

ABSTRACT
Fast and effective methods are needed for sequencing of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome to track genetic mutations and to identify new and emerging variants during the ongoing pandemic. The objectives were to assess the performance of the SARS-CoV-2 AmpliSeq research panel and S5 plug-in analysis tools for whole-genome sequencing analysis of SARS-CoV-2 and to compare the results with those obtained with the MiSeq-based ARTIC analysis pipeline, using metrics such as depth, coverage, and concordance of single-nucleotide variant (SNV) calls. A total of 191 clinical specimens and a single cultured isolate were extracted and sequenced with AmpliSeq technology and analysis tools. Of the 191 clinical specimens, 83 (with threshold cycle [CT] values of 15.58 to 32.54) were also sequenced using an Illumina MiSeq-based method with the ARTIC analysis pipeline, for direct comparison. A total of 176 of the 191 clinical specimens sequenced on the S5XL system and prepared using the SARS-CoV-2 research panel had nearly complete coverage (>98%) of the viral genome, with an average depth of 5,031×. Similar coverage levels (>98%) were observed for 81/83 primary specimens that were sequenced with both methods tested. The sample with the lowest viral load (CT value of 32.54) achieved 89% coverage using the MiSeq method and failed to sequence with the AmpliSeq method. Consensus sequences produced by each method were identical for 81/82 samples in areas of equal coverage, with a single difference present in one sample. The AmpliSeq approach is as effective as the Illumina-based method using ARTIC v3 amplification for sequencing SARS-CoV-2 directly from patient specimens across a range of viral loads (CT values of 15.56 to 32.54 [median, 22.18]). The AmpliSeq workflow is very easily automated with the Ion Chef and S5 instruments and requires less training and experience with next-generation sequencing sample preparation than the Illumina workflow.
KEYWORDS: SARS-CoV-2, whole-genome sequencing, Ion Torrent, AmpliSeq, ARTIC, MiSeq
INTRODUCTION
A novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was first identified in December 2019 (1) and rapidly spread around the world, infecting hundreds of millions and killing hundreds of thousands of people to date (2). The first confirmed case in New York State (NYS) was reported on 1 March 2020, after which NYS quickly became the epicenter of the pandemic in the United States. Rapid methods to track genetic changes have been essential for understanding the evolution and transmission dynamics of the virus (3–5). Currently, there remains a pressing need for increased sequencing efforts to identify the spread of new variants as quickly as possible. Thermo Fisher Scientific AmpliSeq panels have been used effectively in the past for rapid, time-effective, and cost-effective whole-genome sequencing (WGS) when faced with a viral outbreak (6). When combined with the highly automated Ion Chef system and the S5XL sequencing platform, they can provide sequence data with short turnaround time but minimal hands-on time, making it an ideal system for generating information during an active outbreak.
In this study, we evaluated the Thermo Fisher Scientific Ion AmpliSeq SARS-CoV-2 research panel (7) and its ability to sequence the genomes of SARS-CoV-2 directly from patient samples from NYS in the early months of the outbreak. To create the SARS-CoV-2 AmpliSeq panel, amplicon primers were designed in two pools, to cover all lineages of SARS-CoV-2 available at the GISAID site (https://www.gisaid.org) in late January 2020, which reacted with high analytical specificity and no cross talk with other coronaviruses. In silico analysis confirms that the panel should detect all recently identified variants, including B.1.1.7 and B.1.351 (A. Geachy, personal communication). The design covers 99.6% of the total SARS-CoV-2 genome.
As a comparison, a subset of the same clinical samples were sequenced using an Illumina-based method with the ARTIC v3 amplification primers. Shortly after publication of the first SARS-CoV-2 sequence, a group of scientists representing the ARTIC network developed a PCR amplicon enrichment method for sensitive and cost-effective WGS. The first iteration of the v1 primer set for SARS-SoV-2 was designed in January 2020 for use with Nanopore sequencing technology, with the flexibility to be adapted for other next-generation sequencing (NGS) platforms (8). There have been several iterations and improvements of the primer design (9). In this comparative study, we used primer set v3 (https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019), with modifications described in the supplemental material. Here, our aims were to evaluate how effective the AmpliSeq-based approach was in sequencing the SARS-CoV-2 genome and to compare the results with those from an Illumina-based workflow using the ARTIC primer set and analysis pipeline. We report on the depth and coverage metrics over a wide range of viral loads and on the concordance of single-nucleotide variant (SNV) calling and lineage assignment between the two methods.
MATERIALS AND METHODS
Viral RNA and clinical samples.
A total of 192 samples, which had previously tested positive for SARS-CoV-2 with the CDC 2019-novel coronavirus (2019-nCoV) real-time reverse transcription (RT)-PCR diagnostic panel, were selected from the Wadsworth Virology Laboratory archive for sequencing. Of the 192 samples, 191 were primary clinical specimens, including 122 nasopharyngeal swab samples, 7 oropharyngeal swab samples, 52 nasooropharyngeal swab samples, and 10 swab samples of unknown origin, from the initial outbreak of SARS-CoV-2 in NYS. Initial testing for SARS-CoV-2 was performed for diagnostic purposes. All additional testing was performed with approval from the NYS institutional review board under studies 07-022 and 02-054. The threshold cycle (CT) values of these specimens ranged from 15.56 to 36.94, with a median CT value of 22.41. Based on prior experience with AmpliSeq panels for viral targets such as mumps virus, measles virus, and Zika virus, samples with a maximum real-time CT value of 28 were selected, except for cases of special clinical significance. A cultured isolate with a published sequence (SARS-CoV-2/human/USA/WA-CDC-WA1/2020) was included for control purposes. Positive samples that had been collected from patients across multiple counties of NYS during the first 2 months of the outbreak, with collection dates ranging from 29 February 2020 to 29 April 2020, were selected for sequencing to provide maximum geographic and temporal diversity at the time this study was conducted. Complete metadata of clinical specimens are available at GISAID. Viral RNA was extracted on easyMAG instruments (bioMérieux, Raleigh, NC) according to the manufacturer’s instructions, with 110 μl of sample eluted into 110 μl.
Ion Torrent AmpliSeq panel sequencing.
cDNA was synthesized using the SuperScript VILO cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, using 10 μl of RNA extract. Libraries were prepared on the Ion Chef system as described in the Ion AmpliSeq library preparation on the Ion Chef system user guide. Samples were amplified for 17 cycles with a 4-min extension time. The Ion AmpliSeq SARS-CoV-2 research panel, supplied by Thermo Fisher Scientific for this study, contained 247 primer pairs designed to cover the SARS-CoV-2 genome with 125- to 275-bp overlapping amplicons. Amplified samples were then sequenced on the Ion S5XL system (Thermo Fisher Scientific), as described in the Ion S5 and Ion S5XL instrument user guide, using Ion 530 chips (Thermo Fisher Scientific), initially with 16 samples per chip. Later runs were increased to 32 samples per 530 chip without any changes in library preparation.
Illumina MiSeq sequencing.
Amplicon WGS of SARS-CoV-2 was performed using a modified version of the COVID-19 ARTIC v3 Illumina library construction and sequencing protocol V.2 (https://www.protocols.io/view/covid-19-artic-v3-illumina-library-construction-an-be3wjgpe?) in the Applied Genomics Technology Core at the Wadsworth Center. Briefly, cDNA was synthesized by incubating SuperScript IV reverse transcriptase (Invitrogen, Carlsbad, CA, USA) with random hexamers, deoxynucleoside triphosphates (dNTPs), RNase inhibitor, and 5 μl of extracted RNA. The reaction mixture was incubated at 25°C for 5 min, at 42°C for 50 min, and then at 70°C for 10 min, on a SimpliAmp thermal cycler (Thermo Fisher Scientific). Amplicons were generated with two premixed ARTIC v3 primer pools (Integrated DNA Technologies, Coralville, IA, USA). Additional primers to supplement those that showed poor amplification efficiency were identified (https://github.com/artic-network/artic-ncov2019/tree/master/primer_schemes/nCoV-2019) and ordered separately from Integrated DNA Technologies as a 100 μM stock. Modified pools were generated from these primers as described in the supplemental material.
PCR conditions were 98°C for 30 s, followed by 24 cycles of 98°C for 15 s and 63°C for 5 min, with a final 65°C extension for 5 min. Amplicons from pool 1 and pool 2 reactions were combined, purified with AMPure XP beads (Beckman Coulter, Brea, CA, USA) with a 1:1 bead/sample ratio, eluted in 10 mM Tris-HCl (pH 8.0), and quantified with the Quant-IT double-stranded DNA (dsDNA) assay kit on an ARVO X3 multimode plate reader (Perkin Elmer, Waltham, MA, USA). Libraries were generated using the Nextera DNA Flex library preparation kit with Illumina index adaptors and sequenced on a MiSeq instrument (Illumina, San Diego, CA, USA). Samples were sequenced using 300 × 150-bp paired-end reads, with 94 samples and 2 negative-control samples multiplexed per run.
Data analysis.
For Ion Torrent-generated data, reads were processed with the S5 software plug-ins coverageAnalysis, IRMAreport (10), AssemblerTrinity (11, 12), variantCaller (Torrent Suite software v5.12, with germline low-stringency settings according to the TS5.12 user guide), and GenerateConensus (Ion AmpliSeq SARS-CoV-2 Insight research assay user guide). CoverageAnalysis was set to a minimum depth of 50 reads. The consensus sequence was taken directly from the IRMAreport and GenerateConsensus fasta output and compared to sequences manually assembled in Geneious v9.1.8 using the fastq data files. In Geneious, reads were trimmed using bbduk (http://jgi.doe.gov/data-and-tools/bb-tools) for quality and assembled with respect to SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank accession number MN908947) as a reference. Consensus sequences derived from the Torrent Suite software v5.12 (settings according to the TS5.12 user guide) were compared with those generated by Illumina sequencing.
For data generated on the Illumina MiSeq system, reads were processed by the ARTIC Nextflow pipeline (https://github.com/connor-lab/ncov2019-artic-nf/tree/illumina). Briefly, reads were trimmed with TrimGalore (https://github.com/FelixKrueger/TrimGalore) and aligned to the reference assembly (strain Wuhan-Hu-1 [GenBank accession number MN908947]) by BWA (13). Primers were trimmed and consensus sequences were generated with iVar (14). Positions were required to be covered to a minimum depth of 50 reads, and variants were required to be present at a frequency ≥0.75. All consensus sequences were assigned to lineages by Pangolin v.2.0.4 (PangoLEARN v3/16/21) (15).
Data availability.
Raw reads for all samples sequenced in this study have been deposited in GenBank under BioProject accession number PRJNA757011. Individual SRA and GISAID accession numbers and tabulated details of raw and mapped reads are available in Table S1 in the supplemental material.
RESULTS
Of the 191 clinical specimens processed for Ion S5 AmpliSeq sequencing, 176 returned high-quality sequences with at least 98% coverage of the SARS-CoV-2 genome and an average depth of 5,031×. The consensus obtained from the culture isolate was identical to the published sequence (SARS-CoV-2/human/USA/WA-CDC-WA1/2020 [GenBank accession number MN985325]). Four samples had >50× genome coverage ranging from 60.12% to 93.89%. Of the samples that failed to sequence, 3 had low viral loads, with real-time CT values of 32.54 to 36.86, and presumably levels were too low to amplify. Eight samples did not produce any reads because of a failure in the library preparation on the Ion Chef for unknown reasons; the 24 additional samples on that chip were sequenced successfully. The 11 failures were removed from further analysis. All 180 high-quality genome sequences have been submitted to GISAID (see Table S1 in the supplemental material for GISAID accession numbers).
Of the 180 patient samples that produced sequences, 176 had a minimum coverage depth of 50× for more than 98% of the genome, as measured by the coverageAnalysis plug-in. The average read depth of the samples sequenced on 530 chips containing 16 samples was 8,015×, which decreased to 3,413× when the number of samples was increased to 32 samples per chip. However, this did not lead to any decrease in genome coverage (>50×).
For specimens sequenced using the AmpliSeq method, the variantCaller plug-in detected an average of 7.3 mutations in the 180 clinical specimens (collected from counties across NYS from 29 February 2020 to 29 April 2020), compared to the Wuhan-CoV-2 reference genome (GenBank accession number MN908947). Nearly all of the variants were SNVs (Table 1). Deletions were also identified in 6 of the clinical specimens (Table 1), while no insertions were detected. Frameshift deletions were detected in two samples, i.e., a single-nucleotide deletion in open reading frame 3a (ORF3a) and a two-nucleotide deletion in ORF6, both resulting in premature stop codons (Table 2). A total of 19 unique lineages were detected in the 180 samples that produced sequences with the AmpliSeq workflow.
TABLE 1.
Nonsynonymous SNVs detected in clinical specimens of SARS-CoV-2a
| Coding region and genomic position | Codon change | Amino acid change | No. (%) |
|---|---|---|---|
| ORF1ab | |||
| 490 | GAU →GAA | D75E | 6 (3.3) |
| 1059 | ACC→AUC | T265I | 136 (75.1) |
| 3177 | CCU→CUU | P971L | 6 (3.3) |
| 11916 | UCA→UUA | S3884L | 11 (6.1) |
| 14408 | CCU→CUU | P4715L | 158 (87.3) |
| 18736 | UUU→CUU | F6158L | 6 (3.3) |
| 19684 | GUA→UUA | V6474L | 5 (2.8) |
| S | |||
| 23403 | GAU→GGU | D614G | 159 (87.8) |
| ORF3a | |||
| 25563 | CAG→CAU | Q57H | 147 (81.2) |
| 26144 | GGU→GUU | G251V | 6 (3.3) |
| ORF7a | |||
| 27635 | UCA→UUA | S81L | 5 (2.8) |
| ORF8 | |||
| 28077 | GUG→CUG | V62L | 6 (3.3) |
| 28144 | UUA→UCA | L84S | 12 (6.6) |
The nonsynonymous mutations listed are those that were detected in at least 2 of the 181 clinical samples that were sequenced successfully. Specimen collection dates ranged from 29 February 2020 to 29 April 2020.
TABLE 2.
Deletions detected in clinical specimens of SARS-CoV-2a
| Region | Type | Effect | Genomic position | Coverage (×) |
|---|---|---|---|---|
| ORF1ab | Nonframeshift | H81Q; amino acids 82–87 deleted | 508–525 | 2,186 |
| Nonframeshift | Amino acids 140–142 deleted | 686–694 | 5,512 | |
| ORF3a | Frameshift | Premature stop introduced | 26162 | 2,403 |
| ORF6 | Frameshift | Premature stop introduced | 27267–27268 | 4,431 |
| 3′ UTRb | Noncoding | Unknown | 29730–29744 | 1,151, 4,133 |
The deletion mutations shown are those detected with the read coverage of the region from the Ion Torrent AmpliSeq data. Deletions were confirmed following propagation in culture.
The deletion in the 3′ untranslated region (UTR) was detected in two samples; all others were observed only once.
A subset of 83 of the primary patient specimens that were sequenced using the AmpliSeq method were also sequenced with a modified ARTIC protocol on the Illumina MiSeq sequencing platform. The two methods provided similar (>98%) coverage of the SARS-CoV-2 genome except for two specimens, with the specimen with the lowest viral load (real-time CT value of 32.54) failing with the AmpliSeq method (Fig. 1B). The average depth of the samples sequenced with the MiSeq method was 2,810.3×, with 94 samples and 2 negative-control samples multiplexed per run, compared to 8,015× and 3,413× average depth for 16 and 32 samples, respectively, processed on the Ion Torrent (see Table S1 for per-sample metrics for each sequencing method).
FIG 1.
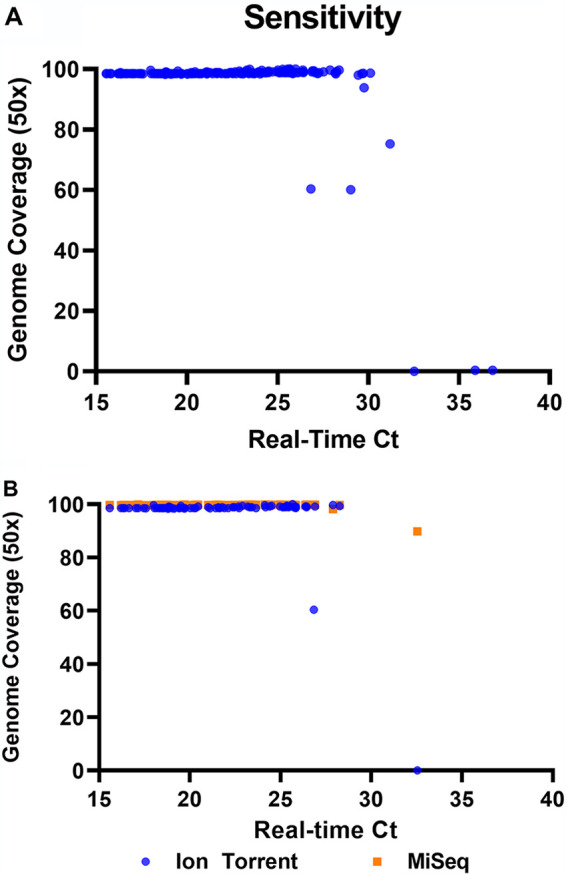
(A) Genome coverage of the 181 primary samples that returned sequences using the AmpliSeq method, compared to the CT value obtained with the CDC 2019-nCoV real-time RT-PCR diagnostic panel as an indication of viral load. The percentage of bases with at least 20× coverage was reported by the CoverageAnalysis plug-in. (B) Genome coverage of the 83 samples that were processed with both the AmpliSeq Ion Torrent and Illumina MiSeq ARTIC methods.
For specimens sequenced with both methods, Pangolin assigned the same lineage in 81/82 cases. For one specimen (IDR2000018637), the AmpliSeq consensus was assigned to B.1, while the MiSeq consensus was assigned to B. This was due to the presence of more ambiguous bases (n = 12 bases) in the MiSeq consensus sequence, rather than discrepancies in SNV calling. Both methods called the same SNVs in 81/82 of the clinical specimens, with a single difference in IDR2000045078, which did not affect lineage assignment. All other areas of equal coverage were identical between the two methods.
DISCUSSION
Monitoring the genomic evolution of viruses is imperative for a full understanding of their mutation rates, diversity, potential for development of drug resistance and vaccine evasion, and potential for pathogenesis or virulence changes. However, NGS methods can be time-consuming and, especially in the face of a major outbreak, there is a need for rapid, simple, high-throughput methods that are convenient and may be readily deployed to multiple sites. We have had past success using the Ion Torrent AmpliSeq panels and S5 platform to sequence viral pathogens such as Zika virus, mumps virus, and measles virus. One major advantage of AmpliSeq panels with automated Ion Chef instruments is that one person can generate all of the libraries with minimal hands-on time. Following cDNA synthesis, the entire library preparation and templating procedure is performed on the Ion Chef. A single Ion Chef run takes about 15 min of hands-on time and about 7 h of run time to create eight libraries. Templating is most efficient when 64 libraries are created, which takes 15 min of hands-on time and 15 h to run. In total, it takes about 79 h to complete library preparation and templating of 64 libraries using a single instrument, with only about 2 h of hands-on time (Fig. 2). The sequencing reaction can be started immediately after templating and takes about 2.5 h per chip, with data analysis beginning immediately after the first chip has finished sequencing and usually taking about 6 to 8 h. In contrast, MiSeq library preparation following cDNA synthesis takes about 7 to 8 h of hands-on time, with 8 h of instrument time and an additional 24 h to run the sequencing reaction on the MiSeq sequencer. Compared to the AmpliSeq protocol, the Illumina workflow used here requires significantly more hands-on time and a trained technician. However, there are ways to automate much of the Illumina library preparation workflow as well, with the addition of third-party instrumentation and programming.
FIG 2.

Workflow for each of the methods described and the associated preparation and incubation times for each step. The ARTIC Illumina workflow is for 94 samples and 2 negative-control samples, and the AmpliSeq workflow is for 64 samples using a single Ion Chef instrument. QC, quality control.
When considering the ability to scale up each protocol, one of the significant hurdles for the Ion Torrent workflow is that the Ion Chef instrument can create only 8 libraries at a time. Higher throughput can be achieved by increasing the number of Ion Chef instruments used to create libraries or by using Thermo Fisher Scientific 96- or 384-well-plate-based AmpliSeq library kits, which can then be sequenced together on a larger chip. While not compatible with the Ion Chef, the plate-based workflow can be automated using additional instrumentation and programming. The Illumina workflow can be readily scaled up for sequencing on NextSeq instruments, following multiple completions of the library preparation described here. The total cost of reagents can change based on the quantity of reagents purchased and other factors. Additionally, test volume and subsequent batch size impact efficiency, which can dramatically affect global costs per sample. Our cost analysis shows that the cost per sample due to reagents would remain higher for the AmpliSeq protocol when operating at maximum efficiency, compared to Illumina methods.
Sequencing on the S5XL system allows for real-time data analysis and the return of sequences within several hours after initiation of the sequencing reaction. The analysis plug-ins are included with the instrument itself. The IRMAreport and GenerateConsensus plug-ins return fasta files of the consensus genomes to further streamline the process. However, compared to manual curation, the IRMAreport output was less accurate near the ends of the genome. GenerateConsensus is a more recent analysis plug-in that allows users to set different thresholds for base calls in homopolymeric regions, separately from the rest of the genome, improving the accuracy of base calling in those areas. These consensus sequences are generated using a previously completed VariantCaller run, allowing users to set their own threshold for SNV calls. This plug-in was run on a subset of the S5 Ion Torrent data produced in this study; results were compared to consensus sequences produced by IRMA with manual editing and were found to be identical (data not shown). The VariantCaller plug-in can also be used to analyze minor allele frequency, which may be of further research interest, such as for measuring intrahost diversity.
The Nextflow-adapted ARTIC bioinformatics pipeline employed to process Illumina-generated reads in the cloud is freely available and is designed to be highly parallelized. The number of virtual machines (VMs) deployed is automatically determined by the number of samples, which should theoretically yield the same processing time for different batch sizes. Consensus sequences were typically generated in 30 min, but processing time can vary according to machine availability in different regions and the time of day. In addition to consensus sequences, the Nextflow pipeline provides quality-trimmed fastq files, primer-trimmed BAM alignment files, and VCF files with information on variants (SNVs and indels), including minor variants. Thus, the ARTIC pipeline offers more transparency than the Ion Torrent plug-in, with intermediate data files being available for additional analyses. However, establishing the necessary accounts in the cloud, properly configuring VMs, and installing and running Nextflow software and the ARTIC pipeline require both information technology and bioinformatics expertise, which might be prohibitive for some laboratories. The ARTIC bioinformatics pipeline can be installed locally with the Conda package manager (https://docs.conda.io) but currently processes only Nanopore MinION-generated reads. Alternatively, the individual components that constitute the Nextflow ARTIC pipeline, such as TrimGalore, BWA, and iVAR, can be freely and locally installed. While this approach negates the need for a cloud account and the expertise for navigating in this environment, it still requires some bioinformatics training.
Of the 15 samples that produced less than 98% genome coverage, 8 were prepared together on a single Ion Chef run and failed to produce usable sequences for unknown reasons, returning less than 50 reads per sample. Four samples produced partial genome coverage, ranging from 60.12% to 93.89%. The remaining 3 patient samples were complete failures and had previously generated CT values in real-time RT-PCR ranging from 32.54 to 36.94, indicating poor specimen quality or viral loads too low to amplify. We hypothesize that increasing the amount of specimen loaded for extraction, reducing the volume eluted, and increasing the cycle number during library preparation may improve the chances of attaining sequences for weakly positive samples. For this reason, our current extraction protocol for sequencing high-CT specimens with clinical significance uses concentrated extraction (1 ml into 25 μl) on an easyMAG system. Previous studies have shown the Swift Biosciences amplicon panel to have a higher sensitivity than that reported here when used with Illumina MiSeq sequencing (16), and it may be better suited for sequencing samples with extremely low viral loads.
Interestingly, we detected five unique deletions in our 181 clinical specimens. These deletions were first identified using the AmpliSeq method but were confirmed using the Illumina MiSeq ARTIC method following propagation in culture, providing strong evidence that these are real deletions. Two unique frameshift deletions were confirmed with both methods, and the effects of these deletions on factors such as growth kinetics and fitness are currently being investigated.
In conclusion, the AmpliSeq chemistry combined with the Ion Chef and S5XL instruments provides an effective, rapid, simple, and highly automated option for generating WGS data for SARS-CoV-2 specimens. Results were highly comparable in sensitivity, SNV calling, and lineage assignment to sequences generated with the ARTIC primer-based Illumina workflow.
ACKNOWLEDGMENTS
We thank the Wadsworth Center Applied Genomics Technology Core for performing MiSeq sequencing and the staff of the Wadsworth Center Virology Laboratory for the initial testing of samples.
This research was supported in part by an appointment to the Infectious Disease Laboratory Fellowship Program (J.P.) administered by the Association of Public Health Laboratories and funded by the Centers for Disease Control and Prevention. Some reagents and supplies were provided by Thermo Fisher Scientific.
J.P., S.G., E.L.-N., N.S., and D.M.L. declare no competing interests. K.S.G. receives research support from Thermo Fisher Scientific for the evaluation of new assays for the detection and characterization of viruses. She also has a royalty-generating collaborative agreement with Zeptometrix.
Footnotes
Supplemental material is available online only.
Contributor Information
Daryl M. Lamson, Email: daryl.lamson@health.ny.gov.
John P. Dekker, National Institute of Allergy and Infectious Diseases
REFERENCES
- 1.Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao Z-W, Tian J-H, Pei Y-Y, Yuan M-L, Zhang Y-L, Dai F-H, Liu Y, Wang Q-M, Zheng J-J, Xu L, Holmes EC, Zhang Y-Z. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong E, Du H, Gardner L. 2020. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis 20:533–534. 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, Zhao C, Zhang Q, Liu H, Nie L, Qin H, Wang M, Lu Q, Li X, Sun Q, Liu J, Zhang L, Li X, Huang W, Wang Y. 2020. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182:1284–1294.e9. 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucey M, Macori G, Mullane N, Sutton-Fitzpatrick U, Gonzalez G, Coughlan S, Purcell A, Fenelon L, Fanning S, Schaffer K. 2021. Whole-genome sequencing to track severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission in nosocomial outbreaks. Clin Infect Dis 72:e727–e735. 10.1093/cid/ciaa1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Speake H, Phillips A, Chong T, Sikazwe C, Levy A, Lang J, Scalley B, Speers DJ, Smith DW, Effler P, McEvoy SP. 2020. Flight-associated transmission of severe acute respiratory syndrome coronavirus 2 corroborated by whole-genome sequencing. Emerg Infect Dis 26:2872–2880. 10.3201/eid2612.203910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arias A, Watson SJ, Asogun D, Tobin EA, Lu J, Phan MVT, Jah U, Wadoum REG, Meredith L, Thorne L, Caddy S, Tarawalie A, Langat P, Dudas G, Faria NR, Dellicour S, Kamara A, Kargbo B, Kamara BO, Gevao S, Cooper D, Newport M, Horby P, Dunning J, Sahr F, Brooks T, Simpson AJH, Groppelli E, Liu G, Mulakken N, Rhodes K, Akpablie J, Yoti Z, Lamunu M, Vitto E, Otim P, Owilli C, Boateng I, Okoror L, Omomoh E, Oyakhilome J, Omiunu R, Yemisis I, Adomeh D, Ehikhiametalor S, Akhilomen P, Aire C, Kurth A, Cook N, Baumann J, Gabriel M, Wölfel R, Di Caro A, Carroll MW, Günther S, Redd J, Naidoo D, Pybus OG, Rambaut A, Kellam P, Goodfellow I, Cotten M. 2016. Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Virus Evol 2:vew016. 10.1093/ve/vew016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai A, Bergna A, Caucci S, Clementi N, Vicenti I, Dragoni F, Cattelan AM, Menzo S, Pan A, Callegaro A, Tagliabracci A, Caruso A, Caccuri F, Ronchiadin S, Balotta C, Zazzi M, Vaccher E, Clementi M, Galli M, Zehender G. 2020. Molecular tracing of SARS-CoV-2 in Italy in the first three months of the epidemic. Viruses 12:798. 10.3390/v12080798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, Burton DR, Lewis-Ximenez LL, de Jesus JG, Giovanetti M, Hill SC, Black A, Bedford T, Carroll MW, Nunes M, Alcantara LC, Sabino EC, Baylis SA, Faria NR, Loose M, Simpson JT, Pybus OG, Andersen KG, Loman NJ. 2017. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 12:1261–1276. 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Itokawa K, Sekizuka T, Hashino M, Tanaka R, Kuroda M. 2020. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS One 15:e0239403. 10.1371/journal.pone.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shepard SS, Meno S, Bahl J, Wilson MM, Barnes J, Neuhaus E. 2016. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genomics 17:708. 10.1186/s12864-016-3030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grubaugh ND, Gangavarapu K, Quick J, Matteson NL, De Jesus JG, Main BJ, Tan AL, Paul LM, Brackney DE, Grewal S, Gurfield N, Van Rompay KKA, Isern S, Michael SF, Coffey LL, Loman NJ, Andersen KG. 2019. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 20:8. 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rambaut A, Holmes EC, O'Toole Á, Hill V, McCrone JT, Ruis C, Du Plessis L, Pybus OG. 2020. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 5:1403–1407. 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Addetia A, Lin MJ, Peddu V, Roychoudhury P, Jerome KR, Greninger AL. 2020. Sensitive recovery of complete SARS-CoV-2 genomes from clinical samples by use of Swift Biosciences' SARS-CoV-2 multiplex amplicon sequencing panel. J Clin Microbiol 59:e02226-20. 10.1128/JCM.02226-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download jcm.00649-21-s0001.xlsx, XLSX file, 0.04 MB (45.9KB, xlsx)
Supplemental methods. Download jcm.00649-21-s0002.pdf, PDF file, 0.07 MB (69.2KB, pdf)
Fig. S1. Download jcm.00649-21-s0003.pdf, PDF file, 0.6 MB (570KB, pdf)
Data Availability Statement
Raw reads for all samples sequenced in this study have been deposited in GenBank under BioProject accession number PRJNA757011. Individual SRA and GISAID accession numbers and tabulated details of raw and mapped reads are available in Table S1 in the supplemental material.