blotting are listed in Supplementary Table S2.
Gene ontology analysis
Gene ontology of the screen hits was performed using the Metacore Genego system. Briefly, significantly enriched or depleted genes from each screen (−log10 P value > 2; Supplementary Table S3) were uploaded to the Genego system (https://portal.genego.com/), and the gene ontology was calculated using the Process Networks function. Top ten pathways ranked by enrichment P values were presented.
RNA sequencing of IFNγ-treated cells
Note that 10 × 106 MOLM13, SEM, and HCC827 cells were treated with 100 ng/mL human IFNγ (Peprotech; Catalog #300–02) and cultured in RPMI with 10% FBS. Cells were lysed in buffer RLT provided from the PureLink RNA Mini Kit (Invitrogen; Catalog #12183018A) at 0, 6, and 24 hours after IFNγ treatment. Samples were prepared in triplicate. The total RNA for each sample was extracted using the PureLink RNA Mini Kit. The RNA quality was assessed using the Bioanalyzer 2100 (Agilent) to ensure RNA Integrity Number (RIN) scores for all samples were above 9.0. RNA samples were diluted to 4 ng/μL for RNA sequencing (RNA-seq). The RNA-seq data were analyzed by in-house proprietary pipelines; P values are derived from the Student t test. The differentially expressed genes were listed in Supplementary Table S4. The RNA-seq datasets have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE159547.
LCA pulldown of core fucosylated protein and mass cytometry
Note that 10 × 106 MOLM13 cells of different genotypes were lysed in buffer containing 0.05 mol/L Tris-HCl, pH 7.5 (Sigma), 0.5% NP-40 (Sigma), and 150 mmol/L NaCl (Sigma), and supplemented with 1% n-Dodecyl-β-D-Maltopyranoside (Sigma), 1% Triton X-100 (Sigma), 10% Glycerol (Sigma), and protease inhibitor (Invitrogen; Catalog #78437). Note that 1 mmol/L CaCl2, MnCl2, or MgCl2 (Sigma) was added to 2 mg protein from each group. LCA-Agarose beads (Vector Labs; Catalog #AL-1043) were used to pack the purification columns (Thermo Fisher; Catalog #89896). The proteins were incubated in column in 4°C for 1 hour. Next, proteins were washed 2 times with 50 mmol/L Tris-HCl and 2 times with 50 mmol/L Tris-HCl and 300 mmol/L NaCl. Finally, the proteins were eluted with buffer containing 50 mmol/L Tris-HCl, 300 mmol/L NaCl, 400 mmol/L mannoside, and 400 mmol/L glycoside. The pulldown samples were collected in three biological repeats. The complexity of proteins of the eluted samples was assessed by silver staining (Invitrogen; Catalog #LC6070). Sample preparation and LC-MS analysis were outsourced to IQ Proteomics. Eluted protein samples were reduced with 20 mmol/L dithiothreitol, and cysteines were alkylated with 60 mmol/L iodoacetamide, followed by methanol–chloroform precipitation. Purified proteins were digested with trypsin (Promega) and labeled with Tandem Mass Tags (Pierce). Pooled labeled peptides were separated into three fractions using high pH reversed-phase spin column (Pierce; Catalog #84868) containing 10%, 20%, and 50% acetonitrile in 0.1% triethylamine, and desalted with lab-made Stage-Tips prior to mass spec analysis. LC-MS (nanoLC-1000, Thermo Fisher) was performed using the SPS mode on Lumos instrument (Thermo Fisher). See ref. 18, page S101–102, for detailed sample preparation and LC-MS methods. Resulting data were processed by ProteomeDiscoverer 2.1 (Thermo Fisher) and internally developed scripts (proprietary scripts written in python and R programming languages) to calculate protein fold changes and P values to compare individual sample groups using LIMMA package (Bioconductor). The protein list was filtered first by P value of less than 0.005 for both Scramble versus FUT8-g1 and Scramble-FUT8-g3 comparisons, then by filtering out common contaminants, and finally by enriching to cell membrane proteins. The protein list for this study is listed in Supplementary Table S5.
Expression constructs
All constructs were synthesized from GeneArt (Invitrogen) and cloned into proprietary expression vectors. CD123His was designed to fuse 6 × Histidine Tags to the C-terminal of CD123 extracellular domain (ECD). Chimeric CD123 constructs were designed to swapping human CD123 ECD domains with mouse CD123 ECD domains: huCD123-ECD contains the full-length ECD of human CD123; muCD123-ECD contains the full-length mouse CD123 ECD; muNTD-huD2-huD3 contains mouse CD123 NTD fused with human CD123 D2 and D3 at C-terminal; huNTD-muD2-huD3 contains mouse CD123 D2 fused with human CD123 NTD at N-terminal and human CD123 D3 at C-terminal; huNTD-huD2-muD3 contains mouse CD123 D3 fused with human CD123 NTD and human CD123 D2 at N-terminal. The protein sequences for each expression cassette are listed in Supplementary Table S6.
CD123 protein expression and surface plasmon resonance analysis
Ribonucleoprotein (RNP) knockout of FUT8 was performed in FreeStyle 293-F cells (Gibco; Catalog #R79007); control (Scramble) and FUT8KO FreeStyle 293 cells were further sorted based on the LCA staining. The CD123His expression plasmids were transfected into the modified FreeStyle 293 cells using the 293fectin Transfection Reagent (Gibco; Catalog #12347500). The culture supernatant was collected 5 days after transfection. The supernatants were purified using the AKTA system (GE Life Sciences) and eluted in 50 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, and 250 mmol/L Imidazole. The purified CD123His proteins were dialyzed (Thermo Scientific; Catalog #87730) in PBS and concentrated (Thermo Scientific; Catalog #88517). The quality of purified samples was assessed by Western blot and analytical SEC.
The surface plasmon resonance (SPR) assay was performed on the Biacore T200 (GE Life Sciences). The CD123His proteins were immobilized on different flow cells in a Sensor Chip CM5 (GE Life Sciences; Catalog #29149603). The regeneration condition was manually tested, and the optimal regeneration condition for antibody binding was glycine, pH 1.7, whereas no regeneration was needed for IL3 binding. The sample flow rate was set to 30 μL/sec. The binding kinetics was analyzed using the Biacore T200 Evaluation Software (GE Life Sciences).
Capillary electrophoresis analysis of CD123 afucosylation status
Afucosylation of purified CD123-His6 samples produced in HEK293-Scramble or HEK293-FUT8KO cell line, respectively, was analyzed on Lab Chip GXII Touch instrument (PerkinElmer, Software version 1.8.915.0) using the Profiler Pro Glycan Kit (PerkinElmer, Part No. 760523) according to the manufacturer’s instructions. Briefly, protein samples in a concentration range of 0.8 to 1.9 mg/mL were denatured (PerkinElmer, Denaturation Buffer, Glycan Release and Labeling Kit, Part No. 760523) and incubated at 70°C for 10 minutes (Thermomixer, 600 rpm). N-linked glycans were released from the purified and reduced protein samples by enzymatic digestion with PNGaseF at 37°C for 90 minutes (PerkinElmer, PNGaseF-plate, Glycan Release and Labeling Kit, Part No. 760523). Content of deglycosylation of the protein samples was verified via analyzing unlabeled protein samples before and after PNGaseF digestion on LabChip HT Protein Express Chip (PerkinElmer, Part No. 760499, HT Protein Expression Kit, Part No. CLS960008) under reducing conditions according to the manufacturer’s guidelines. Released glycans were fluorescently labeled in the presence of released proteins (PerkinElmer, Dye Plate, Glycan Release and Labeling Kit, Part No. 760523) and incubated at 55°C for 2 hours or until dry. After sample reconstitution in water (according to manufacturer’s instructions), samples were separated by microchip-based capillary electrophoresis (High-Resolution Lab Chip, PerkinElmer, Part No. 760524, Glycan Profiling Assay Kit, PerkinElmer, Part No. 760525) and subsequently detected by laser-induced fluorescence in comparison with glycan standards (Agilent, Man5 Part No. GKM-002500, G0 Part No. GKC-004300, G0F Part No. GKC-004301, G1 Part No. GKC-014300, G1F Part No. GKC-014301, G2 Part No. GKC-024300, G2F Part No. GKC-024301) using Lab Chip GXII instrument and Lab Chip Reviewer Software (Version 5.6.2411.0).
The percentage of afucosylation was calculated on the measured relative chromatographic peak area ratios
Ribonucleoprotein Cas9/crRNA delivery in human CD34+ stem cells
Human cord blood CD34+ stem cells were purchased from Lonza (Catalog #2C-101). The cells were thawed and cultured in stem cell maintenance media [StemSpan SFEM (STEMCELL Technologies), supplied with IL6 (50 ng/mL; Peprotech), TPO (50 ng/mL; Peprotech), SCF (50 ng/mL; Peprotech), and Flt-3L (50 ng/mL; Peprotech)]. After 2 days of culture, the cells were subjected to RNP Cas9/crRNA delivery. The crRNAs for knockout are listed in Supplementary Table S1. Briefly, the custom crRNA was synthesized using the Alt-R platform (IDT Technologies). Note that 5 μL of 100 μmol/L crRNA was annealed with 5 μL of 100 μmol/L tracrRNA and assembled with 6.5 μL of recombinant Cas9 protein (2.5 mg/mL). The mixture was added to 1 × 106 CD34+ stem cells. The transfection was performed using the Neon Transfection System (Thermo Scientific; Catalog #MPK10025) and Neon device (Thermo Fisher). Specifically, the cells were resuspended in buffer T (as provided in the kit), and the electroporation was programmed to 1,600 V, 10 ms, and 3 pulses. After Cas9/crRNA delivery, the cells were recovered in the stem cell maintenance media for 3 days, followed by knockout validation by flow cytometry or Western blot. The antibodies used for knockout validation are listed in Supplementary Table S2.
Methocult colony formation and plasmacytoid dendritic cell differentiation assay
For the colony formation assay, CD34+ stem cells of different genotypes were collected and resuspended in the Methocult H4100 media (STEMCELL Technologies; Catalog #04100). This incomplete media were supplemented with IL3 (50 ng/mL; Peprotech), SCF (50 ng/mL; Peprotech), and Flt-3L (50 ng/mL; Peprotech) diluted in StemSpan SFEM (STEMCELL Technologies). Three thousand cells were seeded in one well of a 6-well plate (STEMCELL Technologies; Catalog #27371). The colony formation was visualized using the StemVision (STEMCELL Technologies) 10 days after initial seeding. The colonies were quantified with ImageJ (https://imagej.nih.gov/ij/). Flow cytometry results were analyzed by FlowJo (BD Biosciences).
For plasmacytoid dendritic cell (pDC) differentiation, 8 × 104 CD34+ stem cells were cultured in StemSpan SFEM (STEMCELL Technologies) supplied with TPO (50 ng/mL; Peprotech), Flt-3L (100 ng/mL; Peprotech), IL3 (20 ng/mL; Peprotech), and 1 μmol/L SR-1 (Sigma). The cells were split by half every week for 3 weeks, and on day 21, pDC differentiation was assessed by flow cytometry. Antibodies used for pDC staining (CD123, CD303) are listed in Supplementary Table S2. Flow cytometry results were analyzed by FlowJo (BD Biosciences).
FACS competition assay
MOLM13 and SEM cells were cultured in RPMI1640 (Invitrogen) with 10% FBS (Lonza). The FACS competition assay was performed to quantify the competitive binding of different CD123 antibodies to CD123 on MOLM13 and SEM cell. 6H6 (Biolegend; Catalog #306002), CD123-DARTIgG (in-house proprietary), and isotype (in-house proprietary) antibodies were labeled with AF647 using the antibody labeling kit (Thermo Fisher; Catalog # A20186). In the FACS competition assay, 2.5 μg of unlabeled antibody was first added to 5 × 105 cells and incubated at 4°C for 30 minutes, followed by the addition of 0.5 μg AF647-labeled antibody and incubation at 4°C for 60 minutes. The cells were then washed 3 times with autoMACS running buffer and resuspended in autoMACS running buffer (Miltenyi) for FACS analysis. The AF647 fluorescence was read out using MACSQuant instrument (Miltenyi). The competition assay was performed for each antibody pairs, and the %inhibition was calculated by normalizing mean fluorescence intensity (MFI) signals to the isotype/6H6 group.
Mixed RTCC assay
MOLM13 or SEM cells with Scramble, JAK1KO, or FUT8KO modifications were mixed at 8:1:1 ratio. Note that 2 × 105 mixed cells were cocultured with 2 × 105 T cells in 24-well plates in complete RPMI. On day 7, an extra 2 × 105 T cells were added to the coculture. In the CD123-DART continuous treatment group, cell mixture was dosed with 5 pmol/L CD123-DART on days 0 and 7. In the CD33-BiTE switch group, cell mixture was dosed with 5 pmol/L CD123-DART on day 0 and switched to 5 pmol/L CD33-BiTE on day 7. On days 0, 3, 5, 7, 10, 12, 14, and 17, 10% of the cells were collected and used for flow cytometry analysis. The results were analyzed by FlowJo (BD Biosciences) and presented by the proportion of each genotype.
Statistical analysis
Heatmaps were generated using the Morpheus program (https://software.broadinstitute.org/morpheus/). Statistical analysis was performed using GraphPad Prism software. Two-tailed Student t tests were conducted in CRISPR screen analysis, RNA-seq analysis, CD34+ stem cell colony formation analysis, pDC differentiation analysis, and in vivo tumor growth analysis, as indicated in the corresponding figures. P < 0.05 indicated statistically significant differences.
Results
Cancer cell–intrinsic pathways associated with CD3-bispecific antibody resistance
We initiated screening efforts by engineering relevant target-positive cell lines from the Cancer Cell Line Encyclopedia to stably express Cas9 from a ubiquitous promoter. Subsequently, cells were infected with a pooled lentivirus library of sgRNAs and cocultured with T cells in the presence of CD3-bispecific antibodies to enrich for cells that had acquired resistance or sensitivity to the CD3-bispecific mechanism of action by their knockout genotype (Fig. 1A). To understand the breadth of resistance or sensitization mechanisms, we employed MOLM13 (an AML cell line) and SEM (an ALL line), both with high CD123 expression (19). The CD123-DART based on the published sequence of flotetuzumab was used for the screen in these two lines (4). HCC827, a P-cadherin+ non–small cell lung adenocarcinoma-derived line, was screened with a PCAD-DART derived from the sequence of PF-06671008, which is currently in clinical trials for multiple solid tumor indications (10). The first screen was conducted using the CD123-DART antibody in MOLM13 cells. To optimize the screen conditions, we knocked out CD123 in MOLM13 cells (CD123KO) to mimic antigen loss–induced resistance (Supplementary Fig. S1A). We also developed a companion qPCR assay that allowed molecular quantification of sgRNA-transduced cells with high specificity (Supplementary Fig. S1B). In a pilot test, CD123KO cells were mixed 1:9 with wild-type Scramble cells followed by an RTCC assay using the CD123-DART antibody. By quantifying the relative ratio between CD123KO and Scramble cells, we found that the best differential was achieved at 1 pmol/L of CD123-DART treatment and a T-cell to target cell ratio (E:T) of 1:1 (Supplementary Fig. S1C). From the actual CRISPR screen, CD123-DART treatment at a higher dose (10 pmol/L) performed significantly worse than the lower dose (1 pmol/L; Supplementary Fig. S1D), which echoed the results of our qPCR-based optimization (Supplementary Fig. S1C). These data indicated that increasing the selection pressure did not necessarily enhance screen performance.
We tracked T-cell and tumor cell dynamics during the screen by analyzing the proportions of pooled MOLM13 cells (RFP+) and T cells (CD3+) by flow cytometry (Fig. 1B). Compared with the nontargeting isotype antibody treatment, CD123-DART treatment eradicated over 90% of the target cells by day 3; at day 7, resistant target cells gradually recovered; from day 11 on, most T cells likely underwent activation-induced cell death, and resistant tumor cells began to take over the population. Following next-generation sequencing of the sgRNA sequences from the genomic DNA, the enrichment of each gene was calculated by first normalizing the treatment groups to the pretreatment groups, then averaging the values from the ten guides for each gene. In parallel, a t test was performed between CD3-bispecific and isotype antibody groups to identify statistically significant hits (Fig. 1C). Next, we optimized the screen conditions for SEM and HCC827 cells using the qPCR assay (Supplementary Fig. S1E and S1F) and performed the genome-wide CRISPR screens in the same format. The results from all three screens were plotted by each gene’s enrichment score with CD3-bispecific antibody treatment versus isotype antibody treatment, and the hits with statistical significance were highlighted (Supplementary Table S3; Fig. 1D). We classified hits into two categories: resistance hits were the genes with higher enrichment following bispecific antibody treatment, and sensitizing hits were the genes with higher enrichment following isotype antibody treatment. Gene ontology analysis suggested that the resistance hits were primarily enriched in the inflammation and IFN signaling pathways (Supplementary Fig. S1G and S1H). JAK1 and PTPN2 were the common hits from the resistance side and sensitizer side, respectively (Fig. 1E and F).
IFNγ signaling regulates cancer cell resistance to CD3-bispecific antibodies
Our CRISPR screen identified JAK1 and PTPN2 as general regulators of cancer cell resistance/sensitivity to multiple CD3-bispecific antibodies (Fig. 1E and F). Both molecules are key components of IFNγ signaling: JAK1 is the primary tyrosine kinase in the IFNγ–JAK1–STAT1 signaling axis (20); PTPN2 is one of the phosphatases that negatively regulate this pathway (21), whose deficiency generally enhances T-cell–mediated killing (22). We next validated whether IFNγ signaling was required for CD3-bispecific antibody–induced killing. We first blocked IFNγ engagement to its receptor using an anti-IFNGR antibody in the RTCC assay and noticed consistent inhibition of killing activity across all three lines used in the screen (Fig. 2A). Next, we knocked out JAK1 (JAK1KO) and STAT1 (STAT1KO) individually in these cells (Supplementary Fig. S2A and S2B) and found that it reduced the potency of CD123-DART activity in MOLM13 cells and SEM cells by 5-fold and 3-fold, respectively (Fig. 2B and C). Although knocking out JAK1 or STAT1 did not affect the EC50 of the PCAD-DART in HCC827 cells, it lowered the maximum killing efficacy by 40% (Fig. 2D). When we knocked out PTPN2 in the MOLM13 cells (PTPN2KO; Supplementary Fig. S2C), it not only amplified downstream STAT1 phosphorylation (Fig. 2E), but also increased cell sensitivity to the CD123-DART in the RTCC assay by 5-fold (Fig. 2F), consistent with the literature (22). Together, these data confirmed our findings in the CRISPR screen that loss of IFNγ signaling renders tumor cells resistant to killing by CD3-bispecific antibodies, and conversely, augmenting this pathway could sensitize cells to treatment.
Figure 2.
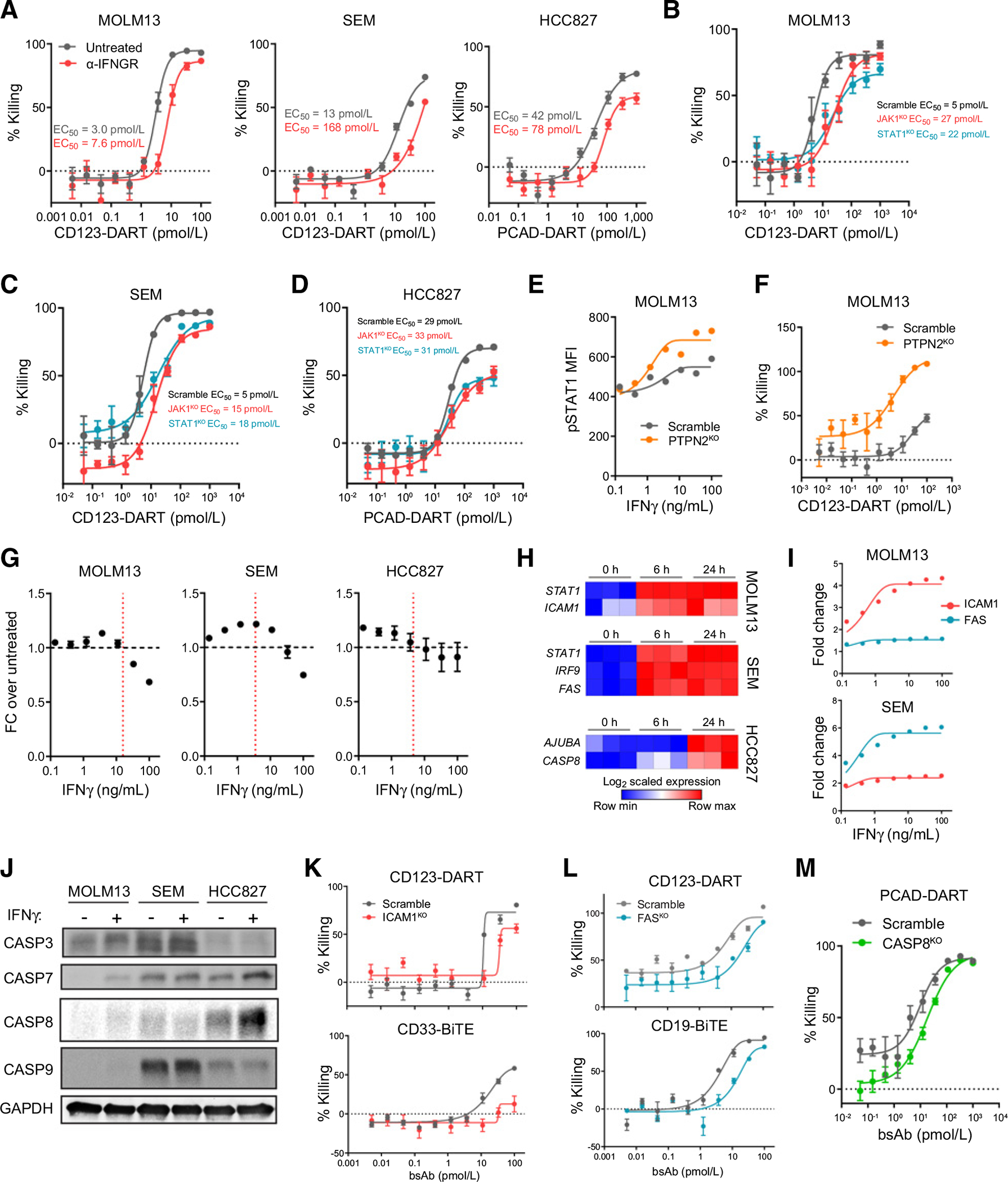
The IFNγ signaling pathway is a central regulator of cancer cell resistance. A, MOLM13, SEM, and HCC827 cells were treated with a blocking antibody against IFNGR1 (α-IFNGR) and applied in the RTCC assay with corresponding CD3-bispecific antibodies (n = 6). The RTCC assay used T-cell:target cell ratio of 0.5:1, and target cell killing was measured 48 hours after treatment. B–D, MOLM13, SEM, and HCC827 cells were knocked out for JAK1 (JAK1KO) and STAT1 (STAT1KO), and killing by CD3-bispecific antibodies was compared with control groups (Scramble; n = 4). The RTCC assay used T-cell:target cell ratio of 0.5:1, and target cell killing was measured 48 hours after treatment. E, Scramble and PTPN2KO cells were starved in RPMI1640 overnight and treated with IFNγ for 5 minutes. IFNγ signaling was reported by STAT1 phosphorylation (pSTAT1) in Scramble and PTPN2KO MOLM13 cells. F, RTCC assay of PTPN2KO MOLM13 cells (n = 4). The RTCC assay used T-cell:target cell ratio of 0.5:1, and target cell killing was measured 24 hours after treatment. G, The growth of MOLM13 (n = 3), SEM (n = 5), and HCC827 (n = 8) cells 72 hours after IFNγ treatment. The red dashed line indicates the maximum IFNγ release in the corresponding RTCC assay (referring to Supplementary Fig. S2D). H, Heatmap of the overlapped hits from the IFNγ treatment RNA-seq and CRISPR screen datasets of each individual cell line. I, Flow cytometric analysis of ICAM1 and FAS expression in the MOLM13 and SEM cells with IFNγ titration (n = 3). J, Western blot of caspase family protein expression in the three cell lines 24 hours after IFNγ (100 ng/mL) treatment (n = 1). K, RTCC assay of ICAM1KO MOLM13 cells using the CD123-DART or CD33-BiTE (n = 6). The RTCC assay used T-cell:target cell ratio of 0.5:1, and target cell killing was measured 24 hours after treatment. bsAb, bispecific antibody. L, RTCC assay of FASKO SEM cells using the CD123-DART or CD19-BiTE (n = 4). The RTCC assay used T-cell:target cell ratio of 0.5:1, and target cell killing was measured 48 hours after treatment. M, RTCC assay of CASP8KO HCC827 cells using the PCAD-DART (n = 4). The RTCC assay used T-cell:target cell ratio of 1:1, and target cell killing was measured 48 hours after treatment. For each plot, error bars represent SEM.
IFNγ signaling plays a role in cancer cell sensitivity to T-cell–based immunotherapies (23, 24). Activated T cells are a major source of IFNγ release, and IFNγ is reported to inhibit cancer cell growth directly (25). When we treated these three target cells with recombinant IFNγ, however, the observed impact on cell growth was limited (Fig. 2G). Although a partial inhibition was observed when IFNγ was dosed at 100 ng/mL, this concentration was far beyond the maximum IFNγ concentration achieved in the RTCC assay (Supplementary Fig. S2D). This insensitivity to direct IFNγ treatment indicated that IFNγ activity alone did not drive the potent cytotoxicity observed in these three cell lines. The loss of IFNγ signaling could suppress antigen presentation pathways to induce inefficient T-cell killing (24). To test whether antigen presentation was involved, we then knocked out B2M (B2MKO), a structural component of the antigen presentation complex in the cell lines. However, B2MKO cells showed no significant resistance (Supplementary Fig. S2E), indicating MHC class I and CD1d antigen presentation were irrelevant in the context of CD3 bispecifics.
We next speculated that IFNγ signaling could function secondarily by inducing the expression of one or more downstream effector molecules that facilitate T-cell cytotoxicity. To further elucidate this possibility, we treated MOLM13, SEM, and HCC827 cells with IFNγ and analyzed global transcriptional changes by RNA-seq (Supplementary Fig. S2F and Supplementary Table S4). In our analysis, we focused on genes that were induced by IFNγ treatment while being defined as resistance hits in the CRISPR screens. From this analysis, we identified multiple hits that were potentially involved in T-cell–dependent cytotoxicity. Foremost among these genes were ICAM1 in MOLM13 cells, FAS in SEM cells, and CASP8 in HCC827 cells. ICAM1 is an adhesion molecule that ligates LFA-1 on T cells to form the immunologic synapse, which is crucial for T-cell–mediated cytotoxicity (26). Both FAS and CASP8 are effector molecules in the apoptosis pathway, which play a role in T-cell–induced apoptosis (27, 28). ICAM1, FAS, and CASP8 were upregulated in a cell line–specific manner by IFNγ treatment (Fig. 2I and J).
Next, we validated the resistance phenotype when these effector molecules were individually depleted in the corresponding cell lines. We found that the ICAM1KO MOLM13 cells not only showed resistance to CD123-DART treatment, but also to another CD3-bispecific antibody, AMG330 (CD33-BiTE), which targets the AML antigen CD33 (Fig. 2K; ref. 11). On the other hand, SEM cells showed resistance to the CD123-DART and CD19-BiTE when they lost FAS expression (Fig. 2L); CASP8KO HCC827 cells were less potently killed by PCAD-DART (Fig. 2M). To provide further mechanistic insight, we found that knocking out JAK1 significantly inhibited ICAM1 upregulation by IFNγ (Supplementary Fig. S2G) and that overexpressing ICAM1 in JAK1KO MOLM13 cells rescued the resistance phenotype (Supplementary Fig. S2H). For SEM cells, codosing the FAS ligand (FasL) and IFNγ led to increased killing of target cells (Supplementary Fig. S2I), suggesting a potential synergistic effect between the FAS pathway and IFNγ pathway. Taken together, our data suggested that deficiency in IFNγ signaling was a generalizable resistance mechanism to CD3-bispecific antibody treatment, but that the specific function of IFNγ signaling may differ by cell type.
Deficiency in the core fucosylation pathway leads to resistance to CD123-DART
Although JAK1 is the only resistance hit shared among all three CRISPR screens (Fig. 1E), a greater number of shared hits were identified between the MOLM13 and SEM screens that employed the CD123-DART (Fig. 3A). Based on ontology analysis, these genes were significantly enriched in the core fucosylation pathway, in which GMDS, SLC35C1, and FUT8 constituted the top hits (Fig. 3B; Supplementary Table S3). Along this pathway, FUT8 is the terminal fucosyltransferase that specifically catalyzes α-(1,6)-fucosylation or core fucosylation of proteins (29). Knocking out FUT8 resulted in total loss of core fucosylation, measured by LCA staining (Fig. 3C; ref. 30). FUT8KO MOLM13 and SEM cells also showed resistance to the CD123-DART in the RTCC assays. This resistance effect was as strong as target antigen loss (CD123KO)–induced resistance (Fig. 3C). However, we observed that at a high CD123-DART dosage, both CD123KO and FUT8KO cells were still depleted (Fig. 3C; Supplementary Fig. S1A). The nonspecific killing could be attributed to potential nonspecific binding of the CD123-DART molecule to target-negative cells, especially at high concentrations. We also generated GMDSKO MOLM13 cells and observed a similar degree of resistance against CD123-DART (Fig. 3D). Because GMDS could be functionally replaced by the parallel fucose salvage pathway (Fig. 3B; ref. 31), when we treat GMDSKO cells with excess L-fucose (100 mmol/L), it not only restored core fucosylation in the cells, but also led to a full rescue of the resistance phenotype in RTCC (Fig. 3D). Conversely, we wondered whether excess L-fucose would boost wild-type cell sensitivity to the CD123-DART. However, core fucosylation was minimally increased with doses of exogenous L-fucose (Supplementary Fig. S3A), and accordingly, L-fucose failed to enhance the efficacy of the CD123-DART in wild-type MOLM13 and SEM cells (Supplementary Fig. S3B). Overexpression of FUT8 was also unable to elevate core fucosylation or improve killing efficacy by RTCC (Supplementary Fig. S3C and S3D), suggesting that FUT8 was nonlimiting in the wild-type setting.
Figure 3.
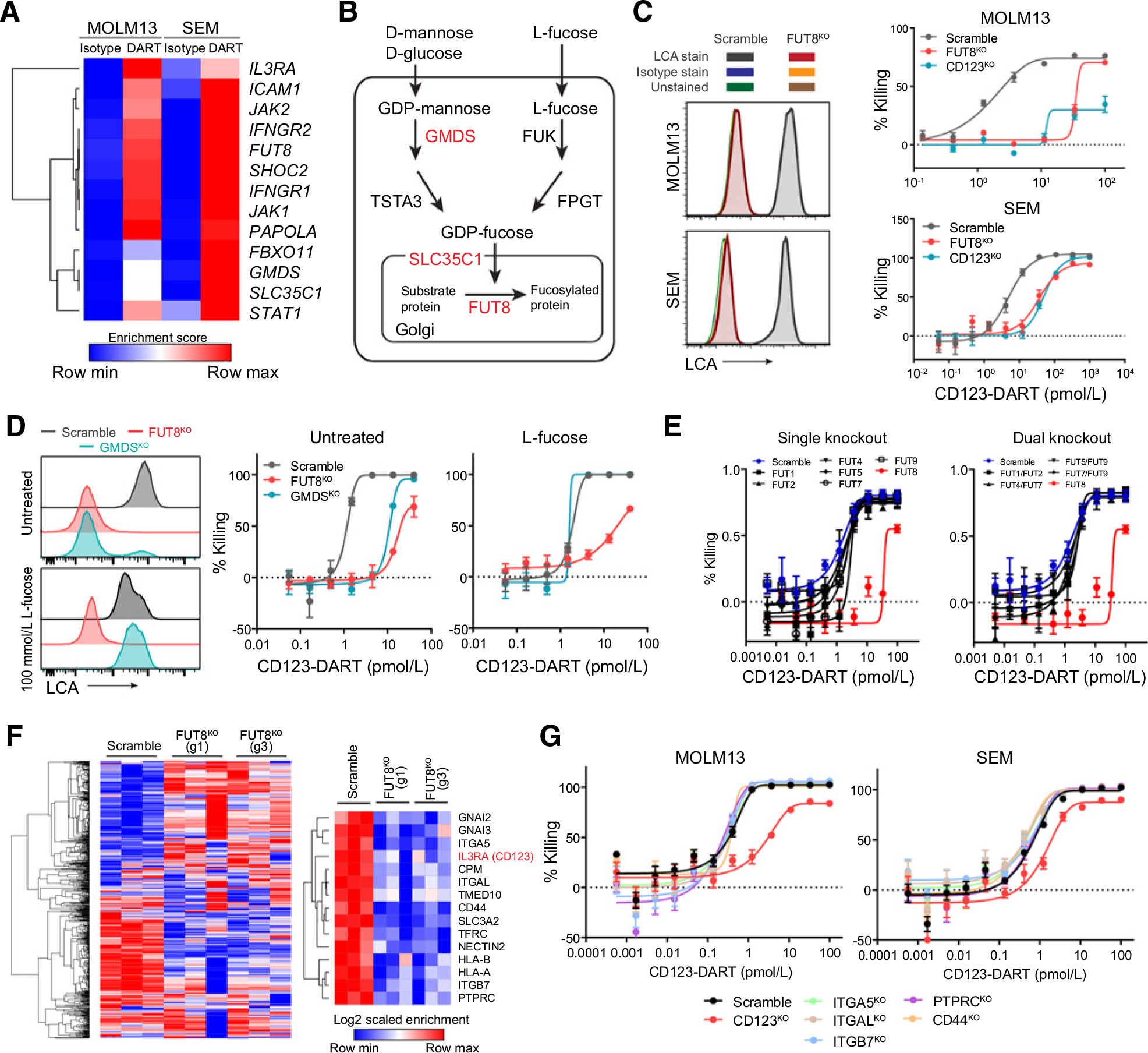
Deficiency in the core fucosylation pathway constitutes a novel resistance mechanism to CD123-DART treatment. A, Heatmap showing the enrichment scores of the shared hits from the MOLM13 and SEM CRISPR screen. DART represents CD123-DART. B, Depiction of the core fucosylation pathway. The hits from the screens are highlighted in red. C, RTCC assay of FUT8KO and CD123KO cells with CD123-DART treatment (n = 4). The RTCC assay used T-cell:target cell ratio of 1:1, and target cell killing was measured 72 hours after treatment. Knockout of FUT8 was confirmed by the loss of LCA staining (left plots). Data represent two independent experiments. The isotype antibody staining and unstained groups overlapped with the histogram of FUT8KO cells. D, Scramble, FUT8KO, and GMDSKO cells were cultured in media supplemented with 100 mmol/L L-fucose, and core fucosylation was measured by LCA staining (left plots). The cells were then applied to the RTCC assay using CD123-DART (n = 4). The RTCC assay used T-cell:target cell ratio of 1:1, and target cell killing was measured 72 hours after treatment. E, RTCC assay (n = 4) of MOLM13 cells with single fucosyltransferase knockout (left plot) or dual fucosyltransferase knockout (right plot) compared with the killing of the FUT8KO group (shown in red; n = 3). The RTCC assay used T-cell:target cell ratio of 1:1, and target cell killing was measured 72 hours after treatment. F, Quantification of core fucosylated proteins by LCA pulldown in concatenation with mass spectrometry (n = 3). Cell-surface proteins with significant enrichment (see Materials and Methods; Supplementary Table S5) in the Scramble group are listed (right plot). G, RTCC assay (n = 4) of MOLM13 cells with knockout of selected genes from the protein list in F. The RTCC assay used T-cell:target cell ratio of 1:1, and target cell killing was measured 72 hours after treatment. For each plot, error bars represent SEM.
Fucosyltransferases (FUT) are a large family of enzymes encompassing at least nine members. Although they catalyze different fucosylation reactions, functional redundancies are also present in many of the FUTs (32). To determine whether the resistance is uniquely attributed to FUT8-mediated core fucosylation, we knocked out other FUTs, as well as functionally redundant FUT pairs. Neither single knockout nor dual knockouts of the FUTs elicited a resistance phenotype as prominent as FUT8KO (Fig. 3E), suggesting that the observed resistance phenotype was dependent on core fucosylation. To better understand the targets of FUT8 modification, we used lectin LCA to pulldown total core fucosylated proteins from wild-type or FUT8KO MOLM13 cells, and subjected isolated proteins to bottoms-up mass spectrometry (Supplementary Fig. S3E). This analysis revealed 603 protein species, of which we chose to focus on 15 candidates that were significantly enriched in the Scramble pulldown group relative to the FUT8KO control (Fig. 3F; Supplementary Table S5). Within this list, adhesion molecules such as ITGA5, ITGAL, and ITGB7 are confirmed FUT8 targets (33) and speculated to participate in immune synapse formation (34). However, knocking out these molecules individually failed to induce resistance in the RTCC assay (Fig. 3G). IL3RA (CD123) was among the identified proteins (Fig. 3F) and could elicit a resistance response in the RTCC assay validation when knocked out (Fig. 3G). These data suggested that CD123 was core fucosylated by FUT8, and that this modification was the primary determinant for cell resistance to the CD123-DART. Beyond CD123-DART, the efficacy of other bispecific antibodies was not affected by FUT8 deficiency (Supplementary Fig. S3F).
Core fucosylation of CD123 is required for efficient binding of the CD123-DART
Next, we aimed to determine how core fucosylation of CD123 mediates sensitivity to CD123-DART treatment. We applied the Scramble and FUT8KO cells in FACS assays with a widely reported anti-CD123 antibody 6H6 (35). No significant difference was found between the two groups, indicating that loss of core fucosylation did not alter CD123’s surface expression (Fig. 4A). For a fair comparison, we converted the CD123-DART into a bivalent full-length human IgG1 format (CD123-DARTIgG). FUT8KO cells lost binding to the CD123-DARTIgG as judged by flow cytometry (Fig. 4A). A second CD123 therapeutic antibody, CSL362 (36), was also found to have reduced binding to FUT8KO cells (Fig. 4A).
Figure 4.
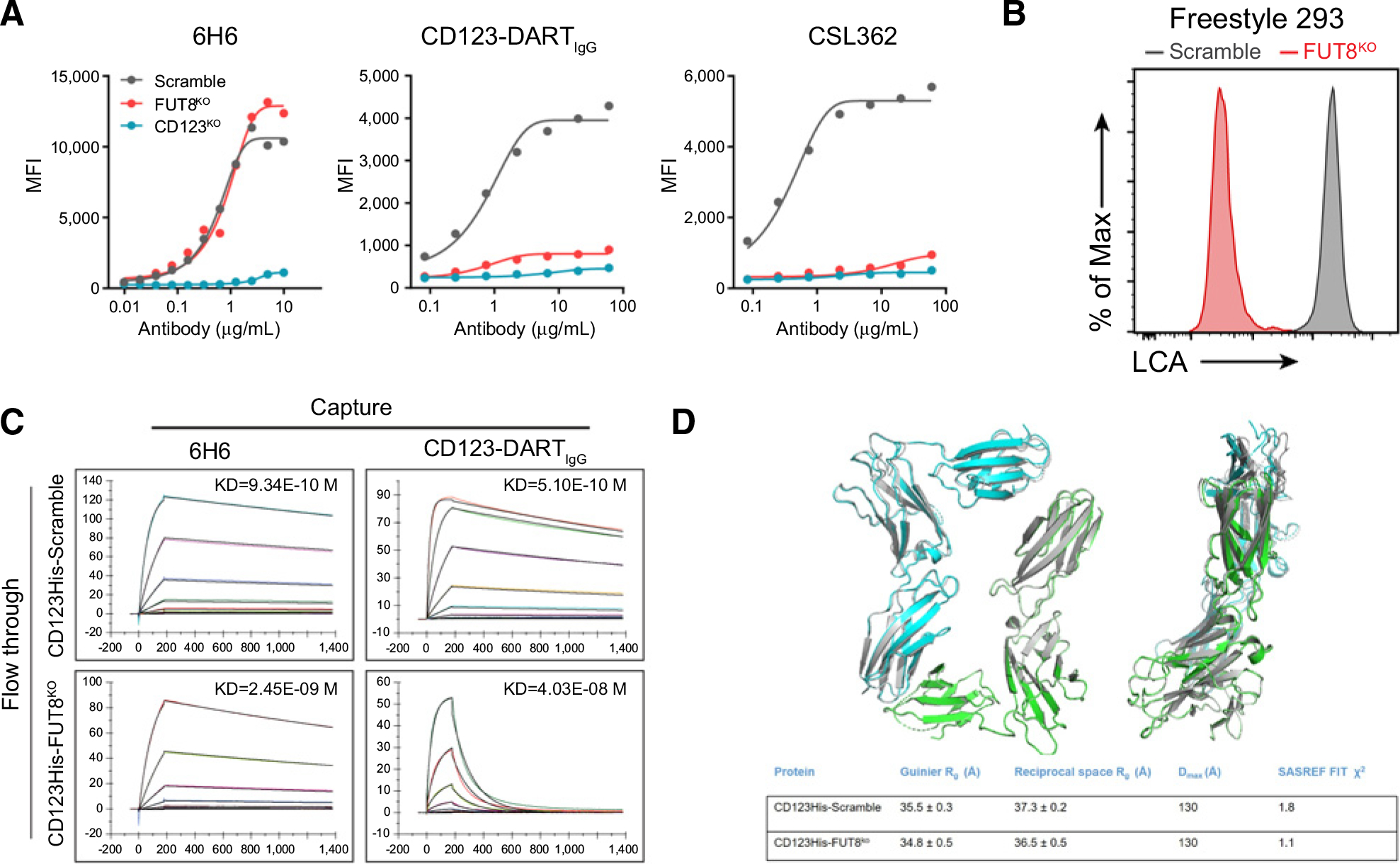
Loss of core fucosylation impedes the binding of CD123 to CD123-DART. A, Flow cytometric analysis of 6H6, CD123-DARTIgG, and CSL362 binding to Scramble, FUT8KO, and CD123KO MOLM13 cells. Cells were incubated with the antibodies for 30 minutes at 4°C, followed by flow cytometry analysis. Data represent three independent experiments. B, Freestyle 293 cells with Scramble and FUT8KO were generated by RNP knockout, and the knockout efficiency was evaluated by LCA staining. Data represent three independent experiments. C, SPR analysis on the binding of purified CD123His-Scramble and CD123His-FUT8KO proteins to 6H6 and CD123-DARTIgG. The antibodies were captured on the chip, and the proteins were subsequently flowed through. The association and dissociation times were set up as indicated in the plots. D, Models generated using SASREF for CD123His-Scramble (blue and green chains) and CD123His-FUT8KO (gray) superimposed using SUPCOMB as described in the Materials and Methods. Rg, Dmax, and χ2 values are displayed in the table (bottom). Chains A and G were from the dimeric 5UV8 structure used to generate the SASREF models.
To investigate this binding defect, we assessed the biophysical properties of CD123 with or without FUT8-dependent core fucosylation. We expressed and purified the His-tagged CD123 ECD from Scramble or FUT8KO freestyle 293 cells (CD123His-Scramble, CD123His-FUT8KO; Fig. 4B). We assessed the afucosylation status of the produced CD123 materials (CD123His-Scramble and CD123His-FUT8KO) by analyzing the cleaved glycan species after PNGase treatment. This assay suggested that the CD123His-FUT8KO protein had significantly higher afucosylation compared with the CD123His-Scramble protein (59.52% vs. 9.66%; Supplementary Fig. S4A). SPR was then used to evaluate the binding affinity of these two proteins to 6H6 and CD123-DARTIgG. We found at least a 100-fold reduction in the binding affinity toward CD123-DARTIgG, largely driven by an increased off-rate when CD123 was devoid of core fucosylation. In contrast, the affinity drop for 6H6 binding was not prominent (Fig. 4C). This finding was recapitulated with an inverted SPR setup (Supplementary Fig. S4B). In this setup, instead of capturing the 6H6/CD123-DARTIgG on the chip and flowing the CD123 antigen, we captured CD123 antigen on the chip and flowed the relevant antibodies. This setup not only reciprocally validated the difference in binding but was also useful in assessing the affinity between CD123 and the nonconverted CD123-DART which lacks the Fc domain (Supplementary Fig. S4B, right plot). Together, these data provided biophysical evidence that core fucosylation of CD123 was essential for proper binding by the CD123-DART.
The epitope of either 6H6 or CD123-DART has not been documented. The ECD of CD123 can be segregated into three domains, namely the N-terminal domain (NTD), domain 2 (D2), and domain 3 (D3), and each domain was predicted to have an N-glycosylation site (Supplementary Fig. S4C, left plot). We therefore constructed chimeric variants of human CD123 by swapping out each domain with the mouse counterpart. We found that when the NTD was swapped, both 6H6 and CD123-DARTIgG completely lost binding, suggesting that both antibodies bind to the NTD of CD123 (Supplementary Fig. S4C, right plot). Next, we conducted antibody competition assays to assess the epitope overlap between these two antibodies. There was competition between 6H6 and the CD123-DARTIgG, indicating both antibodies targeted similar regions in the NTD of CD123 (Supplementary Fig. S4D). These data implied that the core fucosylated residue in NTD was required for CD123-DARTIgG binding.
Alternatively, because posttranslational modifications (PTM) are known to affect protein conformation (37, 38), it is possible that the loss of core fucosylation could affect the general structure of CD123 to selectively prohibit its access to CD123-DART. We performed SAXS analysis (39) of CD123His-Scramble and CD123His-FUT8KO proteins to understand their conformation in solution state. Fitting of the background-subtracted scattering profiles of each protein (Supplementary Fig. S4E) to the pair distance distribution p(r) function revealed that both proteins adopt compact globular conformations, with radius of gyration values (Rg) of 35.5 ± 0.3 Å for CD123His-Scramble and 35.8 ± 0.5 Å for CD123His-FUT8KO (Fig. 4D; Supplementary Fig. S4F). The pair-distance distribution functions of the two proteins were comparable and were both approximately 130 Å (Supplementary Fig. S4E). We compared Rg values obtained from Guinier approximation for both proteins with the published crystal structure of CD123 in complex with IL3 (PDB: 5UV8 chain A) and found that the calculated Rg for 5UV8 (26.1 Å) was significantly smaller than the Rg measurements in this study. In contrast, a dimeric CD123 from 5UV8 had a calculated Rg (32.6 Å), which is in closer agreement to the SAXS-derived data (Fig. 4D). In order to explore potential differences in the relative orientation of the two subunits in the homodimer, we performed SASREF (40). The theoretical SAXS curves from dimers generated by SASREF fit well against the corresponding experimental data (χ2 of 1.8 and 1.1 respectively for CD123His-Scramble and CD123His-FUT8KO). The P(r) curves suggested the dimeric SASREF models for the two constructs were virtually identical and superimposable (Fig. 4D).
Loss of core fucosylation impairs IL3 signaling through CD123
Aside from being a target in AML/ALL, CD123 plays pivotal roles in IL3 signaling. The IL3 receptor complex contains two subunits, with CD123 being the engaging receptor for IL3 and CSF2RB being the signaling coreceptor (41). We wondered whether the fucosylation loss could interrupt CD123/IL3 interactions. In the SPR analysis, we found that removing core fucosylation led to a measurable decrease (10-fold) in the binding affinity between CD123 and IL3 (Supplementary Fig. S5A). Next, we assessed the biological consequences of FUT8 deficiency. We knocked out FUT8 in an IL3 signaling–sensitive cell line, TF-1 (Supplementary Fig. S5B). These cells were kept in culture with GM-CSF, which shares the same CSF2RB signaling subunit as IL3 but a unique alpha subunit. No defect in cell growth was observed in FUT8KO TF-1 cells maintained in GM-CSF (Supplementary Fig. S5C). In comparison, when the culture was switched to IL3, FUT8 deficiency led to a partial reduction in cell growth (Fig. 5A). Accordingly, downstream STAT5 phosphorylation (pSTAT5) was impeded in FUT8KO TF-1 cells (Fig. 5C; Supplementary Fig. S5D).
Figure 5.
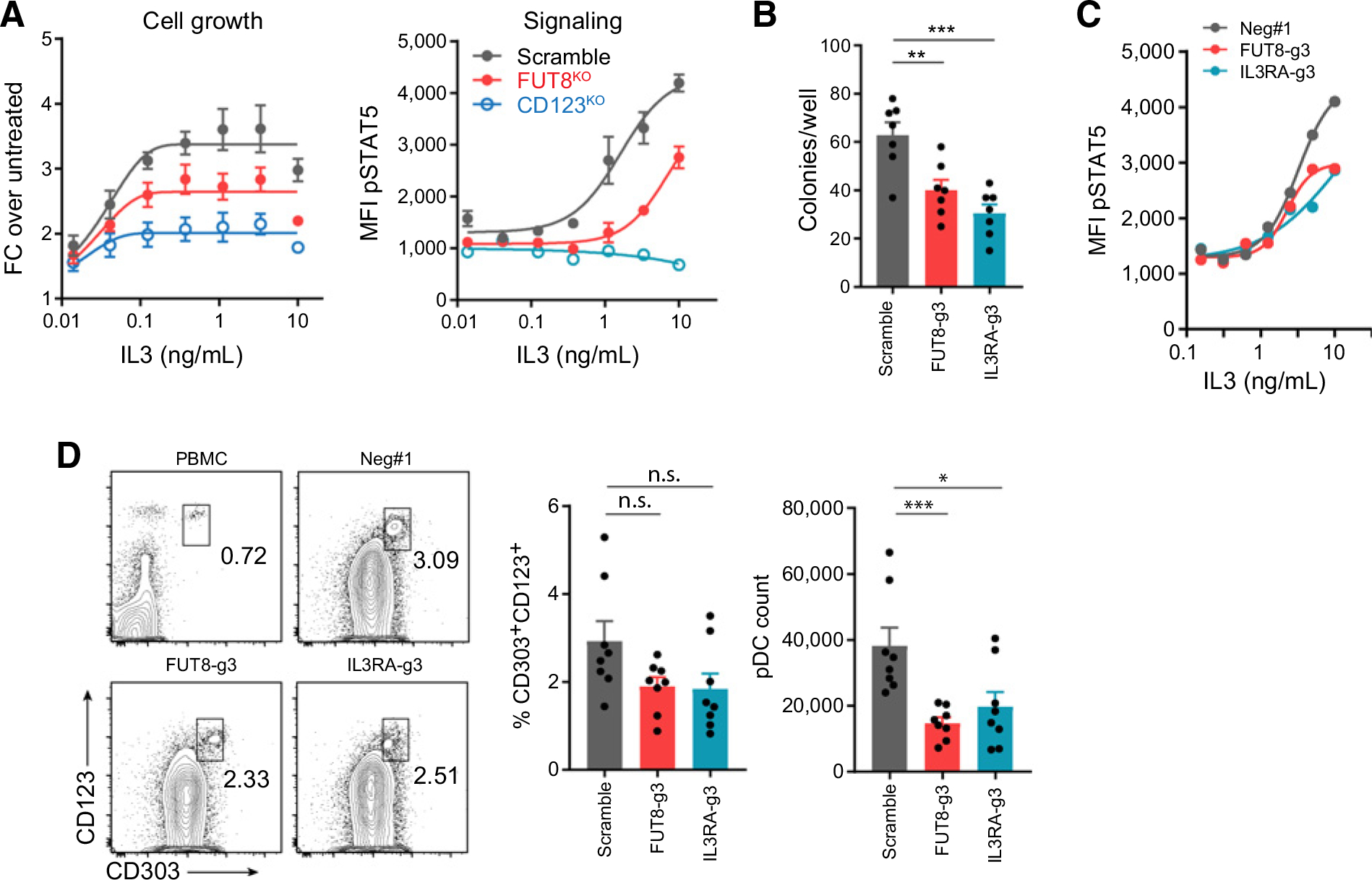
Loss of core fucosylation impairs IL3 signaling and its biological functions. A, Growth and IL3 signaling of Scramble, FUT8KO, and CD123KO TF-1 cells. The cell growth was measured by cell titer glow 3 days after IL3 treatment (left plot; n = 3), and the IL3 signaling was assessed by STAT5 phosphorylation 15 minutes after IL3 treatment (right plot; n = 3). B, Colony formation of Scramble, FUT8-g3, and IL3RA-g3 stem cells cultured in Methocult media for 10 days, supplied with IL3, SCF, and Flt3L (n = 7). C, IL3 signaling was reported by pSTAT5. CD34+ stem cells of different genotypes were treated by titrated dose of IL3 for 30 minutes, and pSTAT5 was measured by flow cytometry (n = 1). D, pDC differentiation of CD34+ stem cells. The proportion and absolute count of pDCs (CD303+CD123+) were measured at day 21 (n = 8). *, P < 0.05; **, P < 0.01; ***, P < 0.001; and n.s., not significant. For each plot, error bars represent SEM.
IL3 signaling is generally involved in hematopoiesis. One function is to support the expansion of hematopoietic stem cells (42). Therefore, we knocked out FUT8 in human cord blood CD34+ stem cells using RNP-mediated Cas9/crRNA delivery (FUT8-g3, Supplementary Fig. S5E). To define a condition that best reflected IL3-dependent stem cell expansion, we treated the wild-type (Scramble) and CD123 knockout (IL3RA-g3) cells with various cytokine cocktails. The best differential in cell growth was achieved with IL3/SCF/Flt3L cocktails (Supplementary Fig. S5F). Next, we used this cytokine combination to test stem cell expansion in colony formation assays. FUT8-g3 stem cells had significantly reduced colony formation capacity in this condition (Fig. 5B). Accordingly, FUT8-g3 stem cells also showed partially impaired IL3 signaling (Fig. 5C). Apart from driving CD34+ stem cell expansion, IL3 can participate in pDC differentiation (43). We found that FUT8-g3 stem cells generated significantly less pDCs quantity-wise (Fig. 5D).
Dual CD3-bispecific antibody treatment overcomes antigen loss–induced resistance
In our CRISPR screen, we found that deficiency of either IFNγ signaling or the core fucosylation pathway induced resistance to the CD123-DART. To understand which pathway contributed more to resistance, we designed a mixed RTCC assay in which JAK1KO and FUT8KO were spiked into a major population of Scramble cells (Supplementary Fig. S6A). During continued CD123-DART dosing, antigen loss–driven resistance by FUT8KO had a dominant advantage in both MOLM13 and SEM models (Fig. 6A and B), indicating that antigen loss–driven resistance was the major determinant in response to a single bispecific antibody. However, when we switched the treatment to a CD33-BiTE, whose efficacy is dependent on an alternative antigen, the FUT8KO population diminished, whereas JAK1KO cells preferentially expanded in MOLM13 cells (Fig. 6A).
Figure 6.
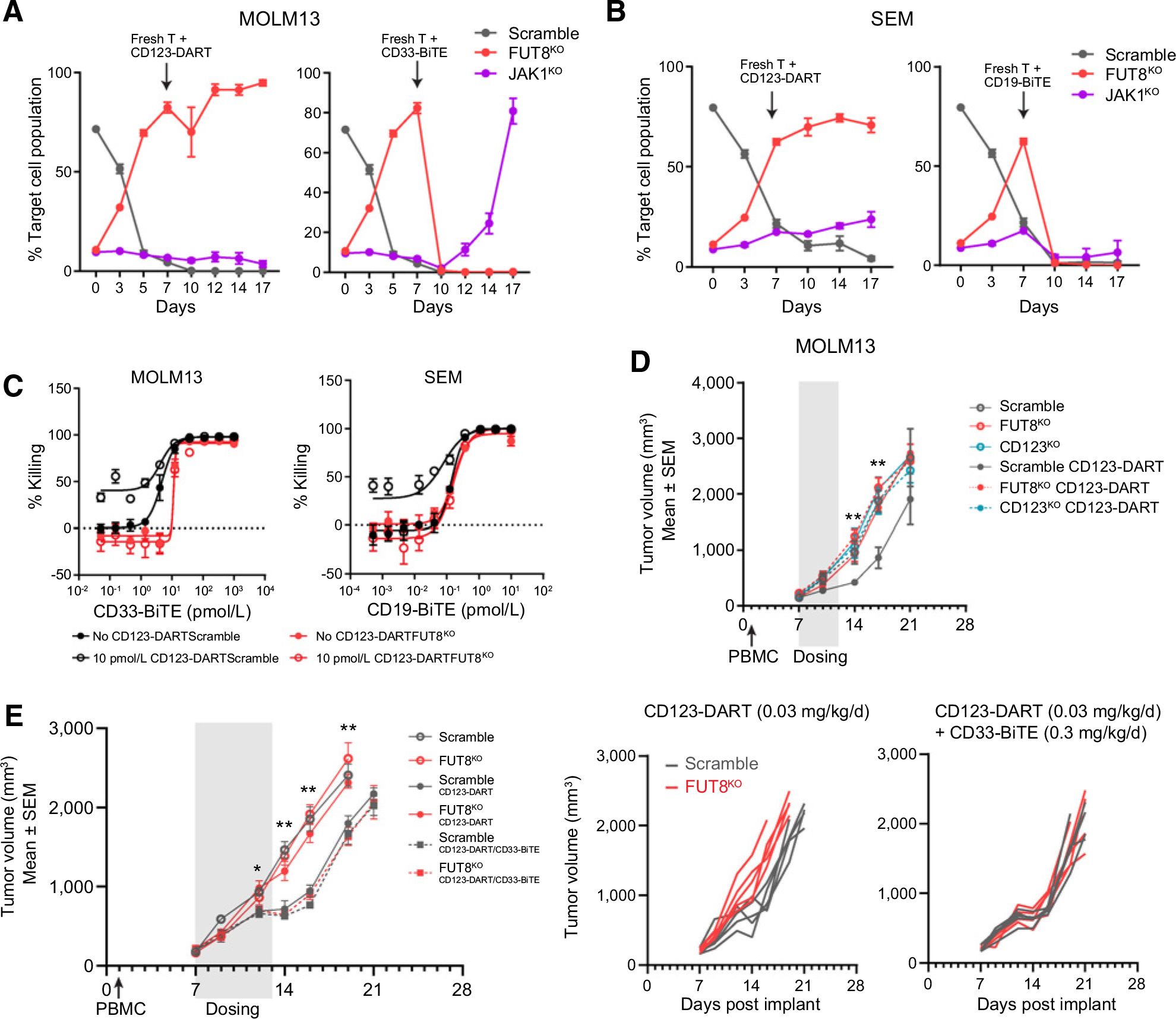
FUT8KO-induced resistance can be managed by dual CD3-bispecific antibody treatment. A and B, Mini-pool validation of MOLM13 (A) and SEM (B) cells in response to CD123-DART and CD33-BiTE/CD19-BiTE. Cells of different genotypes were spiked into the Scramble cells, all with different fluorescent labels (as shown in Supplementary Fig. S6A). The cell mixture was dosed with CD123-DART for the initial 7 days, followed by continued CD123-DART treatment or switched to CD33-BiTE (for MOLM13) or CD19-BiTE (for SEM) for another 10 days. The relative proportion of each genotype was tracked by flow cytometry along the time course (n = 4). C, RTCC assay with single or dual CD3-bispecific antibody. Scramble and FUT8KO MOLM13 or SEM cells were treated with 10 pmol/L CD123-DART in combination with titrated doses of CD33-BiTE or CD19-BiTE, respectively. The killing was normalized to untreated groups (n = 4). The RTCC assay used T-cell:target cell ratio of 1:1, and target cell killing was measured 72 hours after treatment. D, In vivo efficacy study of CD123-DART. MOLM13 of different genotypes (Scramble/FUT8KO/CD123KO) was implanted, and the mice were treated with CD123-DART for 6 consecutive days. The tumor volume was measured on indicated days. E, Comparison of single-agent (CD123-DART) and dual-agent (CD123-DART/CD33-BiTE) bispecific antibody efficacy in vivo (left plot). The tumor growth in each individual mouse implanted with either Scramble or FUT8KO tumors is presented in the right plots. T test was performed comparing FUT8KO tumors with single- and dual-agent treatment. n = 5 for each group. *, P < 0.05 and **, P < 0.01. For each plot, error bars represent SEM.
With these results, we sought to understand if a CD3-bispecific combination strategy could overcome antigen loss–dependent resistance. Combination therapies have been widely appreciated in treating relapsed cancers due to target modulation (44, 45). In the RTCC assay, we found that the CD123-DART as a single agent had no therapeutic effect in FUT8KO MOLM13 cells, whereas combination with the CD33-BiTE effectively eradicated resistant cells. A similar result was observed in FUT8KO SEM cells treated in combination with the CD19-BiTE (Fig. 6C). To validate the therapeutic effect of a dual CD3-bispecific antibody approach in vivo, we first investigated FUT8KO-induced resistance in a MOLM13 tumor model. We found that in accordance with the in vitro RTCC results (Fig. 3C), FUT8KO cells showed marked resistance to CD123-DART treatment in vivo (Fig. 6D). Next, we compared the efficacy of single-agent with dual-agent bispecific antibody treatment. Whereas FUT8KO tumors showed significant resistance to single-agent CD123-DART, their growth was more effectively suppressed by the combination therapy of CD123-DART/CD33-BiTE (Fig. 6E; Supplementary Fig. S6B).
Discussion
Advances in CRISPR-Cas9 technology have enabled genome-wide knockout screens in diverse biological settings, serving as a powerful tool to uncover novel mechanisms of cancer resistance and next-generation therapeutic strategies (13, 22, 46). Our screen with CD3-bispecific antibodies provided another example of genome-wide CRISPR screens in the context of immunotherapy. Different from the conventional tumor cell–intrinsic screens, our screen featured the coculture of CRISPR-engineered target cells, primary T cells, and therapeutic bispecific antibodies. We also developed a companion qPCR assay to optimize screen parameters in small scale. Our data suggested that selection pressure could negatively influence screen outcomes, indicating that optimized screening conditions are needed to obtain robust results in complex systems. Because we aimed to understand cancer resistance mechanisms, the screen conditions depicted in this study were biased toward revealing resistance phenotypes, and optimization toward the discovery of sensitizing hits may require alternative screening conditions.
From the CRISPR screens, we found that loss of IFNγ signaling represented a general mechanism of cancer cell resistance to CD3-bispecific antibodies. The central role of IFNγ signaling in immunotherapy has been well-established. This includes both a direct impact on tumor cell viability and a role in antigen presentation that promotes T-cell activity. In our study, both the direct cytotoxic activity of IFNγ and the promotion of antigen presentation proved to be dispensable for the antitumor effects of CD3 bispecifics. Nevertheless, we identified several additional mechanisms by which IFNγ signaling mediated sensitivity to cytotoxic T cells in the context of CD3 bispecifics, including promotion of a productive immune synapse and increased tumor cell apoptosis. IFNγ signaled through different effector molecules in different cell line models. A more sophisticated understanding of the IFN-stimulated gene transcriptional landscape in different cellular contexts will be important to expand our understanding of IFNγ’s role in immunotherapy.
Independent of IFNγ signaling, we identified core fucosylation as a novel resistance mechanism, particularly in response to CD123-DART treatment. Core fucosylation by FUT8 plays a prometastatic role in some cancer types (47), and fucosylated alpha-fetoprotein is a biomarker for hepatocarcinoma (48). A CRISPR screen in T cells demonstrates that core fucosylation has a role in the surface presentation of PD-1, which mediates T-cell exhaustion in antitumor responses (49). In our study, core fucosylation was critical for responsiveness to the CD123-DART, arguing against inhibiting this pathway in patients under CD123-DART therapy. Currently, there is no reported mutation of this pathway from patients with AML or ALL, but such mutations could occur in patients who develop acquired resistance to CD123-DART treatment. Because flotetuzumab is progressing through clinical development, it will be interesting to monitor whether alterations in the core fucosylation pathway correlate with efficacy.
In our biophysical analysis, we determined that core fucosylation of CD123 was mandatory for efficient CD123-DART binding. The FACS competition assay with 6H6 suggested that CD123-DART binding may depend on the core fucosylated residues. However, CD123-DART was not specifically designed to target the glycosylated form of CD123. Although our SEC–SAXS analysis of wild-type and afucosylated CD123 proteins suggested that core fucosylation loss did not induce a general conformational change, it could still affect the local conformation in the NTD which was not identifiable by SAXS. The fact that FUT8 deficiency impaired CD123’s affinity toward IL3 provided additional evidence to support the conformational change model because IL3 and CD123-DART likely engage different domains on CD123. We have also found that FUT8 loss impaired IL3 signaling through CD123. The associated biological consequence, especially in hematopoiesis, was evident. Fucose-deficient hematopoietic stem cells show abnormal homing, whereas enforced fucosylation improves the engraftment of certain lineages during stem cell transplant (50, 51). In addition to IL3 signaling, core fucosylation is also required for signaling through multiple surface receptors, including the TGFβ receptor, EGF receptor, and IGF receptor (52–54). The structural basis for these interactions was unclear, but it could represent a broader regulatory role of core fucosylation in cell signaling.
Existing and acquired resistance is an investigated area of study for targeted therapies and immunotherapies in oncology (55). Our data confirm that antigen loss was a primary mechanism of resistance to a specific CD3-bispecific antibody, whereas other mechanisms likely have an enhancing or synergistic role. Antigen loss is typically derived from silenced expression or alternative splicing of the target antigen (56–58). Therefore, most drug resistance studies have been focused on the transcriptional or epigenetic regulation of the target antigen. However, our data identified a PTM as the primary mechanism of target loss by a biologic therapy. Hence, it could be beneficial to consider the PTM status of the target antigen when resistance occurs. To treat the antigen loss–induced resistance, we propose the use of an additional molecule targeting a different antigen. This approach has already been established in the clinic for other modalities and will likely be necessary for CD3 bispecifics going forward. For example, a CD22-targeting antibody–drug conjugate (inotuzumab-ozogamicin) was used in combination with CD19-BiTE and achieved encouraging clinical outcomes (59). Other modalities, such as biologics targeting two antigens at once, are also being employed to combat resistance from single antigen loss (60, 61). As our understanding of resistance mechanisms advances, we expect that addressing this therapeutic challenge by rational therapeutic designs including combination and dual-antigen approaches will become more common.
Supplementary Material
Acknowledgments
The authors thank A. Kapoor and R. deBeaumont for contribution to the CRISPR screen, A. Oka for supporting T-cell expansion, P. Ting for providing protocols for RNP knockout in CD34+ stem cells, P. Zhang for helping with cell sorting, and G. Kerr and G. Elliott for bioinformatics analysis.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Authors’ Disclosures
G. Dranoff reports personal fees from Novartis (employee) during the conduct of the study, as well as personal fees and other from Novartis (stock ownership) outside the submitted work. No disclosures were reported by the other authors.
Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
References
- 1.Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther 2018;12:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu Rev Med 2019;70:437–50. [DOI] [PubMed] [Google Scholar]
- 3.Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med 2017;376:836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uy GL, Godwin J, Rettig MP, Vey N, Foster M, Arellano ML, et al. Preliminary results of a phase 1 study of flotetuzumab, a CD123 × CD3 bispecific dart (R) protein, in patients with relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Blood 2017;130 Suppl 1:637. [Google Scholar]
- 5.Aldoss I, Song J, Stiller T, Nguyen T, Palmer J, O’Donnell M, et al. Correlates of resistance and relapse during blinatumomab therapy for relapsed/refractory acute lymphoblastic leukemia. Am J Hematol 2017;92: 858–65. [DOI] [PubMed] [Google Scholar]
- 6.Braig F, Brandt A, Goebeler M, Tony HP, Kurze AK, Nollau P, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 2017;129: 100–4. [DOI] [PubMed] [Google Scholar]
- 7.Junttila TT, Li J, Johnston J, Hristopoulos M, Clark R, Ellerman D, et al. Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res 2014;74:5561–71. [DOI] [PubMed] [Google Scholar]
- 8.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hannon M, Lechanteur C, Lucas S, Somja J, Seidel L, Belle L, et al. Infusion of clinical-grade enriched regulatory T cells delays experimental xenogeneic graft-versus-host disease. Transfusion 2014;54:353–63. [DOI] [PubMed] [Google Scholar]
- 10.Fisher TS, Hooper AT, Lucas J, Clark TH, Rohner AK, Peano B, et al. A CD3-bispecific molecule targeting P-cadherin demonstrates T cell-mediated regression of established solid tumors in mice. Cancer Immunol Immunother 2018;67:247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laszlo GS, Gudgeon CJ, Harrington KH, Dell’Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014;123:554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goebeler ME, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma 2016; 57:1021–32. [DOI] [PubMed] [Google Scholar]
- 13.Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med 2019;25:95–102. [DOI] [PubMed] [Google Scholar]
- 14.Kirby N, Cowieson N, Hawley AM, Mudie ST, McGillivray DJ, Kusel M, et al. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallogr D Struct Biol 2016;72:1254–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopkins JB, Gillilan RE, Skou S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr 2017;50:1545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr 2012;45:342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr 2001;34:33–41. [Google Scholar]
- 18.Geri JB, Oakley JV, Reyes-Robles T, Wang T, McCarver SJ, White CH, et al. Microenvironment mapping via Dexter energy transfer on immune cells. Science 2020;367:1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva CM, Lu H, Weber MJ, Thorner MO. Differential tyrosine phosphorylation of JAK1, JAK2, and STAT1 by growth hormone and interferon-gamma in IM-9 cells. J Biol Chem 1994;269:27532–9. [PubMed] [Google Scholar]
- 21.Kleppe M, Soulier J, Asnafi V, Mentens N, Hornakova T, Knoops L, et al. PTPN2 negatively regulates oncogenic JAK1 in T-cell acute lymphoblastic leukemia. Blood 2011;117:7090–8. [DOI] [PubMed] [Google Scholar]
- 22.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017;547:413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016;167:397–404 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burke F, East N, Upton C, Patel K, Balkwill FR. Interferon gamma induces cell cycle arrest and apoptosis in a model of ovarian cancer: enhancement of effect by batimastat. Eur J Cancer 1997;33:1114–21. [DOI] [PubMed] [Google Scholar]
- 26.Azuma A, Yagita H, Matsuda H, Okumura K, Niitani H. Induction of intercellular adhesion molecule 1 on small cell lung carcinoma cell lines by gamma-interferon enhances spontaneous and bispecific anti-CD3 × antitumor antibody-directed lymphokine activated killer cell cytotoxicity. Cancer Res 1992; 52:4890–4. [PubMed] [Google Scholar]
- 27.Simon MM, Waring P, Lobigs M, Nil A, Tran T, Hla RT, et al. Cytotoxic T cells specifically induce Fas on target cells, thereby facilitating exocytosis-independent induction of apoptosis. J Immunol 2000;165:3663–72. [DOI] [PubMed] [Google Scholar]
- 28.Medema JP, Toes RE, Scaffidi C, Zheng TS, Flavell RA, Melief CJ, et al. Cleavage of FLICE (caspase-8) by granzyme B during cytotoxic T lymphocyte-induced apoptosis. Eur J Immunol 1997;27:3492–8. [DOI] [PubMed] [Google Scholar]
- 29.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science 2001; 291:2364–9. [DOI] [PubMed] [Google Scholar]
- 30.Tateno H, Nakamura-Tsuruta S, Hirabayashi J. Comparative analysis of core-fucose-binding lectins from Lens culinaris and Pisum sativum using frontal affinity chromatography. Glycobiology 2009;19:527–36. [DOI] [PubMed] [Google Scholar]
- 31.Becker DJ, Lowe JB. Fucose: biosynthesis and biological function in mammals. Glycobiology 2003;13:41r–53r. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Morales-Levy M, Rose J, Mackey LC, Bodary P, Eitzman D, et al. alpha (1,3)-Fucosyltransferases FUT4 and FUT7 control murine susceptibility to thrombosis. Am J Pathol 2013;182:2082–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan Z, Yin H, Nie S, Lin Z, Zhu J, Ruffin MT, et al. Large-scale identification of core-fucosylated glycopeptide sites in pancreatic cancer serum using mass spectrometry. J Proteome Res 2015;14:1968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sims TN, Dustin ML. The immunological synapse: integrins take the stage. Immunol Rev 2002;186:100–17. [DOI] [PubMed] [Google Scholar]
- 35.Lee CS, Kim KY, Im JB, Choi JW, Kim HK, Park JS, et al. bPAK-interacting exchange factor may regulate actin cytoskeleton through interaction with actin. Exp Mol Med 2004;36:582–7. [DOI] [PubMed] [Google Scholar]
- 36.Lee EM, Yee D, Busfield SJ, McManus JF, Cummings N, Vairo G, et al. Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica 2015;100:914–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaszuba K, Grzybek M, Orlowski A, Danne R, Rog T, Simons K, et al. N-Glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc Natl Acad Sci U S A 2015;112:4334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakae Y, Satoh T, Yagi H, Yanaka S, Yamaguchi T, Isoda Y, et al. Conformational effects of N-glycan core fucosylation of immunoglobulin G Fc region on its interaction with Fcgamma receptor IIIa. Sci Rep 2017;7:13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malaby AW, Chakravarthy S, Irving TC, Kathuria SV, Bilsel O, Lambright DG. Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering. J Appl Crystallogr 2015; 48:1102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J 2005;89:1237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stomski FC, Sun Q, Bagley CJ, Woodcock J, Goodall G, Andrews RK, et al. Human interleukin-3 (IL-3) induces disulfide-linked IL-3 receptor alpha- and beta-chain heterodimerization, which is required for receptor activation but not high-affinity binding. Mol Cell Biol 1996;16:3035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossmanith T, Schroder B, Bug G, Muller P, Klenner T, Knaus R, et al. Interleukin 3 improves the ex vivo expansion of primitive human cord blood progenitor cells and maintains the engraftment potential of scid repopulating cells. Stem Cells 2001;19:313–20. [DOI] [PubMed] [Google Scholar]
- 43.Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 2015;15:471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer 2018;17:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shah NN, Maatman T, Hari P, Johnson B. Multi targeted CAR-T cell therapies for B-cell malignancies. Front Oncol 2019;9:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, et al. A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 2015;162:675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agrawal P, Fontanals-Cirera B, Sokolova E, Jacob S, Vaiana CA, Argibay D, et al. A systems biology approach identifies FUT8 as a driver of melanoma metastasis. Cancer Cell 2017;31:804–19 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aoyagi Y, Isemura M, Yosizawa Z, Suzuki Y, Sekine C, Ono T, et al. Fucosylation of serum alpha-fetoprotein in patients with primary hepatocellular carcinoma. Biochim Biophys Acta 1985;830:217–23. [DOI] [PubMed] [Google Scholar]
- 49.Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of core fucosylation reduces cell-surface expression of PD-1 and promotes anti-tumor immune responses of T cells. Cell Rep 2017;20:1017–28. [DOI] [PubMed] [Google Scholar]
- 50.Myers J, Huang Y, Wei L, Yan Q, Huang A, Zhou L. Fucose-deficient hematopoietic stem cells have decreased self-renewal and aberrant marrow niche occupancy. Transfusion 2010;50:2660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popat U, Mehta RS, Rezvani K, Fox P, Kondo K, Marin D, et al. Enforced fucosylation of cord blood hematopoietic cells accelerates neutrophil and platelet engraftment after transplantation. Blood 2015;125:2885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu M, Cui X, Wang H, Liu J, Qin H, Liu S, et al. FUT8 drives the proliferation and invasion of trophoblastic cells via IGF-1/IGF-1R signaling pathway. Placenta 2019;75:45–53. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Gu J, Miyoshi E, Honke K, Taniguchi N. Phenotype changes of Fut8 knockout mouse: core fucosylation is crucial for the function of growth factor receptor(s). Methods Enzymol 2006;417:11–22. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Gu J, Ihara H, Miyoshi E, Honke K, Taniguchi N. Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J Biol Chem 2006;281:2572–7. [DOI] [PubMed] [Google Scholar]
- 55.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 2015;5:1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braig F, Marz M, Schieferdecker A, Schulte A, Voigt M, Stein A, et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget 2015; 6:12035–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mishima Y, Terui Y, Takeuchi K, Matsumoto-Mishima Y, Matsusaka S, Utsubo-Kuniyoshi R, et al. The identification of irreversible rituximab-resistant lymphoma caused by CD20 gene mutations. Blood Cancer J 2011;1:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jabbour E, Sasaki K, Ravandi F, Huang X, Short NJ, Khouri M, et al. Chemoimmunotherapy with inotuzumab ozogamicin combined with mini-hyper-CVD, with or without blinatumomab, is highly effective in patients with Philadelphia chromosome-negative acute lymphoblastic leukemia in first salvage. Cancer 2018;124:4044–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kugler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M, et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol 2010;150:574–86. [DOI] [PubMed] [Google Scholar]
- 61.Schubert I, Kellner C, Stein C, Kugler M, Schwenkert M, Saul D, et al. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. MAbs 2011;3: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.