

Summary
Secreted proteins and peptides hold large potential both as therapeutics and as enzyme catalysts in biotechnology. The high stability of many secreted proteins helps maintain functional integrity in changing chemical environments and is a contributing factor to their commercial potential. Disulphide bonds constitute an important post‐translational modification that stabilizes many of these proteins and thus preserves the active state under chemically stressful conditions. Despite their importance, the discovery and applications within this group of proteins and peptides are limited by the availability of synthetic biology tools and heterologous production systems that allow for efficient formation of disulphide bonds. Here, we refine the design of two DisCoTune (Disulphide bond formation in E. coli with tunable expression) plasmids that enable the formation of disulphides in the highly popular Escherichia coli T7 protein production system. We show that this new system promotes significantly higher yield and activity of an industrial protease and a conotoxin, which belongs to a group of disulphide‐rich venom peptides from cone snails with strong potential as research tools and pharmacological agents.
Short abstract
Secreted proteins and peptides hold large potential both as therapeutics and as enzyme catalysts in biotechnology. Despite their importance, the discovery and applications within this group of proteins and peptides are limited by the availability of synthetic biology tools and heterologous production systems that allow for efficient formation of disulfide bonds. Here, we refine the design of two plasmids that enable the formation of disulfides in the highly popular Escherichia coli T7 protein production system.
Introduction
The most widely used microbial chassis for protein production is the Escherichia coli T7/pET vector system (Rosenberg et al., 1987; Studier et al., 1990; Shilling et al., 2020). The first pET expression vector was introduced by Studier and co‐workers more than 30 years ago, and the vector collection now has more than 220 000 entries in publication records (Shilling et al., 2020). The more than 100 available pET vectors are equipped with strong T7 promoters driving transcription when introduced into E. coli strains modified to express T7 RNA polymerase (T7 RNApol). This setup has been employed in thousands of laboratories worldwide and has been a foundation for many of our scientific advancements in understanding proteins, molecular systems, and biophysics. Thus, any improvements of the T7 expression systems will likely have broad implications for the advancement of the biological sciences in general.
Production of disulphide‐containing proteins requires oxidative folding. Using this process, proteins fold into their native structure while forming disulphide bonds between pairs of cysteine residues (Rabenstein, 2009). The efficiency of oxidative folding is dependent on the protein itself and the cellular redox environment including assistance by enzyme catalysts. Due to the reducing environment of the E. coli cytoplasm, structural protein disulphides very rarely form in this compartment (Østergaard et al., 2001; Østergaard et al., 2004). Oxidative folding of disulphide‐containing proteins produced in E. coli has therefore usually been accomplished by secretion into the periplasm (Yoon et al., 2009). Disulphide bond formation in the periplasm of E. coli is driven by the membrane protein DsbB that oxidizes catalytic cysteine residues of the soluble thiol‐disulphide oxidoreductase DsbA, which in turn transfers disulphides to substrate proteins (Landeta et al., 2018). In parallel with this oxidative pathway, a pathway for reduction and isomerization exists – here the membrane protein DsbD functions to reduce periplasmic DsbC that then reduces or isomerizes non‐native substrate disulphides (Landeta et al., 2018).
Early this century, Beckwith and co‐workers did pioneering work on modifying the cytoplasm of E. coli to allow oxidative folding. To push the balance towards more oxidizing conditions in the cytoplasm, the genes trxB and gor, encoding thioredoxin reductase and glutaredoxin reductase, were disrupted (Prinz et al., 1997). These mutant strains were supplemented with reducing agents in the media to support growth. A suppressor mutation in the gene encoding the cytosolic peroxidase AhpC, which ablates the peroxidase activity and instead allows the enzyme to transfer electrons into the glutathione/glutaredoxin pathway, restored viability of the trxB and gor‐deficient strains (Ritz et al., 2001; Yamamoto et al., 2008) in the absence of an exogenous reducing agent. Based on this work, Novagen introduced the Origami™ B(DE3) strain for T7‐based expression and New England Biolabs introduced the SHuffle® strains, which also feature constitutive cytoplasmic expression of DsbC to further improve native disulphide bond formation (Lobstein et al., 2012).
More recently, Ruddock and co‐workers created an alternative approach for modifying the reducing environment in the cytoplasm of E. coli to allow oxidative folding of disulphide‐containing proteins (Hatahet et al., 2010; Nguyen et al., 2011). Their system, referred to as CyDisCo (Cytoplasmic Disulphide bond formation in E. coli) (Alanen et al., 2015), is based on co‐ or pre‐expression of the yeast mitochondrial thiol oxidase Erv1p and a human protein disulphide isomerase (hPDI) in the cytoplasm of E. coli. In this system, Erv1p uses molecular oxygen to provide oxidizing equivalents for generating disulphide bonds de novo, while hPDI catalyses protein folding and reconfigures incorrectly formed disulphide bonds in substrate proteins. To further extend this system, we previously created a variant of the CyDisCo system by supplementing a conotoxin‐specific PDI (Safavi‐Hemami et al., 2016) (csPDI) in addition to Erv1p and hPDI (Nielsen et al., 2019). This variant is designed to specifically assist folding of conotoxins, a group of disulphide‐rich venom peptides from cone snails with strong potential as research tools and therapeutics (Terlau and Olivera, 2004).
In the original CyDisCo vectors, the polycistronic fragment carrying the genes of the folding factors was either cloned into a pET vector together with the target gene or provided on a separate plasmid based on the pLysS backbone (Nguyen et al., 2011; Gaciarz et al., 2016). The auxiliary nature of the pLysS‐based CyDisCo vectors makes them easy to implement in a variety of standard production scenarios and strains and thereby enables comparison of production efficiency in different chassis such as BL21(DE3), K12 strains, Shuffle T7 or Origami(DE3) (Nativel et al., 2016). The pLysS plasmid was originally designed to facilitate expression of T7 lysozyme (T7lys) that inhibits the T7 RNApol to lower transcription and thereby relieve potential stress from the T7 production system (Studier, 1991). The T7lys control of T7 RNApol was refined by putting expression of T7lys under the control of the titratable, rhamnose‐inducible rhaBAD promotor (Schlegel et al., 2012). This tool, referred to as pLemo (LEss is MOre), was used to titrate, or ‘tune’, expression and aided in increasing fractions of properly folded membrane proteins.
Here, we combine the features of the tunable expression from pLemo with two variants of the CyDisCo system for disulphide bond formation in the E. coli cytoplasm. By doing this, we achieve a more stable CyDisCo system for tunable expression (DisCoTune), while eliminating features in the pLysS backbone that potentially impact expression of T7lys and the CyDisCo folding factors. In one scenario, we show that the DisCoTune system enables titration of T7 RNApol repression to find the optimal level for producing active protein, possibly due to better resource allocation in the complex synthetic biology system. In a different scenario, the DisCoTune system produces more correctly folded protein than the CyDisCo system in the absence of T7 RNApol repression.
Results
Design and construction of the pDisCoTune and pcsDisCoTune plasmids
Two pLysS‐derived CyDisCo vectors for T7‐based production of disulphide‐containing proteins in E. coli were previously constructed: pMJS205 (Gaciarz et al., 2016) and pLE577, where pLE577 hosts an additional conotoxin‐specific PDI (csPDI) to assist the production of conotoxins (Nielsen et al., 2019). We will from here on refer to these plasmids as pCyDisCo and pcsCyDisCo respectively. Both versions are based on the same pLysS backbone (Fig. S1), typically recommended for expression of toxic proteins within a T7‐based system. However, some modifications should offer advantages: Firstly, we previously showed that adding additional features (such as more genes) onto the original pLysS plasmid may have unintentional negative effects on bacterial growth and on the performance of plasmid features (Søgaard and Nørholm, 2016). For example, presence and/or expression of the folding factors in the CyDisCo plasmids could affect the expression of T7lys and vice versa. This could lead to unbalanced transcription, protein misfolding and toxicity (Fig. 1A). Secondly, having expression of T7lys controlled by the highly titratable rhaBAD promoter would allow more control of T7 RNApol activity for optimal system performance (Fig. 1B). To this end, inspired by the pLemo system for tunable expression of toxic proteins, we assembled the folding factor genes from pCyDisCo and pcsCyDisCo onto a new vector backbone that utilizes a rhamnose PrhaBAD‐controlled T7lys resulting in two new constructs pDisCoTune and pcsDisCoTune (Fig. S1).
Fig. 1.
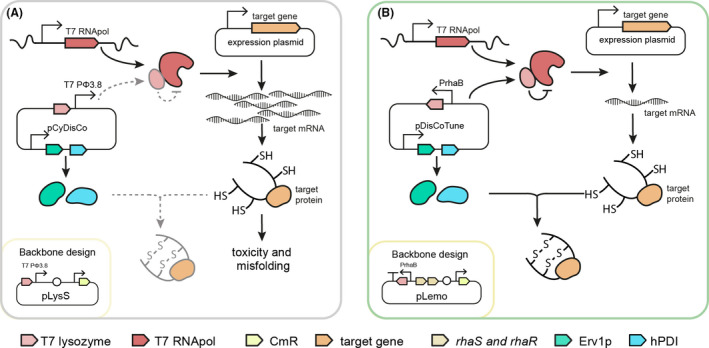
Schematic illustration of the central elements in the CyDisCo and DisCoTune expression systems for heterologous expression of disulphide‐containing proteins.
A. In the pLysS‐based CyDisCo plasmid, T7 lysozyme is controlled by the downstream T7 Pϕ3.8 promoter. The unknown effect this design has on the efficiency of the production system is illustrated by dashed arrows.
B. On pDisCoTune, the CyDisCo system is introduced on a new backbone that allows for accurate titration of T7 lysozyme expression from PrhaB with activators RhaS and RhaR to control transcription of the target gene by T7RNApol. The chloramphenicol resistance gene (CmR) is indicated and genetic features like promoters, terminators and origins of replication are illustrated according to the SBOL standard (Madsen et al., 2019).
Testing the effect of pDisCoTune on the production of proteinase K
In a first protein production test scenario, aimed at exploring the properties of pDisCoTune, we expressed a temperature‐sensitive variant of the enzyme proteinase K (Liao et al., 2007). Proteinase K is a broad‐spectrum proteinase that is used for many different applications in molecular biology as well as in industry (Pähler et al., 1984). The protein contains five cysteine residues of which four form the two Cys34—Cys123 and Cys178—Cys249 disulphide bonds (Betzel et al., 1988). The gene encoding the proteinase K variant without a signal peptide was cloned into the expression vector pET28 in frame with sequences for the natural N‐terminal propeptide, since this can function as an intramolecular chaperone in proteases (Gunkel and Gassen, 1989; Winther and Sorensen, 1991; Kojima et al., 1997), and a C‐terminal His6‐tag. Next, we co‐transformed E. coli BL21(DE3) with pET28‐protK and either pLemo, pCyDisCo or pDisCoTune. pLemo served as a control, allowing for T7 RNApol inhibition similar to the one enabled by pDisCoTune, but without co‐expression of the CyDisCo folding factors.
We initially screened protease activity from the different E. coli strains directly on skim milk agar plates, supplemented with different concentrations of rhamnose (Fig. 2A and B). Loss of opacity surrounding the bacterial growth, indicative of active protease released from lysed cells, was only observed in the presence of pDisCoTune. The clearance zone was present already in the absence of rhamnose and increased with the rhamnose concentration, but peaked and declined again at concentrations above 100 μM (Fig. 2B). Rhamnose‐based titration of T7lys levels was confirmed by western blotting (Fig. 2C).
Fig. 2.
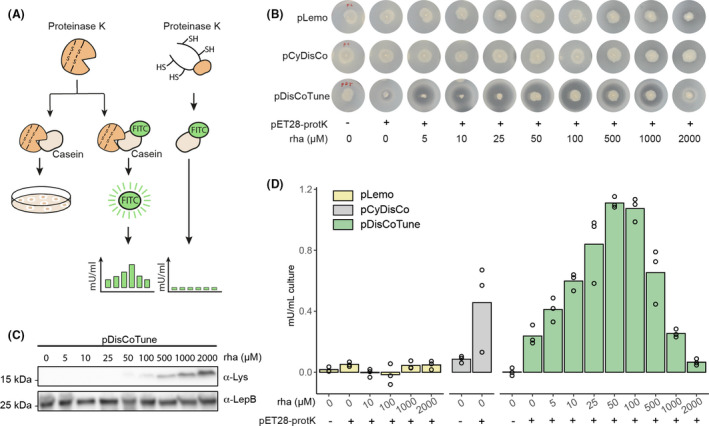
Screening of proteinase K activity produced in E. coli facilitated by pDisCoTune.
A. Proteinase K activity was screened based on casein cleavage either directly on skim milk agar plates by loss of opacity or based on activity in lysates measured by release of the fluorophore FITC.
B. Colonies of BL21(DE3) harbouring either pLemo, pCyDisCo or pDisCoTune were stabbed on to skim milk agar plates with different concentrations of rhamnose (rha).
C. Western blot against lysozyme showing expressing levels from pDisCoTune at the relevant rhamnose concentrations. An antibody (anti‐LepB) against E. coli leader peptidase was used as a loading control.
D. Graph showing the protease activity in mU ml‐1 of culture based on release of FITC. The activity was measured from lysates of E. coli carrying either pLemo (yellow), pCyDisCo (grey) or pDisCoTune (green). Proteinase K was expressed from pET28‐protK, and cultures were supplemented with relevant concentrations of rhamnose. Circles denote results obtained from three independent experiments.
We next measured proteinase activity with a more quantitative assay based on proteolytic release of the fluorophore FITC from Casein, a substrate of proteinase K (Fig. 2A and D). Overnight cultures were back diluted 1:100 into LB media with different rhamnose concentrations and grown to mid‐exponential phase, when protein production was induced with IPTG followed by overnight incubation with shaking. Expression cultures were harvested by centrifugation and activity monitored directly from the soluble fraction of crude lysates. As expected, no active proteinase K was detected in the strains containing the pLemo control. In contrast, active proteinase K was produced in the presence of the CyDisCo folding factors either supplied on pCyDisCo or pDisCoTune. However, the data from the pCyDisCo variant varied greatly between replicates. With pDisCoTune, the same was true at some of the rhamnose concentrations (Fig. 2D). In strains containing pDisCoTune, we observed a dose dependency between rhamnose concentration and active proteinase K produced, with maximal activity between 50 to 100 µM rhamnose. This observation is in agreement with the initial screening on skim milk. Activity increased by three‐fold going from 0 to 100 µM rhamnose. When T7lys was induced with more than 500 µM rhamnose, the T7 RNApol inhibition appeared to limit the production of active proteinase K. The pDisCoTune background at 100 µM rhamnose produced proteins with two‐fold higher activity compared to the pCyDisCo background. These observations show that T7‐based expression can be fine‐tuned with rhamnose from the pDisCoTune plasmid and that this can be favourable for optimizing the active fraction of a disulphide‐containing enzyme.
To investigate the underlying mechanism of the result shown in Fig. 2D, we repeated the proteinase K production in quadruplicate (Fig 3) with pCyDisCo and pDisCoTune in the presence and absence of 100 µM rhamnose (Fig. 3A). We again observed a large spread in activity between replicates harbouring the pCyDisCo plasmid [Fig. 3B, labelled CyDisCo (C) 1‐4]. Fractions of the expression cultures were pelleted, and the peptide content assessed by LC‐MS/MS‐based targeted proteomics (Fig. 3A). From the proteomics analysis, we were able to quantify the most relevant proteins in the production system. These include Erv1p, hPDI, proteinase K, T7lys and T7 RNApol (Fig. 3C–G respectively). Proteinase K peptides were identified in greater abundance without rhamnose in the DisCoTune background (Fig. 3E). This observation is in agreement with the T7lys‐dependent inhibition of the T7 RNApol activity. In turn, this indicated accumulation of inactive proteinase K and an unsuccessful folding machinery without T7 RNApol repression. The DisCoTune samples with optimized rhamnose induction and proteinase K activity were recognized by increased accumulation of lysozyme (Fig. 3F), confirming that T7 RNApol activity can be tuned with the pDisCoTune plasmid and that this can facilitate the optimization of the activity levels of a disulphide‐containing enzyme.
Fig. 3.
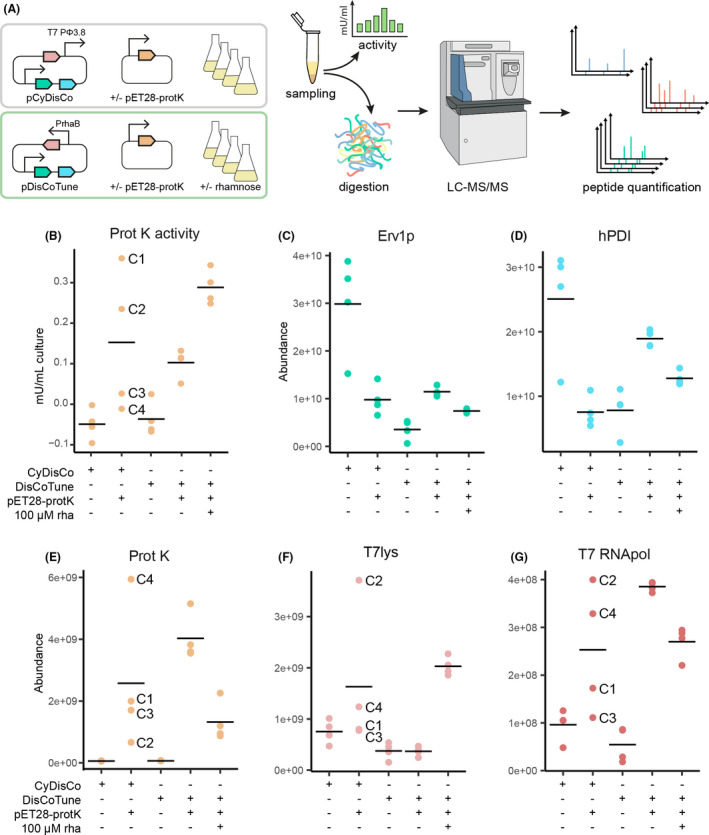
Functional assaying and quantitative proteomics analysis of proteinase K expressions.
A. Schematic diagram of experimental procedure in the quantitative proteomics analysis. Expression of proteinase K was compared between pCyDisCo and pDisCoTune with or without 100 µM rhamnose (rha). Details on the experimental procedure of the quantitative proteomics are described in the experimental procedures.
B. Graph showing the protease activity in mU ml−1 of culture based on release of FITC.
C–G. Quantification of peptide abundance for relevant proteins: Erv1p, hPDI, proteinase K(ProtK), T7 lysozyme and T7RNApol respectively. Individual samples from the combination of pCyDisCo and proteinase K are marked with C1‐4.
The proteomics analysis of the individual CyDisCo samples showed highly different accumulation of proteinase K, T7lys, and T7 RNApol with no obvious correlation between the different factors. For example, CyDisCo sample C4 shows low activity together with high proteinase K and low lysozyme peptide abundance. In contrast, C1 shows high activity, but both low proteinase K and lysozyme abundance. We interpret this as evidence of an unstable production system possibly due to resource competition and, as a result of this, mutations may accumulate. Resource competition could also explain the much higher abundance of the Erv1p and hPDI folding factors expressed from pCyDisCo in the absence rather than in the presence of proteinase K. The low levels of T7lys expressed from pCyDisCo is also in agreement with the western blot (Fig. 4B) and could be a consequence of adding additional DNA between T7lys and its downstream promoter ϕ3.8 as previously shown for the similar pRARE plasmid (Søgaard and Nørholm, 2016).
Fig. 4.
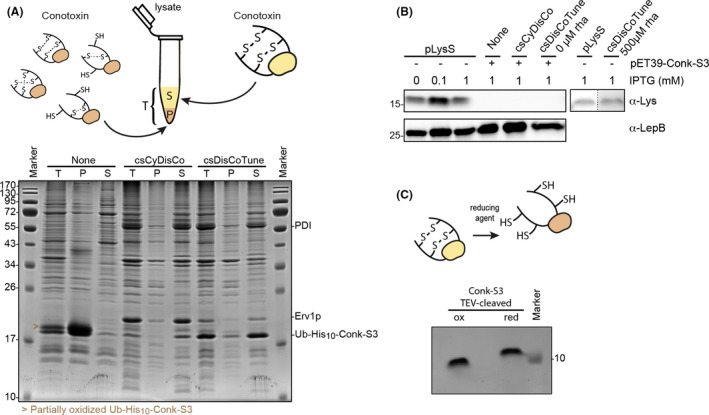
Conk‐S3 constitutes a correctly folded and oxidized protein.
A. Comparison of Ub–His10–Conk‐S3 expression without or with the csCyDisCo or csDisCoTune system. 15% SDS‐PAGE gel stained with Coomassie Brilliant Blue showing the total cell extract (T), resuspended pellet after lysis and centrifugation (P), and the soluble fraction (S) from cells expressing Ub–His10–Conk‐S3. The arrowhead denotes a semi‐oxidized form of Ub–His10‐Conk‐S3 (see illustration above gel and Fig. S2B). Expression was performed as described under Experimental procedures. Protein levels are comparable between lanes, and the gel represents three independent experiments.
B. Western blot against lysozyme in samples from Ub–His10–Conk‐S3 expressions compared to expression level of lysozyme from pLysS with varying IPTG induction and in the absence and presence of rhamnose. The anti‐LepB signal was used as a loading control.
C. 15% Tris‐Tricine SDS‐PAGE gel of Conk‐S3 in the oxidized (ox) and reduced (red; treated with 0.5 mM TECP and 5 mM DTT) state.
Correctly folded Conk‐S3 is efficiently produced from pcsDisCoTune
As a second test production scenario, we chose the conotoxin peptide Conk‐S3 from Conus striatus. Conk‐S3 belongs to a family of conotoxins called conkunitzins (Conks), comprising a Kunitz domain similar to the fold observed in Kunitz‐type protease inhibitors such as BPTI (Ranasinghe and McManus, 2013). The sequence of Conk‐S3 was identified from the venom gland transcriptome of C. striatus (Li et al., 2018). The predicted mature Conk‐S3 peptide comprises 62 residues including four cysteines that form two disulphide bonds. The connectivity of these bonds is predicted to be Cys8—Cys58 and Cys33—Cys54 as previously shown for the related peptides, conkunitzin‐S1 (Bayrhuber et al., 2005) and conkunitzin‐S2. Conk‐S3 was produced from a pET vector as a His10‐tagged fusion protein with the solubility‐enhancing ubiquitin (Ub), Ub–His10‐Conk‐S3, in E. coli BL21(DE3). In this set of experiments, we performed a direct gel‐based comparison between the level and distribution of reduced and oxidized protein in the soluble and insoluble fractions when the peptide was produced in the absence or presence of either the original conotoxin‐specific pcsCyDisCo plasmid or the new pcsDisCoTune variant.
Protein expression was assessed by SDS‐PAGE analysis of total protein, as well as the pellet and soluble fractions (Fig. 4A). We observed that the total amount of Ub–His10‐Conk‐S3 was higher when expressed in the absence of either of pcsCyDisCo or pcsDisCoTune, but the large majority was observed in the pellet fraction. In contrast, the protein was expressed mainly in the soluble fraction when produced in the presence of pcsCyDisCo or pcsDisCoTune. These observations closely matched our results obtained for another conotoxin, H‐Vc7.2, when expressed in the absence and presence of the pcsCyDisCo plasmid (Nielsen et al., 2019). When including the thiol‐alkylating agent N‐ethylmaleimide (NEM) in the lysis buffer, we moreover observed that in the pcsCyDisCo system, Ub–His10‐Conk‐S3 predominantly folded post‐translationally, while this was not the case with pcsDisCoTune (Fig. S2A). Importantly, the level of Ub–His10‐Conk‐S3 was clearly higher in the pcsDisCoTune system, both in terms of total and soluble protein, as compared with the pcsCyDisCo system. Specifically, based on densitometric analysis of SDS‐PAGE gels from five independent experiments, we observed 2.4–4.1 times higher levels of Ub–His10‐Conk‐S3 in the soluble fraction when expressed in the pcsDisCoTune system compared with the pcsCyDisCo system. Concomitantly, we consistently observed a lower expression level of Erv1p in the pcsDisCoTune system (Fig. 4A). We also confirmed by Western blotting low to no expression of T7lys not only in case of pcsDisCoTune, but also in the pcsCyDisCo system (Fig. 4B). The expression of Ub–His10‐Conk‐S3 supported by pcsDisCoTune was investigated with titration of rhamnose (Fig. S3). No beneficial effect was observed on abundance of Ub–His10‐Conk‐S3. On the contrary, and in agreement with our proteinase K expression experiments, at concentrations at and above 500 µM rhamnose, the level of Conk‐S3 in the soluble fraction decreased. Additionally, we observed expression of T7lys from pcsDisCoTune when induced with 500 µM rhamnose, but not in the absence of rhamnose (Fig. 4B).
We next purified and characterized structural features of Conk‐S3 to verify that the protein produced in the soluble fraction constitutes a correctly folded and oxidized protein, and to investigate whether producing the protein in the presence of either pcsDisCoTune or pcsCysDisCo resulted in conformational differences. Ub–His10‐Conk‐S3 was purified on Ni‐NTA material before TEV protease cleavage to release Conk‐S3 from its fusion partner. An additional reverse Ni‐NTA agarose purification step followed by reversed‐phase high‐performance liquid chromatography (RP‐HPLC) on a C18 column completed the purification. The folding status of purified Conk‐S3 was first assessed by looking at disulphide bond formation using SDS‐PAGE analysis on a Tris‐Tricine gel (Fig. 4C). Here, a clear mobility shift was observed upon treating the sample with a strong reducing agent, indicating disulphide bond formation (Fig. 4C). Moreover, under non‐reducing conditions, we observed only the oxidized form of the protein, indicating complete (or near‐complete) oxidation. The formation of the two expected disulphide bonds in Conk‐S3 was verified by matrix‐assisted laser desorption ionization‐mass spectrometry (MALDI‐MS) after tryptic digestion, which showed peptide masses representing tryptic peptides with disulphides between Cys8 and Cys58, and between Cys33 and Cys54 respectively. These masses were not observed after reduction and alkylation (Table S4).
We next used far‐UV circular dichroism (CD) spectroscopy to assess the overall folding status of Conk‐S3. CD spectra of Conk‐S3 produced either in the pcsCyDisCo or pcsDisCoTune background, purified from the soluble fraction and showed near‐identical spectra (Fig. 5A). The overall features of the spectra with a global minimum at ˜ 201 nm and a local minimum at ˜ 220 nm clearly indicated a properly folded polypeptide. This conclusion was supported by deconvolution of the spectrum obtained for Conk‐S3 produced in the presence of pcsDisCoTune (Fig. 5B), which showed a high degree of coincidence between the predicted secondary structure element content (helix: 9.2%, sheet: 23.2%, other: 67.7%) and the known secondary structure content based on the crystal structure of Conk‐S2 (Korukottu et al., 2006) (PDB: 2J6D)(helix: 11.9%, sheet: 23.3%, other: 64.8%). Overall, Conk‐S3 produced in the soluble fraction from cells transformed with either plasmid showed features typical of a correctly folded and oxidized protein. The quality of Conk‐S3 produced from pcsDisCoTune thus remained the same while being produced at higher levels.
Fig. 5.
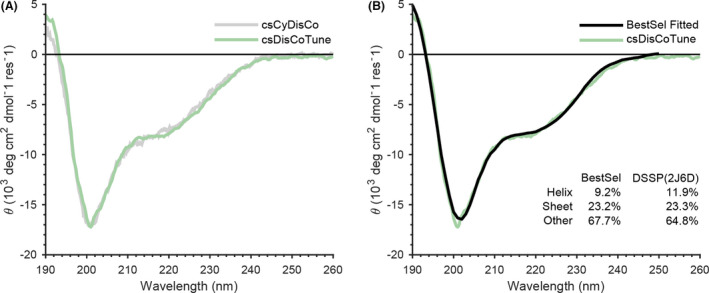
Conk‐S3 constitutes a correctly folded peptide.
A. CD spectra of Conk‐S3 produced in the presence of either pcsCyDisCo (grey) or pcsDisCoTune (green).
B. Overlay of the experimental CD spectrum obtained for Conk‐S3 produced in the presence of pcsDisCoTune (green) with the deconvoluted spectrum (black). The secondary structure element contributions obtained from the deconvolution are listed along with those calculated from the crystal structure using the Define Secondary Structure of Proteins (DSSP) program (Kabsch and Sander, 1983; Touw et al., 2015). Deconvolution was performed using the BestSel web server (Micsonai et al., 2015).
Discussion
According to the statistics available in the PDB protein structure database, E. coli BL21(DE3) is by far the most frequently used host for recombinant protein production and more than 1000 structures have been solved using proteins produced with the pLysS plasmid present. In principle, pLysS can work as an RNApol autoregulation system as a result of T7lys being under the control of the T7 promotor ϕ3.8. Hence, increased T7 RNApol activity will lead to an increased T7lys expression that in turn will decrease the T7 RNApol activity. pLysS has previously been used as a backbone for adding more features to the T7 toolbox, such as co‐expression of tRNA genes to facilitate expression of rare codon containing genes. However, we previously reported an apparent design flaw caused by using pLysS as a backbone for such systems (Søgaard and Nørholm, 2016): In the original pLysS design, the T7 promotor ϕ3.8 is placed downstream from the gene of T7lys (Fig. 1A). As a consequence, the transcript from ϕ3.8 includes the entire backbone of pLysS before reaching the gene of T7lys. Consequently, when pLysS is modified with additional features downstream from the ϕ3.8 promotor, such as in the pRARE, pCyDisCo and pcsCyDisCo plasmids, it is likely that there is a significant impact on the expression of T7lys and T7 RNApol activity. We previously demonstrated this was the case for the pRARE plasmids that showed reduced expression of T7lys, likely due to the added genetic features in between the ϕ3.8 promoter and the T7lys gene (Søgaard and Nørholm, 2016). Similarly, we here see that T7lys expression is not detected in western blots from the pCyDisCo plasmids, but from the pDisCoTune variant in the presence of rhamnose. Thus, a clear and simple benefit of the new plasmids designed is the ability to control T7 RNApol activity. We further demonstrate that this control can be key to optimizing production titres for a disulphide‐containing enzyme‐like proteinase K. It would be interesting to test the effect of pDisCoTune in another bacterial chassis, such as Bacillus subtilis.
Synthetic biology increasingly deals with complex designs such as multigene biosynthetic pathways or advanced genetic information circuits. For living systems to cope with such complex recombinant genetics, resource balancing is key for optimal performance. Although we were not able to pinpoint the exact underlying mechanism, the apparent instability of the pCyDisCo system detected by our proteomics analysis, leading to uncontrolled expression levels of the different factors involved in producing correctly folded proteinase K, provides a good example of this. In this case, putting just one of the genes under control of a titratable promoter provided sufficient stabilization of the entire system for multiple factors to express stable levels. We did not directly compare the performance of pDisCoTune and pcsDisCoTune, as the latter is designed for production of conotoxins. Potentially, the pcsDisCoTune system may also be favourable for the production of other small animal venom toxins. For larger disulphide‐containing peptides, it is likely beneficial to use the simpler pDisCoTune based on our findings on resource competition.
A consequence of the DNA assembly strategy we used in this study was that the ribosome binding site of Erv1p was modified. It is thereby likely that pDisCoTune displays a different expression level of Erv1p in comparison with the original pCyDisCo. Indeed, both our proteomics and SDS‐PAGE analyses could indicate lower expression of Erv1p from the two DisCoTune variants. It is possible that different expression levels of the folding factors affect the cellular fitness. To our knowledge, the effect of different expression levels of the folding factors on efficiency of disulphide bond formation has not previously been explored. Thus, further optimization efforts could target the expression of these factors.
Proteinase K is mostly produced in eukaryotic chassis due to its fungal origin. Finding simpler and cheaper production platforms like E. coli would therefore make the enzyme more accessible, for example for protein engineering. This is demonstrated here, by showing successful production of a heat‐sensitive proteinase K variant in the cytoplasm of E. coli with the aid of the DisCoTune system. A heat‐sensitive proteinase K could find use as a ‘molecular eraser’ in multi‐step enzymatic high‐throughput molecular biology applications such a DNA sequencing.
Conotoxins are venom peptides produced by predatory marine cone snails. Peptides from venomous animals often bind their targets with high specificity and potency, making them excellent research tools and promising candidates for the development of selective biopharmaceuticals. In fact, several animal venom peptides, such as the conotoxin ω‐MVIIA from Conus magus (Miljanich, 2004), have already been developed as approved drugs (Pennington et al., 2018). A large proportion of animal venom peptides contain disulphide bonds for stabilization, and while new sequencing technologies have made available a very large number of venom‐peptide sequences, the production of these often complex molecules remains a substantial challenge of decisive importance for our ability to fully explore their potential. Here, we investigated the production of Conk‐S3, a conotoxin that belongs to a class of peptides that bind to certain voltage‐gated potassium channels of the Kv1 type (Finol‐Urdaneta et al., 2020). The facile production of toxins, which can be used as research tools in the investigation of ion channels, will help further the development of therapeutics for the treatment of diseases related to ion‐channel dysfunction.
We showed that the introduction of pcsDisCoTune resulted in the production of correctly folded Conk‐S3 with the large majority of protein present in the soluble fraction. In contrast, when expressing the highly similar (72% sequence identity) Conk‐S1 in BL21(DE3) or Origami(DE3) cells, the majority of the protein was found in the insoluble fraction (Bayrhuber et al., 2006). Expression in the periplasm or together with the molecular chaperones trigger factor and GroEL/GroES did not improve solubility (Bayrhuber et al., 2006). Thus, for NMR or X‐ray structure determination, Conk‐S1 was either refolded from inclusion bodies (Bayrhuber et al., 2005) or chemically synthesized (Dy et al., 2006). Likewise, when following the protocol for Conk‐S1 production, Conk‐S2, which shares 92% sequence identity with Conk‐S3, as well as Conk‐C3 (Saikia et al., 2021), were expressed in the insoluble fraction and had to be refolded to obtain a protein sample for structure determination (Korukottu et al., 2007).1 While sometimes applicable, these methods are often inefficient and/or costly. The introduction of the DisCoTune system holds the potential to bypass the need for refolding from a denatured and reduced state, and instead allow the efficient production of correctly folded, disulphide‐bonded proteins and peptides in the cytoplasm of E. coli.
Experimental procedures
DisCoTune plasmid generation
The cloning of the polycistronic fragment into the new backbone was performed with uracil excision cloning as described previously (Cavaleiro et al., 2015). Polymerase chain reactions (PCR) were carried out using Phusion U Hot Start polymerase (Thermo Fisher Scientific, Waltham, MA, USA), according to manufacturer’s instructions. We experienced problems cloning the entire CyDisCo operon when controlled by the Ptac promotor. Initially, the Ptac promoter in CyDisCo and csCyDisCo was therefore exchanged for the medium and low strength promoters J23105 and J23112, respectively, from the Anderson collection. This was achieved using oligonucleotides J23105_CyDisCo_rev and J231xx_CyDisCo_fwd for CyDisCo and J231xx_CyDisCo_fwd and J23112_pCyDisCo_rev for csDysCisCo(oligonucleotide sequences are listed in Table S3). The polycistronic fragments from CyDisCo versions were PCR amplified with oligonucleotides CyDisCo_fragment_J231xx_fwd and CyDisCo_fragment_rrnC_rev, and from csCyDisCo with CyDisCo_fragment_J231xx_fwd and csCyDisCo_fragment_rrnC_rev. This PCR exchanged the previous T7 terminator for the BBa_B0062‐R. The new backbone was ordered from Invitrogen (Thermo Fisher Scientific) and PCR amplified using oligonucleotides CyDisCo‐in‐pLemo_rev and CyDisCo‐in‐pLemo_fwd. A successful clone was isolated and sequence verified. The Ptac promotor was now reintroduced by PCR amplification using oligonucleotides pTac_pDisCoTune _fwd and pTac_pDisCoTune _rev. Following this PCR, the region upstream of the start codon of Erv1p was modified from TTGTTTAACTTTAAGAAGGAGATACATATG to CAGGACGCACTGACCAGGAGGTACATATG. The resulting two plasmids are referred to as pDisCoTune and pcsDisCoTune. They carry the genes for Erv1p, hPDI and csPDI in the case of csDisCoTune controlled by the Ptac promotor and terminated by the rrnC terminator (Fig. S1). The pLemo control plasmid was purchased from Xbrane Bioscience (Stockholm, Sweden).
Proteinase K expression plasmid generation
The sequence of proteinase K with propeptide was codon‐optimized for E. coli and ordered from Invitrogen (Thermo Fisher Scientific). The fragment of proteinase K with propeptide was amplified with oligonucleotides ProtK_fwd and ProtK_rev and cloned by uracil excision into the pET28 vector amplified with oligonucleotides ProtK‐in‐pET28_BB _fwd and ProtK‐in‐pET28_BB _rev (Table S3). The resulting plasmid was sequence verified.
Conk‐S3 expression plasmid generation
The full‐length sequence of Conk‐S3 was retrieved from the venom gland transcriptome of Conus striatus (Li et al., 2018) (NCBI Sequence Read Archive ID SRX5015022). The predicted sequence of the mature toxin (KDRPSYCNLPADSGSGTKPEQRIYYNSAKKQCVTFTYNGKGGNGNNFSRTNDCRQTCQYPA) shares 72% and 92% sequence identity with the two other conkunitzins previously identified from C. striatus, Conk‐S1 (Bayrhuber et al., 2005) and Conk‐S2 (Korukottu et al., 2006) respectively. The sequence of Conk‐S3 was codon‐optimized for bacterial expression using the CodonOpt tool (http://eu.idtdna.com/CodonOpt). The codon‐optimized sequence was used as a template for primer design of Conk‐S3_fwd and Conk‐S3_rev (Table S3) for subsequent USER cloning into the pET39_Ub19 expression construct (Rogov et al., 2012). The resulting plasmid pET39‐Conk‐S3 was sequence verified. The fusion protein produced from pET39‐Conk‐S3 was named Ub–His10–Conk‐S3 and contains a TEV protease recognition site following the Ub–His10‐tag. Upon TEV protease‐mediated release, the Conk‐S3 protein contains an additional N‐terminal glycine residue, inserted to ensure efficient TEV protease cleavage, as compared with the predicted native protein.
Protein expression of proteinase K
The pET28‐protK plasmid was transformed into chemically competent E. coli BL21(DE3) cells carrying pLemo, pCyDisCo or pDisCoTune, and plated on LB agar containing kanamycin and chloramphenicol (50 and 30 µg ml‐1 respectively). A single colony was transferred into 2 ml LB medium containing appropriate antibiotics and grown overnight in an orbital shaker at 37°C at 200 rpm. The following day, expression cultures of 2 ml LB were set up in a 24‐deepwell plate with appropriate antibiotics and a dilution series of rhamnose. For the rhamnose titration, following concentrations were used: 5, 10, 25, 50, 100, 500, 1000 and 2000 µM. The expression cultures were inoculated with 20 µl of the overnight culture and incubated at 37°C, 250 rpm until the optical density at 600 nm (OD600) reached 0.5–0.8. Protein expression was induced with 1 mM IPTG, and cultures were grown overnight at 30°C before harvesting.
Protease activity measurements
For screening proteinase K activity on skim milk agar plates, a colony for each of the expression systems pLemo, pCyDisCo and pDisCoTune with and without pET28‐ProtK was picked and stabbed with a sterile toothpick onto LB agar supplemented with appropriate antibiotics, 1 mM IPTG, 1.5% skim milk and rhamnose in varying concentrations. For the rhamnose titration, following concentrations were used: 5, 10, 25, 50, 100, 500, 1000 and 2000 µM. The plates were incubated at 37°C for 5 days. For measuring proteinase K activity using FITC labelled Casein, for each culture condition, 1 ml of expression culture was harvested by centrifugation at 5,000 g, 4°C for 10 min. The remaining pellet was resuspended in 100 µl lysis buffer [CelLytic™ B (Sigma‐Aldrich, Saint Louis, MO, USA)] in 50 mM Tris‐HCl pH 8, 10 mM Imidazole, 150 mM NaCl) and incubated on ice for 30 min before the soluble fraction was isolated by centrifugation at 15 000 g for 10 min. Enzyme activity was quantified directly from the soluble lysates using the protease activity assay kit ab111750 (Abcam, Cambridge, UK) according to manufacturer’s protocol. Lysates were diluted 1:10 in the protease assay buffer for analysis.
Protein expression of Conk‐S3
The pET39‐Conk‐S3 plasmid was transformed into chemically competent E. coli BL21(DE3) cells either alone, or together with pcsCyDisCo or pcsDisCoTune, and plated on an LB agar plate containing solely kanamycin (50 µg ml−1; when transforming only with pET39‐Conk‐S3), or kanamycin and chloramphenicol (30 µg ml−1) when performing co‐transformations. A single colony was transferred into 10 ml LB medium containing appropriate antibiotics and grown overnight in an orbital shaker at 37°C at 200 rpm. 0.5 ml of the overnight culture was inoculated into 50 ml of LB medium containing appropriate antibiotics and incubated at 37°C until the optical density at 600 nm (OD600) reached 0.8‐1. Protein expression was induced by 1 mM IPTG, and cultures were grown overnight at 25°C before cells were harvested by centrifugation for 20 min at 5000 g.
SDS‐PAGE sample preparation of cell extracts
Harvested cells were resuspended in 1 ml 50 mM Tris, 300 mM NaCl, 20 mM imidazole, pH 8 (resuspension buffer). The cell suspensions were sonicated using 5 × 30‐sec pulses, with 30‐sec resting periods on ice. To generate a total protein sample, a fraction of the lysate was mixed with 4x SDS‐PAGE loading buffer to an OD600 equivalent of 20 (based on the final OD600 of the cell cultures). The lysate was then centrifuged for 45 min at 20 000 g at 4°C. A fraction of the supernatant was mixed with 4× SDS‐PAGE loading buffer to an OD600 = 20 to generate a sample representing the soluble fraction. Finally, the pellet was resuspended in 1 ml resuspension buffer containing 8 M Urea before mixing a fraction of the suspension with 4× SDS‐PAGE loading buffer to an OD600 = 20 in order to generate a sample representing the insoluble fraction.
To trap free thiols and thus capture the thiol/disulphide status of Ub–His10–Conk‐S3, harvested cells were treated as above with the addition of 100 mM N‐ethylmaleimide (NEM). Likewise, the pellet was resuspended in 1 ml resuspension buffer supplemented with 8 M urea and containing 100 mM NEM before mixing with 4x SDS‐PAGE loading buffer to an OD600 = 20.
Western blot analysis
Samples were mixed 1:1 with sample buffer (8 M urea, 0.0105% (w/v) bromophenol blue, 5 mM EDTA, 100mM Tris‐HCl pH 6.8, 4% (w/v) SDS and 25% (v/v) glycerol) and heated to 98°C for 10 min. Samples were normalized to OD 0.1 when loaded on a 4−20% Mini‐PROTEAN‐TGX gel (Bio‐Rad, Hercules, CA, USA) and run at 180 V for 45 min. Proteins were transferred to a nitrocellulose membrane using an iBlot Dry Blotting System (Invitrogen, Thermo Fisher Scientific) at 20V for 7 min. The membrane was blocked with 5% skim milk in TBS‐T(20 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.1% (v/v) Tween‐20) for at least 30 min, followed by incubation overnight at 4°C with primary antibody (anti‐T7 lysozyme from rabbit, 1:5000 dilution as previously described (Søgaard and Nørholm, 2016) or (anti‐Lep from rabbit, 1:1000, a generous gift from IngMarie Nilsson and Gunnar von Heijne, Stockholm University) diluted in 5% skim milk TBS‐T. The membrane was washed three times for 5 min in TBS‐T and incubated with the secondary antibody (anti‐Rabbit‐HRP IgG, 1:10 000; Sigma‐Aldrich) diluted in TBS‐T for 1 h. The washing steps were repeated again, and the protein:antibody complexes were visualized using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Chicago, IL, USA).
Protein purification of Conk‐S3
For purification of Ub–His10–Conk‐S3, the protein was expressed as described above in 1 l of LB medium. Protein purification was performed as previously described (Nielsen et al., 2019) with a few modifications. Briefly, harvested cells were resuspended in resuspension buffer and lysed by sonication performed for 5 min at half duty and full amplitude on ice, followed by centrifugation at 30 000 g for 30 min at 4°C. Ub–His10–Conk‐S3 was purified from the cleared lysate using gravity flow on a column packed with 5 ml Qiagen Superflow Ni‐NTA resin pre‐equilibrated with resuspension buffer. Upon sample application, the column was washed with resuspension buffer containing 20 mM imidazole before elution in resuspension buffer containing 400 mM imidazole.
SDS‐PAGE was used to analyse eluted fractions. The fractions containing Ub–His10–Conk‐S3 were diluted 4× in 50 mM Tris, 300 mM NaCl, pH 8, before adding recombinant TEV protease containing an N‐terminal His‐tag (His6–TEV), which was expressed and purified as previously described (Nielsen et al., 2019). TEV cleavage was carried out overnight at room temperature using a 1:20 molar ratio of His6–TEV to Ub–His10–Conk‐S3. Next, uncleaved protein, free Ub–His10‐tag and His6–TEV were removed by applying the cleavage mixture to a 5 ml Qiagen Superflow Ni‐NTA resin pre‐equilibrated with 50 mM Tris, 300 mM NaCl, pH 8. The flow‐through and first 10 ml of column wash with resuspension buffer contained cleaved Conk‐S3, as determined by SDS‐polyacrylamide gel analysis. Final purity was reached by reversed‐phase high‐performance liquid chromatography using a Kromasil C18 300 4.6 × 250 mm column and a linear gradient from 0.1% TFA to 75% of 0.1% TFA:70% acetonitrile over 7.5 column volumes at 1 ml min−1. Fractions containing Conk‐S3 were pooled and lyophilized.
Concentration determination
The concentration of Conk‐S3 was determined by measuring the absorbance at 280 nm using a theoretical extension coefficient, ɛ = 7700 M‐1 cm‐1, calculated using the ProtParam tool (Walker, 2005) at the Expasy bioinformatics resource portal.
Circular dichroism (CD) spectroscopy
For CD spectroscopy, lyophilized Conk‐S3 was dissolved in water and diluted to a final concentration of 20 μM in a volume of 200 μl. CD spectra were recorded on a Jasco J‐810 CD spectropolarimeter in a 0.1 cm cuvette at 25°C in the wavelength range of 190–260 nm, using a data interval of 0.2 nm and a bandwidth of 1 nm. The final spectra were obtained after averaging over 5 spectra recorded at a scan rate of 100 nm min−1, and baseline (water only, same conditions) was subtracted. Finally, the measured ellipticity was converted to residual molar ellipticity and the data were visualized using Matlab. The deconvolution of the CD spectrum was calculated using BestSel (Micsonai et al., 2015).
MALDI mass spectrometry
Conk‐S3 was digested with sequencing‐grade modified trypsin (Merck, NJ, USA) using an enzyme‐to‐substrate ratio of 1:50 (w/w), in 50 mM ammonium bicarbonate, at 37°C overnight. For reduction of disulphides, peptides were incubated in 50 mM ammonium bicarbonate containing 15 mM DTE (1,4‐dithioerythritol) at 56°C for 45 min, and subsequently, free cysteines were alkylated by addition of iodoacetamide (to 50 mM) and incubated for 30 min in the dark at room temperature. Prior to MALDI‐MS analyses, peptides were desalted and concentrated on a Zip‐tip column containing C18 reversed‐phase material (Merck). MALDI‐MS was performed using a Bruker Autoflex III instrument (Bruker Daltonics, Bremen, Germany). The analysis was performed using a saturated solution of α‐cyano‐4‐hydroxycinnamic acid. The instrument was operated in linear and reflected positive ionization mode and calibrated in the mass range 1000 to 3200 Da using a peptide calibration standard (Bruker Daltonics). The theoretical peptide masses were calculated using the General Protein/Mass Analysis for Windows software (Lighthouse Data, Odense, Denmark).
Sample preparation for proteomic analysis
Cells from 500 µl expression culture were pelleted and frozen on dry ice. Pellets were kept at −80°C, before they were thawed on ice and 100 μl 95°C Guanidinium HCl (6 M Guanidinium hydrochloride (GuHCl), 5 mM tris(2‐carboxyethyl)phosphine (TCEP), 10 mM chloroacetamide (CAA), 100 mM Tris–HCl pH 8.5) were added to the samples together with two 3 mm zirconium oxide beads (Glen Mills, Clifton, NJ, USA). Cells were disrupted using a Mixer Mill (MM 400 Retsch, Haan, Germany) for 5 min at 25 Hz. The samples were placed in a thermo mixer at 95°C for 10 min at 2000 rpm. After this, the samples were centrifuged at 15 000 g for 10 min, and 50 μl of supernatant was collected and diluted with 50 μl of 50 mM ammonium bicarbonate. Protein concentrations were measured using BSA as a standard, and 100 μg were used for tryptic digestion. Tryptic digestion was carried out for 12 h, after which 10 μl of 10% TFA was added and samples were StageTipped using C18 (Empore, 3M, Saint Paul, MN, USA).
After stagetipping, the samples were analysed using a CapLC system (Thermo Fisher Scientific) coupled to a 15 cm C18 easy spray column (PepMap RSLC C18 2 μm, 100 Å, 150 μm × 15 cm). Initially, the samples were trapped on a precolumn (μ‐precolumn C18 PepMap 100, 5 μm, 100 Å) after which the peptides were separated using a gradient from 4% acetonitrile in water to 76% over 60 min at a constant flow rate of 1.2 μl min−1. The samples were sprayed into an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific). MS‐level scans were performed from 375 to 1500 m/z with Orbitrap resolution set to 120 000; AGC Target 300%; maximum injection time set to auto; intensity threshold 5.0 × 103; dynamic exclusion 20 s. Data‐dependent MS2 selection was performed in Top 20 Speed mode with HCD collision energy set to 40% (AGC target 70%, maximum injection time 30 ms, Isolation window 1.3 m/z).
Proteomics data analysis
The raw files were analysed using Proteome Discoverer (version 2.4; Thermo Fisher Scientific) in order to obtain protein identifications and quantification. While analysing the data the following settings were used: fixed modifications, carbamidomethyl (C); and variable modifications, oxidation of methionine residues. First search mass tolerance 20 ppm and a MS/MS tolerance of 0.5 Da. Trypsin as enzyme and allowing two missed cleavages. Target false discovery rate (FDR) was set at 0.01. The retention time alignment was set to ∆RT 0.2. For quantification, top N 3 was used only allowing quantification based on unique peptides. Intensities were normalized to total peptide level. For the searches, a protein database consisting of the reference E. coli proteome UP000002032 combined with the sequences of proteinase K, Enlys, ERV1 and PDIA1 was used.
Funding Information
This work was supported by Danish Council for Independent Research Technology and Production Sciences Grant 7017‐00288 (L.E.) and by a grant from the Novo Nordisk Foundation NNF20CC0035580 (M.H.H.N).
Conflict of interest
None declared.
Author contributions
The manuscript was written by ABB, LE and MN. The design and cloning of pDisCoTune and pcsDisCoTune was performed by ABB, MR and MN. The experiments on the production of proteinase K was performed by ABB and CB. The following experimental proteomics was performed by TW. The design and performance of the experiments on the production and validation of Conk‐S3 structure were performed by CMH, LDK, HSH and LE. MS validation of disulphide connectivity was performed by BC and ESS. All authors read and approved the content of the manuscript.
Supporting information
Fig. S1. Schematic overview of plasmid relations. The diagram shows the relations between the pLysS backbone and pMJS205 and pLE577(top)(Here referred to as pCyDisCo and pcsCyDisCo, respectively), and the pLemo backbone and pDisCoTune and pcsDisCoTune (bottom). In the original pLysS plasmid, the transcript that is initiated from the T7 PΦ3.8 has to cover the entire plasmid before reaching the open reading frame for T7 lysozyme. The PrhaB promoter allows for titratable control the transcription of T7 lysozyme with the activators rhaS and rhaR.
{kind=link}
Fig. S2. A. Comparison of Ub–His10–Conk‐S3 expression without or with the csCyDisCo or pDisCoTune system. 15% SDS‐PAGE gel stained with Coomassie Brilliant Blue showing the total cell extract (T), resuspended pellet after lysis and centrifugation (P), and the soluble fraction (S) from cells expressing Ub–His10–Conk‐S3. Cells were lysed in the absence (‐NEM) or presence (+NEM) of the thiol‐alkylating agent N‐ethylmaleimide (NEM). The arrowhead denotes a semi‐oxidized form of Ub–His10‐Conk‐S3. Expression was performed as described under Materials and Methods. Protein levels are comparable between lanes and the gel represents three independent experiments. Note that in the presence of NEM very little Ub–His10–Conk‐S3 is observed in the soluble fraction, indicating that in the absence of NEM, protein folding primarily takes place post‐translationally (i.e. upon cell lysis). B. Analysis of the solubilized pellet from cells expressing Ub–His10–Conk‐S3 in the absence of helper plasmid under non‐reducing (ox) and reducing (red) conditions. The result shows that Ub–His10–Conk‐S3 in the pellet comprises a mixture of oxidized and semi‐oxidized forms (as illustrated in Fig. 4).
{kind=link}
Fig. S3. Screen of Conk‐S3 production supported by pcsDisCoTune at different rhamnose concentrations. 15% SDS‐PAGE gel stained with Coomassie Brilliant Blue showing the total cell extract (T), resuspended pellet after lysis and centrifugation (P), and the soluble fraction (S) from cells expressing Ub–His10–Conk‐S3 at the indicated concentrations of rhamnose. Expression was performed as described under Materials and Methods.
{kind=link}
Table S1. Strains used in this study.
Table S2. Plasmids used in this study.
Table S3. Oligonucleotides used in this study.
Table S4. Mapping of disulfide bonds in Conk‐S3 by MALDI‐MS.
Acknowledgements
We thank Cecilie L. Søltoft for expert technical assistance, and Dr. Lloyd Ruddock, University of Oulu, and Lina Dahlberg, University of Copenhagen, for useful discussions and critical reading of the manuscript.
Microb. Biotechnol. (2021) 14(6), 2566–2580
Footnotes
Personal communication: Dr. Stefan Becker, Max Planck Institute for Biophysical Chemistry, Dept. for NMR based Structural Biology, Göttingen, Germany.
Contributor Information
Lars Ellgaard, Email: lellgaard@bio.ku.dk.
Morten H. H. Nørholm, Email: morno@biosustain.dtu.dk.
References
- Alanen, H.I. , Walker, K.L. , Lourdes Velez Suberbie, M. , Matos, C.F.R.O. , Bönisch, S. , Freedman, R.B. , et al. (2015) Efficient export of human growth hormone, interferon α2b and antibody fragments to the periplasm by the Escherichia coli Tat pathway in the absence of prior disulfide bond formation. Biochim Biophys Acta ‐ Mol Cell Res 1853: 756–763. 10.1016/j.bbamcr.2014.12.027 [DOI] [PubMed] [Google Scholar]
- Bayrhuber, M. , Graf, R. , Ferber, M. , Zweckstetter, M. , Imperial, J. , Garrett, J. E. , et al. (2006) Production of recombinant Conkunitzin‐S1 in Escherichia coli . Protein Expr Purif 47: 640–644. 10.1016/j.pep.2006.01.019 [DOI] [PubMed] [Google Scholar]
- Bayrhuber, M. , Vijayan, V. , Ferber, M. , Graf, R. , Korukottu, J. , Imperial, J. , et al. (2005) Conkunitzin‐S1 is the first member of a new kunitz‐type neurotoxin family. J Biol Chem 280: 23766–23770. 10.1074/jbc.C500064200 [DOI] [PubMed] [Google Scholar]
- Betzel, C. , Pal, G.P. , and Saenger, W. (1988) Three‐dimensional structure of proteinase K at 0.15‐nm resolution. Eur J Biochem 178: 155–171. 10.1111/j.1432-1033.1988.tb14440.x [DOI] [PubMed] [Google Scholar]
- Cavaleiro, A.M. , Kim, S.H. , Seppälä, S. , Nielsen, M.T. , and Nørholm, M.H.H. (2015) Accurate DNA assembly and genome engineering with optimized uracil excision cloning. ACS Synth Biol 4: 1042–1046. 10.1021/acssynbio.5b00113 [DOI] [PubMed] [Google Scholar]
- Dy, C.Y. , Buczek, P. , Imperial, J.S. , Bulaj, G. , and Horvath, M.P. (2006) Structure of conkunitzin‐S1, a neurotoxin and Kunitz‐fold disulfide variant from cone snail. Acta Crystallogr Sect D Biol Crystallogr 62: 980–990. 10.1107/S0907444906021123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finol‐Urdaneta, R.K. , Belovanovic, A. , Micic‐Vicovac, M. , Kinsella, G.K. , McArthur, J.R. , and Al‐Sabi, A. (2020) Marine toxins targeting KV1 channels: pharmacological tools and therapeutic scaffolds. Mar Drugs 18: 173. 10.3390/md18030173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaciarz, A. , Veijola, J. , Uchida, Y. , Saaranen, M.J. , Wang, C. , Hörkkö, S. , and Ruddock, L.W. (2016) Systematic screening of soluble expression of antibody fragments in the cytoplasm of E. coli . Microb Cell Fact 15: 1–10. 10.1186/s12934-016-0419-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunkel, F.A. , and Gassen, H.G. (1989) Proteinase K from tritirachium album limber characterization of the chromosomal gene and expression of the cDNA in Escherichia coli . Eur J Biochem 179: 185–194. 10.1111/j.1432-1033.1989.tb14539.x [DOI] [PubMed] [Google Scholar]
- Hatahet, F. , Nguyen, V.D. , Salo, K.E.H. , and Ruddock, L.W. (2010) Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of E. coli . Microb Cell Fact 9:67. 10.1186/1475-2859-9-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. , and Sander, C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22: 2577–2637. 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- Kojima, S. , Minagawa, T. , and Miura, K.I. (1997) The propeptide of subtilisin BPN’ as a temporary inhibitor and effect of an amino acid replacement on its inhibitory activity. FEBS Lett 411: 128–132. 10.1016/S0014-5793(97)00678-9 [DOI] [PubMed] [Google Scholar]
- Korukottu, J. , Bayrhuber, M. , Montaville, P. , Vijayan, V. , Jung, Y.‐S. , Becker, S. , and Zweckstetter, M. (2006) Conkunitzin‐S2 ‐ cone snail neurotoxin. Published online. 10.1002/ANIE.200603213. [DOI]
- Korukottu, J. , Bayrhuber, M. , Montaville, P. , Vijayan, V. , Jung, Y.‐S. , Becker, S. , and Zweckstetter, M. (2007) Fast high‐resolution protein structure determination by using unassigned NMR data. Angew Chemie Int Ed 46: 1176–1179. 10.1002/anie.200603213 [DOI] [PubMed] [Google Scholar]
- Landeta, C. , Boyd, D. , and Beckwith, J. (2018) Disulfide bond formation in prokaryotes. Nat Microbiol 3: 270–280. 10.1038/s41564-017-0106-2 [DOI] [PubMed] [Google Scholar]
- Li, Q. , Watkins, M. , Robinson, S. , Safavi‐Hemami, H. , and Yandell, M. (2018) Discovery of novel conotoxin candidates using machine learning. Toxins (Basel) 10: 503. 10.3390/toxins10120503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, J. , Warmuth, M.K. , Govindarajan, S. , Ness, J.E. , Wang, R.P. , Gustafsson, C. , and Minshull, J. (2007) Engineering proteinase K using machine learning and synthetic genes. BMC Biotechnol 7: 1–19. 10.1186/1472-6750-7-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobstein, J. , Emrich, C.A. , Jeans, C. , Faulkner, M. , Riggs, P. , and Berkmen, M. (2012) SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb Cell Fact 11: 1. 10.1186/1475-2859-11-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen, C. , Goñi Moreno, A. , Palchick, Z. , Roehner, N. , Atallah, C. , Bartley, B. , et al. (2019) Synthetic biology open language (SBOL) Version 2.3. J Integr Bioinform 16: 1–149. 10.1515/jib-2019-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai, A. , Wien, F. , Kernya, L. , Lee, Y.‐H. , Goto, Y. , Réfrégiers, M. , and Kardos, J. (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci USA 112: E3095–E3103. 10.1073/pnas.1500851112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljanich, G. (2004) Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr Med Chem 11: 3029–3040. 10.2174/0929867043363884 [DOI] [PubMed] [Google Scholar]
- Nativel, B. , Figuester, A. , Andries, J. , Planesse, C. , Couprie, J. , Gasque, P. , Viranaicken, W. , et al. (2016) Soluble expression of disulfide‐bonded C‐type lectin like domain of human CD93 in the cytoplasm of Escherichia coli . J Immunol Methods 439: 67–73. 10.1016/j.jim.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Nguyen, V.D. , Hatahet, F. , Salo, K.E.H. , Enlund, E. , Zhang, C. , and Ruddock, L.W. (2011) Pre‐expression of a sulfhydryl oxidase significantly increases the yields of eukaryotic disulfide bond containing proteins expressed in the cytoplasm of E.coli . Microb Cell Fact 10:1. 10.1186/1475-2859-10-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, L.D. , Foged, M.M. , Albert, A. , Bertelsen, A.B. , Søltoft, C.L. , Robinson, S.D. , et al. (2019) The three‐dimensional structure of an H‐superfamily conotoxin reveals a granulin fold arising from a common ICK cysteine framework. J Biol Chem 294: 8745–8759. 10.1074/jbc.RA119.007491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østergaard, H. , Henriksen, A. , Hansen, F.G. , and Winther, J.R. (2001) Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J 20: 5853–5862. 10.1093/emboj/20.21.5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østergaard, H. , Tachibana, C. , and Winther, J.R. (2004) Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol 166: 337–345. 10.1083/jcb.200402120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pähler, A. , Banerjee, A. , Dattagupta, J.K. , Fujiwara, T. , Lindner, K. , Pal, G.P. , et al. (1984) Three‐dimensional structure of fungal proteinase K reveals similarity to bacterial subtilisin. EMBO J 3: 1311–1314. 10.1002/j.1460-2075.1984.tb01968.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington, M.W. , Czerwinski, A. , and Norton, R.S. (2018) Peptide therapeutics from venom: current status and potential. Bioorganic Med Chem 26: 2738–2758. 10.1016/j.bmc.2017.09.029 [DOI] [PubMed] [Google Scholar]
- Prinz, W.A. , Åslund, F. , Holmgren, A. , and Beckwith, J. (1997) The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem 272: 15661–15667. 10.1074/jbc.272.25.15661 [DOI] [PubMed] [Google Scholar]
- Rabenstein, D.L. (2009) Chapter 3. Redox Potentials of Cysteine Residues in Peptides and Proteins: Methods for their Determination. Published online. 220‐235. 10.1039/9781847559265-00220 [DOI]
- Ranasinghe, S. , and McManus, D.P. (2013) Structure and function of invertebrate Kunitz serine protease inhibitors. Dev Comp Immunol 39: 219–227. 10.1016/j.dci.2012.10.005 [DOI] [PubMed] [Google Scholar]
- Ritz, D. , Lim, J. , Reynolds, C.M. , Poole, L.B. , and Beckwith, J. (2001) Conversion of a peroxiredoxin into a disulfide reductase by a triplet repeat expansion. Science (80‐ ) 294: 158–160. 10.1126/science.1063143 [DOI] [PubMed] [Google Scholar]
- Rogov, V.V. , Rozenknop, A. , Rogova, N.Y. , Löhr, F. , Tikole, S. , Jaravine, V. , et al. (2012) A universal expression tag for structural and functional studies of proteins. ChemBioChem 13: 959–963. 10.1002/cbic.201200045 [DOI] [PubMed] [Google Scholar]
- Rosenberg, A.H. , Lade, B.N. , Dao‐shan, C. , Lin, S.‐W. , Dunn, J.J. , and Studier, F.W. (1987) Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene 56: 125–135. 10.1016/0378-1119(87)90165-X [DOI] [PubMed] [Google Scholar]
- Safavi‐Hemami, H. , Li, Q. , Jackson, R.L. , et al. (2016) Rapid expansion of the protein disulfide isomerase gene family facilitates the folding of venom peptides. Proc Natl Acad Sci USA 113: 3227–3232. 10.1073/pnas.1525790113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia, C. , Dym, O. , Altman‐Gueta, H. , Gordon, D. , Reuveny, E. , and Karbat, I. (2021) A molecular lid mechanism of K+ channel blocker action revealed by a cone peptide. J Mol Biol 433: 166957. 10.1016/j.jmb.2021.166957 [DOI] [PubMed] [Google Scholar]
- Schlegel, S. , Löfblom, J. , Lee, C. , Hjelm, A. , Klepsch, M. , Strous, M. , et al. (2012) Optimizing membrane protein overexpression in the Escherichia coli strain Lemo21(DE3). J Mol Biol 423: 648–659. 10.1016/j.jmb.2012.07.019 [DOI] [PubMed] [Google Scholar]
- Shilling, P.J. , Mirzadeh, K. , Cumming, A.J. , Widesheim, M. , Köck, Z. , and Daley, D.O. (2020) Improved designs for pET expression plasmids increase protein production yield in Escherichia coli . Commun Biol 3: 1–8. 10.1038/s42003-020-0939-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søgaard, K.M. , and Nørholm, M.H.H. (2016) Side effects of extra tRNA supplied in a typical bacterial protein production scenario. Protein Sci 25: 2102–2108. 10.1002/pro.3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W. , Rosenberg, A.H. , Dunn, J.J. , and Dubendorff, J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 185: 60–89. 10.1016/0076-6879(90)85008-C [DOI] [PubMed] [Google Scholar]
- Studier, F.W. (1991) Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J Mol Biol 219: 37–44. 10.1016/0022-2836(91)90855-Z [DOI] [PubMed] [Google Scholar]
- Terlau, H. , and Olivera, B.M. (2004) Conus venoms: a rich source of novel ion channel‐targeted peptides. Physiol Rev 84: 41–68. 10.1152/physrev.00020.2003 [DOI] [PubMed] [Google Scholar]
- Touw, W.G. , Baakman, C. , Black, J. , te Beek, T.A.H. , Krieger, E. , Joosten, R.P. , and Vriend, G. (2015) A series of PDB‐related databanks for everyday needs. Nucleic Acids Res 43: D364–D368. 10.1093/nar/gku1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, J.M. (ed). (2005) The Proteomics Protocols Handbook. Totowa, NJ: Humana Press. 10.1385/1592598900 [DOI] [Google Scholar]
- Winther, J.R. , and Sorensen, P. (1991) Propeptide of carboxypeptidase Y provides a chaperone‐like function as well as inhibition of the enzymatic activity. Proc Natl Acad Sci USA 88: 9330–9334. 10.1073/pnas.88.20.9330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, Y. , Ritz, D. , Planson, A.G. , Jönsson, T.J. , Faulkner, M.J. , Boyd, D. , Beckwith, J. , and Poole, L.B. (2008) Mutant AhpC peroxiredoxins suppress thiol‐disulfide redox deficiencies and acquire deglutathionylating activity. Mol Cell 29: 36–45. 10.1016/j.molcel.2007.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, S. , Kim, S. , and Kim, J. (2009) Secretory production of recombinant proteins in Escherichia coli . Recent Pat Biotechnol 4: 23–29. 10.2174/187220810790069550 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Schematic overview of plasmid relations. The diagram shows the relations between the pLysS backbone and pMJS205 and pLE577(top)(Here referred to as pCyDisCo and pcsCyDisCo, respectively), and the pLemo backbone and pDisCoTune and pcsDisCoTune (bottom). In the original pLysS plasmid, the transcript that is initiated from the T7 PΦ3.8 has to cover the entire plasmid before reaching the open reading frame for T7 lysozyme. The PrhaB promoter allows for titratable control the transcription of T7 lysozyme with the activators rhaS and rhaR.
Fig. S2. A. Comparison of Ub–His10–Conk‐S3 expression without or with the csCyDisCo or pDisCoTune system. 15% SDS‐PAGE gel stained with Coomassie Brilliant Blue showing the total cell extract (T), resuspended pellet after lysis and centrifugation (P), and the soluble fraction (S) from cells expressing Ub–His10–Conk‐S3. Cells were lysed in the absence (‐NEM) or presence (+NEM) of the thiol‐alkylating agent N‐ethylmaleimide (NEM). The arrowhead denotes a semi‐oxidized form of Ub–His10‐Conk‐S3. Expression was performed as described under Materials and Methods. Protein levels are comparable between lanes and the gel represents three independent experiments. Note that in the presence of NEM very little Ub–His10–Conk‐S3 is observed in the soluble fraction, indicating that in the absence of NEM, protein folding primarily takes place post‐translationally (i.e. upon cell lysis). B. Analysis of the solubilized pellet from cells expressing Ub–His10–Conk‐S3 in the absence of helper plasmid under non‐reducing (ox) and reducing (red) conditions. The result shows that Ub–His10–Conk‐S3 in the pellet comprises a mixture of oxidized and semi‐oxidized forms (as illustrated in Fig. 4).
Fig. S3. Screen of Conk‐S3 production supported by pcsDisCoTune at different rhamnose concentrations. 15% SDS‐PAGE gel stained with Coomassie Brilliant Blue showing the total cell extract (T), resuspended pellet after lysis and centrifugation (P), and the soluble fraction (S) from cells expressing Ub–His10–Conk‐S3 at the indicated concentrations of rhamnose. Expression was performed as described under Materials and Methods.
Table S1. Strains used in this study.
Table S2. Plasmids used in this study.
Table S3. Oligonucleotides used in this study.
Table S4. Mapping of disulfide bonds in Conk‐S3 by MALDI‐MS.