

Abstract
Background and Objectives
AP-4-associated hereditary spastic paraplegia (AP-4-HSP: SPG47, SPG50, SPG51, SPG52) is an emerging cause of childhood-onset hereditary spastic paraplegia and mimic of cerebral palsy. This study aims to define the spectrum of brain MRI findings in AP-4-HSP and to investigate radioclinical correlations.
Methods
We performed a systematic qualitative and quantitative analysis of 107 brain MRI studies from 76 individuals with genetically confirmed AP-4-HSP and correlation with clinical findings including surrogates of disease severity.
Results
We define AP-4-HSP as a disorder of gray and white matter and demonstrate that abnormal myelination is common and that metrics of reduced white matter volume correlate with severity of motor symptoms. We identify a common diagnostic imaging signature consisting of (1) a thin splenium of the corpus callosum, (2) an absent or thin anterior commissure, (3) characteristic signal abnormalities of the forceps minor (“ears of the grizzly sign”), and (4) periventricular white matter abnormalities. The presence of 2 or more of these findings has a sensitivity of ∼99% for detecting AP-4-HSP; the combination of all 4 is found in ∼45% of cases. Compared to other HSPs with a thin corpus callosum, the absent anterior commissure appears to be specific to AP-4-HSP. Our analysis identified a subset of patients with polymicrogyria, underscoring the role of AP-4 in early brain development. These patients displayed a higher prevalence of seizures and status epilepticus, many at a young age.
Discussion
Our findings define the MRI spectrum of AP-4-HSP, providing opportunities for early diagnosis, identification of individuals at risk for complications, and a window into the role of the AP-4 complex in brain development and neurodegeneration.
The hereditary spastic paraplegias (HSPs) are a group of more than 80 monogenic, neurodegenerative conditions1 and the most common cause of inherited spasticity and associated disability, affecting about 2–8:100,000 individuals worldwide.2 Despite the increasing availability of genetic testing, there is often a diagnostic delay, particularly in pediatric cases.3 MRI pattern recognition can shorten the diagnostic process and can inform genetic testing. Bi-allelic loss of function variants in the genes encoding the subunits of the adaptor protein complex 4 (AP-4) cause childhood-onset HSP, termed SPG47 (AP4B1), SPG50 (AP4M1), SPG51 (AP4E1), and SPG52 (AP4S1).4 While the clinical spectrum of AP-4–associated HSP (AP-4-HSP) is increasingly understood,3 a comprehensive qualitative and quantitative analysis of brain imaging findings has not been performed. From recent work, AP-4-HSP has emerged as a common subtype of complex HSP with a thin corpus callosum and an important genetic mimic of cerebral palsy.3,5-7 Here, we systematically review 107 MRI studies from 76 individuals with genetically confirmed AP-4-HSP. We define AP-4-HSP as a disorder that affects both gray and white matter in the majority of cases and identify a common imaging signature.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Institutional Review Board at Boston Children's Hospital (IRB-P00033016).
Patient Ascertainment and Clinical Characterization
Patients with a genetically confirmed diagnosis of AP-4-HSP were recruited from the AP-4-HSP International Registry (ClinicalTrials.gov Identifier: NCT04712812). Clinical information was collected using the AP-4-HSP Natural History Study Questionnaire3 (n = 76), 4-Stage Functional Mobility Score2 (n = 72), and the Spastic Paraplegia Rating Scale (SPRS)8 (n = 50). Clinical data and a limited set of qualitative imaging findings from 56 individuals were reported previously.3 Clinical data of 20 individuals are reported for the first time.
Brain MRI and Analysis
Brain MRI studies acquired as part of clinical care were collected from participating centers. Studies with insufficient image quality were removed. A total of 107 brain MRI studies from 76 individuals were systematically reviewed by an experienced, board-certified neuroradiologist (E.Y.). Key findings were independently confirmed by a second experienced, board-certified neuroradiologist blinded to the initial assessment (S.P.P.). Where qualitative assessments diverged, cases were reanalyzed and a consensus was reached among the investigators (D.E.-F., S.P.P., and E.Y.). For 21 cases, 2 or more MRI studies were available for review and assessment of longitudinal changes. For quantitative and qualitative assessments of gray and white matter structures, at a minimum, T1-weighted (axial and sagittal), T2-weighted (axial), and fluid-attenuated inversion recovery (FLAIR) (axial) images were reviewed. Diffusion-weighted images, apparent diffusion coefficient, and susceptibility-weighted images (SWI) were also available for the majority of studies. Contrast-enhanced images and magnetic resonance spectroscopy (MRS) were available for a subset of cases. Age-dependent myelination was scored as described by Plecko et al.9 with extension of this scoring system to T2-weighted imaging (eTable 1, links.lww.com/WNL/B549) to evaluate a wider range of myelination maturity. Myelination scores more than 2 points from age-expected values were scored as delayed. In our qualitative analysis of white matter structures, we differentiated between reduced or delayed myelination and “dysmyelination.”
We use the term dysmyelination to refer to focal or diffuse signal abnormalities on a background of otherwise normal brain myelination. Of note, this is an imaging pattern that does not differentiate between irreversible injury and unstable myelination. Frontal occipital horn ratios and measures of atrophy adapted from X-linked adrenoleukodystrophy were calculated.10 For scoring corpus callosum and brainstem biometry, age-matched reference data were used.11-13 Corpus callosum measurements were obtained following methods reported by Garel et al.11 In cases of suspected hypogenesis of the corpus callosum (reduced anterior-posterior dimension), the most posterior parts were treated as the splenium. Cortical thickness was measured in the precentral gyrus using axial T1- or T2-weighted imaging and caudate nucleus thickness was measured in the coronal plane at the level of the lamina terminalis. For assessment of the anterior commissure, this structure was scored as absent if it could not be identified separate from the lamina terminalis in the sagittal and axial plane, and it was scored as thin if it could only be identified in 1 plane. For assessing iron accumulation, we relied on a qualitative assessment of SWI/T2* sequences in comparison to the age-expected appearance. In 5 individuals, MRI scans of the entire spine were available for review as well.
Statistical Analysis
Statistical analysis was performed with IBM SPSS Statistics v27 and GraphPad Prism v9.0. Frequency counts and percentages are provided for categorical variables. Mean, SD, ranges, or the median and interquartile range (IQR) were calculated for continuous variables. Linear regression analysis and Pearson correlation coefficient were employed to determine statistical correlation. The Venn diagram shown in Figure 4 was generated using Venny 2.1 (bioinfogp.cnb.csic.es/tools/venny/index.html).
Figure 4. Radioclinical Correlations in AP-4-HSP.

Several metrics of white matter (WM) volume inversely correlate with Spastic Paraplegia Rating Scale (SPRS) scores as an indicator of motor impairment and associated complications. This includes (A) the thickness of the splenium of the corpus callosum (Pearson correlation coefficient: r2 = 0.14, p = 0.02) and (B) the width of the periatrial WM (Pearson correlation coefficient: r2 = 0.11, p = 0.03). (C) No such correlation is found for the fronto-occipital horn ratio (FOHR) as a surrogate of ventriculomegaly, indicating that this metric lacks sensitivity (Pearson correlation coefficient: r2 = 0.02, p = 0.32). By contrast, indicators of gray matter volume such as (D) the thickness of the precentral gyrus or (E) diameter of the head of the caudate did not correlate with SPRS scores. (F) Venn diagram of key MRI findings in adaptor protein complex 4–associated hereditary spastic paraplegia (AP-4-HSP). Key imaging findings in AP-4-HSP include (1) reduced thickness of the splenium of the corpus callosum, (2) an absent or abnormally thin anterior commissure (AC), (3) abnormal signal of the forceps minor, and (4) periventricular WM (PVWM) signal abnormalities. All 4 features were present in 45% (33/73) of cases. A thin splenium and an absent or thin anterior commissure covered ∼84% (61/73) and more than 1 of the 4 findings was present in the large majority of cases (72/73; 99%).
Data Availability
Data are available from the corresponding authors upon reasonable request.
Results
Demographic Information and Clinical Characterization
This study included 76 patients with AP-4-HSP from 63 families. Demographic and clinical data are presented in Table 1. The majority of patients presented with developmental delay and subsequent intellectual disability, progressive spasticity, and impaired ambulation. About two-thirds of patients developed unprovoked seizures and 63% had seizures in the setting of fever. A history of status epilepticus was found in 44% of patients with seizures. A total of 65% of individuals qualified for a diagnosis of epilepsy, and of these, 31% were classified as medically refractory with continuation of seizures despite treatment with 2 appropriately dosed antiseizure medications. Most individuals ambulated with assistance or using a wheelchair, as indicated by 4-Stage Functional Mobility Scores of III (39%) and IV (53%). The average SPRS score was 31.1 ± 9.9 (SD). SPG47 (AP4B1) and SPG50 (AP4M1) were the most common subtypes, and most cases were caused by homozygous nonsense or frameshift variants with higher rates of splice variants in SPG52 (AP4S1) (Table 1). Major clinical features were distributed evenly between subtypes with some differences likely accounted for by a difference in the average age at last follow-up (Table 1). The median age at which brain MRI was performed was 5.6 years (median; IQR 1.7–11.2 years). A total of 21 individuals had more than 1 study performed, with the cohort covering an age range from 2 weeks to 45 years.
Table 1.
Demographic and Clinical Information

Qualitative and Quantitative Assessment of White Matter and Gray Matter Volume
Whereas AP-4-HSP is thought to be a CNS disorder resulting from AP-4 deficiency in neurons,14-16 the pattern of signal changes and relative involvement of gray matter and white matter remains to be established. To address this question, we performed a qualitative and quantitative analysis of gray and white matter structures (Table 2). Focusing on the corpus callosum first, we analyzed sagittal midline T1 images and found a hypoplastic corpus callosum in 94% of cases, including in some of the youngest patients in our cohort (Figure 1A). This further establishes thinning of the corpus callosum as an early and prevalent finding of reduced supratentorial white matter development in AP-4-HSP. To detail this finding, we used age-matched normative data11 and discovered that 94% of patients with AP-4-HSP had a splenium thickness below the third percentile, whereas the thickness of the genu was similarly thin in only 42% of cases. The anterior-posterior diameter was reduced below the third percentile in 58% of cases, indicating a significant shortening that had not been appreciated previously3,6,17 (Figure 1A). Reviewing the large white matter tracts further, we discovered that the anterior-posterior diameter of the pons was significantly reduced in 45% of patients. Assessing global cerebral volume qualitatively, 53% were classified as having a severe or moderate reduction of volume (11% and 42%, respectively). This occurred predominantly in a pattern that affected posterior areas more than anterior areas (Figure 1, B and C). The Loes global atrophy score10 was 0 in the majority of cases (81%), with scores between 1 and 3 in the remaining cases. Volume reduction disproportionally affected white matter structures, where we found that volume was severely depressed in 14% and moderately depressed in 40% (Figure 1, B and C). By contrast, gray matter volume was moderately depressed in only 23% of cases, with the majority having normal or minimally reduced volume. This was also reflected in the volume of the basal ganglia, which was reduced in a subset of patients but normal in the majority (Table 2). Cerebellar atrophy was rare, occurring in only 2 patients (3%) (Figure 1D).
Table 2.
Quantitative and Qualitative Assessment of White Matter and Gray Matter Volume

Figure 1. AP-4-HSP Is a Disorder of Gray and White Matter.

(A) T1-weighted sagittal image at the level of the midline (patient with AP4B1-related hereditary spastic paraplegia [HSP]: AP4B1 [NM_001253852.3]: c.1345A>T [p.Arg449Ter]/c.1160_1161delCA [p.Thr387ArgfsTer30]; age: 11 months). Underdevelopment of the corpus callosum is one of the defining features of adaptor protein complex 4–associated HSP (AP-4-HSP), present in 94% of patients. This includes prominent thinning of the splenium and a decreased anterior-posterior diameter. (B) T2-weighted axial image demonstrating severe reduction in white matter volume, particularly in the posterior, periventricular white matter that, in combination with the thinning of the posterior corpus callosum, gives rise to ex vacuo ventriculomegaly in a colpocephalic configuration (patient with AP4B1-related HSP: AP4B1 [NM_001253852.3]: c.664delC [p.Leu222CysfsTer31]/c.664delC [p.Leu222CysfsTer31]; age: 6 years). (C) T2-weighted axial image demonstrating mild reduction in white matter volume as is the case in the majority of patients (patient with AP4B1-related HSP: AP4B1 [NM_001253852.3]: c.530_531insA [p.Asn178GlufsTer20]/c.114-2A>C; age: 4 years). (D) T1-weighted sagittal image showing cerebellar atrophy, which is overall a rare finding, present in only 3% of cases and mainly in patients with advanced disease (patient with AP4B1-associated HSP: AP4B1 [NM_001253852.3]: c.1177C>T [p.Arg393Ter]/c.1177C>T [p.Arg393Ter]; age: 19 years).
A quantitative analysis of the intracranial CSF spaces revealed ex vacuo ventriculomegaly with an increase in the size of the lateral and third ventricles and a mean frontal-occipital horn ratio (FOHR) of 0.42 ± 0.04. The most common ventricular configuration was asymmetric colpocephaly with prominent enlargement of the occipital horns likely due to thinning of the posterior parts of the corpus callosum and reduced white matter volume in this region (Figure 1, B and C).
Corresponding with rates of low gray matter volume, the subarachnoid spaces were found to be abnormally enlarged in 19% of cases.
Patterns of Myelination
Clinically, AP-4-HSP is thought to be both a neurodevelopmental and a neurodegenerative disorder, with early-onset developmental delay, hypotonia, microcephaly, and early-onset epilepsy being a result of altered early CNS development. White matter signal abnormalities are commonly noted,3 but remain ill-defined. One important question remains whether these white matter signal changes are the result of delayed development of myelin with subsequent incomplete myelination or represent a progressive loss of myelin. To approach this, we first assessed age-dependent myelination. We found that 34% of cases showed evidence of delayed myelination upon qualitative assessment (Table 2). Employing an adapted version of myelination staging system established by Plecko et al.,9 we assessed 18 areas and found that 19% (based on T1 sequence) and 41% (based on T2 sequence) had a deviation of ≥2 from the age-expected score, indicating reduced myelination (Table 2). Taking advantage of serial MRI in a subset of patients, we evaluated for longitudinal changes. In 11/17 (65%) cases, the pattern of reduced myelination remained unchanged, while 5 cases showed evidence of ongoing myelination with improved myelination (Figure 2, A and B), and only 1 individual displayed progressive loss of myelin. It is important to point out that the median age at the first MRI scan obtained for the group with no longitudinal change in myelination was significantly higher (2.3 years [median], IQR 1.0–3.8 years) than in that with catch-up myelination (0.8 years [median], IQR 0.3–1.5, Mann-Whitney test: p = 0.04). This underscores that myelination progresses but plateaus at a low level in the majority of cases. Looking at the pattern of dysmyelination, which was overall present in 80%, we found that this was most common in the periventricular white matter (55%; Figure 2, A and B) and extended beyond that region in only 25% (Table 2).
Figure 2. AP-4-HSP Presents With Abnormal Periventricular White Matter.

(A, B) T2-weighted axial image showing delayed myelination at age 1.5 years (A) with evidence of continued myelination at age 9 years (B) (patient with AP4E1-related hereditary spastic paraplegia [HSP]: AP4E1 [NM_007347.5]: c.652_653delGA [p.Asp218CysfsTer13]/c.652_653delGA [p.Asp218CysfsTer13]; age: 9 years). (C, D) T1-weighted axial image (C) and T2-weighted axial image (D) showing hypointense signal on T1 and hyperintense signal on T2 images in the forceps minor consistent with the ears of the lynx sign, found in 11% of cases (patient with AP4S1-associated SPG52: AP4S1 [NM_007077.4]: c.289C>T [p.Arg97Ter]/c.289C>T [p.Arg97Ter]; age: 12 months). (E, F) T1-weighted axial image (E) and T2-weighted axial image (F) showing short and round T1 hypointense and T2 hyperintense signal in the forceps minor. Based on this appearance and in reference to the ears of the lynx sign, we term this the ears of the grizzly sign, which is present in 48% of cases (patient with AP4B1-related HSP: AP4B1 [NM_001253852.3]: c.664delC [p.Leu222CysfsTer31]/c.664delC [p.Leu222CysfsTer31]; age: 6 years). (G–J) T1-weighted sagittal (G, H) image demonstrating an absent anterior commissure in adaptor protein complex 4–associated HSP (AP-4-HSP), contrasting findings in other forms of complex HSP with a thin corpus callosum such as SPG15. (I, J) The anterior commissure is confirmed to be absent in the axial plane on T2-weighted axial images. Overall a thin or absent anterior commissure is found in 96% of AP-4-HSP cases (G and I: patient with AP4S1-related HSP: AP4S1 [NM_007077.4]: c.138+3_138+6delAAGT/c.138+3_138+6delAAGT; age: 12 years; H and J: patient with SPG15: ZFYVE26 [NM_015346.4]: c.4312C>T [p.Arg1438Ter]/c.837T>G [p.Tyr279Ter]; age: 19 years).
Focal signal changes of the forceps minor of the corpus callosum have been described in SPG11 and SPG15, 2 forms of HSP with thin corpus callosum18 (eTable 2, links.lww.com/WNL/B550). These have been termed the “ears of the lynx” sign, which consists of hypointense signal on T1-weighted and hyperintense signal on FLAIR images, which, on axial views, resembles the shape of the ears of a lynx with their characteristic apical hair tuft.19 A detailed review of the frontal periventricular white matter in our cohort revealed signal abnormalities in the majority of cases, which included a classic appearance of the ears of the lynx sign (in 11%; Figure 2, C and D) but also a distinct and novel pattern consisting of a more voluminous yet short and round T1-hypointense and T2/FLAIR-hyperintense signal change, which we term the “ears of the grizzly sign,” in 35/73 patients (48%, Figure 2, E and F; eFigure 1, links.lww.com/WNL/B547).
Encouraged by the near-universal finding of a thin corpus callosum, we explored other midline structures embryologically linked to corpus callosum development.20 We discovered that the anterior commissure was absent in nearly 82% of studies (Figure 2, G and I) and was abnormally thin in another 14%. To explore the specificity of this finding for AP-4-HSP, we reviewed cases of genetically confirmed SPG15 (n = 4) and SPG49 (n = 2) for the presence of a similarly abnormal anterior commissure. We did not detect an absent anterior commissure in any of these cases (Figure 2, H and J; eTable 3, links.lww.com/WNL/B551) or published reports of SPG11, SPG15, and SPG49.18,21-26 Similarly, we discovered no such deficiency of the anterior commissure structures in MRI scans of 16 infants with a history of prematurity. Therefore, we conclude that an absent or thin anterior commissure is a finding more specific to AP-4-HSP than other HSP.
Developmental Brain Malformations and Evidence of Brain Iron Deposition
There is emerging evidence that loss of AP-4 function impairs neurite outgrowth and axon guidance.14-16,27,28 To expand on previous reports of polymicrogyria in AP-4-HSP,3,29 we systematically reviewed all MRI studies for developmental brain malformations. In our cohort, 19/73 patients (26%) were found to have polymicrogyria, classified as definite polymicrogyria (11/19) or possible polymicrogyria or dysgyria (8/19). This mainly consisted of bilateral perisylvian polymicrogyria (Figure 3, A and B). Other developmental brain malformations consisted of periventricular nodular heterotopia seen in 1 case (Figure 3, C and D).
Figure 3. Malformations of Cortical Development in AP-4-HSP.
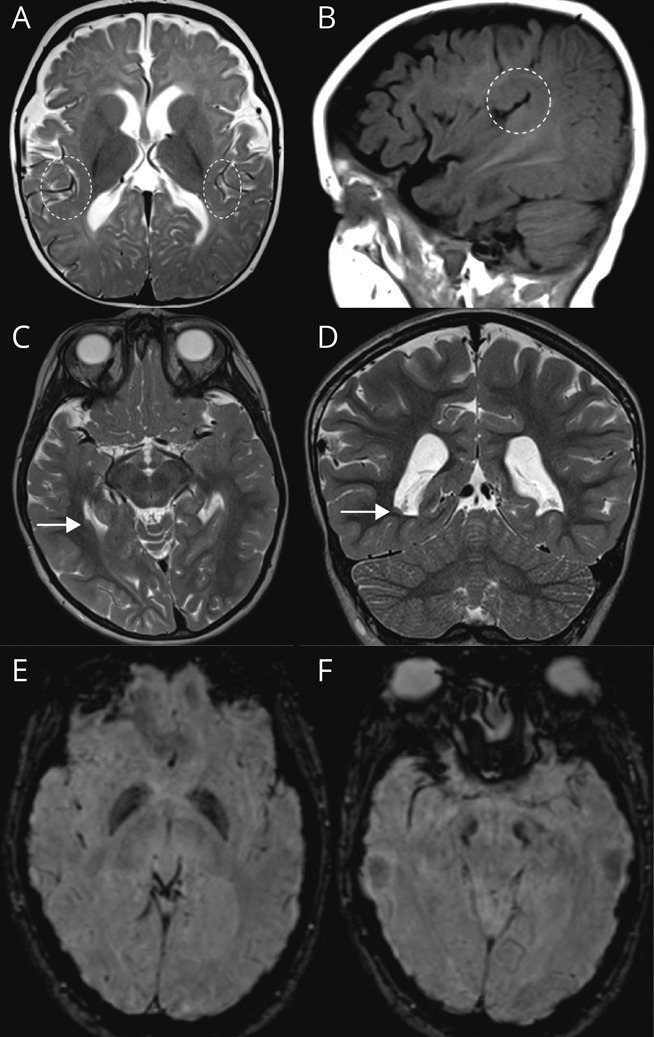
(A, B) T2-weighted axial (A) and T1-weighted sagittal (B) images demonstrating perisylvian polymicrogyria, found in up to 25% of cases (shown is a patient with AP4S1-related hereditary spastic paraplegia [HSP]: AP4S1 [NM_007077.4]: c.294+1G>T/c.294+1G>T). (C, D) Periventricular nodular heterotopia in 1 patient with adaptor protein complex 4–associated HSP (AP-4-HSP) (patient with AP4B1-related HSP: AP4B1 [NM_001253852.3]: c.530_531insA [p.Asn178GlufsTer20]/c.114-2A>C; age: 4 years). (E, F) Susceptibility-weighted axial images showing evidence of iron accumulation in the globus pallidus (E) and substantia nigra (F) in a patient with AP4M1-related HSP, previously reported by Roubertie et al.30 in 2018 (patient with AP4M1-related HSP: AP4M1 [NM_004722.4]: c.916C>T [p.Arg306Ter]/c.916C>T [p.Arg306Ter]).
Prior reports of iron deposition in the basal ganglia30 prompted us to systematically review SWI/T2* sequences, yielding 6 cases with findings suggestive of iron deposition in the globus pallidus and substantia nigra (Figure 3, E and F). These cases include 2 patients with SPG50 and a single case of SPG52 reported previously,30,31 as well as 3 individuals with SPG50 identified here for the first time.
Other Structures and Spinal Imaging
A systematic review of other structures including the olfactory and optic apparatus, pituitary gland, and a review of all available spinal MRI studies showed no significant structural or signal abnormalities. In 3 cases, a fatty filum terminale was noted. Available MRS studies (n = 17) showed a normal metabolic profile.
Radioclinical Correlations
To explore correlations between clinical manifestations and brain MRI findings, we employed SPRS scores as a marker of motor impairment and spasticity. We found that indicators of white matter volume, including the thicknesses of the splenium and the periventricular white matter (average thinnest periatrial diameter), had a moderate inverse correlation with SPRS scores, with a reduced thickness being associated with higher degrees of motor impairment (Figure 4, A and B). We found no correlation of the FOHR, as a surrogate for the degree of ex vacuo ventriculomegaly, with SPRS scores (Figure 4C). Turning to gray matter structures, we found no correlation of average precentral gyrus thickness (measured on T1), as an indicator of cortical atrophy, or of the thickness of the head of the caudate, as a marker for deep gray matter structures, with motor symptom severity (Figure 4, D and E). In aggregate, these patterns establish that indicators of white matter volume may correlate inversely with the degree of motor impairment. This association is possibly underestimated as SPRS scores tend to increase in an age-dependent manner,3 as is the case for white matter volume.
Investigating the subset of patients with polymicrogyria for distinct clinical manifestations, we found that these individuals had high rates of febrile seizures (75%) and unprovoked seizures (78%) compared to patients without polymicrogyria (Table 3). A history of status epilepticus was common (53%), whereas rates of medically refractory epilepsy were not (Table 3). Taken together with the overall younger age of the patients with polymicrogyria, these data indicate higher rates of provoked and unprovoked seizures as well as status epilepticus, highlighting that these clinical manifestations should lead to a careful review for malformations of cortical development.
Table 3.
Clinical Characteristics of Individuals With and Without Polymicrogyria and Iron Deposition

To determine the clinical characteristics specific to the small subgroup of patients with AP-4-HSP with evidence of iron accumulation on SWI/T2* sequences, we compared their clinical profiles to patients without iron accumulation or developmental brain malformations (Table 3). The 6 patients with brain iron accumulation were older, with an average age at last brain MRI of 28.6 ± 13.2 (SD) years. Five individuals from 2 families had variants in the AP4M1 (SPG50) gene and all had advanced disease with spastic tetraplegia, wheelchair dependency, and high average SPRS scores (Table 3). Seizures were relatively common although none had medically refractory epilepsy. Based on these data, we conclude that brain iron accumulation may be a rare manifestation in patients with advanced disease, particularly in SPG50.
Synopsis and Sensitivity
We found that a combination of the 4 findings of (1) reduced thickness of the splenium of the corpus callosum, (2) an absent or abnormally thin anterior commissure, (3) abnormal signal of the forceps minor, and (4) periventricular white matter signal abnormalities was present in ∼45% (33/73) of cases (Figure 4F). A thin splenium and an absent or thin anterior commissure covered ∼84% (61/73), and more than 1 of the 4 findings was present in the vast majority of cases (72/73%, 99%). A flowchart summarizing the approach to these key imaging findings is shown in eFigure 2, links.lww.com/WNL/B548.
Discussion
AP-4-HSP has emerged as a form of complex HSP with a thin corpus callosum. While the finding of a thin corpus callosum can be helpful for distinguishing HSP from cerebral palsy, the differential diagnosis for genetic disorders that present with corpus callosum hypoplasia or dysgenesis is broad.20,32 Recent work has begun to delineate the clinical spectrum associated with AP-4-HSP, establishing clinical features of both a neurodevelopmental and neurodegenerative disorder.3,4,6,17,33-35 Understanding the MRI spectrum of AP-4-HSP provides an opportunity for early diagnosis, identification of individuals at risk for certain clinical manifestations (i.e., status epilepticus), and a window into the role of the AP-4 complex in brain development and neurodegeneration.
In this first systematic analysis of the brain MRI characteristics of the largest cohort of patients with AP-4-HSP assembled to date, we establish a common brain MRI signature and demonstrate both gray and white matter involvement. Analyzing >100 MRI scans from more than 75 patients, we conclude that the combination of a thin splenium of the corpus callosum and an absent or thin anterior commissure is present in the majority of cases (∼84%). The presence of 1 of these 2 findings or an abnormal signal of the forceps minor or periventricular white matter increases the sensitivity to ∼99%. While the specificity of these findings, particularly in comparison with other forms of HSP with a thin corpus callosum, remains to be established, our analysis in a small number of patients with SPG15 and SPG49 and a review of the literature18,21,23,24,36 indicates that a thinning or absence of the anterior commissure is more specific to AP-4-HSP. This and other differences among the major forms of complex HSP with a thin corpus callosum might aid clinicians in their differential diagnosis (eTable 2, links.lww.com/WNL/B550).
Focal signal changes of the forceps minor have been a defining feature of SPG11 and SPG15.18,19 A detailed review of our cohort of patients with AP-4-HSP revealed signal abnormalities in the region of the forceps minor in about 60%. In a subset of cases (∼11%), these were consistent with a classic ears of the lynx appearance. In the majority, however, the signal changes consisted of a more voluminous yet short and round T1 hypointense and T2/FLAIR hyperintense signal. Based on this characteristic appearance and in reference to the ears of the lynx sign, we term this the ears of the grizzly sign (eFigure 1, links.lww.com/WNL/B547). This finding is present early and thus may be a useful radiologic feature to suggest a genetic etiology in cases of undifferentiated developmental delay and spasticity. The specificity of this finding remains to be established in future studies.
From a neurobiological perspective, it is interesting to speculate what the most commonly affected structures in AP-4-HSP, namely the corpus callosum, anterior commissure, forceps minor, and periventricular white matter, may have in common and how they may be linked in neurodevelopment. Perhaps the first step in the development of all these midline structures is patterning of the commissural plate, where all telencephalic commissures initially cross.37 Perturbed development of subdomains of the commissural plate results in disruption of the corresponding projections passing through. It is thus conceivable that disruption in midline patterning signaling pathways or impaired axon guidance and targeting might account for the abnormalities seen in multiple midline structures. Supportive of the notion of a midline crossing defect early in development is the presence of similar findings in other conditions caused by loss-of-function variants that affect functions closely related to vesicle formation and trafficking, such as the actin (i.e., CTNNA238) and tubulin cytoskeleton (i.e., TUBB339,40 or MACF141) or transcription factors that control neuronal differentiation and migration (i.e., TBR142).
Of note, recent work identified the 2-arachidonoylglycerol producing enzyme diacylglycerol lipase-beta (DAGLB) as a cargo of AP-4,27 potentially linking loss of AP-4 function with abnormal endocannabinoid signaling and therefore to impaired axon growth, axon guidance, and myelination.43 ATG9A, a second cargo of AP-4,14,44,45 provides another compelling molecular link to impaired development of structures that arise from the commissural plate. Whereas ATG9A's role in autophagy possibly accounts for some of the neurodegenerative features and the progressive axonopathy seen in AP-4-HSP, it is important to note that ATG9A has roles outside of the autophagy pathway and that CNS-specific Atg9a knockout mice display a thin corpus callosum and an absent anterior commissure,46 with striking similarity to what is described here in AP-4-HSP. Supportive of a role of autophagy in corpus callosum development, thinning of the corpus callosum is also a notable feature of several of the congenital disorders of autophagy47,48 and thinning of the anterior commissure is a described feature of neurodegenerative disease with frontal and temporal atrophy, such as frontotemporal dementia,49 a condition again linked to autophagy.50
As another indicator of impaired early CNS development in AP-4-HSP, we found a subset of patients with polymicrogyria. The prevalence of polymicrogyria in our cohort was around 25%, higher than what we expected to find based on prior studies3,29 and indicating that polymicrogyria is an often subtle finding that requires a careful review. While the presence of polymicrogyria bears clinical significance in that this subset of patients was prone to epilepsy and anecdotally often presented to clinical attention after a prolonged first seizure, this pathology may also substantiate the role that AP-4 and its putative cargoes play in neuronal migration and cortical folding.
Our qualitative and quantitative analysis of gray and white matter involvement establishes AP-4-HSP as a disorder that affects both, though the loss of white matter is more prominent. This is important as it helps define the brain areas that would have to be targeted by therapies, that is, gene replacement approaches would likely have to target cortical areas and the corticospinal tracts, whereas other areas such as the deep gray nuclei or the cerebellum seem less involved at least at the level detected by clinical MRI. Low white matter volume occurred predominantly in the posterior periventricular region, giving rise to ex vacuo ventriculomegaly with a typical colpocephalic configuration. Assessing the long white matter tracts further, we found a significant reduction in pontine diameter, implicating transverse pontine and corticospinal tract involvement. Interestingly, cerebellar atrophy was rare in our cohort, occurring in 2 patients with advanced disease only. The low prevalence of cerebellar atrophy may be a feature that distinguishes AP-4-HSP from other corpus callosum development syndromes20 and from other forms of complex HSP.
Assessing myelination in detail, we discovered that about a third of patients had evidence of delayed myelination, raising the question of whether loss of AP-4 function in neurons or glia may be the driving force behind this finding. To our surprise, a longitudinal assessment of myelination in 21 cases with serial imaging found that the reduction in myelin remained static in most but not all cases. Only 1 case demonstrated clear loss of myelin over time. These findings classify the white matter signal changes in AP-4-HSP as a disorder of myelination rather than a progressive demyelinating disorder, with myelination remaining reduced in ∼80% of patients.
Our systematic review of brain MRI features, in addition to key clinical characteristics and surrogates of disease severity, allowed us to explore radioclinical correlations. White matter volume, that is, thickness of the splenium and the periventricular white matter, had an inverse correlation with SPRS scores, indicating that patients with reduced white matter volume show a greater degree of corticospinal tract dysfunction and associated complications. The size of the lateral ventricles, quantified as the FOHR, did not show such correlation and thus may lack sensitivity as it may represent an indirect sign of white matter loss. Similarly, thickness of the precentral gyrus thickness, as a surrogate of cortical atrophy, or global cerebral, deep gray matter, or cerebellar gray matter volumes did not correlate with SPRS scores. This may be due in part to the fact that the SPRS largely measures symptoms that arise from corticospinal tract dysfunction and the limited sensitivity of unidimensional measures of cortical volume (i.e., thickness). Automated volumetry may provide a means of investigating the relationship of gray and white matter volume to clinical severity with greater sensitivity in future studies.
We provide a detailed, systematic analysis of the brain MRI findings in AP-4-HSP. We define a set of core features, shared by the majority of patients, which include (1) reduced thickness of the splenium of the corpus callosum, (2) an absent or abnormally thin anterior commissure, (3) abnormal signal of the forceps minor, and (4) periventricular white matter signal abnormalities. The high sensitivity of these findings and the possible specificity of some (i.e., the absent anterior commissure and the ears of the grizzly sign) in conjunction with clinical findings may facilitate an early diagnosis. The high prevalence of developmental abnormalities of the midline structures and the presence of polymicrogyria in some support a role of AP-4 in early CNS development and the classification of AP-4-HSP as both a neurodevelopmental and neurodegenerative disorder.
Acknowledgment
The authors thank the patients and their families for participating in this study and Robert Smigiel (Wroclaw Medical University) and Rosa Pasquariello (IRCCS Fondazione Stella Maris) for help with patient enrollment.
Glossary
- AP-4
adaptor protein complex 4
- AP-4-HSP
adaptor protein complex 4–associated hereditary spastic paraplegia
- FLAIR
fluid-attenuated inversion recovery
- FOHR
frontal-occipital horn ratio
- HSP
hereditary spastic paraplegia
- IQR
interquartile range
- MRS
magnetic resonance spectroscopy
- SPRS
Spastic Paraplegia Rating Scale
- SWI
susceptibility-weighted image
Appendix 1. Authors

Appendix 2. Coinvestigators
Study Funding
D.E.F. received support from the CureAP4 Foundation, the CureSPG50 Foundation, the Spastic Paraplegia Foundation, the Manton Center for Orphan Disease Research, and the NIH/National Institute of Neurologic Disorders and Stroke (2R25NS070682 and 1K08NS123552-01). J.E.A. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; 270949263/GRK2162), the German National Academic Foundation, and the Max Weber Program of the State of Bavaria. The Rosamund Stone Zander Translational Neuroscience Center at Boston Children's Hospital is supported by the Massachusetts Life Sciences Center, J.P. Fletcher Foundation, and the BCH Intellectual and Developmental Disabilities Research Center Clinical/Translational Core is supported by the NIH (BCH IDDRC, 1U54HD090255).
Disclosure
D. Ebrahimi-Fakhari and M. Sahin received research funding through a joint research agreement between Boston Children's Hospital and Astellas Pharmaceuticals Inc./Mitobridge Inc. D. Ebrahimi-Fakhari received an honorarium for speaking at the Movement Disorder International Congress in 2019, royalties from Cambridge University Press as Editor of Movement Disorders and Inherited Metabolic Disorders, and a consulting fee from Alcimed Inc. J.E. Alecu, M. Ziegler, G. Geisel, C. Jordan, A. D'Amore, R.C. Yeh, S.K. Akula, A. Saffari, and S.P. Prabhu report no disclosures relevant to the manuscript. M. Sahin reports grant support from Novartis, Roche, Pfizer, Biogen, Ipsen, LAM Therapeutics, Bridgebio, and Quadrant Biosciences and has served on Scientific Advisory Boards for Sage, Roche, Novartis, Celgene, Aeovian, Alkermes, Regenxbio, and Takeda. E. Yang reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136-1146. [DOI] [PubMed] [Google Scholar]
- 2.Erichsen AK, Koht J, Stray-Pedersen A, Abdelnoor M, Tallaksen CM. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain. 2009;132(Pt 6):1577-1588. [DOI] [PubMed] [Google Scholar]
- 3.Ebrahimi-Fakhari D, Teinert J, Behne R, et al. Defining the clinical, molecular and imaging spectrum of adaptor protein complex 4-associated hereditary spastic paraplegia. Brain. 2020;143(10):2929-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ebrahimi-Fakhari D, Behne R, Davies AK, Hirst J. AP-4-associated hereditary spastic paraplegia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. University of Washington; 2018. [PubMed] [Google Scholar]
- 5.Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52(10):1046-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ebrahimi-Fakhari D, Cheng C, Dies K, et al. Clinical and genetic characterization of AP4B1-associated SPG47. Am J Med Genet A. 2018;176(2):311-318. [DOI] [PubMed] [Google Scholar]
- 7.Ebrahimi-Fakhari D, Saffari A, Pearl PL. Childhood-onset hereditary spastic paraplegia and its treatable mimics. Mol Genet Metab. Epub 2021 Jun 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schüle R, Holland-Letz T, Klimpe S, et al. The Spastic Paraplegia Rating Scale (SPRS): a reliable and valid measure of disease severity. Neurology. 2006;67(3):430-434. [DOI] [PubMed] [Google Scholar]
- 9.Plecko B, Stöckler-Ipsiroglu S, Gruber S, et al. Degree of hypomyelination and magnetic resonance spectroscopy findings in patients with Pelizaeus Merzbacher phenotype. Neuropediatrics. 2003;34(3):127-136. [DOI] [PubMed] [Google Scholar]
- 10.Loes DJ, Stillman AE, Hite S, et al. Childhood cerebral form of adrenoleukodystrophy: short-term effect of bone marrow transplantation on brain MR observations. AJNR Am J Neuroradiol. 1994;15(9):1767-1771. [PMC free article] [PubMed] [Google Scholar]
- 11.Garel C, Cont I, Alberti C, Josserand E, Moutard ML, Ducou le Pointe H. Biometry of the corpus callosum in children: MR imaging reference data. AJNR Am J Neuroradiol. 2011;32(8):1436-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jandeaux C, Kuchcinski G, Ternynck C, et al. Biometry of the cerebellar vermis and brain stem in children: MR imaging reference data from measurements in 718 children. AJNR Am J Neuroradiol. 2019;40(11):1835-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology. 2005;64(12):2050-2055. [DOI] [PubMed] [Google Scholar]
- 14.Behne R, Teinert J, Wimmer M, et al. Adaptor protein complex 4 deficiency: a paradigm of childhood-onset hereditary spastic paraplegia caused by defective protein trafficking. Hum Mol Genet. 2020;29(2):320-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Pace R, Skirzewski M, Damme M, et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLOS Genet. 2018;14(4):e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivankovic D, Drew J, Lesept F, et al. Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy. 2019;16(3):391-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tüysüz B, Bilguvar K, Koçer N, et al. Autosomal recessive spastic tetraplegia caused by AP4M1 and AP4B1 gene mutation: expansion of the facial and neuroimaging features. Am J Med Genet A. 2014;164A(7):1677-1685. [DOI] [PubMed] [Google Scholar]
- 18.Pensato V, Castellotti B, Gellera C, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain. 2014;137(Pt 7):1907-1920. [DOI] [PubMed] [Google Scholar]
- 19.Pascual B, de Bot ST, Daniels MR, et al. “Ears of the lynx” MRI sign is associated with SPG11 and SPG15 hereditary spastic paraplegia. AJNR Am J Neuroradiol. 2019;40(1):199-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain. 2014;137(Pt 6):1579-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faber I, Martinez ARM, de Rezende TJR, et al. SPG11 mutations cause widespread white matter and basal ganglia abnormalities, but restricted cortical damage. Neuroimage Clin. 2018;19:848-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kara E, Tucci A, Manzoni C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016;139(Pt 7):1904-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuser S, Brechmann B, Heimer G, et al. Clinical, neuroimaging and molecular spectrum of TECPR2-associated hereditary sensory and autonomic neuropathy with intellectual disability. Hum Mutat. 2021;42(6):762-776. [DOI] [PubMed] [Google Scholar]
- 24.Franca MC, Jr., Yasuda CL, Pereira FR, et al. White and grey matter abnormalities in patients with SPG11 mutations. J Neurol Neurosurg Psychiatry. 2012;83(8):828-833. [DOI] [PubMed] [Google Scholar]
- 25.Chrestian N, Dupré N, Gan-Or Z, et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol Genet. 2017;3(1):e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goizet C, Boukhris A, Maltete D, et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology. 2009;73(14):1111-1119. [DOI] [PubMed] [Google Scholar]
- 27.Davies AK, Ziegler M, Jumo H, Saber WA, Ebrahimi-Fakhari D, Borner GHH. AP-4 mediates vesicular transport of the 2-AG endocannabinoid producing enzyme DAGLB. bioRxiv. Preprint posted online October 25, 2020. doi: 10.1101/2020.10.25.353995. [DOI]
- 28.D'Amore A, Tessa A, Naef V, et al. Loss of ap4s1 in zebrafish leads to neurodevelopmental defects resembling spastic paraplegia 52. Ann Clin Transl Neurol. 2020;7(4):584-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmona S, Marecos C, Amorim M, et al. AP4S1 splice-site mutation in a case of spastic paraplegia type 52 with polymicrogyria. Neurol Genet. 2018;4(5):e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roubertie A, Hieu N, Roux CJ, et al. AP4 deficiency: a novel form of neurodegeneration with brain iron accumulation? Neurol Genet. 2018;4(1):e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vill K, Müller-Felber W, Alhaddad B, et al. A homozygous splice variant in AP4S1 mimicking neurodegeneration with brain iron accumulation. Mov Disord. 2017;32(5):797-799. [DOI] [PubMed] [Google Scholar]
- 32.Al-Hashim AH, Blaser S, Raybaud C, MacGregor D. Corpus callosum abnormalities: neuroradiological and clinical correlations. Dev Med Child Neurol. 2016;58(5):475-484. [DOI] [PubMed] [Google Scholar]
- 33.Abou Jamra R, Philippe O, Raas-Rothschild A, et al. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 2011;88(6):788-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verkerk AJ, Schot R, Dumee B, et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 2009;85(1):40-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreno-De-Luca A, Helmers SL, Mao H, et al. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet. 2011;48(2):141-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebrahimi-Fakhari D, Alecu JE, Blackstone C. Spastic paraplegia 15. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. University of Washington; 2021. [PubMed] [Google Scholar]
- 37.Raybaud C. The corpus callosum, the other great forebrain commissures, and the septum pellucidum: anatomy, development, and malformation. Neuroradiology. 2010;52(6):447-477. [DOI] [PubMed] [Google Scholar]
- 38.Schaffer AE, Breuss MW, Caglayan AO, et al. Biallelic loss of human CTNNA2, encoding alphaN-catenin, leads to ARP2/3 complex overactivity and disordered cortical neuronal migration. Nat Genet. 2018;50(8):1093-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chew S, Balasubramanian R, Chan WM, et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal beta-tubulin isotype 3. Brain. 2013;136(Pt 2):522-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitman MC, Andrews C, Chan WM, et al. Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am J Med Genet A. 2016;170A(2):297-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobyns WB, Aldinger KA, Ishak GE, et al. MACF1 mutations encoding highly conserved zinc-binding residues of the GAR domain cause defects in neuronal migration and axon guidance. Am J Hum Genet. 2018;103(6):1009-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nambot S, Faivre L, Mirzaa G, et al. De novo TBR1 variants cause a neurocognitive phenotype with ID and autistic traits: report of 25 new individuals and review of the literature. Eur J Hum Genet. 2020;28(6):770-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oudin MJ, Hobbs C, Doherty P. DAGL-dependent endocannabinoid signalling: roles in axonal pathfinding, synaptic plasticity and adult neurogenesis. Eur J Neurosci. 2011;34(10):1634-1646. [DOI] [PubMed] [Google Scholar]
- 44.Davies AK, Itzhak DN, Edgar JR, et al. AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun. 2018;9(1):3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziegler M, Russell BE, Eberhardt K, et al. Blended phenotype of Silver-Russell syndrome and SPG50 caused by maternal isodisomy of chromosome 7. Neurol Genet. 2021;79(1):e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamaguchi J, Suzuki C, Nanao T, et al. Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy. 2018;14(5):764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ebrahimi-Fakhari D, Saffari A, Wahlster L, et al. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain. 2016;139(Pt 2):317-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teinert J, Behne R, Wimmer M, Ebrahimi-Fakhari D. Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J Inherit Metab Dis. 2019;43(1):51-62. [DOI] [PubMed] [Google Scholar]
- 49.Moon WJ, Kim HJ, Roh HG, Han SH. Atrophy measurement of the anterior commissure and substantia innominata with 3T high-resolution MR imaging: does the measurement differ for patients with frontotemporal lobar degeneration and Alzheimer disease and for healthy subjects? AJNR Am J Neuroradiol. 2008;29(7):1308-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walker C, El-Khamisy SF. Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain. 2018;141(5):1247-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the corresponding authors upon reasonable request.