

PURPOSE
Approximately 10%-40% of patients with lung cancer report no history of tobacco smoking (never-smokers). We analyzed whole-exome and RNA-sequencing data of 160 tumor and normal lung adenocarcinoma (LUAD) samples from never-smokers to identify clinically actionable alterations and gain insight into the environmental and hereditary risk factors for LUAD among never-smokers.
METHODS
We performed whole-exome and RNA-sequencing of 88 and 69 never-smoker LUADs. We analyzed these data in conjunction with data from 76 never-smoker and 299 smoker LUAD samples sequenced by The Cancer Genome Atlas and Clinical Proteomic Tumor Analysis Consortium.
RESULTS
We observed a high prevalence of clinically actionable driver alterations in never-smoker LUADs compared with smoker LUADs (78%-92% v 49.5%; P < .0001). Although a subset of never-smoker samples demonstrated germline alterations in DNA repair genes, the frequency of samples showing germline variants in cancer predisposing genes was comparable between smokers and never-smokers (6.4% v 6.9%; P = .82). A subset of never-smoker samples (5.9%) showed mutation signatures that were suggestive of passive exposure to cigarette smoke. Finally, analysis of RNA-sequencing data showed distinct immune transcriptional subtypes of never-smoker LUADs that varied in their expression of clinically relevant immune checkpoint molecules and immune cell composition.
CONCLUSION
In this comprehensive genomic and transcriptome analysis of never-smoker LUADs, we observed a potential role for germline variants in DNA repair genes and passive exposure to cigarette smoke in the pathogenesis of a subset of never-smoker LUADs. Our findings also show that clinically actionable driver alterations are highly prevalent in never-smoker LUADs, highlighting the need for obtaining biopsies with adequate cellularity for clinical genomic testing in these patients.
INTRODUCTION
Approximately 10%-40% of lung cancers are diagnosed in never-smokers (individuals smoking fewer than 100 cigarettes during their lifetime).1,2 The most common histological subtype of lung cancer in never-smokers is lung adenocarcinoma (LUAD). The demographics of LUAD in never-smokers are distinct compared with smokers, with a greater proportion of women, Asian or Pacific islanders, and individuals of younger age. Although environmental factors such as passive exposure to cigarette smoke, radon, air pollutants, and genetic polymorphisms in genes including TERT, GPC5, DNA repair genes, glutathione S-transferase, and EGFR and ERBB2 have been implicated as risk factors, causative factors for lung cancer in never-smokers are poorly understood.1,3-11
CONTEXT
Key Objective
To gain a better understanding of the risk factors that might be related to lung cancer in never-smokers, whole-exome and transcriptome data from 160 never-smoker lung cancer samples were examined, in conjunction with whole-exome sequencing data from corresponding normal tissue samples.
Knowledge Generated
Results from this analysis show that a small subset of never-smoker lung cancer samples contain pathogenic and likely pathogenic germline alterations in DNA repair genes. Additionally, in some instances, tumor cells exhibited mutational signatures characteristic of exposure to cigarette smoke (possibly passive). Never-smoker lung cancers also showed a high prevalence of oncogenic driver alterations and heterogeneity in the expression of immune checkpoint molecules and tumor immune composition.
Relevance
These results provide an insight into germline alterations associated with never-smoker lung cancers. Furthermore, these results emphasize the need for comprehensive molecular analyses of never-smoker lung cancers in the clinic, given the high prevalence of targetable alterations in these samples.
Next-generation sequencing studies by various groups including The Cancer Genome Atlas (TCGA) have provided valuable insights into the genomic landscape of lung cancer. However, the majority of these samples were acquired from smokers.12-16 We performed whole-exome sequencing (WES) of 88 tumors and their corresponding germline DNA, and RNA-sequencing of 69 tumor samples from never-smokers with LUAD from three institutions in the United States (Institutional cohort). We also used WES of tumor and germline and RNA-sequencing data from never-smoker LUADs sequenced by TCGA and the Clinical Proteomic Tumor Analysis Consortium (CPTAC), comprising the External cohort.15,17 We analyzed data from these never-smoker LUAD samples (never-smoker cohort) along with sequencing data from smoking-associated LUADs (smoker cohort) sequenced by TCGA and CPTAC to gain an insight into the environmental and hereditary risk factors that could predispose to never-smoker lung cancer and identify clinically actionable alterations.
METHODS
Clinical Information
Lung cancer samples from 88 self-reported never-smokers, diagnosed between 2007 and 2014, were collected from Washington University School of Medicine, MD Anderson Cancer Center, and New York University, through Institutional Review Board–approved protocols in compliance with ethical guidelines and following informed consent. Clinical information and demographic data were available for 85 of 88 patients in the Institutional cohort (Data Supplement, online only). WES and RNA-sequencing data for 513 TCGA and 110 CPTAC samples comprised the External cohort. In addition to self-reported smoking status, a bioinformatic scoring model was applied to infer smoking status for all samples in the Institutional and External cohorts (Fig 1A, Data Supplement). In total, 160 never-smoker (84 from the Institutional cohort and 76 from the External cohort) and 299 smoker LUAD samples that passed the bioinformatic scoring model were used for analyses. Self-reported never-smoker and smoker samples that did not pass the bioinformatic scoring model were excluded from further analyses. Among Institutional cohort samples, the majority (n = 75, 89%) were fresh frozen specimens and the rest were formalin-fixed archival specimens. Driver alterations were defined as known activating mutations or fusions in RTK/RAS/RAF pathway genes.18
FIG 1.
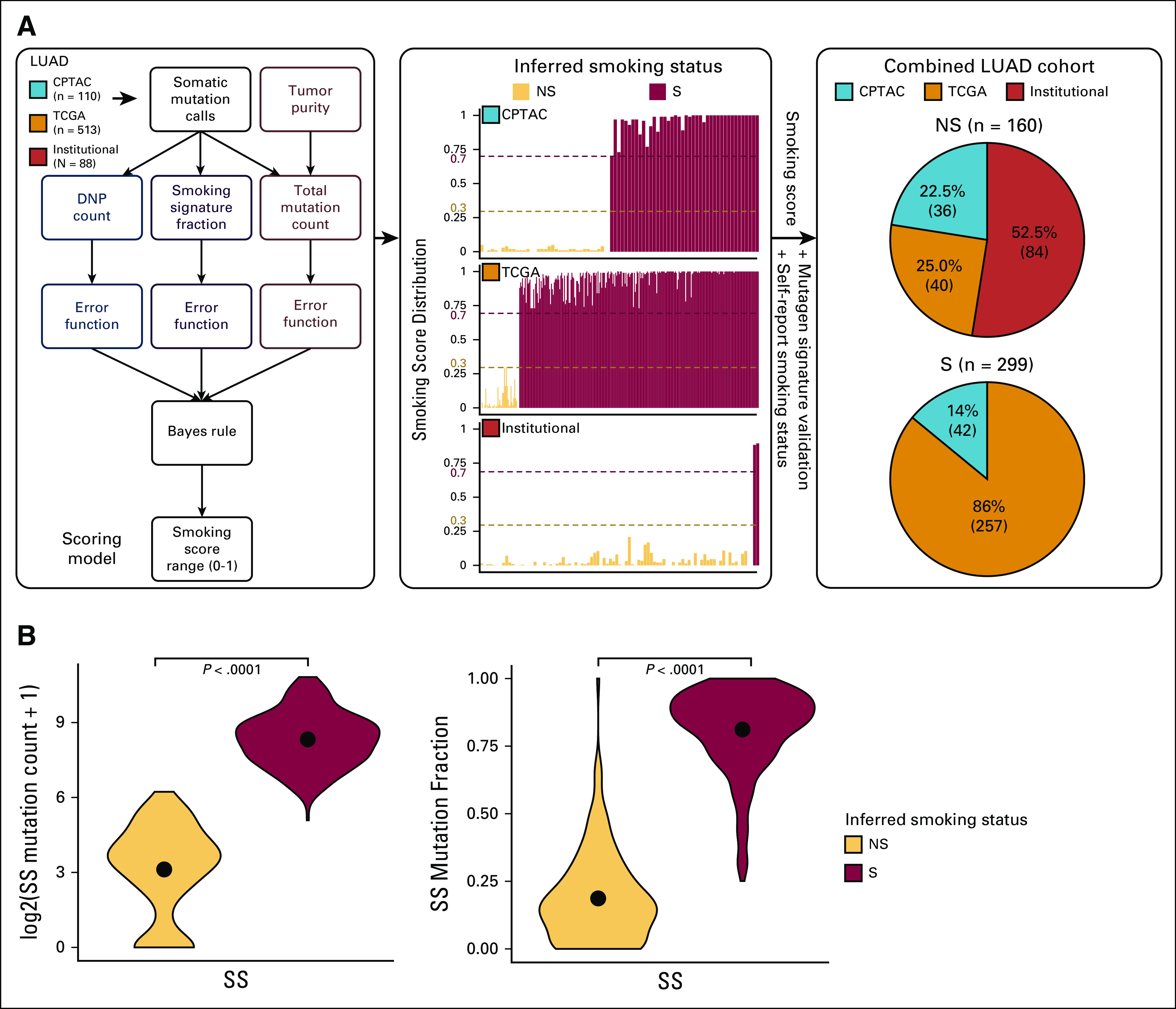
The data-driven smoking classification in the LUAD cohorts (Institutional, CPTAC, and TCGA). (A) The flowchart presents the analysis steps included in deriving the scoring model (continuous smoking score range from 0 to 1). The bar charts show the smoking score distribution in each of the three cohorts. The smoking score (0.3 as the lower-bound cutoff and 0.7 as the upper-bound cutoff) along with the self-reported smoking status, and mutagen signature validation was used to infer smoking status. The pie charts represent the NS and S composition of each of the three cohorts on the basis of the inferred smoking status. In total, the NS group consisted of 160 samples and the S group contained 299 samples. (B) The violin plot shows the comparisons of log2-scaled total mutation counts and mutation fractions that contributed to the SS between NS and S samples (as inferred from steps described in Fig 1A). CPTAC, Clinical Proteomic Tumor Analysis Consortium; DNP, dinucleotide polymorphism; LUAD, lung adenocarcinoma; NS, never-smokers; S, smokers; SS, smoking signature; TCGA, The Cancer Genome Atlas.
Molecular Analysis
We performed WES of tumor samples to an average depth of 20-30× and germline (peripheral blood mononuclear cells) samples from 88 patients from the Institutional cohort. In addition, RNA-sequencing was performed on tumor samples from 69 patients from this cohort. Sequencing data were initially analyzed with the objective of identifying germline variants that could predispose to lung cancer, mutational signatures in tumor tissue related to environmental carcinogen exposure, and clinically targetable alterations in tumor specimens. RNA-sequencing data were analyzed for validating variant calls and studying the tumor microenvironment. We additionally analyzed targeted exome sequencing and clinical molecular test results conducted in a Clinical Laboratory Improvement Amendments–certified laboratory for 17 patients for the Institutional cohort.19 Samples for which we failed to identify driver alterations through WES or clinical test results were subject to WES at higher sequencing depth of nearly 400× when adequate DNA was available (n = 13). The sequencing, molecular, and clinical data for the External cohorts (Data Supplement) were obtained from Genomic Data Commons (GDC).20 A comprehensive description of bioinformatics methods, algorithms, and workflow has been provided in the Data Supplement.
RESULTS
Mutational Landscape
Since self-reported smoking status was difficult to directly verify for patients from whom specimens were collected in both cohorts, we developed a scoring model to infer smoking status (inferred smoking status) with a high degree of confidence. This model used tumor mutation burden (TMB; number of somatic mutations per million base pairs of sequenced DNA) and mutation signatures characteristic of tobacco smoke exposure (smoking signature), apart from documented self-reported smoking status (Fig 1A). This scoring system identified a subset of 160 high-confidence never-smoker samples from Institutional (n = 84 of 88 samples) and External (n = 76) cohorts and 299 high-confidence smoker samples from the External cohort for additional analyses (Data Supplement).
The median age at diagnosis was comparable between never-smoker and smoking-related LUAD cohorts (median: 67 v 65 years; P = .1). Compared with the smoker cohort, females were over-represented in the never-smoker cohort (69.4% v 48.5%; P < .001). Since ancestry information was unavailable for all the samples in the Institutional and External cohorts, this was inferred from germline information (Data Supplement). Ancestry information for TCGA LUADs was obtained through the PanCan Atlas Ancestry Informative Markers working group. The majority of never-smokers were of European ancestry (62%), followed by Asian (25%), African (5.6%), and Admixed American (5.6%) ancestries. In the smoker cohort, 79.6%, 13%, 5.7%, and 1.7% of smokers were of European, African, Asian, and Admixed American ancestries, respectively, suggesting an over-representation of Asian ancestry in never-smoker LUAD.
A median of 2.93 mutations per megabase (Mb) (range: 0.5-5.5) was observed per sample in the Institutional cohort. TMB was comparable between never-smoker samples from Institutional and External cohorts (median TMB of 1.25 per Mb and 1.95 per Mb in the CPTAC and TCGA cohorts, respectively). Smoker samples showed a median TMB of 9.1/Mb and 10.83 per Mb in the CPTAC and TCGA cohorts, respectively. Given that smoker and never-smoker samples were classified on the basis of a scoring system incorporating TMB, and in line with what has previously been reported, TMBs were much higher in smoker compared with never-smoker samples (P < .0001; Fig 1B). A comparison of the mutation frequency of 20 genes, which are recurrently altered in LUAD, between smoker and never-smoker samples showed a significantly higher frequency of alterations in EGFR, CTNNB1, SETD2, MET, and RB1 in never-smoker and KRAS, TP53, STK11, NF1, BRAF, and KEAP1 in smoker LUAD21 (Data Supplement). EGFR (51%; n = 82 of 160) and KRAS (35%; n = 103 of 299) were the most frequent driver alterations among never-smoker and smoker samples, respectively (Data Supplement).
Clinically Actionable Alterations
We were able to readily identify a known RTK/RAS/RAF pathway driver alteration in 65% (55 of 84) of never-smoker samples in the Institutional cohort through WES and RNA-sequencing. Driver alterations were not readily identifiable in 35% (n = 29) of Institutional cohort samples (termed oncogene negative [ON] by WES). A novel kinase domain mutation in EGFR (p.A955R) and an activating mutation in AKT1 (p.E17K) were observed in two samples, raising the possibility that these are the driver alterations in these samples. Further analysis of available clinical sequencing and fluorescent in situ hybridization test results allowed us to identify driver alterations in nine additional samples (including fusions in ALK [n = 2] and ROS1 [n = 1] through fluorescent in situ hybridization, and mutations in EGFR [n = 3], ERBB2 [n = 2], and KRAS [n = 1] through clinical sequencing; Data Supplement). A comparison of bioinformatically estimated sample purity between these nine reclassified samples and the 57 samples in which a driver was readily identified suggested a relatively lower predicted median tumor cellularity in the nine ON by WES samples (46% v 60%; P = .006; Data Supplement). Suspecting that low tumor cellularity could have similarly precluded the detection of driver alterations in the other ON by WES samples, we pursued additional deep WES for 13 of the 18 samples from which sufficient DNA was available. This allowed us to identify exon 14 skipping mutations in MET in two samples. Taken together, we were able to identify a driver alteration in nearly 81% (68 of 84) of never-smoker LUADs from the Institutional cohort, the vast majority of which are targetable with currently available therapies (Fig 2A). Similarly, a high prevalence of driver alterations was also observed in never-smoker LUADs from the External cohort (78% of TCGA and 92% of CPTAC samples). In contrast, driver alterations were observed only in 49.5% (n = 148 of 299) of smoker samples (P < .0001). Co-occurrence and mutual exclusivity analyses showed alterations in EGFR to be co-occurring with alterations in tumor suppressors such as TP53, CDKN2A, and RB1 (P < .05), whereas mutations in KRAS co-occurred with SETD2 alterations and showed a trend (P < .1) toward mutual exclusivity with TP53 (Fig 2B).
FIG 2.
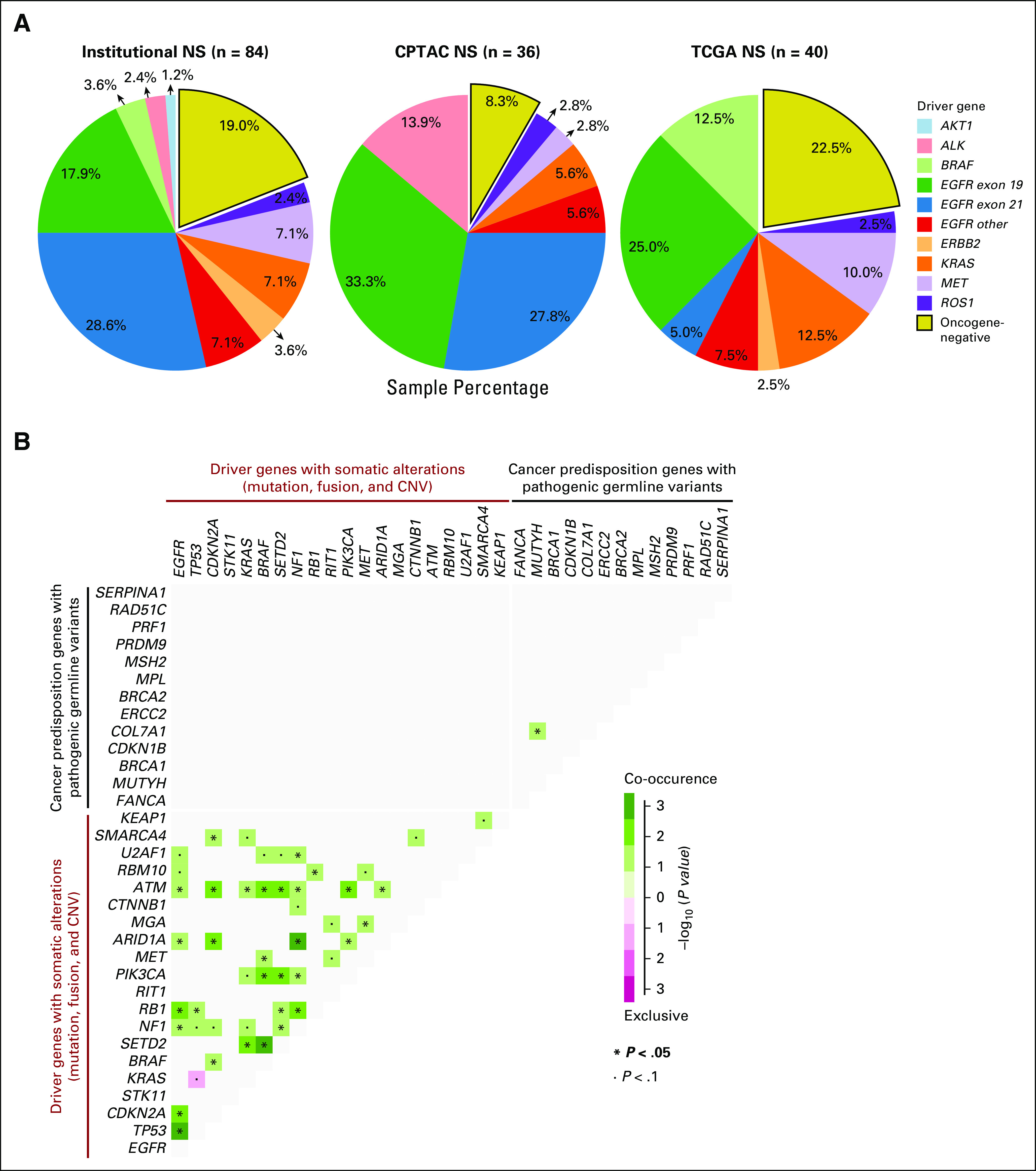
Driver alterations in NS LUAD samples. (A) Distribution of driver alterations in NS samples across the three LUAD cohorts (Institutional, CPTAC, and TCGA). (B) The mutually exclusive or co-occurring set of somatic alterations in LUAD-related genes21 (somatic mutations + gene fusions + CNVs) and cancer predisposition genes with pathogenic and likely pathogenic germline variants (genes altered with germline variants in at least two samples in the Data Supplement) detected by performing pairwise Fisher's exact test. CNV, copy number variation; CPTAC, Clinical Proteomic Tumor Analysis Consortium; LUAD, lung adenocarcinoma; NS, never-smokers; TCGA, The Cancer Genome Atlas.
Germline Variant Analysis
To understand the contribution of germline variants in cancer predisposition to never-smoker LUAD, we used WES data from normal samples in both cohorts for germline variant calling. Variants were processed and classified based on guidelines from the American College of Medical Genetics and the Association for Molecular Pathology into pathogenic, likely pathogenic, or prioritized variants of undetermined significance (VUS), as described in the Data Supplement.22,23 Through these analyses, we prioritized 198, 128, and 284 rare variants (minor allele frequency ≤ 0.05% in gnomAD and/or 1,000 Genomes) in the Institutional, CPTAC, and TCGA cohorts, respectively (Data Supplement). Among these, 14, 24, and 11 manually reviewed variants were found affecting one of the 152 well-described cancer predisposition genes (Fig 3A, Data Supplement).
FIG 3.

Discovery of rare germline predisposition variants in NS. (A) Number of manually reviewed rare germline pathogenic, likely pathogenic, and prioritized VUS identified by using the CharGer tool for each data set. Variants were considered pathogenic, on the basis of their classification in ClinVar or other curated databases; likely pathogenic if their CharGer score was ≥ 9; and prioritized VUS if their CharGer score was ≥ 5. Only variants affecting one of 152 cancer predisposition genes are included in these counts. (B and C) Percentage of S and NS carrying rare pathogenic and likely pathogenic germline variants in cancer predisposition genes in our combined cohort (B) and in each individual data set (C). (D and E) Distribution of rare pathogenic and likely pathogenic germline events in cancer predisposition genes in S and NS. (D) The variant counts for each gene. (E) The variant types. (F) Burden test results for the affected genes in S and NS against the gnomAD noncancer data set. The numbers in each box indicate the percentage of carriers of pathogenic and likely pathogenic variants in each gene in the specified cohort. Gray outlines indicate suggestive enrichment (FDR ≤ 0.15) for pathogenic and likely pathogenic variants in that gene. No significant (FDR ≤ 0.05) enrichment was observed. (G) Pathogenic, likely pathogenic, and prioritized VUSs undergoing LOH in the NS subset. Dots represent individual variants, and the diagonal line indicates neutral selection of the germline variant, where the normal and tumor VAFs are the same. Only genes carrying suggestive and/or significant LOH events in cancer predisposition genes are labeled. (H) All variants undergoing significant or suggestive LOH (depicted in G) were checked for WT allele loss using somatic copy number results from GISTIC2. LOH events were classified as deletion of WT allele if somatic copy number results showed lower ploidy below threshold in the gene region (shown in red), amplification of alternate allele when CNV results showed higher ploidy above threshold in the gene region (no events in the three genes shown in H), or unclassified LOH if the event was not classifiable as either of these two (shown in blue). Events in other samples, apart from those significant or suggestive for LOH, are represented as well and labeled as none (shown in gray). CNV, copy number variation; CPTAC, Clinical Proteomic Tumor Analysis Consortium; FDR, false discovery rate; LOH, loss of heterozygosity; NS, never-smokers; S, smokers; TCGA, The Cancer Genome Atlas; VAF, variant allele frequency; VUS, variants of undetermined significance; WT, wild-type.
Overall, pathogenic and likely pathogenic germline variants were observed in 6.4% of smokers and 6.9% of never-smokers (Figs 3B and 3C). Among these, we observed variants in cancer predisposition genes such as BRCA1, BRCA2, FANCG, FANCM, HMBS, MSH6, NF1, POLD1, TMEM127, and WRN, exclusively among never-smokers (Figs 3D-3F). We also investigated the potential enrichment of pathogenic and likely pathogenic variants at the gene level in both smokers and never-smokers, in comparison with a noncancer cohort from the Genome Aggregation Database (gnomAD), using a total frequency test (Data Supplement).24 Burden test results showed suggestive enrichment (0.05 < false discovery rate ≤ 0.15) of pathogenic and likely pathogenic variants in FANCG and TMEM127 in never-smokers compared with gnomAD noncancer data set, indicating a potential role for these genes in LUAD predisposition in never-smokers. TMEM127 is a negative regulator of mammalian target of rapamycin signaling, and germline variants in this gene have been reported in pheochromocytomas and paragangliomas.25,26 FANCG encodes for a core protein of the Fanconi anemia pathway, which plays an important role in repairing DNA damage. Germline mutations in FANCG have been reported in the context of young-onset pancreatic cancers.27
To further explore the impact of these and other germline variants, we examined if pathogenic, likely pathogenic, and prioritized VUSs observed in the normal sample underwent loss of heterozygosity (LOH) in the tumor sample. Through this analysis, we observed two prioritized VUSs undergoing significant LOH in tumors in BRCA2 and FANCA, as well as two prioritized VUSs in BRCA2 and CHEK2 undergoing suggestive (false discovery rate ≤ 0.15) LOH (Fig 3G). To validate the allele specificity of these prioritized VUS LOH events, we analyzed read-count data from both tumor and normal samples, as well as somatic copy number variation calls (Data Supplement).28 Through this analysis, we were able to identify that the deletion of the wild-type allele in the cancer sample contributed to LOH in BRCA2 (Fig 3H), indicating that this VUS was likely a true pathogenic event. Notably, we did not identify either co-occurring or mutually exclusive interaction between somatic alterations in genes frequently altered in LUAD and any of the pathogenic or likely pathogenic germline variants in cancer predisposing genes (Fig 2B).
Mutational Signatures
Never-smoker samples from both cohorts predominantly demonstrated single base substitution mutation signatures (SBS) 1, 2, 6, and 13 (Data Supplement).29,30 SBS 1 is characterized by C to T transitions in the context of CpG (cytosine followed by guanine sequences) and is likely a result of spontaneous or enzymatic deamination of 5-methylcytosine to thymine. SBS 2 and 13 are related to APOBEC mutagenesis. SBS 6 has been observed in mismatch repair deficient tumors. To investigate the contribution of environmental agents to in never-smoker LUADs, we determined the extent to which the pattern of mutations observed in each sample resembled mutation signatures associated with known environmental carcinogens (Fig 4, Data Supplement).31 As anticipated, these analyses showed that the somatic mutation signatures from 84% of samples from smokers demonstrated a strong correlation (R2 > 0.3) with smoking-related mutagen (polycyclic aromatic hydrocarbon) signatures. Notably, 5.9% (n = 9) of never-smoker samples also demonstrated the presence of smoking-related mutagen signatures. The most frequent driver alteration in these samples was still EGFR (five of nine samples; one sample showed a BRAF G466V mutation and three were ON by WES) implying a role for environmental carcinogens, possibly from passive exposure to cigarette smoke, in the pathogenesis of a subset of never-smoker lung cancers.
FIG 4.
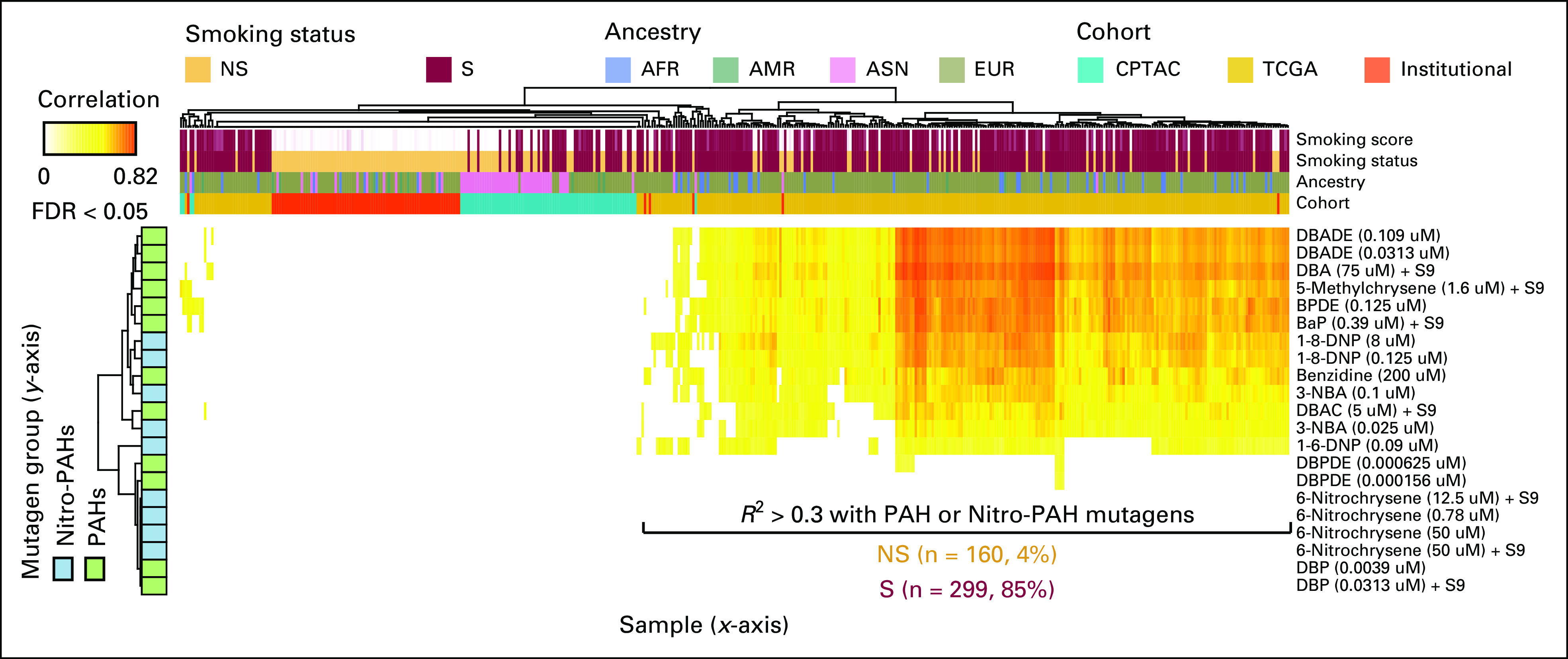
The contributions of smoking-related mutagens to the phenotypes. The heatmap shows the correlation coefficient between the mutational signatures identified 160 NS and 299 S (x-axis) samples and smoking-related mutagen signatures (y-axis) described by Kucab et al.31 AFR, African; AMR, Admixed American; ASN, Asian; CPTAC, Clinical Proteomic Tumor Analysis Consortium; EUR, European; FDR, false discovery rate; NS, never-smoker; PAH, polycyclic aromatic hydrocarbon; S, smoker; TCGA, The Cancer Genome Atlas.
Immune Landscape
Unlike smoker LUADs, never-smoker LUADs typically do not demonstrate durable responses with immune checkpoint blockade despite high programmed cell death ligand 1 (PD-L1) immunohistochemical scores.32,33 Therefore, to gain a better understanding of the immunobiology of never-smoker LUADs, their tumor microenvironment was studied using RNA-sequencing data.34 Consensus clustering on the basis of the presence of various immune and stromal cell types and expression of immune markers identified three clusters of never-smoker LUADs within the Institutional cohort (Fig 5A, Data Supplement). Compared with tumors belonging to cluster 1 (IM-1), cluster 2 (IM-2) and cluster 3 (IM-3) tumors were overall relatively depleted for the presence of various types of immune cells (Fig 5B). IM-1 tumors also demonstrated a higher level of expression of immune markers such as PD-L1, PD-L2, TIM3, CTLA4, and SIGLEC15. In contrast to IM-1 tumors, IM-3 tumors contained tumors with the lowest proportion of immune cells and were relatively depleted in the expression of immune checkpoint molecules. IM-2 tumors appeared to consist of a mix of IM-1 and IM-3 tumors, with the level of expression of immune checkpoint molecules and percentage of immune cells in these tumors varying across a continuum.
FIG 5.
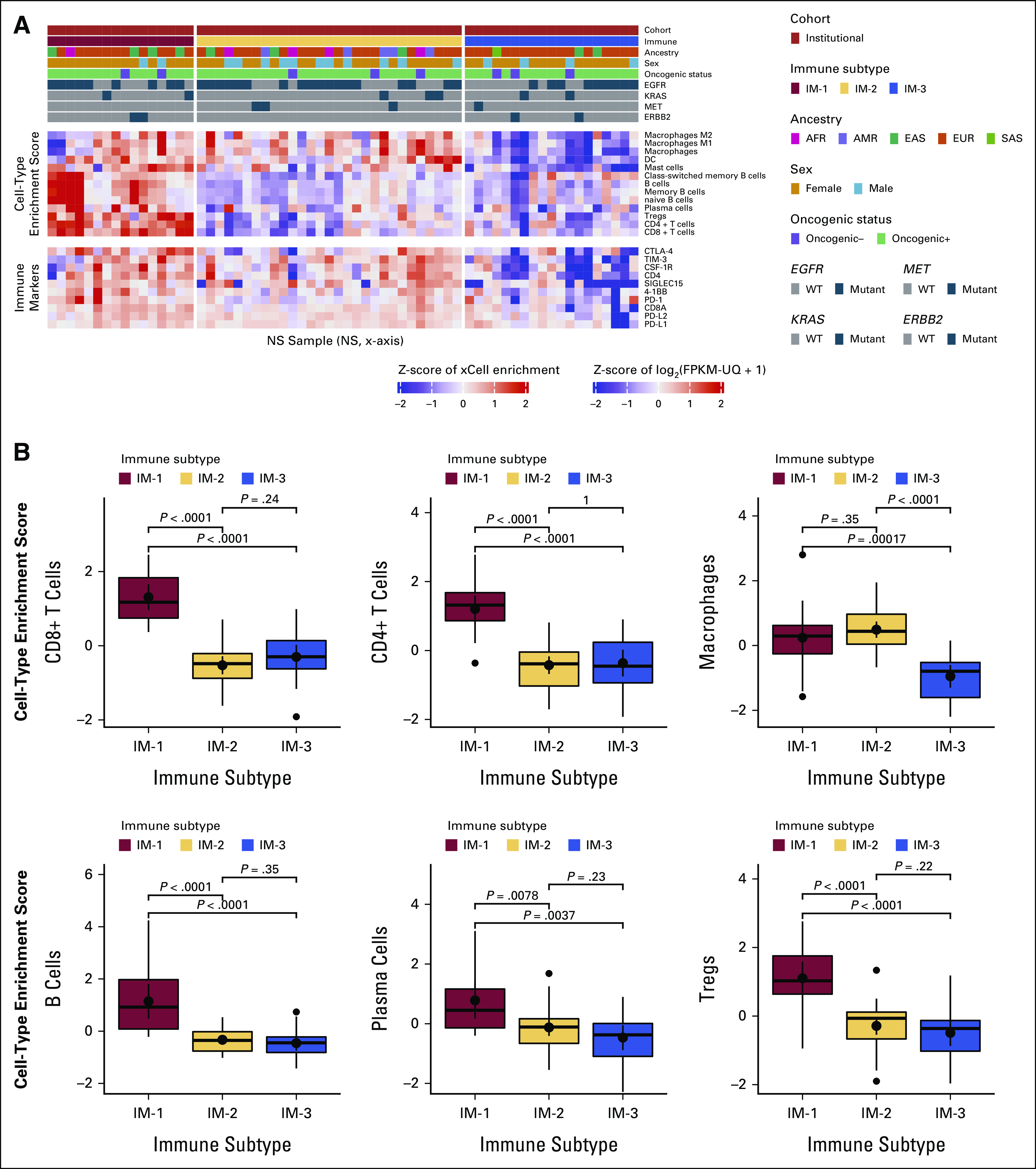
Identification of immune subtypes in NS by RNA-sequencing–based cell-type enrichment. (A) Consensus clustering of cell-type enrichment data and gene-level expression patterns of immune checkpoints and potential immunotherapy target genes in NS samples from the Institutional LUAD cohort (discovery cohort) shows three immune clusters—IM-1, IM-2, and IM-3. Each sample and the attributes (summarized in legend on top right), immune cell composition, and checkpoint molecule expression level (shown as Z-scores) are represented in individual columns on the x-axis. (B) Individual box plots showing differences in immune cell composition with the associated level of statistical significance between Institutional cohort samples from the three immune clusters. AFR, African; AMR, Admixed American; EUR, European; IM, immune subtype; LUAD, lung adenocarcinoma; NS, never-smoker; SAS, South Asian.
A similar pattern of clustering was observed for never-smoker samples in the External cohort, validating the observations made in the Institutional cohort (Data Supplement). The frequencies of driver mutations in EGFR (55.9%, 53.8%, and 51.9% in IM-1, IM-2, and IM-3 clusters, respectively) and KRAS (11.8%, 9.6%, and 7.4% in IM-1, IM-2, and IM-3 clusters, respectively), and TMB (median of 2/Mb, 1.97/Mb, and 2.03/Mb in IM-1, IM-2, and IM-3 clusters, respectively) were not particularly different between these clusters. A direct comparison of immune landscapes of smoker and never-smoker samples using RNA-sequencing data was not performed since such analyses are expected to be confounded by batch effects.
DISCUSSION
Our analyses indicate that the genomic features of smoking-related and never-smoker LUAD are largely comparable with activation of RTK/RAS/RAF signaling as a hallmark feature of LUAD.35 However, our results show that LUADs arising in never-smokers demonstrate a much higher prevalence of driver alterations in this pathway than those from smokers. Most importantly, the vast majority of these alterations are targetable. We were able to identify a driver alteration in nearly 80% of our never-smoker samples using a combination of WES and targeted deep sequencing. These findings support a role for comprehensive molecular testing to identify a driver alteration in never-smoker lung cancers. Furthermore, these results emphasize the need to procure biopsies with adequate tumor cellularity for sequencing, considering that patients with lung cancer often undergo fine needle aspirations that do not yield samples with adequate tumor cellularity for sequencing.
We observed pathogenic and likely pathogenic germline variants in cancer predisposing genes for approximately 7% of patients with never-smoker LUAD in the Institutional and External cohorts. Pathogenic or likely pathogenic germline alterations in the DNA repair genes such as BRCA1, BRCA2, FANCG, FANCM, MSH6, and POLD1 were exclusively mutated in never-smokers. Among these, BRCA2 mutations also demonstrated LOH in a tumor sample, further supporting a contributory role for the dysregulation of DNA repair in a subset of never-smoker LUADs. Germline alterations in DNA repair genes have previously been implicated as a risk factor for lung cancer by multiple groups.36 For instance, in a large study that used cell-free DNA testing on a limited gene panel and reported incidental germline alterations in 0.7% (33 of 4,459) of patients with lung cancer, mutations in BRCA2 were the most frequent (17 of 33 patients).37 Similar results were also reported by Mukherjee et al,38 who observed an increased frequency of germline alterations in patients with lung cancer with a family history of other cancers, early age at onset, or carrying a diagnosis of multiple cancers. Although these studies did not specifically examine the relationship between never-smoker lung cancer and alterations in DNA repair, findings from other studies have reported germline alterations in TP53, BRCA1, and BRCA2 in young patients (age < 45 years) with never-smoker lung cancer.39 Nevertheless, the overall prevalence of germline alterations was, surprisingly, comparable between never-smokers and smokers in our analysis. These results suggest that although germline alterations in known cancer predisposing genes may contribute to lung cancer in a very small subset of never-smokers—germline predisposition alone is unlikely to explain the pathogenesis of this disease. In this context, it is worth noting from our results that environmental exposures such as that of passive exposure to cigarette smoking—as inferred from mutation signature analysis—are also likely to predispose to lung cancer in never-smokers. However, analyses of this nature are limited by the fact that mutational signatures of different environmental carcinogens can show a considerable degree of overlap.31 Additionally, these results can also be biased by misclassification of smoker samples as never-smoker samples, and artifacts introduced by sample fixation and age.40
We also show that it is possible to categorize never-smoker LUADs into relatively immune cold (IM-3 and select IM-2 cluster samples) and hot (IM-1 cluster) subtypes. Compared with immunologically hot tumors, cold never-smoker tumors appeared to lack expression of immune markers that predict for response to immunotherapy, such as PD-L1, and were also depleted for the presence of immune cells, implying immune evasion through mechanisms that are not very well characterized. For instance, never-smoker LUADs in our study, overall, showed relatively lower TMBs and a higher frequency of mutations in genes such as CTNNB1, which participates in WNT signaling, compared with smoker samples. Activation of WNT signaling has been shown to facilitate immune evasion and contribute to immunotherapy resistance across cancers.41 Together, these results possibly explain the relatively lower response rates associated with immunotherapy targeting programmed cell death 1 and PD-L1 in never-smoker LUADs compared with smokers. Classifying never-smoker tumors into immune subtypes in future clinical trials is likely to provide a better understanding of biomarkers that predict for response to different types of immunotherapies in this patient population.
Overall, in this comprehensive analysis of never-smoker lung cancer samples, we report that the key genomic alterations between LUADs arising in smokers and never-smokers are largely similar, although they tend to differ in terms of their prevalence, mutational patterns, and immune cell infiltrates. Although a subset of never-smoker LUADs are likely to be driven by germline alterations or exposure to environmental carcinogens, the etiological underpinnings of this disease continue to remain unclear and will require additional studies using larger samples possibly through multiomic approaches involving tumor tissues and germline DNA analyses.
ACKNOWLEDGMENT
The authors would also like to thank Dr Elaine R. Mardis for reviewing and providing insights on an earlier version of this manuscript.
Humam Kadara
Research Funding: Johnson and Johnson
Irena Lanc
Employment: Gyroscope Therapeutics, Arch Oncology (I)
Saiama N. Waqar
Research Funding: Spectrum Pharmaceuticals, Lilly, Pfizer, Genentech/Roche, Daiichi Sankyo, Newlink Genetics, EMD Serono, Puma Biotechnology, Novartis, Xcovery, Synermore Biologics, Celgene, Vertex, Bristol Myers Squibb, Stem CentRx, Hengrui Therapeutics, Checkpoint Therapeutics, Ignyta, AstraZeneca, ARIAD, Roche, Merck
Daniel Morgensztern
Consulting or Advisory Role: Bristol Myers Squibb, AbbVie, Takeda, PharmaMar, Gilead Sciences, G1 Therapeutics, Lilly Medical
Research Funding: Heat Biologics, Merck, Celgene, AstraZeneca, Baxter, Incyte, AbbVie, Bristol Myers Squibb, EpicentRx, Pfizer, Roche, Lilly, Altum Pharmaceuticals, Array BioPharma, Surface Oncology
Jeffrey Ward
Employment: Millipore
Consulting or Advisory Role: Novocure, Guidepoint Inc
Travel, Accommodations, Expenses: Halozyme
Ashiq Masood
Honoraria: Bristol Myers Squibb, Boehringer Ingelheim
Speakers' Bureau: Bristol-Myers Squibb, Boehringer Ingelheim
Research Funding: Boston Biomedical, Ipsen, Seattle Genetics, Novocure, Macrogenics, Merck, Genentech/Roche, Exelixis, Astellas Pharma, Debiopharm Group, PRA Health, CytomX Therapeutics, Calithera Biosciences, Proteus Digital Health, Tempus
Shankha Satpathy
Employment: FOGPharma
Stock and Other Ownership Interests: FOGPharma
Patents, Royalties, Other Intellectual Property: Proteogenomic Methods for Diagnosing Cancer
Steven A. Carr
Stock and Other Ownership Interests: SEER, Kymera
Honoraria: Biogen
Consulting or Advisory Role: Kymera, SEER, PTM Biolabs
Patents, Royalties, Other Intellectual Property: I have several patents related to use of HLA peptides as vaccine candidates
Ignacio Wistuba
Consulting or Advisory Role: Genentech/Roche, Bristol Myers Squibb, HTG Molecular Diagnostics, Asuragen, Pfizer, AstraZeneca/MedImmune, GlaxoSmithKline, Guardant Health, Merck, MSD Oncology, Bayer, OncoCyte, Flame Biosciences
Speakers' Bureau: Pfizer, MSD Oncology, Roche, Merck, AstraZeneca
Research Funding: Genentech, Merck, HTG Molecular Diagnostics, Silicon Biosytems, Adaptimmune, EMD Serono. Pfizer, MedImmune, OncoPlex Diagnostics, Takeda, Karus Therapeutics, Amgen, 4D Molecular Therapeutics, Bayer, Novartis, Guardant Health, Adaptive Biotechnologies, Johnson & Johnson, Iovance Biotherapeutics, Akoya Biosciences
Harvey Pass
Honoraria: Genentech/Roche
Consulting or Advisory Role: Genentech/Roche, Novartis
Research Funding: Biodesix, Micronoma, NanoString Technologies, Celsius Therapeutics
Patents, Royalties, Other Intellectual Property: Patent Pending, use of fibulin for the diagnosis of mesothelioma; Patent Pending, use of HMGB1 for the diagnosis of mesothelioma, with University of Hawaii; Patent Pending, use of osteopontin for the diagnosis of mesothelioma, with Wayne State University
Travel, Accommodations, Expenses: Genentech/Roche
Ramaswamy Govindan
Honoraria: Genentech/AbbVie, AbbVie, Geneplus
Consulting or Advisory Role: Genentech/Roche, AbbVie, AstraZeneca/MedImmune, Pfizer, Bristol Myers Squibb, Nektar, Jounce Therapeutics, Roche, Janssen, Amgen, Achilles Therapeutics
No other potential conflicts of interest were reported.
SUPPORT
Supported by NIH-NHGRI (RKW, 2U54HG00307910). C.G. and H.P. are supported by funding from NCI-EDRN (U01CA214195) The Mesothelioma Biomarker Discovery Laboratory.
S.D., Y.L., F.M.R., R.K.W., L.D., and R.G. contributed equally to this work.
DATA SHARING STATEMENT
Deidentified data are provided in the Data Supplement. Sequencing data for external cohort samples, ie, TCGA and CPTAC, are available through previous publications (Campbell JD, Alexandrov A, Kim J, et al: Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607-616, 2016; and Gillette MA, Satpathy S, Cao S, et al: Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182:200-225.e35, 2020). Sequencing data for institutional cohort samples will be uploaded to dbGaP.
AUTHOR CONTRIBUTIONS
Conception and design: Siddhartha Devarakonda, Yize Li, Robert Fulton, Harvey Pass, Richard K. Wilson, Li Ding, Ramaswamy Govindan
Financial support: Richard K. Wilson, Li Ding
Administrative support: Richard K. Wilson, Ramaswamy Govindan
Provision of study materials or patients: Siddhartha Devarakonda, Saiama N. Waqar, Daniel Morgensztern, Jeffrey Ward, Ignacio Wistuba, Harvey Pass, Ramaswamy Govindan
Collection and assembly of data: Siddhartha Devarakonda, Yize Li, Fernanda Martins Rodrigues, Humam Kadara, Kymberlie Pepin, Daniel Morgensztern, Ashiq Masood, Robert Fulton, Ignacio Wistuba, Harvey Pass, Richard K. Wilson, Ramaswamy Govindan
Data analysis and interpretation: Siddhartha Devarakonda, Yize Li, Fernanda Martins Rodrigues, Sumithra Sankararaman, Humam Kadara, Chandra Goparaju, Irena Lanc, Saiama N. Waqar, Daniel Morgensztern, Jeffrey Ward, Michael A. Gillette, Shankha Satpathy, Steven A. Carr, Richard K. Wilson, Li Ding, Ramaswamy Govindan
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genomic Profiling of Lung Adenocarcinoma in Never-Smokers
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I =Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Humam Kadara
Research Funding: Johnson and Johnson
Irena Lanc
Employment: Gyroscope Therapeutics, Arch Oncology (I)
Saiama N. Waqar
Research Funding: Spectrum Pharmaceuticals, Lilly, Pfizer, Genentech/Roche, Daiichi Sankyo, Newlink Genetics, EMD Serono, Puma Biotechnology, Novartis, Xcovery, Synermore Biologics, Celgene, Vertex, Bristol Myers Squibb, Stem CentRx, Hengrui Therapeutics, Checkpoint Therapeutics, Ignyta, AstraZeneca, ARIAD, Roche, Merck
Daniel Morgensztern
Consulting or Advisory Role: Bristol Myers Squibb, AbbVie, Takeda, PharmaMar, Gilead Sciences, G1 Therapeutics, Lilly Medical
Research Funding: Heat Biologics, Merck, Celgene, AstraZeneca, Baxter, Incyte, AbbVie, Bristol Myers Squibb, EpicentRx, Pfizer, Roche, Lilly, Altum Pharmaceuticals, Array BioPharma, Surface Oncology
Jeffrey Ward
Employment: Millipore
Consulting or Advisory Role: Novocure, Guidepoint Inc
Travel, Accommodations, Expenses: Halozyme
Ashiq Masood
Honoraria: Bristol Myers Squibb, Boehringer Ingelheim
Speakers' Bureau: Bristol-Myers Squibb, Boehringer Ingelheim
Research Funding: Boston Biomedical, Ipsen, Seattle Genetics, Novocure, Macrogenics, Merck, Genentech/Roche, Exelixis, Astellas Pharma, Debiopharm Group, PRA Health, CytomX Therapeutics, Calithera Biosciences, Proteus Digital Health, Tempus
Shankha Satpathy
Employment: FOGPharma
Stock and Other Ownership Interests: FOGPharma
Patents, Royalties, Other Intellectual Property: Proteogenomic Methods for Diagnosing Cancer
Steven A. Carr
Stock and Other Ownership Interests: SEER, Kymera
Honoraria: Biogen
Consulting or Advisory Role: Kymera, SEER, PTM Biolabs
Patents, Royalties, Other Intellectual Property: I have several patents related to use of HLA peptides as vaccine candidates
Ignacio Wistuba
Consulting or Advisory Role: Genentech/Roche, Bristol Myers Squibb, HTG Molecular Diagnostics, Asuragen, Pfizer, AstraZeneca/MedImmune, GlaxoSmithKline, Guardant Health, Merck, MSD Oncology, Bayer, OncoCyte, Flame Biosciences
Speakers' Bureau: Pfizer, MSD Oncology, Roche, Merck, AstraZeneca
Research Funding: Genentech, Merck, HTG Molecular Diagnostics, Silicon Biosytems, Adaptimmune, EMD Serono. Pfizer, MedImmune, OncoPlex Diagnostics, Takeda, Karus Therapeutics, Amgen, 4D Molecular Therapeutics, Bayer, Novartis, Guardant Health, Adaptive Biotechnologies, Johnson & Johnson, Iovance Biotherapeutics, Akoya Biosciences
Harvey Pass
Honoraria: Genentech/Roche
Consulting or Advisory Role: Genentech/Roche, Novartis
Research Funding: Biodesix, Micronoma, NanoString Technologies, Celsius Therapeutics
Patents, Royalties, Other Intellectual Property: Patent Pending, use of fibulin for the diagnosis of mesothelioma; Patent Pending, use of HMGB1 for the diagnosis of mesothelioma, with University of Hawaii; Patent Pending, use of osteopontin for the diagnosis of mesothelioma, with Wayne State University
Travel, Accommodations, Expenses: Genentech/Roche
Ramaswamy Govindan
Honoraria: Genentech/AbbVie, AbbVie, Geneplus
Consulting or Advisory Role: Genentech/Roche, AbbVie, AstraZeneca/MedImmune, Pfizer, Bristol Myers Squibb, Nektar, Jounce Therapeutics, Roche, Janssen, Amgen, Achilles Therapeutics
No other potential conflicts of interest were reported.
REFERENCES
- 1.Samet JM, Avila-Tang E, Boffetta P, et al. : Lung cancer in never smokers: Clinical epidemiology and environmental risk factors. Clin Cancer Res 15:5626-5645, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subramanian J, Govindan R: Lung cancer in never smokers: A review. J Clin Oncol 25:561-570, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Broderick P, Webb E, et al. : Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet 40:1407-1409, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Broderick P, Matakidou A, et al. : Role of 5p15.33 (TERT-CLPTM1L), 6p21.33 and 15q25.1 (CHRNA5-CHRNA3) variation and lung cancer risk in never-smokers. Carcinogenesis 31:234-238, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Hsiung CA, Lan Q, Hong Y-C, et al. : The 5p15.33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS Genet 6:e1001051, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Sheu C-C, Ye Y, et al. : Genetic variants and risk of lung cancer in never smokers: A genome-wide association study. Lancet Oncol 11:321-330, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonner MR, Bennett WP, Xiong W, et al. : Radon, secondhand smoke, glutathione-S-transferase M1 and lung cancer among women. Int J Cancer 119:1462-1467, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Wenzlaff AS, Cote ML, Bock CH, et al. : GSTM1, GSTT1 and GSTP1 polymorphisms, environmental tobacco smoke exposure and risk of lung cancer among never smokers: A population-based study. Carcinogenesis 26:395-401, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Bell DW, Gore I, Okimoto RA, et al. : Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 37:1315-1316, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Ohtsuka K, Ohnishi H, Kurai D, et al. : Familial lung adenocarcinoma caused by the EGFR V843I germ-line mutation. J Clin Oncol 29:e191-e192, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto H, Higasa K, Sakaguchi M, et al. : Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst 106:djt338, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Govindan R, Ding L, Griffith M, et al. : Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150:1121-1134, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research Network : Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543-550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network : Comprehensive genomic characterization of squamous cell lung cancers. Nature 489:519-525, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell JD, Alexandrov A, Kim J, et al. : Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607-616, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imielinski M, Berger AH, Hammerman PS, et al. : Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150:1107-1120, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillette MA, Satpathy S, Cao S, et al. : Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182:200-225.e35, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrot-Zhang J, Yao X, Devarakonda S, et al. : Whole-genome characterization of lung adenocarcinomas lacking the RTK/RAS/RAF pathway. Cell Rep 34:108707, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagemann IS, Devarakonda S, Lockwood CM, et al. : Clinical next-generation sequencing in patients with non–small cell lung cancer. Cancer 121:631-639, 2015 [DOI] [PubMed] [Google Scholar]
- 20.Genomic Data Commons (GDC) : https://gdc.cancer.gov/
- 21.Bailey MH, Tokheim C, Porta-Pardo E, et al. : Comprehensive characterization of cancer driver genes and mutations. Cell 173:371-385.e18, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, et al. : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405-424, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott AD, Huang K-L, Weerasinghe A, et al. : CharGer: Clinical characterization of germline variants. Bioinformatics 35:865-867, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karczewski KJ, Francioli LC, Tiao G, et al. : The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434-443, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin Y, Yao L, King EE, et al. : Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 42:229-233, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao L, Schiavi F, Cascon A, et al. : Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 304:2611-2619, 2010 [DOI] [PubMed] [Google Scholar]
- 27.van der Heijden MS, Yeo CJ, Hruban RH, et al. : Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res 63:2585-2588, 2003 [PubMed] [Google Scholar]
- 28.Mermel CH, Schumacher SE, Hill B, et al. : GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12:R41, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov LB, Kim J, Haradhvala NJ, et al. : The repertoire of mutational signatures in human cancer. Nature 578:94-101, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kucab JE, Zou X, Morganella S, et al. : A compendium of mutational signatures of environmental agents. Cell 177:821-836.e16, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazieres J, Drilon A, Lusque A, et al. : Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann Oncol 30:1321-1328, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gainor JF, Rizvi H, Jimenez Aguilar E, et al. : Clinical activity of programmed cell death 1 (PD-1) blockade in never, light, and heavy smokers with non-small-cell lung cancer and PD-L1 expression ≥50%. Ann Oncol 31:404-411, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aran D, Hu Z, Butte AJ: xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18:220, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desai TJ, Brownfield DG, Krasnow MA: Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507:190-194, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukuya T, Takahashi K: Germline mutations in lung cancer. Respir Investig 57:201-206, 2019 [DOI] [PubMed] [Google Scholar]
- 37.Slavin TP, Banks KC, Chudova D, et al. : Identification of incidental germline mutations in patients with advanced solid tumors who underwent cell-free circulating tumor DNA sequencing. J Clin Oncol 36:JCO1800328, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukherjee S, Zauderer MG, Ravichandran V, et al. : Frequency of actionable cancer predisposing germline mutations in patients with lung cancers. J Clin Oncol 36, 2018. (suppl 15; abstr 1504) [Google Scholar]
- 39.Donner I, Katainen R, Sipilä LJ, et al. : Germline mutations in young non-smoking women with lung adenocarcinoma. Lung Cancer 122:76-82, 2018 [DOI] [PubMed] [Google Scholar]
- 40.Do H, Dobrovic A: Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin Chem 61:64-71, 2015 [DOI] [PubMed] [Google Scholar]
- 41.Luke JJ, Bao R, Sweis RF, et al. : WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 25:3074-3083, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data are provided in the Data Supplement. Sequencing data for external cohort samples, ie, TCGA and CPTAC, are available through previous publications (Campbell JD, Alexandrov A, Kim J, et al: Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet 48:607-616, 2016; and Gillette MA, Satpathy S, Cao S, et al: Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182:200-225.e35, 2020). Sequencing data for institutional cohort samples will be uploaded to dbGaP.