

Summary
Purple non‐sulfur photosynthetic bacteria (PNSB) such as Rhodobacter capsulatus serve as a versatile platform for fundamental studies and various biotechnological applications. In this study, we sought to develop the class II RNA‐guided CRISPR/Cas12a system from Francisella novicida for genome editing and transcriptional regulation in R. capsulatus. Template‐free disruption method mediated by CRISPR/Cas12a reached ˜ 90% editing efficiency when targeting ccoO or nifH gene. When both genes were simultaneously edited, the multiplex editing efficiency reached > 63%. In addition, CRISPR interference (CRISPRi) using deactivated Cas12a was also evaluated using reporter genes egfp and lacZ, and the transcriptional repression efficiency reached ˜ 80%. In summary, our work represents the first report to develop CRISPR/Cas12a‐mediated genome editing and transcriptional regulation in R. capsulatus, which would greatly accelerate PNSB‐related researches.
CRISPR/Cas12a‐mediated genome editing was developed in purple non‐sulfur photosynthetic bacteria; CRISPR interference based on deactivated Cas12a was established in purple non‐sulfur photosynthetic bacteria; CRISPR/Cas12a‐mediated genome editing/transcriptional repression would greatly accelerate in purple non‐sulfur photosynthetic bacteria related biotechnological applications.
Introduction
Purple non‐sulfur photosynthetic bacteria (PNSB) such as Rhodobacter capsulatus are facultative anaerobic gram‐negative bacteria and are non‐pathogenic (Heck and Drepper, 2017; Troost et al., 2019). These bacteria have the versatile metabolic abilities to grow in a variety of habitats (Zannoni, 1995). For instance, R. capsulatus could use a wide spectrum of carbon sources including short‐chain fatty acids, dicarboxylic acids, sugars, agricultural and food wastes for chemotrophic growth under aerobic to microaerobic conditions (Adessi et al., 2017). It can also utilize the organic and inorganic compounds (such as S2O3 2−, H2S and Fe2+) as electron donors for photoheterotrophic growth (Romagnoli and Tabita, 2009; Jaschke et al., 2011). Moreover, when the nitrogen source is scarce, R. capsulatus can perform the nitrogen fixation and hydrogen production via the action of nitrogenases (Zhang et al., 2016).
To survive under such diverse living conditions, PNSB have evolved a complex metabolic network with highly specialized enzyme complexes and regulatory mechanisms. Recently, PNSB have attracted special attention as a platform microorganism for biotechnological applications such as H2 production as an alternative renewable energy to fossil fuels (Zhang et al., 2017a, 2017b, 2017c, 2016,2017a, 2017b, 2017c, 2016,2017a, 2017b, 2017c, 2016) and polyhydroxybutyrate as a biodegradable plastic (Kranz et al., 1997; Merugu et al., 2012), bioremediation of wastewater (Idi et al., 2014) and heavy metal (Feng et al., 2007), fixation of CO2 and N2 (Wahlund et al., 1996; Atsumi et al., 2009), expression of membrane proteins (Berry et al., 2004; Han and Perner, 2016) and metalloenzymes (Vignais et al., 2000; Kappler and McEwan, 2002). As the synthesis of photopigments in PNSB relies on the isoprenoid biosynthetic pathway, it provides a robust isoprenoid metabolism that makes PNSB as a promising host for isoprenoid biosynthesis (Heck and Drepper, 2017; Loeschcke et al., 2017; Troost et al., 2019).
Currently, the genetic tools in PNSB heavily rely on traditional homologous recombination using suicide plasmids (Zhang et al., 2017a, 2017b, 2017c,2017a, 2017b, 2017c,2017a, 2017b, 2017c), interposon‐mutagenesis using gene transfer agent (GTA) that leads to gene knockout via the insertion of a kanamycin cassette (Daldal et al., 1986), and gene mutation using transposon (Zhang et al., 2016). The synthetic biology tools for engineering PNSB are still limited, which significantly slows the progress in PNSB‐related biotechnological applications. Recently, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR‐associated protein (Cas) systems (Marraffini, 2015) have been developed as versatile tools for genome editing in a variety of organisms (Savitskaya et al., 2016; Pickar‐Oliver and Gersbach, 2019). For instance, the class II type II CRISPR/Cas9‐mediated genome editing was recently established in PNSB such as R. sphaeroides (Mougiakos et al., 2019; Luo et al., 2020).
Cas12a, a class II type V‐A endonuclease, is characterized as a dual nuclease referring to CRISPR RNA (crRNA) processing, target‐site recognition and DNA cleavage (Zetsche et al., 2015). To date, the CRISPR/Cas12a system has also been engineered into a genome editing tool in many species such as Escherichia coli, Mycobacterium smegmatis and mammalian cells (Yan et al., 2017; Zetsche et al., 2017). Compared with CRISPR/Cas9 that recognizes the target locus close to a guanine‐rich protospacer adjacent motif (PAM) sequence to create blunt ends (Hsu et al., 2014), Cas12a assisted by a mature crRNA binds the protospacer segment flanked by a thymidine‐rich PAM to form staggered ends (Zetsche et al., 2015; Swarts et al., 2017). Considering PNSB normally has high GC content (˜70%), the chance of off‐target is expected to be higher for CRISPR/Cas9 than that of CRISPR/Cas12a. The compact design of crRNA and self‐processing ability by Cas12a make CRISPR/Cas12a a more versatile and robust tool for multiplex genome engineering. To the best of our knowledge, there is no report on CRISPR/Cas12a‐mediated genetic tools in PNSB. In this study, we sought to assess the feasibility of CRISPR/Cas12a‐assisted genome editing in R. capsulatus (Fig. 1). In addition, we also attempted to evaluate CRISPR/dCas12a as an artificial transcriptional factor for CRISPR interference (CRISPRi) applications.
Fig. 1.
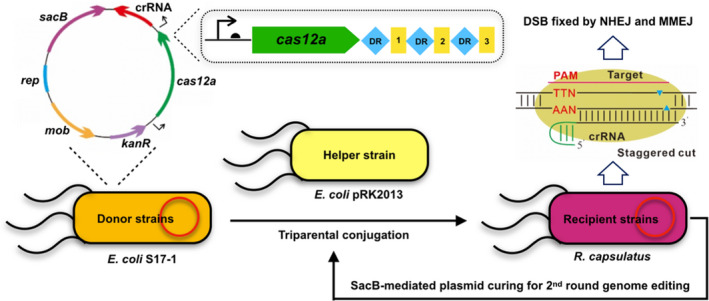
Schematic diagram of CRISPR/Cas12a system for genome editing in R. capsulatus. Both Cas12a and crRNA were expressed from a low copy plasmid with kanamycin marker (kanR), shuttle replicon (rep) and mobilization element (mob) for transferring plasmid from the donor strains E. coli into the recipient strains of R. capsulatus. DSB, double‐strand breaks; NHEJ, non‐homologous end joining; MMEJ, microhomology‐mediated end joining. The sacB cassette was used as the counter‐selection marker for curing plasmid. The CRISPR/Cas12a system was first constructed in E. coli S17‐1, then conjugated into R. capsulatus via the helper strain of E. coli pRK2013. The use of helper strain of E. coli pRK2013 is for the purpose of increasing the conjugation efficiency.
Results
Evaluation of CRISPR/Cas12a‐mediated genome editing in R. capsulatus
We first evaluated the potential toxicity of Cas12a from Francisella novicida in R. capsulatus. The Escherichia coli‐Rhodobacter shuttle vector pBBRdMCS, a multicopy vector derived from pBBR1MCS2 (Kovach et al., 1995), was used to express cas12a gene under the control of P lac promoter, and the resulting plasmid was designated as pBdRCas12a. In particular, we also included a sacB counter‐selection marker from Bacillus subtilis for plasmid curing (Fig. 1) (Link et al., 1997), whereas its expression is lethal when exposed to sucrose. According to the literature, P lac promoter from E. coli was reported to confer constitutive activity in R. capsulatus due to the absence of the lacI repressor gene (Khan et al., 2015). When the plasmid pBdRCas12a in parallel with the control plasmid pBdRSacB was transformed into R. capsulatus, the conjugation efficiency for pBdRCas12a was close to that achieved by the control (Fig. S1a). In addition, we did not observe noticeable growth differences between strains harbouring pBdRCas12a and pBdRSacB, suggesting that Cas12a is non‐toxic to R. capsulatus under the tested condition.
To confirm that Cas12a is functional in R. capsulatus, ccoO gene encoding a subunit of cbb 3‐cytochrome oxidase (cbb 3‐Cox) and nifH gene encoding a subunit of nitrogenase were chosen as the disruption targets. The visualization of cbb3 ‐Cox activity using colorimetric assay was reported in R. capsulatus (Ekici et al., 2012). As nitrogenase is essential for the nitrogen fixation, thus, nifH mutants would abolish the diazotrophic growth under nitrogen depleting conditions in a similar way to a previous report (Ungerer and Pakrasi, 2016). According to the literature, crRNA comprising of a 19‐nt direct repeat and a 23‐nt guide sequence is typically required to achieve the efficient genome editing (Zetsche et al., 2015), and all the crRNAs were designed according to this criteria. The crRNA‐expressing module was assembled into the pBdRCas12a plasmid under the control of the same promoter as Cas12a. We found the conjugation efficiencies declined more than 4 orders of magnitude compared with the control plasmid (Fig. S1a). The generation of double‐strand breaks at the targeted genes by CRISPR/Cas12a typically results in decreased survival rates (Mougiakos et al., 2019). As shown in Fig. 2A and B, ccoO or nifH was successfully mutated by CRISPR/Cas12a‐mediated genome editing. However, the editing efficiency mediated by CRISPR/Cas12a was only 30%˜40%.
Fig. 2.
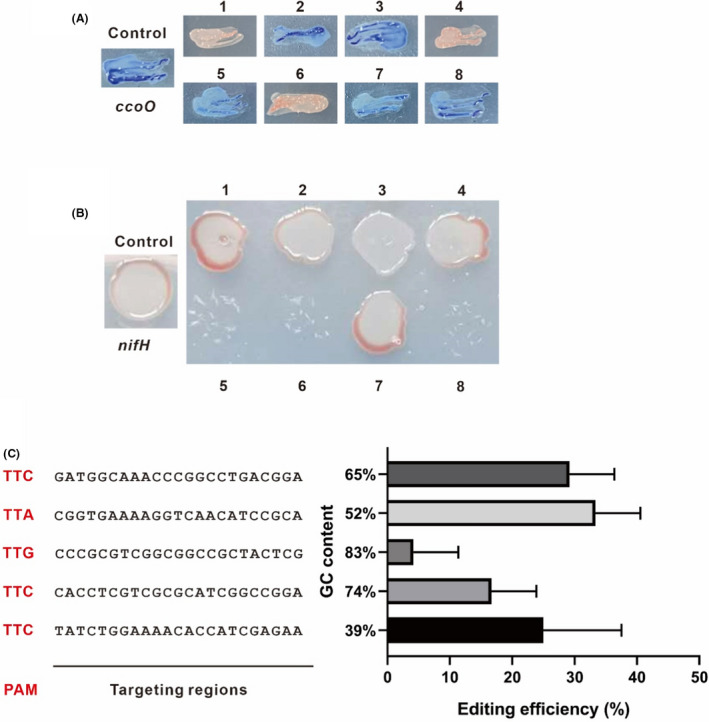
Genome editing using CRISPR/Cas12a in R. capsulatus.
A. Verification of ccoO disruption by NADI staining. The parental R. capsulatus strain containing pBdRCas12a was employed as the control. The disruption of ccoO that abolishes the cbb3 ‐Cox activity would appear background colour instead of blue staining. Eight randomly selected colonies were tested for ccoO disruption.
B. Verification of nifH disruption by the diazotrophic growth under nitrogen depleting conditions. The parental R. capsulatus strain containing pBdRCas12a was employed as the control. The disruption of nifH is expected to abolish the diazotrophic growth under nitrogen depleting conditions. Eight randomly selected colonies were tested for nifH disruption.
C. Comparison of the genome editing efficiency of five crRNAs with different GC contents. Experiments were carried out in triplicate, and the data are presented as mean ± SD.
It was reported that the genome editing efficiency assisted by CRISPR/Cas9 system was observably impacted by GC content of guide RNA and the target positions (Wang et al., 2014). In this study, five crRNAs with different GC contents ranging from 39% to 83% were designed for ccoO. As shown in Fig. 2C, the crRNAs with different GC contents resulted in editing efficiencies ranging from 4% to 33%. In particular, similar efficiencies (25%, 33% and 30% respectively) were achieved for GC contents ranging from 39% to 65%, indicating that Cas12a might be not as sensitive to the GC content as the Cas9 counterpart.
Optimization of CRISPR/Cas12a‐mediated genome editing in R. capsulatus
According to previous reports, efficient expression of Cas12a is also crucial to achieve efficient CRISPR/Cas12a‐assisted genome editing system (Zhao et al., 2019). As the heterologous promoter P lac is a relatively weak promoter, the low editing efficiency might be attributed to the insufficient CRISPR/Cas12a expression. To validate this hypothesis, we sought to search alternative promoters to drive the expression of CRISPR/Cas12a in R. capsulatus. It was reported that promoters of puc operon (encoding light‐harvesting complex II) and puf operon (encoding light‐harvesting complex I) possess relatively high levels of expression under aerobic conditions (Hu et al., 2010). As glycolytic enzymes are typically expressed at high levels in the presence of glucose, promoters of phosphoglycerate kinase gene (pgk) (Piper et al., 1988) and enolase gene (eno) (Toda et al., 2001) were also included in this study. The upstream ˜ 400 bp noncoding regions from puc, puf, pgk and eno genes were cloned for measuring the promoter strengths using eGFP as the reporter. As shown in Fig. 3A, all native promoters exhibited much higher activity than P lac under the tested condition. Among them, P puc showed the highest activity, which corresponds to approximately 5.4‐fold improvement over that of P lac .
Fig. 3.
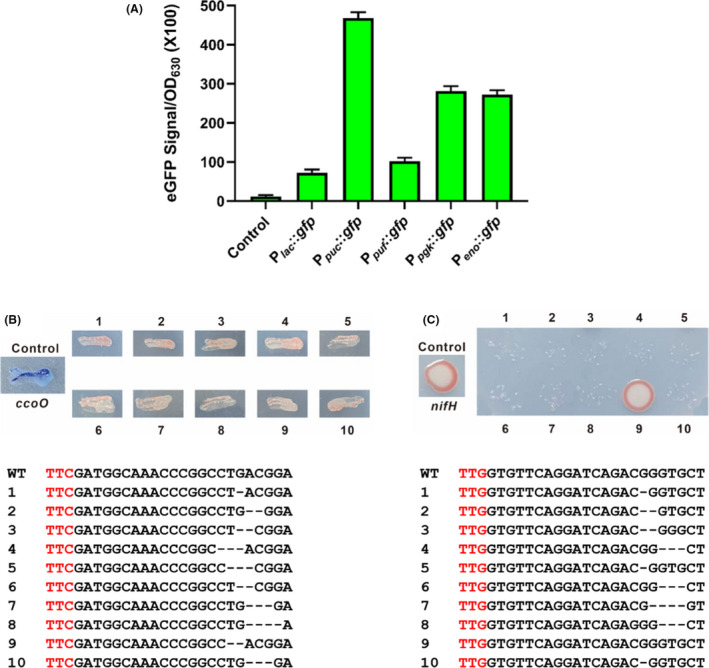
Optimization of CRISPR/Cas12a‐mediated genome editing in R. capsulatus.
A. Transcriptional activities of five promoters in R. capsulatus (P lac , P puc , P puf , P pgk and P eno ) as measured by eGFP fluorescence intensities. The control indicates the strain with the empty plasmid pBBR1MCS2. Experiments were carried out in triplicate, and the data are presented as mean ± SD.
B. NADI staining results of ccoO disruption and sequencing verification. The parental R. capsulatus strain containing pBRucCas12a was employed as the control. Ten randomly picked colonies were tested for ccoO disruption.
C. Results of the diazotrophic growth on nifH disruption and sequencing verification. The parental R. capsulatus strain containing pBRucCas12a was employed as the control. Ten randomly picked colonies were tested for nifH disruption.
Next, the promoter P puc was applied to drive the expression of CRISPR/Cas12a system in the remaining studies. When compared to the first‐generation design, the new version of CRISPR/Cas12a system only differed in the promoter sequences (P puc vs P lac ). These plasmids were transformed into R. capsulatus with pBRPpucSacB as the control plasmid. As depicted in Fig. S1b, the conjugation efficiency of Cas12a expression driven by P puc was close to that of Cas12a driven by P lac , suggesting that higher expression level of Cas12a was not toxic to R. capsulatus. As shown in Table 1, 10 randomly selected transformants on ccoO disruption conferred 93% editing efficiency as verified by α‐naphthol + N′ N′‐dimethyl‐p‐phenylenediamine (NADI) staining. The editing efficiency for the disruption of nifH reached up to 87% (Table 1). And it was observed that short indels (1 to 4 bp) occurred at the protospacer regions by further sequencing validation (Fig. 3B and C). In addition, approximately 63% positive transformants were observed during simultaneous disruption of ccoO and nifH as shown in Table 1. To further investigate the ability to simultaneously edit multiple genes, uracil‐phosphoribosyltransferase (upp) gene was employed as the third target. Since upp disruption results in mutant strains resistant to 5‐fuorouracil, upp mutants could be screened under 5‐fuorouracil‐containing medium in a similar way to a previous report (Mougiakos et al., 2019). It was found that approximately 37% positive transformants were obtained when simultaneously editing ccoO, nifH and upp (Table 1, Figure S2). Taken together, these findings suggested that CRISPR/Cas12a could be an effective genome editing tool in PNSB such as R. capsulatus.
Table 1.
Editing efficiency using CRISPR/Cas12a driven by P puc promoter.
| Target gene | Replicate | Editing efficiency | Average editing efficiency |
|---|---|---|---|
| ccoO | 1 | 100% (10/10) | 93% a |
| 2 | 90% (9/10) | ||
| 3 | 90% (9/10) | ||
| nifH | 1 | 90% (9/10) | 87% a |
| 2 | 80% (8/10) | ||
| 3 | 90% (9/10) | ||
| ccoO, nifH | 1 | 70% (7/10) | 63% a |
| 2 | 60% (6/10) | ||
| 3 | 60% (6/10) | ||
| ccoO, nifH, upp | 1 | 40% (4/10) | 37% b |
| 2 | 40% (4/10) | ||
| 3 | 30% (3/10) |
P < 0.005.
P < 0.01 using t test.
Transcriptional repression by CRISPRi using deactivated Cas12a (dCas12a)
Next, we proceeded to develop CRISPR/dCas12a as an artificial transcriptional factor for targeted gene knockdown. The nuclease activity in the RuvC domain of Cas12a was abolished by introducing the E1006A mutation (Tak et al., 2017). The dCas12a driven by the promoter P puc was cloned into a low copy E. coli‐Rhodobacter shuttle vector pRK415 (Ditta et al., 1985), to obtain plasmid pRKucdCas12a (Fig. 4A). The transcriptional repression efficiency of CRISPRi was evaluated by employing the egfp and lacZ as the reporter genes, which were pre‐integrated at the phbC encoding for poly‐β‐hydroxybutyrate (PHB) synthase and pucBA from the pucBACDE operon respectively. For each reporter gene, three crRNAs targeting at the template strand were designed (g1, g2 and g3 or z1, z2 and z3, as shown in Fig. 4B), and one crRNA targeting at the non‐template strand was examined (g4 or z4, Fig. 4B). The above plasmids harbouring dCas12a with different crRNAs were then transformed into R. capsulatus with chromosomal integrated expression cassettes of egfp and lacZ. As shown in Fig. 4C and D, all the crRNAs targeting the template strand could lead to different degrees of repression of the egfp or lacZ expression. Similar to previous findings that crRNA‐binding position near the translation initiation site on the template strand is more effective for CRISPRi (Kim et al., 2017; Liu et al., 2019), we found the most efficient repression occurred when the crRNAs were designed near the start codon (egfp and lacZ repression, 84% and 82% respectively). However, only ˜ 20% repression was achieved when the non‐template strand was targeted for both reporter genes. To test the capability of multiplex gene regulation, dual crRNA‐expressing plasmid was constructed to simultaneously repress egfp and lacZ expressions. The expression levels of egfp and lacZ were reduced by 80% and 77% respectively. These findings indicated that the CRISPRi system based on dCas12a would have broad utilities to implement multiplex transcriptional repression in R. capsulatus for the future metabolic engineering applications.
Fig. 4.
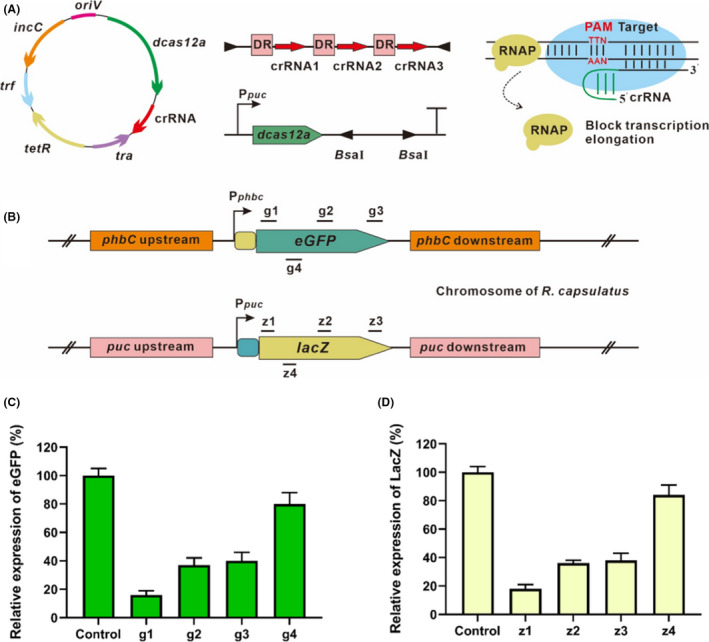
Transcriptional repression of egfp and lacZ by CRISPRi based on dCas12a.
A. dCas12a and crRNA driven by promoter P puc were expressed from a low copy plasmid with tetracycline marker (tetR), shuttle replicon incC, origin of replication oriV, tra combined with trf for transferring plasmid from E. coli to R. capsulatus.
B. crRNAs targeting different positions on the template strand (g1, g2 and g3 or z1, z2 and z3) and the non‐template strand (g4 or z4) of egfp or lacZ gene, which were pre‐integrated into the R. capsulatus chromosome.
C. Comparison of egfp expression level suppressed by dCas12a when targeting at different positions of the template and non‐template strand.
D. Comparison of lacZ expression level suppressed by dCas12a when targeting different positions of the template and non‐template strand. All the above experiments were carried out in triplicate, and the control indicates strains expressing dCas12a without crRNA. The data are presented as mean ± SD.
Discussion
The CRISPR/Cas system is revolutionizing the field of genome editing in various organisms. The type V‐A CRISPR/Cas12a system has the ability to self‐process crRNA, recognize the thymidine‐rich PAM and cleave DNA to give staggered ends. Due to these characteristics, CRISPR/Cas12a‐assisted genome editing is a superior alternative to CRISPR/Cas9 in PNSB. In this work, we developed an efficient genetic tool using Cas12a from F. novicida in R. capsulatus, a promising platform microorganism for PNSB‐related biotechnological applications. Based on previous findings, Cas9 from S. pyogenes (SpCas9) is toxic at high expression in several bacteria such as Corynebacterium glutamicum (Jiang et al., 2017) and Cyanobacteria sp. (Ungerer and Pakrasi, 2016), which limits the application of CRISPR/Cas9 in these microorganisms. In this work, we found that Cas12a expression did not show any noticeable toxicity in R. capsulatus.
As CRISPR/Cas12a creates a staggered end resulting a 5‐nt 5′ overhang rather than a blunt cleavage product, which might promote the double‐strand break repair mediated by non‐homologous end joining (NHEJ) and/or microhomology‐mediated end joining (MMEJ) mechanism (Finger‐Bou et al., 2020). The initial studies revealed that about 30%˜40% editing efficiency was obtained when targeting ccoO or nifH gene, which was not satisfactory when compared to NHEJ‐mediated CRISPR/Cas in Streptomyces coelicolor (Li et al., 2018) and E. coli (Zheng et al., 2017). To further improve the genome editing efficiency in R. capsulatus, a strong promoter of photosynthetic operon puc (P puc ) was identified to drive the CRISPR/Cas12a system, and ˜ 90% editing efficiency was achieved for single gene disruption, whereas 63% editing efficiency was obtained for two gene disruptions. Moreover, the ability of this system to simultaneous multiple genes editing was further investigated, 37% editing efficiency was observed for triple gene disruption. Recently, CRISPR/Cas9 and CRISPR/Cas9‐deaminase have been successfully implemented into R. sphaeroides (Mougiakos et al., 2019; Luo et al., 2020). Considering R. capsulatus has relatively high GC content (˜ 70%), the chance of off‐target is expected to be higher for CRISPR/Cas9 than that of CRISPR/Cas12a.
To the best of our knowledge, CRISPRi using DNase‐deactivated Cas9 (dCas9) or dCas12a has not been reported in any Rhodobacter‐related species. Previous findings suggested that dCas12a exhibits better repression efficiency with crRNAs targeting at the template strand over the non‐template strand, and the closer crRNA near to the translation start site, the higher repression efficiency is obtained (Zhang et al., 2017a, 2017b, 2017c,2017a, 2017b, 2017c,2017a, 2017b, 2017c; Liu et al., 2019; Zhao et al., 2019). In this study, similar results were observed when targeting at egfp or lacZ reporter genes in R. capsulatus using CRISPR/dCas12a. More importantly, dual repression of egfp and lacZ genes reached a similar efficiency to that of single target, indicating that CRISPR/dCas12a might be applicable for multiplex transcription regulation in R. capsulatus.
Experimental procedures
Strains and culture conditions
All plasmids were introduced by heat‐shock into competent cells of Escherichia coli S17‐1 or Top 10 cultured with Luria‐Bertani (LB) medium containing appropriate antibiotics at 37°C. R. capsulatus SB1003 was employed for genetic modifications, and the strain was cultured in Sistrom’s mineral media supplemented with 10 g l−1 glucose (MedA) (Sistrom, 1960) at 35°C under aerobic conditions. Antibiotics were used at the following final concentrations: gentamycin 12 mg ml−1, tetracycline 12.5 mg l−1 and kanamycin 50 mg l−1 for E. coli; gentamycin 12 mg ml−1, tetracycline 2.5 mg l−1 and kanamycin 10 mg l−1 for R. capsulatus. All strains used in this study are listed in Table S1.
Plasmid constructions
All oligonucleotides utilized in this work are listed in Table S2. Plasmids and genomic DNA were extracted by Biospin kits from Bioer Technology (Shanghai, China). PCR amplification was conducted by High Fidelity Phusion DNA polymerase or Taq polymerase from New England Biolab (Ipswich, MA, USA). Plasmids were constructed by standard restriction endonucleases and ligation approach followed by digestion analysis and DNA sequencing verification. The detailed procedures were described in Supplementary Materials and Methods. All the plasmid constructs utilized in this study are listed in Table S3.
Triparental conjugation procedure
Plasmids were transformed into R. capsulatus by triparental conjugation with the helper plasmid of pRK2013 (Ditta et al., 1980). Before the conjugation, R. capsulatus was grown with 10 ml MedA at 35°C and 250 r.p.m. for 48 h under aerobic conditions until optical density at 630 nm (OD630) reached approximately 1.8. The R. capsulatus cells were centrifuged, collected and washed once with fresh MedA and re‐suspended with 400 μl MedA. Meanwhile, E. coli HB101 harbouring the helper plasmid pRK2013 and E. coli S17‐1 carrying the desired plasmid were incubated in 5 ml LB medium supplemented with proper antibiotics at 37°C and 250 r.p.m. for overnight until the respective OD at 600 nm reached approximately 2.8. The E. coli cells were centrifuged, collected and washed once with fresh MedA and re‐suspended with 1 ml MedA respectively. R. capsulatus of 100 μl as a recipient strain, E. coli HB101/pRK2013 of 30 μl as a helper strain and E. coli S17‐1 of 30 μl as a donor strain were mixed and spotted into a MedA agar plate as a ˜ 2 cm diameter spot incubating at 35°C for 24 h. The mixed cells were centrifuged, collected, washed with fresh MedA and re‐suspended with 1 ml MedA. The re‐suspended cell culture of 100 μl was spread on a MedA agar plate with appropriate antibiotic and incubated at 35°C for 2–3 days until colonies appeared.
CRISPR/Cas12a‐mediated genome editing and transcriptional repression in R. capsulatus
For CRISPR/Cas12a‐assisted genome editing, pBdRCas12a or pBRucCas12a derivatives carrying the corresponding crRNAs were transformed into R. capsulatus by triparental conjugation for disrupting the target genes. In order to assess the editing efficiency, single colonies from each plate were randomly selected. Each colony was re‐suspended in 10 μl sterile H2O, which was used as the template for PCR amplification of about 500 bp flanking the region. Then, the PCR products of the tested colonies were sent for sequencing, whereas the parental strain was used as the negative control. Meanwhile, the remaining bacterial mixture was streaked or spread onto the plates for phenotype observation, whereas the parental strain carrying pBdRCas12a or pBRucCas12a was used as the negative control. For curing the CRISPR/Cas12a plasmid, the R. capsulatus mutants were re‐streaked on MedA plates containing 10% sucrose at 35°C for 48 h. Only, these colonies that are kanamycin sensitive were considered to have the CRISPR/Cas12a plasmid removed, and these strains would be used for the subsequent rounds of genetic modifications.
For CRISPR/dCas12a mediated transcriptional repression, egfp and lacZ expression cassettes were integrated at phbC and pucBA sites of the chromosome via the traditional homologous recombination mediated by suicide plasmids, pZJD29c and pRK18mobSacB as previously described (Zhang et al., 2016). Briefly, several recombinants containing pZJ‐ΔphbC::eGFP or pRK18‐ΔpucBA::lacZ were obtained by triparental conjugation, which generated single‐crossover chromosomal integrants. The colonies were then cultivated in MedA medium for overnight so that there was a second homologous recombination event to excise the plasmid DNA. The culture was diluted and spread on the MedA agar plates containing 10% sucrose. Colonies grown on these plates were subjected to diagnostic PCR analysis to confirm the integration of egfp and lacZ cassette into the phbC and pucBA sites. Next, pRKucdCas12a‐derived plasmids carrying the corresponding crRNAs were transformed into R. capsulatus and selected on the MedA agar plate supplemented with tetracycline. The fluorescence intensities and LacZ activities were measured for comparing the transcriptional repression.
In vivo cbb3 ‐cytochrome oxidase activity assay
To test whether the ccoO gene encoding for a subunit of cbb3 ‐cytochrome oxidase (cbb3 ‐Cox) was disrupted by CRISPR/Cas12a system, the in vivo cbb3 ‐Cox activity of R. capsulatus was visualized qualitatively using the NADI reaction (α‐naphthol + N′ N′‐dimethyl‐p‐phenylenediamine → indophenol blue + H2O) by staining the plates with the mixture of 35 mM α‐naphthol and 30 mM N′ N′‐dimethyl‐p‐phenylenediamine dissolved in 1:1 (vol/vol) ethanol and water (Ekici et al., 2012). The randomly selected colonies of R. capsulatus were streaked on MedA plates with kanamycin grown at 35°C for 48 h. When the ccoO gene was successfully disrupted, the colonies lack of cbb3 ‐Cox activity showed no staining (NADI‐) up to 15 min, otherwise, those with cbb3 ‐Cox activity exhibited dark blue staining (NADI+) within 30 s to 1 min.
Nitrogenase activity assay
To test whether the nifH gene encoding for a structural gene of nitrogenase was disrupted by CRISPR/Cas12a system, the nitrogenase activity was visualized qualitatively by the diazotrophic growth with MedA in the absence of nitrogen source (MedA‐N). The randomly selected colonies of R. capsulatus were incubated in MedA with kanamycin for 24 h. Then, 1 ml of cell culture was pelleted by centrifugation, washed once with MedA‐N and re‐suspended in 1 ml MedA‐N. 10 μl of re‐suspended culture was spotted onto MedA‐N agar plates with kanamycin and cultivated for 48 h at 35°C. When the nifH gene was disrupted, the strains would abolish the diazotrophic growth on the MedA‐N plates, while those with intact nifH would grow normally.
5‐Fuorouracil screening for upp disruption
To test whether the upp gene encoding for uracil‐phosphoribosyltransferase was disrupted by CRISPR/Cas12a system, the mutants were visualized qualitatively by the growth with MedA in the presence of 100 mg l−1 5‐fuorouracil (MedAF) using a 50 g l−1 stock solution prepared in dimethyl sulfoxide. The randomly selected colonies of R. capsulatus were incubated in MedA with kanamycin for 24 h. Then, 1 ml of cell culture was pelleted by centrifugation, washed once with MedAF and re‐suspended in 1 ml MedAF. 10 μl of re‐suspended culture was spotted onto MedAF agar plates with kanamycin and cultivated for 48 h at 35°C. When the upp gene was disrupted, the strains grow normally on the MedAF plates, while those with intact upp would not survive.
Fluorescence intensities and β‐galactosidase activity assays
To measure the eGFP fluorescence, R. capsulatus recombinants were cultured in 5 ml MedA medium for 48 h under aerobic conditions. Optical density at 630 nm and fluorescence intensities was monitored by a microplate reader Synergy H1 from BioTek (Winooski, VT, USA). The excitation of eGFP was set at 485 nm and emission at 510 nm. Fluorescence intensities were normalized to culture OD630 for comparing the relative expression level of eGFP. The β‐galactosidase activity assay was performed as previously described (Zhang et al., 2017a, 2017b, 2017c,2017a, 2017b, 2017c,2017a, 2017b, 2017c). In brief, the active protein exacted from R. capsulatus reacted with o‐nitrophenyl‐β‐d‐galactopyranoside (ONPG) at 35°C. When the reaction is over, the mixture was employed to detect β‐galactosidase activity using the following equation:
where GA is the β‐galactosidase activity (μM ONPG‐Hydrolyzed/(min·mg‐protein)), OD420 is the optical density at 420 nm, t is the reaction time (min) and m is the mass of the protein (mg).
Conflict of interests
The authors declare no competing financial interests.
Author contributions
J.Y. conceived the project. Y.Z. performed the experiments and collected the data. J.Y. and Y.Z. interpreted the data and wrote the manuscript.
Supporting information
Table S1. Strains used in this study.
Table S2. Oligonucleotides used in this study.
Table S3. Plasmids used in this study.
Fig. S1. The conjugation efficiency of R. capsulatus using CRISPR/Cas12a system. (a) Colony forming unit (c.f.u) of conjugation mixtures containing Cas12a driven by promoter P lac . The control indicates the conjugation efficiency of R. capsulatus with plasmid pBdRSacB. (b) Colony forming unit obtained after conjugation of empty vector and Cas12a by promoter P puc . The control indicates the conjugation efficiency of R. capsulatus with plasmid pBRPpucSacB. Experiments were carried out in triplicate, and the data are presented as mean ± SD.
Fig. S2. Triple gene deletion in R. capsulatus using CRISPR/Cas12a. (a) NADI staining results of ccoO disruption and sequencing verification. (b) Results of the diazotrophic growth on nifH disruption and sequencing verification. (c) 5‐Fuorouracil screening results of upp disruption and sequencing verification. The parental R. capsulatus strain containing pBRucCas12a was employed as the control. Ten randomly picked colonies were tested for ccoO, nifH, upp disruption.
Acknowledgements
This work was supported by Xiamen University (No. 0660‐X2123310), the Natural Science Foundation of Fujian Province of China (No. 2020J05011), XMU Training Program of Innovation and Entrepreneurship for Undergraduates (No. 2020Y1000 and 202010384194), and ZhenSheng Biotech, China.
Microb. Biotechnol. (2021) 14(6), 2700–2710
Funding information
This work was supported by Xiamen University (No. 0660‐X2123310), the Natural Science Foundation of Fujian Province of China (No. 2020J05011), XMU Training Program of Innovation and Entrepreneurship for Undergraduates (No. 2020Y1000 and 202010384194), and ZhenSheng Biotech, China.
REFERENCES
- Adessi, A. , Corneli, E. , and De Philippis, R. (2017) Photosynthetic purple non sulfur bacteria in hydrogen producing systems: New approaches in the use of well known and innovative substrates. In Modern Topics in the Phototrophic Prokaryotes: Environmental and Applied Aspects. Hallenbeck, P.C. (ed). Cham: Springer International Publishing, pp. 321–350. [Google Scholar]
- Atsumi, S. , Higashide, W. , and Liao, J.C. (2009) Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol 27: 1177–1180. [DOI] [PubMed] [Google Scholar]
- Berry, E.A. , Huang, L.S. , Saechao, L.K. , Pon, N.G. , Valkova‐Valchanova, M. , and Daldal, F. (2004) X‐ray structure of Rhodobacter capsulatus cytochrome bc 1: comparison with its mitochondrial and chloroplast counterparts. Photosynth Res 81: 251–275. [DOI] [PubMed] [Google Scholar]
- Daldal, F. , Cheng, S. , Applebaum, J. , Davidson, E. , and Prince, R.C. (1986) Cytochrome c 2 is not essential for photosynthetic growth of Rhodopseudomonas capsulata . Proc Natl Acad Sci USA 83: 2012–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditta, G. , Schmidhauser, T. , Yakobson, E. , Lu, P. , Liang, X.W. , Finlay, D.R. , et al. (1985) Plasmids related to the broad host range vector, pRK290, useful for gene cloning and for monitoring gene expression. Plasmid 13: 149–153. [DOI] [PubMed] [Google Scholar]
- Ditta, G. , Stanfield, S. , Corbin, D. , and Helinski, D.R. (1980) Broad host range DNA cloning system for gram‐negative bacteria: construction of a gene bank of Rhizobium meliloti . Proc Natl Acad Sci USA 77: 7347–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekici, S. , Yang, H. , Koch, H.G. , and Daldal, F. (2012) Novel transporter required for biogenesis of cbb3‐type cytochrome c oxidase in Rhodobacter capsulatus . MBio 3: e00293‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Yu, Y. , Wang, Y. , and Lin, X. (2007) Biosorption and bioreduction of trivalent aurum by photosynthetic bacteria Rhodobacter capsulatus . Curr Microbiol 55: 402–408. [DOI] [PubMed] [Google Scholar]
- Finger‐Bou, M. , Orsi, E. , van der Oost, J. , and Staals, R.H.J. (2020) CRISPR with a happy ending: Non‐templated DNA repair for prokaryotic genome engineering. Biotechnol J 15: e1900404. [DOI] [PubMed] [Google Scholar]
- Han, Y.C. , and Perner, M. (2016) Sulfide consumption in sulfurimonas denitrificans and heterologous expression of its three sulfide‐quinone reductase homologs. J Bacteriol 198: 1260–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck, A. , and Drepper, T. (2017) Engineering photosynthetic α‐proteobacteria for the production of recombinant proteins and terpenoids. In Modern Topics in the Phototrophic Prokaryotes: Environmental and Applied Aspects. Hallenbeck, P.C. (ed). Cham: Springer International Publishing, pp. 395–425. [Google Scholar]
- Hsu, P.D. , Lander, E.S. , and Zhang, F. (2014) Development and applications of CRISPR‐Cas9 for genome engineering. Cell 157: 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Z.L. , Zhao, Z.P. , Pan, Y. , Tu, Y. , and Chen, G.P. (2010) A powerful hybrid puc operon promoter tightly regulated by both IPTG and low oxygen level. Biochemistry 75: 519–525. [DOI] [PubMed] [Google Scholar]
- Idi, A. , Md Nor, M.H. , Abdul Wahab, M.F. , and Ibrahim, Z. (2014) Photosynthetic bacteria: an eco‐friendly and cheap tool for bioremediation. Rev Environ Sci BioTechnol 14: 271–285. [Google Scholar]
- Jaschke, P.R. , Saer, R.G. , Noll, S. , and Beatty, J.T. (2011) Chapter twenty‐three ‐ Modification of the genome of Rhodobacter sphaeroides and construction of synthetic operons. In Methods in Enzymology. Voigt, C. (ed). Cambridge, MA: Academic Press, pp. 519–538. [DOI] [PubMed] [Google Scholar]
- Jiang, Y. , Qian, F.H. , Yang, J.J. , Liu, Y.M. , Dong, F. , Xu, C.M. , et al. (2017) CRISPR‐Cpf1 assisted genome editing of Corynebacterium glutamicum . Nat Commun 8: 15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappler, U. , and McEwan, A.G. (2002) A system for the heterologous expression of complex redox proteins in Rhodobacter capsulatus: characterisation of recombinant sulphite:cytochrome c oxidoreductase from Starkeya novella . FEBS Lett 529: 208–214. [DOI] [PubMed] [Google Scholar]
- Khan, N.E. , Nybo, S.E. , Chappell, J. , and Curtis, W.R. (2015) Triterpene hydrocarbon production engineered into a metabolically versatile host‐Rhodobacter capsulatus . Biotechnol Bioeng 112: 1523–1532. [DOI] [PubMed] [Google Scholar]
- Kim, S.K. , Kim, H. , Ahn, W.C. , Park, K.H. , Woo, E.J. , Lee, D.H. , and Lee, S.G. (2017) Efficient transcriptional gene repression by Type V‐A CRISPR‐Cpf1 from Eubacterium eligens . ACS Synth Biol 6: 1273–1282. [DOI] [PubMed] [Google Scholar]
- Kovach, M.E. , Elzer, P.H. , Hill, D.S. , Robertson, G.T. , Farris, M.A. , Roop, R.M. 2nd , and Peterson, K.M. (1995) Four new derivatives of the broad‐host‐range cloning vector pBBR1MCS, carrying different antibiotic‐resistance cassettes. Gene 166: 175–176. [DOI] [PubMed] [Google Scholar]
- Kranz, R.G. , Gabbert, K.K. , Locke, T.A. , and Madigan, M.T. (1997) Polyhydroxyalkanoate production in Rhodobacter capsulatus: genes, mutants, expression, and physiology. Appl Environ Microb 63: 3003–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Wei, K. , Zheng, G. , Liu, X. , Chen, S. , Jiang, W. , and Lu, Y. (2018) CRISPR‐Cpf1‐assisted multiplex genome editing and transcriptional repression in Streptomyces. Appl Environ Microb 84: e00827‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link, A.J. , Phillips, D. , and Church, G.M. (1997) Methods for generating precise deletions and insertions in the genome of wild‐type Escherichia coli: application to open reading frame characterization. J Bacteriol 179: 6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Tang, D. , Wang, H. , Lian, J. , Huang, L. , and Xu, Z. (2019) Combined genome editing and transcriptional repression for metabolic pathway engineering in Corynebacterium glutamicum using a catalytically active Cas12a. Appl Environ Microb 103: 8911–8922. [DOI] [PubMed] [Google Scholar]
- Loeschcke, A. , Dienst, D. , Wewer, V. , Hage‐Hulsmann, J. , Dietsch, M. , Kranz‐Finger, S. , et al. (2017) The photosynthetic bacteria Rhodobacter capsulatus and Synechocystis sp. PCC 6803 as new hosts for cyclic plant triterpene biosynthesis. PLoS One 12: e0189816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. , Ge, M. , Wang, B. , Sun, C. , Wang, J. , Dong, Y. , and Xi, J.J. (2020) CRISPR/Cas9‐deaminase enables robust base editing in Rhodobacter sphaeroides 2.4.1. Microb Cell Fact 19: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini, L.A. (2015) CRISPR‐Cas immunity in prokaryotes. Nature 526: 55–61. [DOI] [PubMed] [Google Scholar]
- Merugu, R. , Rudra, M. , Sivadevuni, G. , and Reddy, S. (2012) PHB (Polyhydroxy butyrate) production under nitrogen limitation by Rhodobacter capsulatus KU002 isolated from tannery effluent. Int J Chem Tech Res 4: 1099–1102. [Google Scholar]
- Mougiakos, I. , Orsi, E. , Ghiffary, M.R. , Post, W. , de Maria, A. , Adiego‐Perez, B. , et al. (2019) Efficient Cas9‐based genome editing of Rhodobacter sphaeroides for metabolic engineering. Microb Cell Fact 18: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar‐Oliver, A. , and Gersbach, C.A. (2019) The next generation of CRISPR‐Cas technologies and applications. Nat Rev Mol Cell Biol 20: 490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper, P.W. , Curran, B. , Davies, M.W. , Hirst, K. , Lockheart, A. , Ogden, J.E. , et al. (1988) A heat shock element in the phosphoglycerate kinase gene promoter of yeast. Nucleic Acids Res 16: 1333–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli, S. , and Tabita, F.R. (2009) Carbon dioxide metabolism and its regulation in nonsulfur purple photosynthetic bacteria. In The Purple Phototrophic Bacteria. Hunter, C.N. , Daldal, F. , Thurnauer, M.C. , and Beatty, J.T. (eds). Dordrecht, The Netherlands: Springer, pp. 563–576. [Google Scholar]
- Savitskaya, E.E. , Musharova, O.S. , and Severinov, K.V. (2016) Diversity of CRISPR‐Cas‐mediated mechanisms of adaptive immunity in prokaryotes and their application in biotechnology. Biochemistry 81: 653–661. [DOI] [PubMed] [Google Scholar]
- Sistrom, W.R. (1960) A requirement for sodium in the growth of Rhodopseudomonas spheroides . J Gen Microbiol 22: 778–785. [DOI] [PubMed] [Google Scholar]
- Swarts, D.C. , van der Oost, J. , and Jinek, M. (2017) Structural basis for guide RNA processing and seed‐dependent DNA targeting by CRISPR‐Cas12a. Mol Cell 66: 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak, Y.E. , Kleinstiver, B.P. , Nunez, J.K. , Hsu, J.Y. , Horng, J.E. , Gong, J.Y. , et al. (2017) Inducible and multiplex gene regulation using CRISPR‐Cpf1‐based transcription factors. Nat Methods 14: 1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda, T. , Sano, M. , Honda, M. , Rimoldi, O.J. , Yang, Y. , Yamamoto, M. , et al. (2001) Deletion analysis of the enolase gene (enoA) promoter from the filamentous fungus Aspegillus oryzae . Curr Genet 40: 260–267. [DOI] [PubMed] [Google Scholar]
- Troost, K. , Loeschcke, A. , Hilgers, F. , Ozgur, A.Y. , Weber, T.M. , Santiago‐Schubel, B. , et al. (2019) Engineered Rhodobacter capsulatus as a phototrophic platform organism for the synthesis of plant sesquiterpenoids. Front Microbiol 10: 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerer, J. , and Pakrasi, H.B. (2016) Cpf1 is a versatile tool for CRISPR genome editing across diverse species of Cyanobacteria. Sci Rep 6: 39681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais, P.M. , Dimon, B. , Zorin, N.A. , Tomiyama, M. , and Colbeau, A. (2000) Characterization of the hydrogen‐deuterium exchange activities of the energy‐transducing HupSL hydrogenase and H2‐signaling HupUV hydrogenase in Rhodobacter capsulatus . J Bacteriol 182: 5997–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlund, T.M. , Conway, T. , and Tabita, F.R. (1996) Bioconversion of CO2 to ethanol and other compounds. Abstr Pap Am Chem S 212: 120‐Fuel.
- Wang, T. , Wei, J.J. , Sabatini, D.M. , and Lander, E.S. (2014) Genetic screens in human cells using the CRISPR‐Cas9 system. Science 343: 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, M.Y. , Yan, H.Q. , Ren, G.X. , Zhao, J.P. , Guo, X.P. , and Sun, Y.C. (2017) CRISPR‐Cas12a‐assisted recombineering in bacteria. Appl Environ Microb 83: e00947‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannoni, D. (1995) Aerobic and anaerobic electron transport chains in anoxygenic phototrophic bacteria. In Anoxygenic Photosynthetic Bacteria. Blankenship, R.E. , Madigan, M.T. , and Bauer, C.E. (eds). Dordrecht, The Netherlands: Springer, pp. 949–971. [Google Scholar]
- Zetsche, B. , Gootenberg, J.S. , Abudayyeh, O.O. , Slaymaker, I.M. , Makarova, K.S. , Essletzbichler, P. , et al. (2015) Cpf1 is a single RNA‐guided endonuclease of a class 2 CRISPR‐Cas system. Cell 163: 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche, B. , Heidenreich, M. , Mohanraju, P. , Fedorova, I. , Kneppers, J. , DeGennaro, E.M. , et al. (2017) Multiplex gene editing by CRISPR‐Cpf1 using a single crRNA array. Nat Biotechnol 35: 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.C. , Wang, J.M. , Cheng, Q.X. , Zheng, X. , Zhao, G.P. , and Wang, J. (2017a) Multiplex gene regulation by CRISPR‐ddCpf1. Cell Discov 3: 17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Hu, J. , Ma, H. , Yang, H. , and Guo, L. (2017b) Overexpressing atpXF enhanced photo‐fermentative hydrogen production performance of Rhodobacter sphaeroides . Int J Hydrog Energy 42: 9641–9649. [Google Scholar]
- Zhang, Y. , Li, Q. , Wang, X. , Yang, H. , and Guo, L. (2017c) Enhanced biohydrogen production from cornstalk through a two‐step fermentation: Dark fermentation and photofermentation. Int J Energy Res 41: 2491–2501. [Google Scholar]
- Zhang, Y. , Yang, H. , and Guo, L. (2016) Enhancing photo‐fermentative hydrogen production performance of Rhodobacter capsulatus by disrupting methylmalonate‐semialdehyde dehydrogenase gene. Int J Hydrogen Energy 41: 190–197. [Google Scholar]
- Zhao, R. , Liu, Y. , Zhang, H. , Chai, C. , Wang, J. , Jiang, W. , and Gu, Y. (2019) CRISPR‐Cas12a‐mediated gene deletion and regulation in Clostridium ljungdahlii and its application in carbon flux redirection in synthesis gas fermentation. ACS Syn Biol 8: 2270–2279. [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Li, S.Y. , Zhao, G.P. , and Wang, J. (2017) An efficient system for deletion of large DNA fragments in Escherichia coli via introduction of both Cas9 and the non‐homologous end joining system from Mycobacterium smegmatis . Biochem Biophys Res Commun 485: 768–774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Strains used in this study.
Table S2. Oligonucleotides used in this study.
Table S3. Plasmids used in this study.
Fig. S1. The conjugation efficiency of R. capsulatus using CRISPR/Cas12a system. (a) Colony forming unit (c.f.u) of conjugation mixtures containing Cas12a driven by promoter P lac . The control indicates the conjugation efficiency of R. capsulatus with plasmid pBdRSacB. (b) Colony forming unit obtained after conjugation of empty vector and Cas12a by promoter P puc . The control indicates the conjugation efficiency of R. capsulatus with plasmid pBRPpucSacB. Experiments were carried out in triplicate, and the data are presented as mean ± SD.
Fig. S2. Triple gene deletion in R. capsulatus using CRISPR/Cas12a. (a) NADI staining results of ccoO disruption and sequencing verification. (b) Results of the diazotrophic growth on nifH disruption and sequencing verification. (c) 5‐Fuorouracil screening results of upp disruption and sequencing verification. The parental R. capsulatus strain containing pBRucCas12a was employed as the control. Ten randomly picked colonies were tested for ccoO, nifH, upp disruption.