

PURPOSE
Outcomes in RAS-mutant metastatic colorectal cancer (mCRC) remain poor and patients have limited therapeutic options. Adavosertib is the first small-molecule inhibitor of WEE1 kinase. We hypothesized that aberrations in DNA replication seen in mCRC with both RAS and TP53 mutations would sensitize tumors to WEE1 inhibition.
METHODS
Patients with newly diagnosed mCRC were registered into FOCUS4 and tested for TP53 and RAS mutations. Those with both mutations who were stable or responding after 16 weeks of chemotherapy were randomly assigned 2:1 between adavosertib and active monitoring (AM). Adavosertib (250 mg or 300 mg) was taken orally once on days 1-5 and days 8-12 of a 3-week cycle. The primary outcome was progression-free survival (PFS), with a target hazard ratio (HR) of 0.5 and 80% power with a one-sided 0.025 significance level.
RESULTS
FOCUS4-C was conducted between April 2017 and Mar 2020 during which time 718 patients were registered; 247 (34%) were RAS/TP53-mutant. Sixty-nine patients were randomly assigned from 25 UK hospitals (adavosertib = 44; AM = 25). Adavosertib was associated with a PFS improvement over AM (median 3.61 v 1.87 months; HR = 0.35; 95% CI, 0.18 to 0.68; P = .0022). Overall survival (OS) was not improved with adavosertib versus AM (median 14.0 v 12.8 months; HR = 0.92; 95% CI, 0.44 to 1.94; P = .93). In prespecified subgroup analysis, adavosertib activity was greater in left-sided tumors (HR = 0.24; 95% CI, 0.11 to 0.51), versus right-sided (HR = 1.02; 95% CI, 0.41 to 2.56; interaction P = .043). Adavosertib was well-tolerated; grade 3 toxicities were diarrhea (9%), nausea (5%), and neutropenia (7%).
CONCLUSION
In this phase II randomized trial, adavosertib improved PFS compared with AM and demonstrates potential as a well-tolerated therapy for RAS/TP53-mutant mCRC. Further testing is required in this sizable population of unmet need.
INTRODUCTION
Targeting the cellular DNA damage response (DDR) has been an effective therapeutic strategy in several tumor sites, including ovarian and pancreatic cancer.1,2 These agents can be used as monotherapy in cancers with defective DDR, where we might anticipate a synthetic lethality interaction: two pathways together perform an essential function, and the loss of one pathway (eg, because of mutation) is tolerated but the loss of both pathways leads to cell death.3
CONTEXT
Key Objective
To test if adavosertib, which is a small-molecule inhibitor of the WEE1 kinase, is effective as monotherapy in patients with RAS/TP53-mutant metastatic colorectal cancer (mCRC) as maintenance therapy following induction chemotherapy.
Knowledge Generated
In this phase II randomized trial, adavosertib was well-tolerated and improved progression-free survival in RAS/TP53-mutant mCRC compared with active monitoring. Treatment effect may be affected by primary tumor location and KRAS subtype, with greater benefit seen in left-sided cancers and those with KRAS codon 12/13 mutations. RAS/TP53 subgroup is a distinct moderately poor prognostic population.
Relevance
Adavosertib is a promising therapeutic agent in patients with RAS/P53-mutant mCRC, a poor prognostic population of unmet need, and was well-tolerated. This study demonstrates the potential of targeting the DNA damage response pathway in mCRC, which should be a research priority. Future studies of adavosertib should stratify patient outcomes according to primary tumor location and RAS subtype.
WEE1 is a nuclear tyrosine kinase that has a central role in cell cycle regulation, including being the key regulator of the G2/M checkpoint through actions on CDK1,4 optimizing DNA-histone stoichiometry before mitotic entry4 and modulation of CDK1/2 during the intra-S phase to block replication initiation.5 Inhibition of WEE1 causes unscheduled entry into mitosis, aberrant firing of replication origins leading to dNTP (Dithiobis [5-nitropyridine]) shortage and replication stress,4 and accumulation of DNA damage during S phase, leading to increased reliance on the G1/S checkpoint.4 Adavosertib (AZD1775) is the first small-molecule inhibitor of WEE1 kinase and has been tested in combination with chemotherapy and radiotherapy6,7 but more recently as monotherapy to generate synthetic lethality in tumors with DDR defects.6
There has been limited investigation into agents targeting the DDR in metastatic colorectal cancer (mCRC), mainly because of the lack of systematic identification of alterations in DDR genes.8 Here, we test adavosertib in RAS- and TP53-mutant (RAS/TP53-mut) mCRC, which we hypothesize would be sensitive to WEE1 inhibition. TP53 is a key regulator of the G1/S checkpoint9; loss of function leads to dependence on the intra-S and G2/M checkpoints to detect DNA damage and initiate repair.10 In preclinical studies, AZD1775 possessed preferential killing effect in TP53-deficient compared with TP53 wild-type tumors.11 Mutant RAS, as well as recognized actions through downstream mitogen-activated protein kinase B (MAPK-AKT) pathway signaling, also drives cell cycle progression leading to replication stress during S phase.12 In preclinical studies, mutant RAS drives cells into S phase through regulation of the CDK4 or CDK6 complex and provides sustained mitogenic signals through sustained CDK2 activity. These effects activate the replication stress response including checkpoint activation.13 Theoretically, RAS/TP53-mut tumors will be highly vulnerable to adavosertib, with G1 checkpoint failure, evidence of replication stress, and reliance on the intra-S phase and G2/M checkpoints.
The FOCUS4 trial program was an adaptive molecularly stratified umbrella platform trial that evaluated the safety and efficacy of novel treatments in targeted biomarker subgroups within a phase II/III trial setting in the interval after 16 weeks of first-line therapy of mCRC. The design has been published separately,14 and the trial schema, registration, and biomarker methods are provided in the Data Supplement (online only). Here, we report the findings of FOCUS4-C, which tested the safety and efficacy of adavosertib in patients with RAS/TP53-mut mCRC compared with active monitoring (AM) and has achieved disease stability following induction chemotherapy.
METHODS
Trial Approvals, Patient Eligibility, and Recruitment
The trial and subsequent amendments were approved by the UK National Ethics Committee Oxford—Panel C (reference 13/SC/0111) and by the relevant regulatory body MHRA (CTA No. 20363/0400/001 and EudraCT No. 2012-005111-12).
Patients age more than 18 years with newly diagnosed mCRC were registered into the FOCUS4 trial program, while undergoing induction chemotherapy, from a total of 88 UK hospitals. Following registration, a tumor sample was tested using next generation sequencing platform for stratification into molecular subtypes including BRAF, PIK3CA, TP53, and RAS mutations (Fig 1 and Data Supplement). Patients were required to provide written informed consent for both tissue testing and entry into any of the randomized sub-trials including FOCUS4-C.
FIG 1.

Flowchart of patients through the trial. CR, complete response; ITT, intention-to-treat; PPA, per-protocol analysis; PR, partial response; SD, stable disease.
Patients were randomly assigned into the FOCUS4-C trial in a subset of 25 hospitals between July 2017 and March 2020. Patients were eligible if their tumor had both RAS and TP53 mutations and they had disease stability or response as assessed by computed tomography (CT) scan at the end of 16 weeks of induction chemotherapy, at which point the chemotherapy ceased and the patient was randomly assigned. Patients required a baseline CT scan 4 weeks before random assignment, a minimum 3-week washout period between the last dose of chemotherapy or biologic therapy and the first dose of adavosertib, adequate renal (creatinine clearance > 50 mL/min) and liver function, a WHO performance status of 0-1, and no evidence of prolonged QT interval on ECG.
Trial Procedures
Adavosertib was supplied by AstraZeneca Ltd (Cambridge, UK); packaging, labeling, and distribution were undertaken by Fisher Services (Horsham, UK). Patients randomly assigned to adavosertib continued the drug until disease progression, death, or intolerable toxicity. The first 21 patients received adavosertib 250 mg once daily, on days 1-5 and 8-12 of a 3-week cycle. The next 23 patients received adavosertib 300 mg once daily, on the same schedule. Patients took an oral 5HT3 antagonist with each dose, and oral dexamethasone 4 mg was given on day 1 and day 8 of each cycle unless clinically contraindicated.
Because of the mandatory supportive medication for nausea and vomiting for which a placebo was not available, blinding was not possible, and AM was used as the control arm. Patients randomly assigned to AM followed the same follow-up schedule and remained off any other anticancer treatment until clinical or radiologic evidence of disease progression.
Patient tumor status was assessed at the treating hospital every 8 weeks by CT scan, according to RECIST, version 1.1.15 Toxicities and symptoms were assessed locally every 4 weeks, using the National Cancer Institute's Common Terminology Criteria for Adverse Events (version 3.0). Patients remained on trial until disease progression occurred, at which point the patient was recommended to restart the same chemotherapy that was used in the induction phase. Treatment was stopped in the event of grade 3 or worse toxic effects or persistent toxicities judged medically significant or not tolerated by the patient, until the toxicity resolved to grade 1 or better.
Statistical Methods
A full description of the statistical methods is provided in the Data Supplement. In summary, patients were allocated to either adavosertib or AM, using a 2:1 allocation ratio by minimization with a 20% random element. All analyses were performed according to a predefined statistical analysis plan using Stata (version 16.1; Stata Corporation, TX). The primary outcome measure was progression-free survival (PFS), and the prespecified primary efficacy analysis was a per-protocol analysis (PPA) using Cox regression adjusting for minimization factors. Intention-to-treat (ITT) and unadjusted models were also performed as secondary analyses. Sample size calculations were based upon a target hazard ratio (HR) of 0.5 with 80% power and .025 one-sided alpha requiring a target of 26 PFS events in the control arm for final analysis.
RESULTS
Recruitment and Patient Characteristics
The FOCUS4 trial program ran between January 2014 and March 2020. FOCUS4-C ran between April 2017 and March 2020, during which time 817 patients were registered, of whom 718 underwent successful biomarker profiling (Fig 1 and Data Supplement). Two hundred forty-seven patients (34%) had tumors confirmed with both RAS and TP53 mutations (RAS/TP53-mut). Of these, 151 had stable or responding disease after 16 weeks of first-line treatment and 69 were randomly assigned using a 2:1 ratio: 44 to adavosertib and 25 to AM. Of the remaining eligible 82, two chose to be randomly assigned into the concurrent FOCUS4-N trial and others chose not to be randomly assigned into FOCUS4 for reasons such as toxicity from first-line therapy or patient-clinician choice to seek alternative pathways.
Table 1 summarizes the patient baseline characteristics. There were some minor imbalances, which are corrected for in the adjusted analysis (primary model). There were no differences in the frequency of other molecular alterations between the groups. There were no significant differences between the registration period chemotherapy regimens in the adavosertib and AM arms.
TABLE 1.
Baseline Patient Characteristics by Randomized Group

Primary Analysis: PFS (per-protocol)
Five patients were excluded from the PPA: four did not start treatment (adavosertib arm) and one was subsequently found to have had progressive disease at the point of random assignment (AM arm). One patient was censored early when they received fluorouracil as anticancer treatment before progression (AM arm).
Within the primary PPA (n = 64), there were 40 of 40 PFS events in the adavosertib arm and 22 of 24 in the AM arm. Patients treated with adavosertib had a longer PFS than those on AM (3.61 v 1.87 months). Both unadjusted HR (0.52; 95% CI, 0.30 to 0.89; P = .022) and adjusted HR (0.35; 95% CI, 0.18 to 0.68; P = .0022) were statistically significant. Kaplan-Meier curves are provided in Figure 2.
FIG 2.
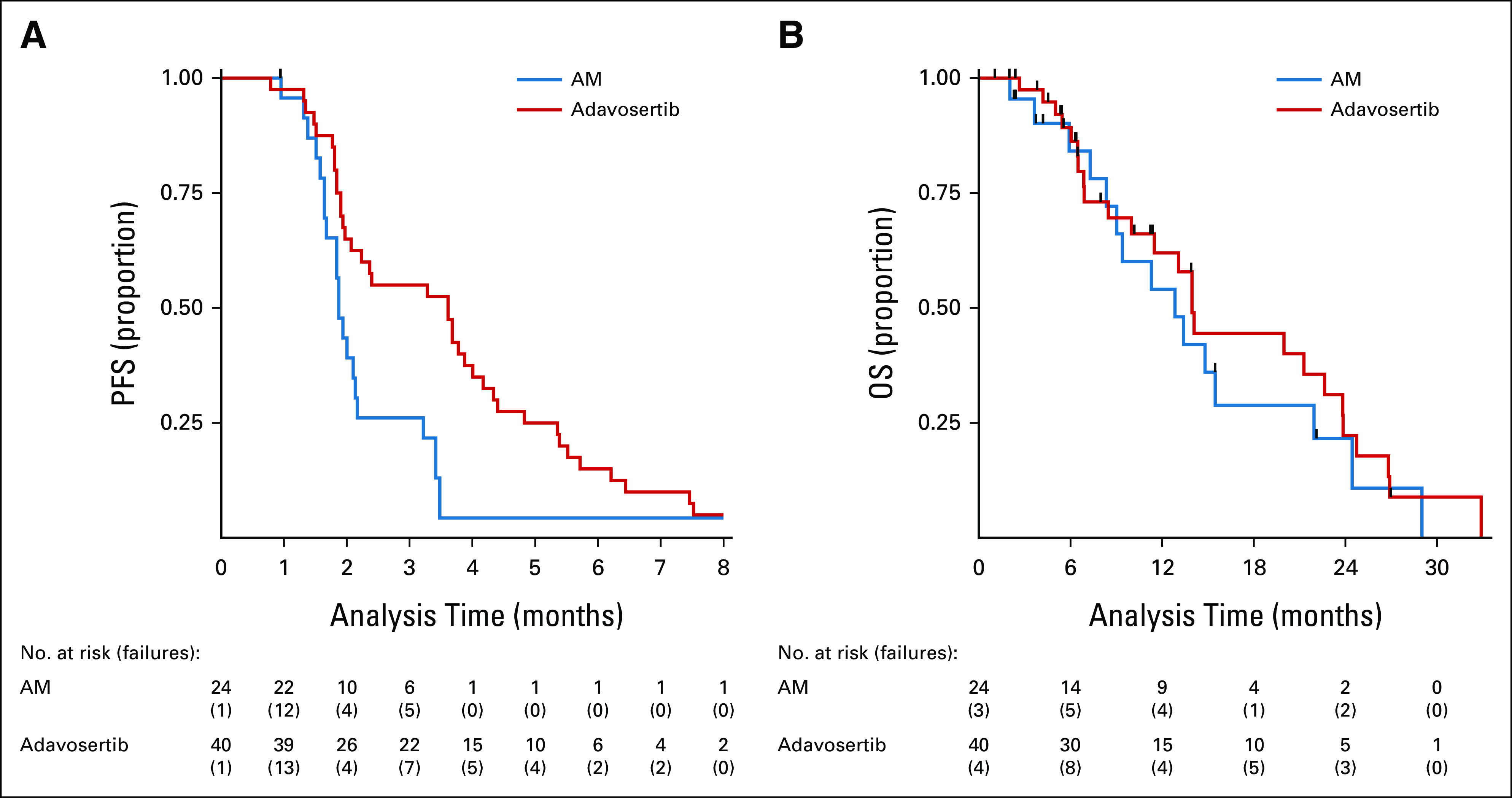
(A) PFS (primary analysis) in PPA population: Cox regression, adjusted for minimization factors—HR = 0.35 (95% CI, 0.18 to 0.68), P = .0022. Minimization factors: location of primary tumor (left, right, and rectum), baseline WHO performance status, baseline disease assessment, number of metastases, and first-line therapy (fluoropyrimidine, oxaliplatin or irinotecan, and monoclonal antibody). (B) OS (secondary analysis) in PPA population: Cox regression, adjusted for minimization factors—HR = 0.86 (95% CI, 0.39 to 1.86), P = .70. Minimization factors: location of primary tumor (left, right, and rectum), baseline WHO performance status, baseline disease assessment, number of metastases, and first-line therapy (fluoropyrimidine, oxaliplatin or irinotecan, and monoclonal antibody). AM, active monitoring; HR, hazard ratio; OS, overall survival; PFS, progression-free survival; PPA, per-protocol analysis.
PFS (ITT)
All patients were included in the ITT analysis, but four patients were censored the day after random assignment: three in the adavosertib arm (two because of patient withdrawal and one without any post–random assignment CT scan assessments) and one in the AM arm without any post–random assignment CT scan assessments.
There were 41 of 44 PFS events in the adavosertib arm and 23 of 25 in the AM arm. Consistent with the PPA, the ITT PFS analysis shows a PFS advantage with adavosertib over AM in both the unadjusted (HR = 0.55; 95% CI, 0.32 to 0.94; P = .032) and adjusted analyses (HR = 0.40; 95% CI, 0.21 to 0.75; P = .0051).
Overall Survival (ITT)
There were 27 of 44 deaths in the adavosertib arm and 16 of 25 in the AM arm. There was no significant overall survival (OS) benefit with adavosertib compared with AM (median survival 14.0 v 12.8 months; unadjusted HR = 0.79; 95% CI, 0.42 to 1.48, P = .47; adjusted HR = 0.92; 95% CI, 0.44 to 1.94, P = .93; Fig 2).
Tumor Control
Adavosertib was associated with a higher proportion of patients with disease control compared with AM (47% v 28% at any time during the trial), including one patient with a documented partial response to adavosertib (Data Supplement).
Subgroup Analyses
The impact of adavosertib versus AM on PFS was explored in prespecified subgroups (Fig 3). The most marked difference in effect was for primary tumor location (PTL): patients with a right PTL had no PFS advantage with adavosertib compared with AM (1.87 v 1.91 months; HR = 1.02; 95% CI, 0.41 to 2.56), whereas those with a left PTL did (3.61 v 1.87 months, HR = 0.24; 95% CI, 0.11 to 0.51; interaction P = .043; Data Supplement).
FIG 3.
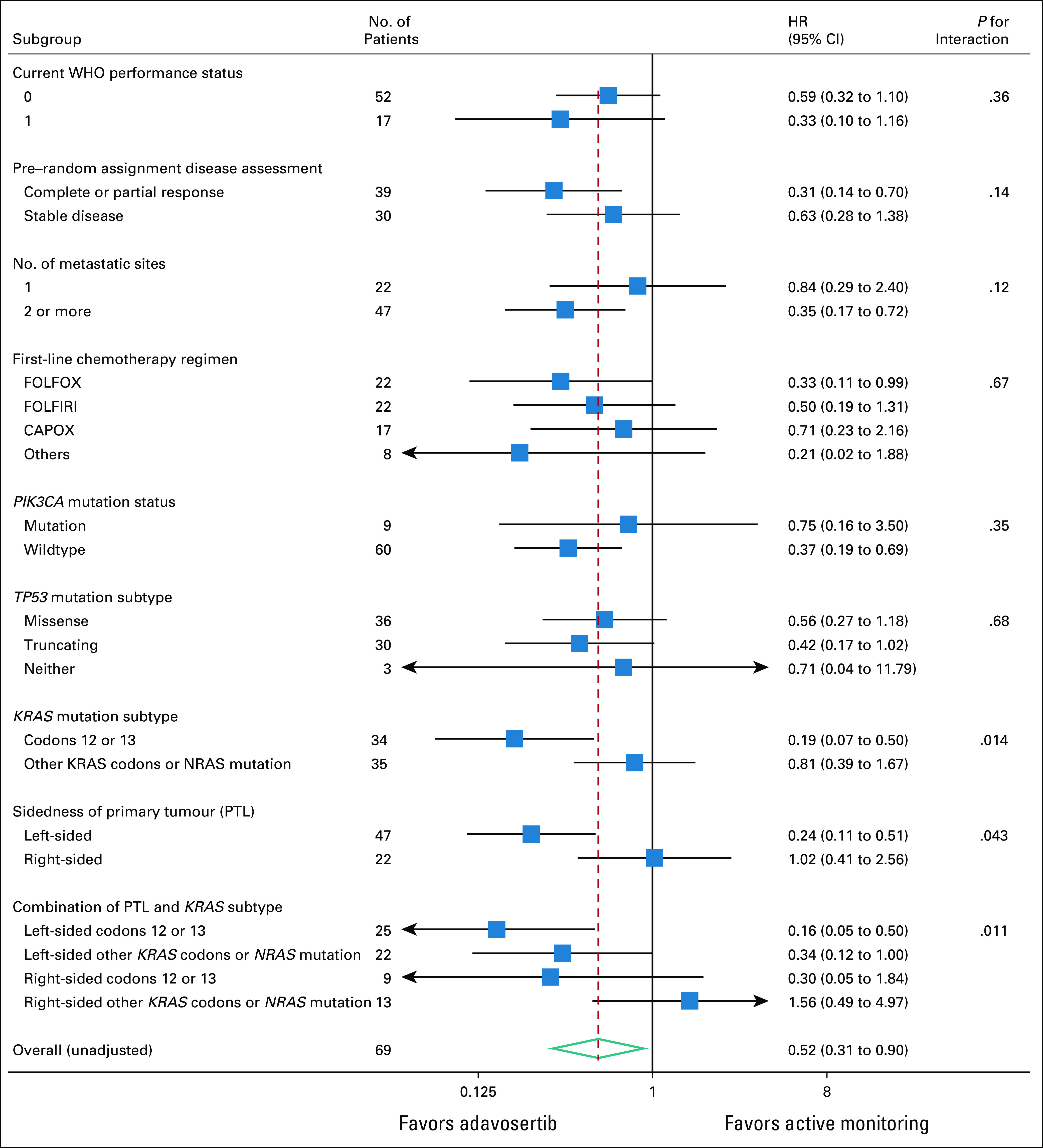
Subgroup analyses for PFS by intention to treat. CAPOX, capecitabine and oxaliplatin; FOLFIRI, fluorouracil, leucovorin, and irinotecan; FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; PFS, progression-free survival; PTL, primary tumor location.
This prompted an unplanned subgroup analysis of PTL on OS, and although the numbers of events were low, the interaction was even more marked (Data Supplement). Median OS was 14.1 versus 11.3 months for adavosertib versus AM in left PTL (adjusted HR = 0.37; 95% CI, 0.15 to 0.87) but was 6.5 versus 15.5 months in right PTL (HR = 6.5; 95% CI, 0.72 to 6.43; interaction P = .0032). In terms of response, 38% of right-sided adavosertib patients versus 42% of right-sided AM patients reported disease stability or response at least once while on trial, whereas for left-sided tumors, the figures were 53% versus 19%.
Patients who had responded to induction chemotherapy (v stable disease) and who had two or more metastatic sites appeared to benefit more from adavosertib, albeit to a lesser degree (interaction P value = .14 for response to induction; P = .12 for number of metastatic sites; Fig 3).
External Analyses to Further Characterize the RAS/TP53-Mut Biomarker Population
The RAS/TP53-mutant population has not been previously described. To understand the prognostic implication of this alteration, we analyzed the outcomes of a subset (n = 438) of patients from the FOCUS trial in whom the S:CORT consortium had analyzed a wider panel of CRC genes including KRAS, NRAS, BRAF, MSI, and TP53. The RAS/RAF wild-type group was the reference population (median OS 21.6 months). The RAS/TP53-mutant population is distinct from either mutation alone (RAS or TP53) and had a worse prognosis than either in isolation with a median OS of 14.9 months (HR = 2.06; 95% CI, 1.08 to 3.93; P = .028; Fig 4). This suggests that the RAS/TP53-mut population is a poor-prognosis subgroup but not as marked as for patients with a BRAF mutation or microsatellite instability-high tumor.
FIG 4.

Prognostic impact of biomarker subgroups on OS in previous FOCUS trial. HR, hazard ratio; MSI-H, microsatellite instability-high; OS, overall survival.
These data are consistent with the finding that during the registration period of FOCUS4, 33% of patients in the RAS/TP53-mut population experienced progression during the first 16 weeks of chemotherapy. This is similar to the rate in the BRAF-mutant group (34% progressed) but higher than that seen in RAS-mutant (24%) and all wild-type (22%) subgroups (Data Supplement).
Effect of RAS and TP53 Mutation Subtypes on Adavosertib Activity
We observed that patients with KRAS codon 12/13 mutations had a significant benefit from adavosertib (P for interaction = .014; Fig 3), whereas no detectable benefit was observed in those with KRAS mutations at other codons or with NRAS mutation. Furthermore, the interaction effects of KRAS subtype and of PTL on PFS may be additive as there is a significant benefit from adavosertib within the subgroup of left PTL KRAS codon 12/13 subtypes (HR = 0.16; 95% CI, 0.05 to 0.50) and a clear disbenefit within the subgroup of right PTL noncodon 12/13 subtypes (HR = 1.56; 95% CI, 0.49 to 4.97; Data Supplement). The subtype of TP53 mutation or the co-occurrence of PIK3CA mutation did not affect outcome.
Toxicity and Compliance
There was good compliance with randomized allocation, and adavosertib was generally well-tolerated (Fig 5 and Data Supplement). Compared with AM, adavosertib was associated with increased reported toxicity (≥ grade 1), most notably increased frequency of diarrhea (61% v 28%), fatigue (75% v 56%), nausea (68% v 32%), and vomiting (41% v 4%). However, the majority of such toxicity was of low grade, with 9% in the adavosertib arm reporting diarrhea of ≥ grade 3, 11% fatigue, 5% nausea, and 2% vomiting, versus none of each in the AM arm. As described, during the trial, there was an increase in the dose of adavosertib from 250 mg to 300 mg. The higher dose was associated with an increased frequency of grade 3 diarrhea (14% v 4%), but otherwise the toxicity profile was similar, and with similar rates of dose modifications and delays.
FIG 5.
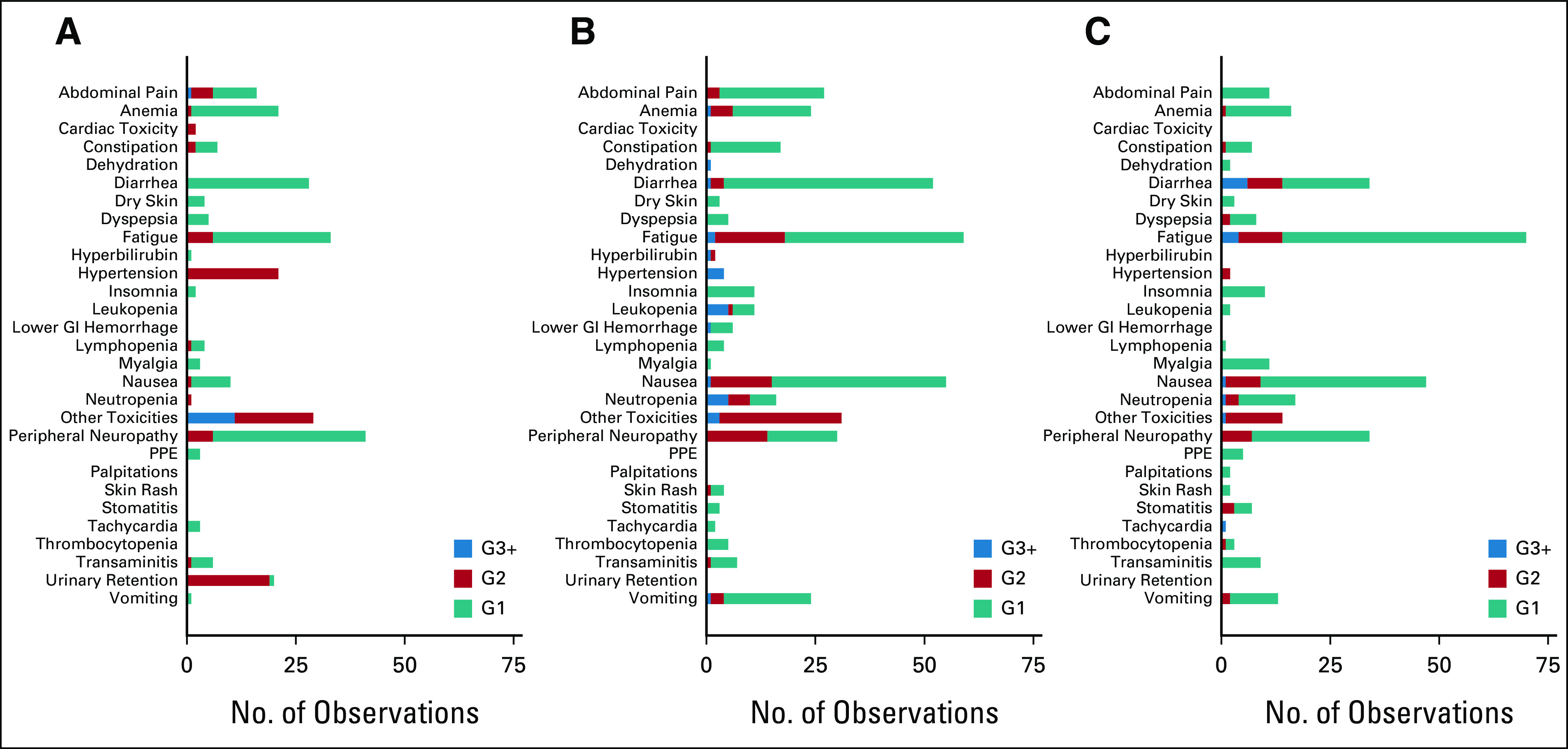
Cumulative reported toxicity, within FOCUS4-C treatment groups and with initial AZD1775 doses separated: (A) active monitoring (n = 25), (B) AZD1775 250 mg (n = 23), and (C) AZD1775 300 mg (n = 21). G, grade; PPE, palmar plantar erythema.
Impact of Adavosertib Dosing
As described, during the trial, there was an increase in the dose of adavosertib from 250 mg to 300 mg. PFS was 2.2 months (HR = 0.58; 95% CI, 0.31 to 1.06) with the 250-mg dose and 3.7 months (HR = 0.47; 95% CI, 0.25 to 0.89) with the 300-mg dose; this difference was nonsignificant (P = .48; Data Supplement). Between the 250-mg and 300-mg doses, there was an increased frequency of grade 3 diarrhea (4% v 14%), but otherwise the toxicity profile was similar. There were similar rates of dose modifications between the 250-mg and 300-mg doses: dose delays (16% v 7%), dose reductions (4% v 5%), and dose omissions (19% v 17%). A swimmer plot integrating the effects of adavosertib dose, randomized group, and PTL on PFS is shown in the Data Supplement.
DISCUSSION
Here, we have reported that FOCUS4-C met its primary end point; patients with RAS/TP53-mutant mCRC had PFS advantage with adavosertib compared with AM following induction chemotherapy. These results are particularly encouraging as RAS/TP53-mutant mCRC is a poor prognostic population with limited treatment options. Adavosertib was well-tolerated at both doses evaluated.
The overarching aim of the FOCUS4 trial program was to test novel agents efficiently with specified biomarker subgroups in mCRC with the multi-arm, multi-stage design allowing for an early signal of drug inactivity14; thus, any demonstrated efficacy would require further confirmatory study to lead to practice change. FOCUS4-C represents a success of this approach, efficiently demonstrating promising activity of adavosertib within patients with RAS/P53-mutant mCRC, and will directly influence research practice in mCRC.
The intermittent treatment strategy used in FOCUS4 follows the demonstration of no detriment in OS in the MRC COIN trial. This is now further substantiated by an individual participant data meta-analysis.16 Thus, AM is an accepted standard of care following a few months of first-line therapy. FOCUS4 was specifically designed to use this window following first-line induction chemotherapy to test novel agents in specified biomarker groups, before the evolution of multiple resistance mechanisms.14
A prespecified analysis demonstrated that adavosertib activity was limited to left colon and rectal PTL, with little activity observed in right PTL. Having observed the significant subgroup effects on PFS, we investigated possible impact on OS. It is provocative to see that in the left-sided tumors, OS was significantly improved with median OS from random assignment increasing from 11.3 months to 14.1 months (HR = 0.40; 95% CI, 0.17 to 0.97). There is also a possibility of adverse effect on outcome in patients with right PTL. However, the number of patients and events was limited and thus, any conclusions need to be cautious in relation to this observed effect on OS in both subgroups. Differences in CRC by PTL are well-documented, in terms of biology, prognosis, and treatment response,17 but the mechanisms for differences of treatment efficacy by PTL are not well-understood.
An exploratory analysis showed that adavosertib had the most PFS effect in patients with KRAS codons 12/13/TP53-mutant tumors, with lesser activity in those with extended KRAS, or NRAS mutations; functional differences between RAS isoforms are documented.18 Despite the small sample sizes in FOCUS4-C, the PTL and RAS subtypes subgroup analyses showed interactions significant at the 5% level.
Although these subgroup analyses provide provocative results, we lack a mechanistic explanation for these differences in adavosertib effect; ongoing translational work shall investigate this. We would recommend that further clinical development of adavosertib in the RAS/TP53-mut mCRC population should not be limited by PTL or RAS subtype but should include close monitoring of patients with right PTL and extended RAS mutations to ensure that neither futility nor detriment are observed.
Although the clinical implications of the RAS/TP53 mutation in mCRC are not well-studied, each alteration is individually well-characterized. Here, we have shown that the double RAS/TP53-mutant subgroup carries a moderately poor prognosis (Fig 4) and appears to confer a worse prognosis than either mutation in isolation. This biomarker subgroup has thus shown distinct prognostic and therapeutic relevance and so merits further study in translational work, existing data sets, and ongoing therapeutic trials in mCRC.
Adavosertib has demonstrated an acceptable safety profile; the main toxicity was diarrhea. Efficacy was noted at both the 250-mg and 300-mg doses, with a suggestion of additional activity with the higher dose. We would therefore recommend the 300-mg dosing to progress to further clinical studies in fit patients. However, in the treatment-refractory setting, the 250-mg dose may be more tolerable.
There are limitations to this study. We considered, and would have preferred, a placebo-controlled design; however, at the time of launching FOCUS4-C, high rates of nausea and vomiting had been observed in other adavosertib trials and high-dose steroid antiemetics were considered necessary. For this reason, both clinicians and patient representatives considered a placebo design unfeasible. Given the favorable safety data for single-agent adavosertib in FOCUS4-C, placebo-controlled design could be considered in the future. It is possible therefore that the PFS effect observed was influenced by investigator and patient preference to restart first-line chemotherapy sooner in the AM arm. However, a marked difference in effect was observed between the right and left PTL groups treated with adavosertib, suggesting a lesser effect on the primary analysis because of this potential bias. Additionally, the PFS end point was not centrally reviewed, but assessed in individual sites by RECIST criteria. A further limitation is that by testing adavosertib in the maintenance setting and requiring stability following induction chemotherapy, we have excluded the RAS/TP53 patients with the worse outcome. We therefore cannot generalize the effect of adavosertib within this entire biomarker group.
In conclusion, adavosertib (AZD1775) has demonstrated promising activity compared with AM in patients with RAS/TP53-mut mCRC. This treatment benefit may relate to PTL and KRAS subtype. Given this clear demonstration of efficacy in an RCT and acceptable toxicity profile, future clinical development of adavosertib is warranted particularly as it may represent a future treatment opportunity in this sizable population of unmet need.
ACKNOWLEDGMENT
The full list of FOCUS4 Trial Investigators can be found in Appendix 1.
APPENDIX 1. Full Authorship List for the FOCUS4 Trial Investigators
Writing Committee for FOCUS4-C Trial
Seligmann J.F., Fisher D.J., Brown L.C., Adams R.A., Graham J., Quirke P., Richman S.D., Butler R., Domingo E., Blake A., Yates E., Braun M., Collinson F., Jones R., Brown E., De Winton E., Humphrey T.C., Parmar M., Kaplan R., Wilson R.H., Seymour M., and Maughan T.S.
Trial Management Group
Maughan T.S. (Chair), Wilson R.H., Adams R.A., Seymour M., Seligmann J.F., Graham J., Wasan H., Pope M., Pope J., Samuel L., Shiu K., Church D., Middleton G., Steward W., Twelves C., Wellman S., Hodgkinson E., Stoner N., Beety J., Duggleby K., Dutton G., MRC CTU team (below), and laboratory staff (see below).
MRC CTU Trial Coordinating Center
Brown L.C., Kaplan R., Parmar M., Fisher D.J., Campos M., Yates E., Santana S., Fiddament A., Harper L., Bara A., Pugh C., Bathia R., Letchemanan K., Przybył B., Bhogal S., Gopalakrishnan G., Purvis C., Diaz-Montana C., Mohamed F., Townsend S., Cragg W., Masters L., Van Looy N., and Rauchenberger M.
Laboratories
Quirke P., Richman S.D., Hemmings G., Davis J., Gallop N., Wilkinson L., Butler R., Roberts H., Jasani B., White R., Dodds R., James M., and Morgan M.
Independent Data-Monitoring Committee
Cameron D. (Chair), Souhami R., Peeters M., Billingham C., Griffiths G., and Brown J.
Trial Steering Committee
Johnson P. (Chair), Rudd R., Whelan J., and Russell A.
Participating Hospitals in Descending Order of the Number of Patients Registered With All Staff Listed (N = 2,076)

Jenny F. Seligmann
Honoraria: Pierre Fabre, Merck Serono, Servier
Consulting or Advisory Role: Pierre Fabre, Roche Molecular Diagnostics
Travel, Accommodations, Expenses: Bristol Myers Squibb/Medarex
Louise C. Brown
Research Funding: AstraZeneca
Richard A. Adams
Honoraria: Merck Serono, Servier, Amgen
Consulting or Advisory Role: Merck Serono, Amgen, Servier, Bayer
Speakers' Bureau: Merck Serono, AstraZeneca, Merck Sharp & Dohme
Travel, Accommodations, Expenses: Servier, Amgen, Merck Serono, AstraZeneca
Janet Graham
Honoraria: Merck Serono, Bristol Myers Squibb, Nucana, Bayer
Consulting or Advisory Role: Merck KGaA
Travel, Accommodations, Expenses: Nucana
Philip Quirke
Honoraria: Roche
Consulting or Advisory Role: Roche, Adlai Nortye, Avacta Life Sciences, Amgen, Bayer
Research Funding: Roche, GeneFirst, ONI
Patents, Royalties, Other Intellectual Property: Roche has filed a patent jointly with my University; I may receive income but none to date
Travel, Accommodations, Expenses: Roche, Bayer, Amgen
Rachel Butler
Speakers' Bureau: Bayer, Pfizer, Roche, AstraZeneca, MSD, Illumina, Lilly, Novartis, Amgen, Boehringer Ingelheim
Michael Braun
Honoraria: Servier, Amgen, Pierre Fabre
Travel, Accommodations, Expenses: Servier
Fiona Collinson
Research Funding: Bristol Myers Squibb
Rob Jones
Honoraria: Astellas Pharma, Janssen, AstraZeneca, MSD Oncology, Bristol Myers Squibb, Pfizer, Novartis, Ipsen, Seattle Genetics, Sanofi, Bayer, Roche/Genentech, EUSA Pharma, Pharmacyclics, Clovis Oncology, Merck Serono, Eisai
Consulting or Advisory Role: Clovis Oncology
Research Funding: Roche, Astellas Pharma, AstraZeneca, Pfizer, Novartis, Exelixis, Clovis Oncology, Bayer
Travel, Accommodations, Expenses: Ipsen, Bayer, Janssen, Astellas Pharma, MSD
Ewan Brown
Research Funding: MSD Oncology
Timothy C. Humphrey
Research Funding: Debiopharm Group
Mahesh Parmar
Research Funding: AstraZeneca, Astellas Pharma, Janssen, Clovis Oncology
Richard Kaplan
Research Funding: AstraZeneca
Richard Wilson
Honoraria: Servier, Amgen, Bristol Myers Squibb
Consulting or Advisory Role: Amgen, CV6 Therapeutics, Pierre Fabre, Amphista Therapeutics, Nucana
Travel, Accommodations, Expenses: Amgen, Pierre Fabre
Matthew Seymour
Research Funding: Amgen, AstraZeneca
Timothy S. Maughan
Consulting or Advisory Role: AstraZeneca, Vertex, Pierre Fabre
Research Funding: AstraZeneca, PsiOxus Therapeutics, Merck KgAA, Almac Diagnostics
No other potential conflicts of interest were reported.
SUPPORT
FOCUS4-C was cofunded by the MRC/NIHR Efficacy and Mechanism Evaluation (EME) program and CRUK with funding commencing in April 2013. Drug supply and distribution of adavosertib and an educational grant were provided by AstraZeneca Ltd. The MRC Clinical Trials Unit at UCL receives core funding from the Medical Research Council within UKRI (Budget: MC_UU_12023/20).
CLINICAL TRIAL INFORMATION
J.F.S., L.C.B., and D.F. contributed equally to this work as first authors.
M.S. and T.S.M. contributed equally to this work as last authors.
DATA SHARING STATEMENT
Individual deidentified participant data (including data dictionaries) can be shared upon appropriate application to the MRC CTU at any time from full publication. Study protocols and statistical analysis plan have been provided in the Data Supplement with this manuscript. Going forward, it is proposed that data will be shared with an appropriate international collaborative repository to enable future IPD meta-analysis.
AUTHOR CONTRIBUTIONS
Conception and design: Jenny F. Seligmann, David J. Fisher, Louise C. Brown, Richard A. Adams, Philip Quirke, Timothy C. Humphrey, Mahesh Parmar, Richard Kaplan, Richard Wilson, Matthew Seymour, Timothy S. Maughan
Administrative support: Emma Yates
Provision of study materials or patients: Jenny F. Seligmann, Janet Graham, Philip Quirke, Susan D. Richman, Michael Braun, Fiona Collinson, Emma de Winton, Richard Wilson
Collection and assembly of data: Jenny F. Seligmann, David J. Fisher, Louise C. Brown, Richard A. Adams, Janet Graham, Susan D. Richman, Rachel Butler, Andrew Blake, Emma Yates, Emma de Winton, Richard Kaplan, Richard Wilson, Matthew Seymour, Timothy S. Maughan
Data analysis and interpretation: Jenny F. Seligmann, David J. Fisher, Louise C. Brown, Richard A. Adams, Janet Graham, Philip Quirke, Enric Domingo, Andrew Blake, Michael Braun, Fiona Collinson, Rob Jones, Ewan Brown, Mahesh Parmar, Richard Kaplan, Richard Wilson, Matthew Seymour, Timothy S. Maughan
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Jenny F. Seligmann
Honoraria: Pierre Fabre, Merck Serono, Servier
Consulting or Advisory Role: Pierre Fabre, Roche Molecular Diagnostics
Travel, Accommodations, Expenses: Bristol Myers Squibb/Medarex
Louise C. Brown
Research Funding: AstraZeneca
Richard A. Adams
Honoraria: Merck Serono, Servier, Amgen
Consulting or Advisory Role: Merck Serono, Amgen, Servier, Bayer
Speakers' Bureau: Merck Serono, AstraZeneca, Merck Sharp & Dohme
Travel, Accommodations, Expenses: Servier, Amgen, Merck Serono, AstraZeneca
Janet Graham
Honoraria: Merck Serono, Bristol Myers Squibb, Nucana, Bayer
Consulting or Advisory Role: Merck KGaA
Travel, Accommodations, Expenses: Nucana
Philip Quirke
Honoraria: Roche
Consulting or Advisory Role: Roche, Adlai Nortye, Avacta Life Sciences, Amgen, Bayer
Research Funding: Roche, GeneFirst, ONI
Patents, Royalties, Other Intellectual Property: Roche has filed a patent jointly with my University; I may receive income but none to date
Travel, Accommodations, Expenses: Roche, Bayer, Amgen
Rachel Butler
Speakers' Bureau: Bayer, Pfizer, Roche, AstraZeneca, MSD, Illumina, Lilly, Novartis, Amgen, Boehringer Ingelheim
Michael Braun
Honoraria: Servier, Amgen, Pierre Fabre
Travel, Accommodations, Expenses: Servier
Fiona Collinson
Research Funding: Bristol Myers Squibb
Rob Jones
Honoraria: Astellas Pharma, Janssen, AstraZeneca, MSD Oncology, Bristol Myers Squibb, Pfizer, Novartis, Ipsen, Seattle Genetics, Sanofi, Bayer, Roche/Genentech, EUSA Pharma, Pharmacyclics, Clovis Oncology, Merck Serono, Eisai
Consulting or Advisory Role: Clovis Oncology
Research Funding: Roche, Astellas Pharma, AstraZeneca, Pfizer, Novartis, Exelixis, Clovis Oncology, Bayer
Travel, Accommodations, Expenses: Ipsen, Bayer, Janssen, Astellas Pharma, MSD
Ewan Brown
Research Funding: MSD Oncology
Timothy C. Humphrey
Research Funding: Debiopharm Group
Mahesh Parmar
Research Funding: AstraZeneca, Astellas Pharma, Janssen, Clovis Oncology
Richard Kaplan
Research Funding: AstraZeneca
Richard Wilson
Honoraria: Servier, Amgen, Bristol Myers Squibb
Consulting or Advisory Role: Amgen, CV6 Therapeutics, Pierre Fabre, Amphista Therapeutics, Nucana
Travel, Accommodations, Expenses: Amgen, Pierre Fabre
Matthew Seymour
Research Funding: Amgen, AstraZeneca
Timothy S. Maughan
Consulting or Advisory Role: AstraZeneca, Vertex, Pierre Fabre
Research Funding: AstraZeneca, PsiOxus Therapeutics, Merck KgAA, Almac Diagnostics
No other potential conflicts of interest were reported.
REFERENCES
- 1. Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moore K, Colombo N, Scambia G, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379:2495–2505. doi: 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 3. Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 4. Beck H, Nähse-Kumpf V, Larsen MS, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aarts M, Sharpe R, Garcia-Murillas I, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 6. Lheureux S, Cristea MC, Bruce JP, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397:281–292. doi: 10.1016/S0140-6736(20)32554-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cuneo KC, Morgan MA, Sahai V, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019;37:2643–2650. doi: 10.1200/JCO.19.00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reilly NM, Novara L, Di Nicolantonio F, et al. Exploiting DNA repair defects in colorectal cancer. Mol Oncol. 2019;13:681–700. doi: 10.1002/1878-0261.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000;33:261–274. doi: 10.1046/j.1365-2184.2000.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–519. [PubMed] [Google Scholar]
- 11. Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murcia L, Clemente-Ruiz M, Pierre-Elies P, et al. Selective killing of RAS-malignant tissues by exploiting oncogene-induced DNA damage. Cell Rep. 2019;28:119–131.e4. doi: 10.1016/j.celrep.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 13. Fikaris AJ, Lewis AE, Abulaiti A, et al. Ras triggers ataxia-telangiectasia-mutated and Rad-3-related activation and apoptosis through sustained mitogenic signaling. J Biol Chem. 2006;281:34759–34767. doi: 10.1074/jbc.M606737200. [DOI] [PubMed] [Google Scholar]
- 14. Kaplan R, Maughan T, Crook A, et al. Evaluating many treatments and biomarkers in oncology: A new design. J Clin Oncol. 2013;31:4562–4568. doi: 10.1200/JCO.2013.50.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumors: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16. Adams R, Goey K, Chibaudel B, et al. Treatment breaks in first line treatment of advanced colorectal cancer: An individual patient data meta-analysis. Cancer Treat Rev. 2021;99:102226. doi: 10.1016/j.ctrv.2021.102226. [DOI] [PubMed] [Google Scholar]
- 17. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumor side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28:1713–1729. doi: 10.1093/annonc/mdx175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Individual deidentified participant data (including data dictionaries) can be shared upon appropriate application to the MRC CTU at any time from full publication. Study protocols and statistical analysis plan have been provided in the Data Supplement with this manuscript. Going forward, it is proposed that data will be shared with an appropriate international collaborative repository to enable future IPD meta-analysis.