

SUMMARY
Myeloid-derived suppressor cells (MDSCs) impede antitumor immunity; however, the precise mechanisms that regulate their suppressive function remain unresolved. Identifying these mechanisms could lead to therapeutic interventions to boost cancer immunotherapy efficacy. Here, we reveal that β2 adrenergic receptor (β2-AR) expression on MDSCs increases with tumor growth and that the β2-AR stress pathway drives the immune suppressive activity of MDSCs by altering their metabolism. We show that β2-AR signaling decreases glycolysis and increases oxidative phosphorylation and fatty acid oxidation (FAO). It also increases expression of the fatty acid transporter CPT1A, which is necessary for the FAO-mediated immunosuppressive function of MDSCs. Moreover, we show that β2-AR signaling increases autophagy and activates the arachidonic acid cycle, both required for increasing the release of the immunosuppressive mediator, PGE2. Our data reveal that β2-AR signaling triggered by stress is an important physiological regulator of key metabolic pathways in MDSCs, driving their immunosuppressive function.
Graphical Abstract
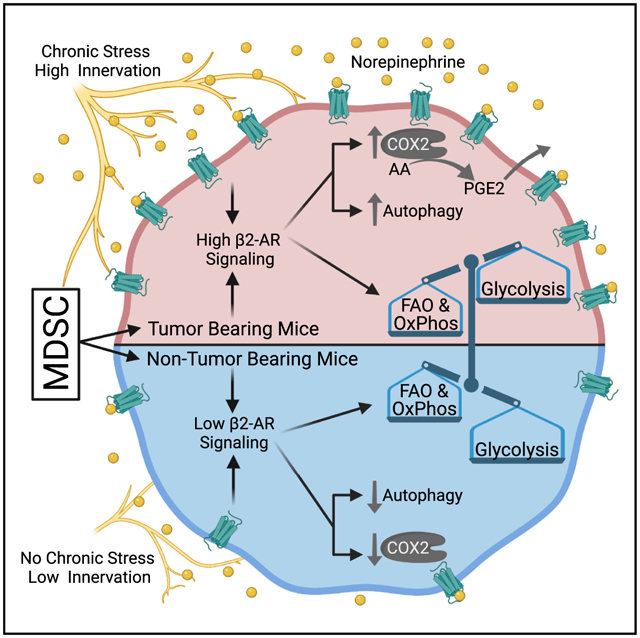
In brief
Mohammadpour et al. show that β2-AR signaling in MDSCs alters their metabolic state and increases their immunosuppressive function. Specific processes found to be increased include fatty acid oxidation, oxidative phosphorylation, and autophagy. In addition, these metabolic alterations facilitate an increase in PGE2 production via elevated COX2 expression.
INTRODUCTION
The production of myeloid-derived suppressor cells (MDSCs) from bone marrow-derived precursors increases during tumor growth (Veglia et al., 2018, 2021) and during repair of normal tissue damage (Nachmany et al., 2019; Ou et al., 2015). In addition to their ability to directly inhibit CD8+ T cell activity, MDSCs can mobilize regulatory T cells (Tregs), contributing further to forming a highly immunosuppressive network within the tumor microenvironment (TME) (Ostrand-Rosenberg and Fenselau, 2018; Pawelec et al., 2021). Enhanced accumulation of MDSCs within the TME is thought to occur due to a change in the complex balance of tumor-promoting factors derived from tumor or stromal cells and those factors that inhibit tumor growth through the modulation of antitumor immunity, such as interferon-γ (Groth et al., 2019). Targeting any one determinant of this highly complex network, however, continues to present significant therapeutic challenges. Therefore, a better understanding of the mechanisms that regulate the immunosuppressive function of MDSCs is needed. While multiple mechanisms are likely at play, considerable interest has been devoted recently to dissecting the role and impact of metabolic pathways in regulating MDSC function (Fleming et al., 2018; Hu et al., 2020; Won et al., 2019). As an example, fatty acid oxidation (FAO) is now known to play an important role in driving the immunosuppressive function of MDSCs (Yan et al., 2019). However, the factors that regulate FAO in MDSCs remain incompletely understood, which could lead to the identification of novel therapeutic interventions to target MDSCs to improve antitumor immunity.
To that end, our prior work revealed a previously unrecognized role for the stress-signaling receptor, β-adrenergic receptor (β-AR), in the antitumor immune response. We previously have shown that mice housed at room temperature (22°C) instead of 30°C (the preferred temperature for a mouse), experience continuous cold stress (Kokolus et al., 2013), resulting in β2-AR activation in different cells, including MDSCs (Mohammadpour et al., 2019). We also showed that β-AR signaling promotes tumor growth in part through enhancing MDSC accumulation and their immunosuppressive function (Mohammadpour et al., 2019). This finding is especially disconcerting, given that patients with cancer frequently experience considerably higher levels of chronic stress following their diagnosis and during treatment (Abuatiq et al., 2020). Since both β-AR signaling and FAO enhance MDSC suppressive function, we sought to determine whether β2-AR signaling alters the metabolic state of MDSCs and whether their immunosuppressive function is regulated through a novel β2-AR-FAO axis.
RESULTS
β2-AR signaling in MDSCs increases with tumor burden
To evaluate this relationship, we sought to further determine the kinetics of β2-AR surface expression on MDSCs using the MDSC-proficient 4T1 mammary tumor model. As expected, MDSC accumulation increased within tumors, as assessed through day 21 (Figure 1A). Interestingly, we discovered that the expression of the β2-AR on splenic polymorphonuclear (PMN)-MDSCs (the prominent MDSC subpopulation in the 4T1 model) also significantly increased with tumor burden (Figure 1B). We also used the EL-4 tumor model and analyzed the expression of the β2-AR on MDSCs. Similar to the 4T1 model, we found that the expression of β2-AR increased on both PMN-MDSCs and M-MDSCs in the spleen and PMN-MDSCs in the blood of tumor-bearing mice compared to naive mice (Figure 1C). Comparing these murine model findings with human data, we found that the frequency of MDSCs significantly increased in the blood of patients with cancer compared to healthy donors (Figure 1D). These data also demonstrated that the level of β2-AR expression on MDSCs from patients with cancer is significantly higher than that of healthy donor controls (Figure 1E). Healthy donor and patient demographics have been summarized in Table S1, and the gating strategy to identify human MDSCs is presented in Figure S1. The increase in β2-AR expression is known to result from increased downstream signaling when high levels of systemic norepinephrine (NE) are also present to promote receptor internalization and signaling cascade activation (Vistein and Puthenveedu, 2013). In addition, it has been shown that an increase in NE results in the heightened expression of the β2-AR (Mohammadpour et al., 2019; Ramos and Arnsten, 2007). Prior data from our laboratory also has shown that systemic levels of NE are increased with tumor burden (Bucsek et al., 2017). Our new analysis demonstrates that an increase in β2-AR expression results in an increase in β2-AR signaling in MDSCs, as well as providing a novel, positive feedback loop, resulting in a further increase in β2-AR signaling.
Figure 1. The expression of β2-AR in MDSCs increases with tumor growth.
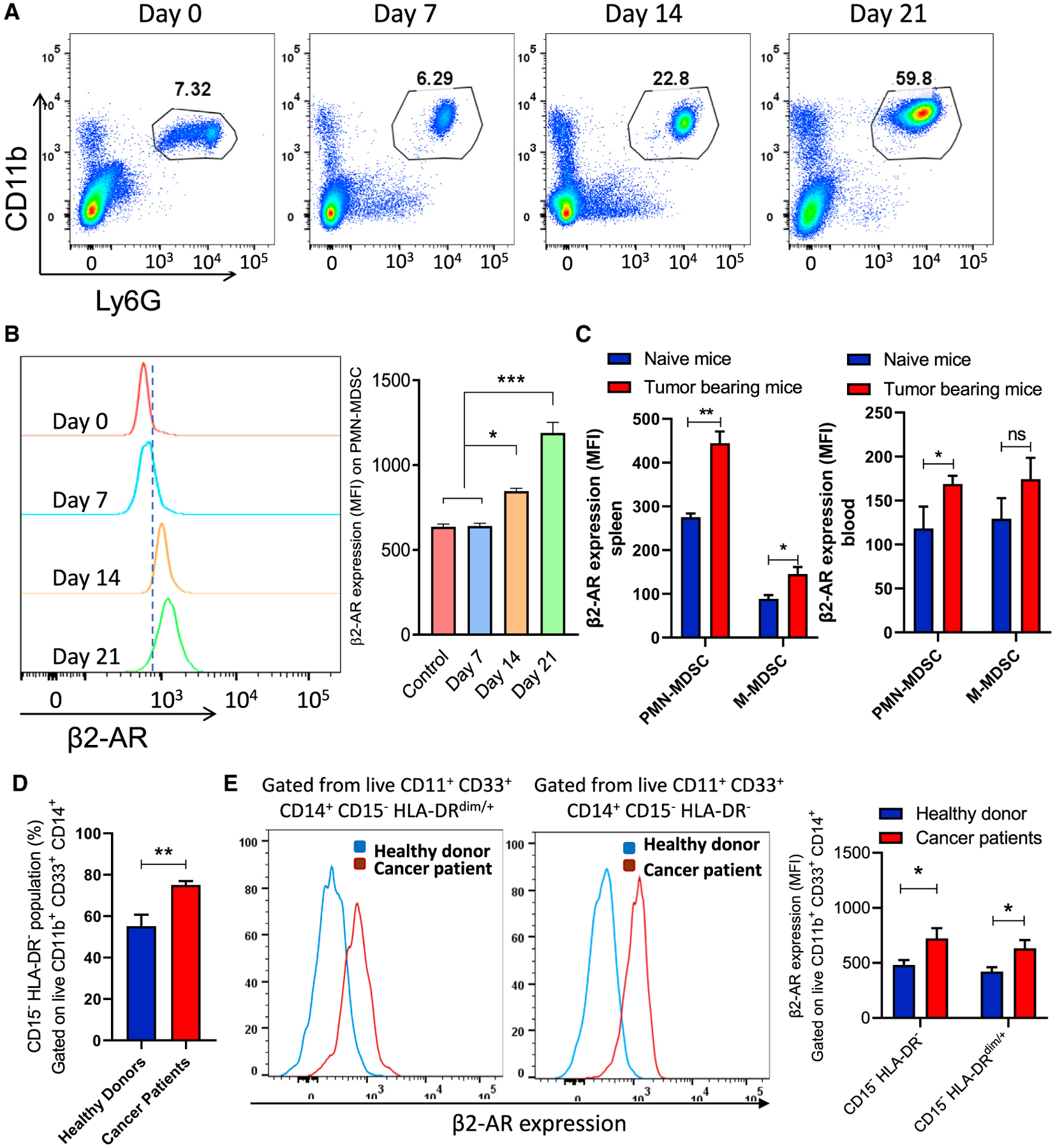
(A) Representative flow cytometry analysis of the PMN-MDSC subpopulation percentage in the spleen of 4T1 tumor-bearing mice at different time points after tumor injection.
(B) Histogram representative of β2-AR expression in PMN-MDSCs (gated on live CD45+ CD11b+ Ly6G+ cells) from spleens of 4T1 tumor-bearing mice in different time points (n = 5).
(C) The expression of β2-AR in MDSCs from the spleen and blood of EL-4 tumor-bearing mice (n = 5) at day 25 after tumor implantation.
(D) The frequency of MDSCs in healthy donors and patients with cancer.
(E) The expression of β2-AR on MDSCs in healthy donors (n = 5) and patients with cancer (n = 5). Mouse data are presented as means ± SDs from 3 biological replicates in all of the graphs, and 1-way ANOVA was used to analyze statistical significance between >2 groups.
In all of the panels, *p < 0.05, **p < 0.01, and ***p < 0.001. p < 0.05 was considered significant.
β2-AR signaling in MDSCs decreases glycolysis and enhances oxidative phosphorylation
We then tested whether β2-AR signaling is linked to altered MDSC metabolism. MDSCs were sorted from tumor-bearing wild-type (WT) or β2-AR−/− mice, and the NanoString Metabolic Pathways Panel was used to evaluate changes in the expression of key genes that inform the metabolic profile of cells (Figure S2). In addition to increasing the expression of genes related to the immunosuppressive function of MDSCs, such as Prkaa2, Aspg, Nos1, Ccl2, Cd276, and Prdx1 (Alshetaiwi et al., 2020; Ouzounova et al., 2017), the metabolic signature between WT and β2-AR−/− MDSCs was notably different. We found that AMPK signaling (Prkaa2), the tricarboxylic acid (TCA) cycle (Acacb, Pck1, Ogdhl), FAO (Cpt1a), fatty acid degradation (Ehhadh, Cyp4a, Adh7, Adh1), and arachidonic acid (Ptgs2) metabolism was significantly higher in WT MDSCs compared to β2-AR−/− MDSCs (Figure S2), suggesting that β2-AR signaling in MDSCs is associated with a shift toward greater oxidative phosphorylation and lipid metabolism.
To gain a better understanding of the role of β2-AR expression in MDSC metabolism, MDSCs were derived from bone marrow with granulocyte macrophage-colony-stimulating factor (GM-CSF) and interleukin-6 (IL-6) in the presence or absence of isoproterenol (ISO) (Figure S3). Extracellular flux analysis was then used to evaluate the metabolic profile of these MDSCs. We measured the extracellular acidification rate (ECAR) and determined that the non-glycolytic acidification rate was increased in response to ISO, while the glycolytic rate and glycolytic capacity were decreased (Figures S4A–S4D), mainly due to the difference in basal levels between ISO-treated MDSCs and control MDSCs. In conjunction with these findings, the oxygen consumption rate (OCR) data indicated that ISO treatment also increased basal respiration, ATP production, and maximum respiration (Figures 2A–2D). To determine the role of β2-AR signaling in FAO, MDSCs were cultured in media containing only palmitate as a source of fatty acid substrate for their metabolism. We found that oxidative consumption of this specific fatty acid was increased by ISO treatment in WT MDSCs (Figures 2E and 2F). Fatty acid uptake and accumulation of neutral lipids were measured by Bodipy FL C16 and Bodipy FL 493/503, respectively, by using flow cytometry, and we found that fatty acid uptake and Bodipy staining of lipid droplets were elevated due to ISO treatment (Figures 2G and 2H). This same OCR analysis was performed using MDSCs isolated from 4T1 tumor-bearing WT and β2-AR−/− mice, and a similar trend was observed. β2-AR−/− MDSCs compared to WT MDSCs showed decreased basal respiration, ATP production, and maximum respiration (Figures 2I–2L). β2-AR−/− MDSCs have decreased levels of fatty acid consumption (Figures 2M and 2N) and fatty acid uptake (Figure 2O) when compared to WT MDSCs (Figures 2M and 2N). We found that the mitochondrial mass and mitochondrial potential were significantly higher in WT MDSCs treated with ISO versus WT MDSCs treated with PBS (Figures S5A and S5B) and decreased in β2-AR−/− MDSCs compared to WT MDSCs (Figures S5C and S5D). These data indicate that β2-AR signaling in MDSCs increases oxidative phosphorylation and FAO in vitro and in vivo, suggesting that blocking β2-AR using an antagonist such as propranolol could be a viable option to prevent the metabolic reprogramming of MDSCs.
Figure 2. β-AR signaling in MDSCs decreases glycolysis and enhances oxidative phosphorylation.
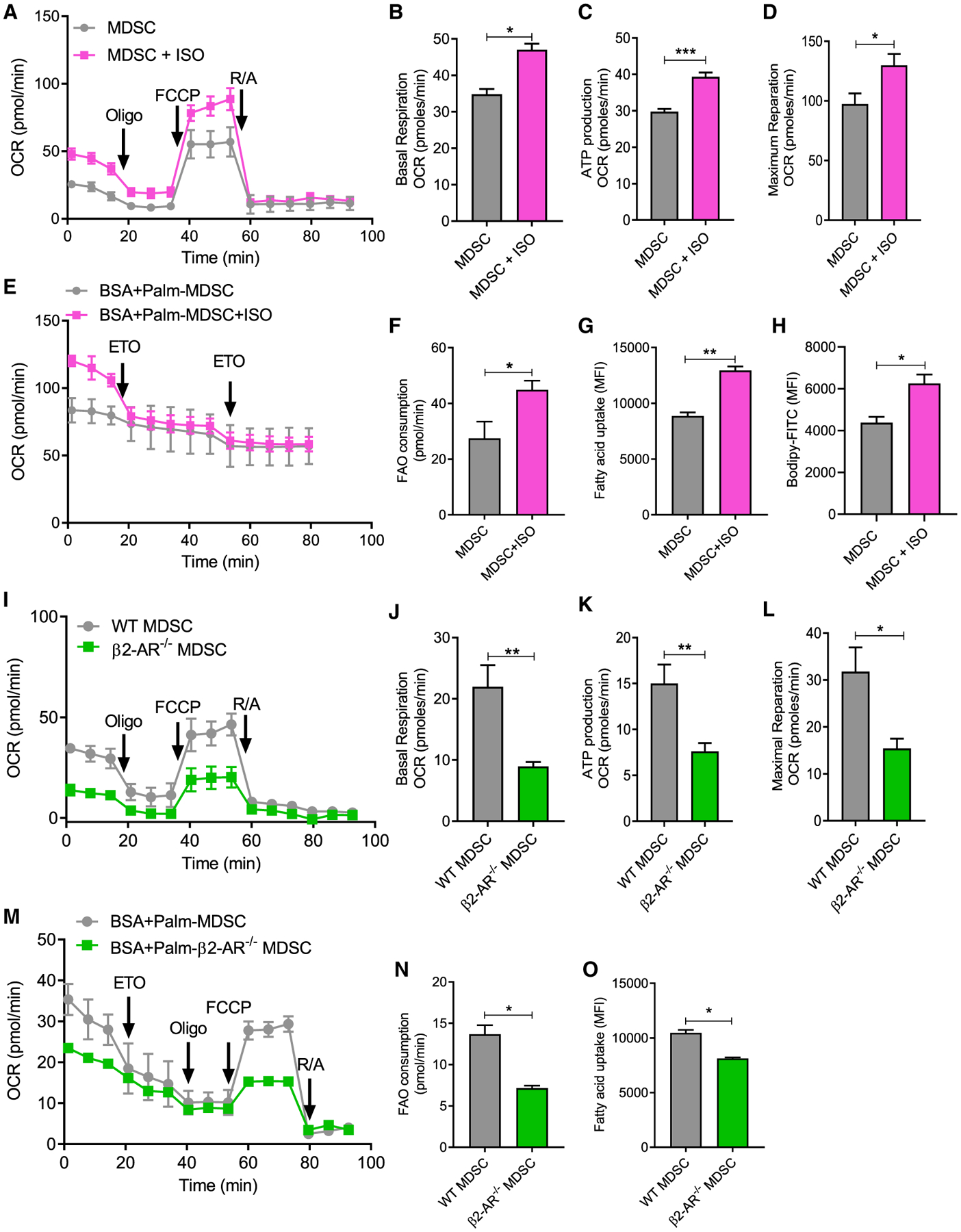
(A–H) MDSCs were derived from bone marrow in the presence of GM-CSF and IL-6, with or without isoproterenol (ISO).
(A) Mitochondrial respiration was measured using a Seahorse Extracellular Flux Analyzer (arrows indicate when reagents were added: (1) oligomycin; (2) FCCP; and (3) antimycin A and rotenone).
(B) Basal respiration levels.
(C) ATP production.
(D) Maximum respiration.
(E) FAO was measured using a Seahorse Extracellular Flux Analyzer in media in which palmitate was the only fatty acid source (arrows indicate when etomoxir [ETO] was added).
(F) FAO consumption.
(G and H) MDSCs were derived from bone marrow in the presence of GM-CSF and IL-6, with or without ISO and (G) fatty acid uptake (Bodipy FL C16) and (H) lipid accumulation (Bodipy FL 493/503) was measured.
(I–O) WT and β2-AR−/− mice were orthotopically implanted with 4T1 tumor cells. At day 25, WT or β2-AR−/− PMN- MDSCs were sorted by flow cytometry and the rates of (I–L) oxidative phosphorylation, (M and N) FAO, and (O) fatty acid uptake were measured. Mitochondrial respiration was measured using a Seahorse Extracellular Flux Analyzer (arrows indicate when reagents were added: [1] oligomycin; [2] carbonyl cyanide p-trifluoromethoxy-phenylhydrazone [FCCP]; and [3] antimycin A and rotenone).
(J) Basal respiration levels.
(K) ATP production.
(L) Maximal respiration.
(M and N) FAO was measured using a Seahorse Extracellular Flux Analyzer in media in which palmitate was the only fatty acid source (arrows indicate when ETO was added). (N) FAO consumption.
(O) WT and β2-AR−/− PMN-MDSCs were sorted from 4T1 tumor-bearing mice, and fatty acid uptake in WT or β2-AR−/− were analyzed by flow cytometry.
These data are presented as means ± SDs from 3 biological replicates in all of the graphs, and the Student’s t test was used to analyze statistical significance between 2 groups. In all of the panels, *p < 0.05, **p < 0.01, and ***p < 0.001. p < 0.05 was considered significant.
These findings are important because oxidative phosphorylation and FAO are key metabolic pathways that drive the immunosuppressive functions of myeloid cells, including tumor-associated macrophages (TAMs) in the TME (Biswas, 2015; Hossain et al., 2015). It has also been reported that blocking FAO dramatically inhibits the immunosuppressive function of MDSCs, and it is partially mediated through AMPK signaling (Hammami et al., 2012). As a potential mechanistic driver of these metabolic changes, we found that AMPK and phosphorylated AMPK levels are elevated in MDSCs that were isolated from the bone marrow of WT 4T1 tumor-bearing mice and treated with ISO, whereas this did not occur when the pan-β-AR antagonist (propranolol) was added to culture to block the effects of ISO (Figure S4E), corroborating mRNA expression data (Figure S2).
FAO inhibition decreases MDSC populations in tumor-bearing mice
Metabolic reprogramming is a critical factor in the generation of MDSC suppressive function (Kumar et al., 2016). Recently, cytoplasmic lipid accumulation was reported in MDSCs from patients with cancer and mice with established tumor burdens, and this was associated with elevated immunosuppressive function (Al-Khami et al., 2017). CPT1A, a mitochondrial enzyme responsible for the formation of acyl carnitines and the transfer of acyl carnitine from the cytosol into the intermembrane space of mitochondria, plays a crucial role in FAO (Qu et al., 2016) and is a key regulator in the immunosuppressive functions of different cells, including MDSCs (Hossain et al., 2015), adipocytes (Xiong et al., 2020), and Tregs (Cluxton et al., 2019). Thus, we sought to determine whether β2-AR signaling in MDSCs affects CPT1A expression, and whether CPT1A-dependent changes in MDSC metabolism affect tumor growth rate and the generation of antitumor immunity.
We found that the PMN-MDSCs from β2-AR−/− 4T1 tumor-bearing mice express lower levels of CPT1A compared to WT mice (Figures 3A and 3B). Using this same 4T1 model in WT mice, treatment with the CPT1A inhibitor etomoxir (ETO) led to a significant decrease in tumor growth (Figure 3C), potentially by limiting MDSC fatty acid metabolism. At the endpoint, we observed a significant decrease in MDSC accumulation in the tumors and spleens of the WT mice treated with ETO (Figures 3F and 3G). This effect was not seen in severe combined immuno-deficient (SCID) mice or β2-AR−/− mice (Figures 3D and 3E), suggesting that the antitumor effects of ETO are dependent upon β2-AR signaling in immune cells. To confirm that the antitumor effects of ETO are mediated by MDSCs, we depleted MDSCs in tumor-bearing mice using anti-GR-1 antibody treatments. We found that ETO did not change the tumor growth rate in mice treated with anti-GR-1, while ETO significantly decreased the tumor growth rate in mice treated with the isotype control antibody (Figure 3H). These data strongly suggest that MDSCs mediate the effect that ETO has on decreasing tumor growth, rather than effects of ETO directly on tumor cells.
Figure 3. Fatty acid oxidation (FAO) inhibition decreases MDSC populations in tumor-bearing mice.
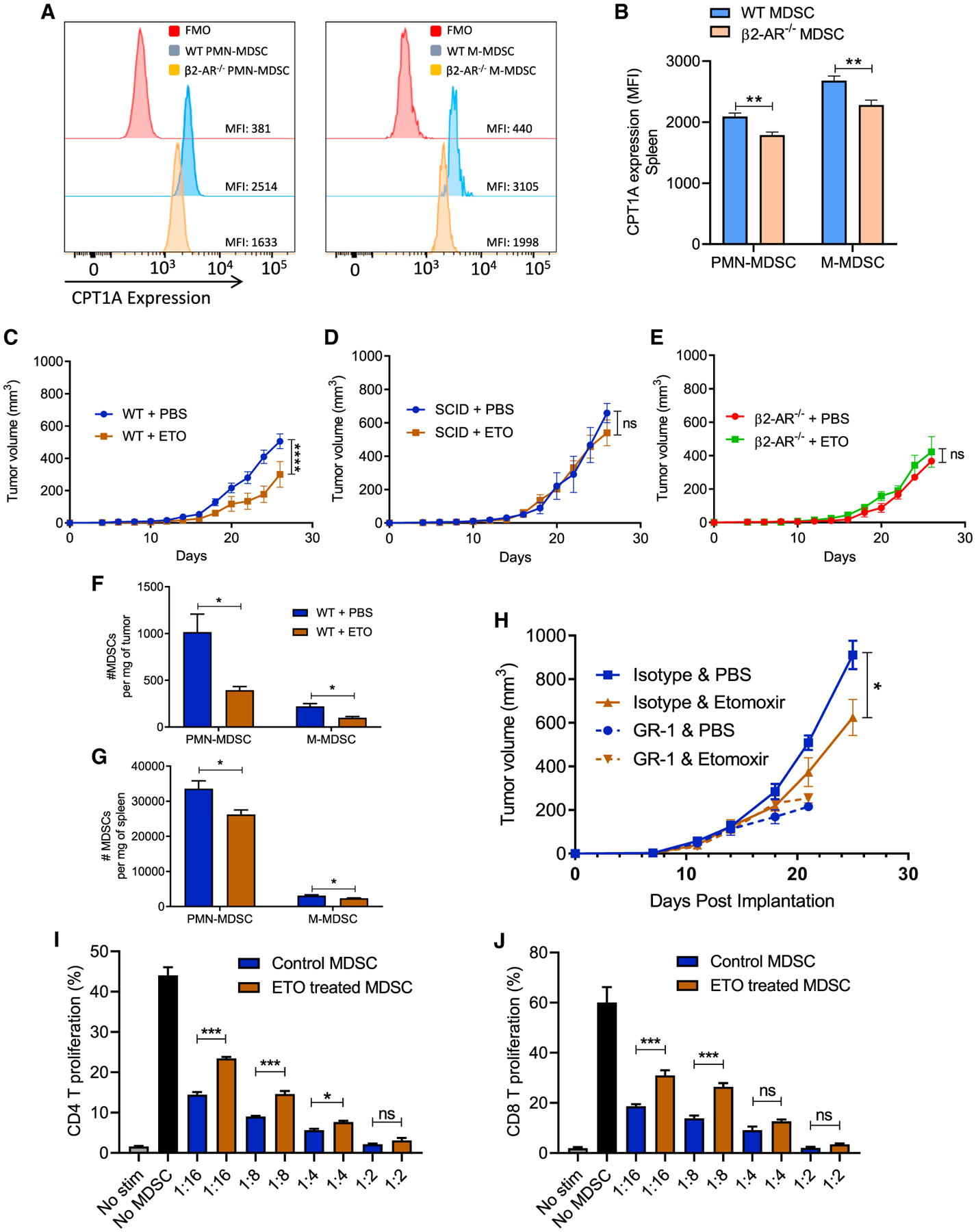
(A) Histogram presentation of CPT-1 expression in WT versus β2-AR−/− MDSC isolated from WT and β2-AR−/− 4T1 tumor-bearing mice.
(B) CPT1A expression in splenic MDSCs isolated from WT or β2-AR−/− 4T1 tumor-bearing mice.
(C) Tumor growth kinetics in WT or β2-AR−/− mice orthotopically injected with 4T1 tumor cells receiving PBS or ETO (intraperitoneal [i.p.] daily injection) (n = 10).
(D) Tumor growth kinetics in SCID mice orthotopically injected with 4T1 tumor cells receiving PBS or ETO (i.p. daily injection) (n = 5).
(E) Tumor growth kinetics in β2-AR−/− mice orthotopically injected with 4T1 tumor cells receiving PBS or ETO (i.p. daily injection) (n = 10).
(F and G) Absolute number of PMN-MDSCs and M-MDSCs in the tumors and spleens of tumor-bearing mice (4T1) on day 25 after tumor injection treated with PBS or ETO.
(H) 4T1-bearing WT mice were injected with isotype or anti-Gr-1 antibodies (i.p. 200 μg per mouse every 4 days), in combination with PBS or ETO, and tumor growth was monitored. The data presented are from groups of 5–10 mice from 2 replicate studies.
(I and J) WT mice orthotopically injected with 4T1 cells. Mice were treated with PBS or etomoxir for 25 days. On day 25, PMN-MDSCs were sorted from the spleen using an MDSC isolation kit. Isolated MDSCs were cultured with T cells in the presence of CD3/CD18 beads and IL-2 in various ratios, and CD4 and CD8 proliferation were measured (n = 3). Two-way ANOVA was used to analyze statistical significance between tumor growth in different groups.
Data are presented as means ± SDs. The Student’s t test was used to analyze statistical significance between 2 groups. In all of the panels, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. p < 0.05 was considered significant.
Although changes in the metabolic profiles and accumulation of MDSCs are important findings, we next examined the effects of ETO on the functional ability of MDSCs to inhibit CD4+ and CD8+ T cell proliferation. MDSCs were isolated from the spleens of tumor-bearing WT mice treated with ETO or PBS. The MDSCs were cocultured with T cells, and T cell proliferation was evaluated (Figures 3I and 3J). We found that MDSCs from ETO-treated mice were less capable of suppressing CD4+ and CD8+ T cell proliferation compared to MDSCs from PBS-treated mice (Figures 3I and 3J). These data suggest that signaling through the β2-AR on MDSCs is a key driver of the intracellular events that lead to immune suppression and elevated tumor growth rates.
β2-AR signaling in MDSCs increases autophagy
Autophagy also plays a critical role in the immunosuppressive function of MDSCs in the TME by promoting MDSC survival (Parker et al., 2016), lysosomal function (Alissafi et al., 2018), and metabolic reprogramming (Li et al., 2018). Autophagy is known to be enhanced when there is a metabolic shift toward oxidative phosphorylation and FAO to increase mitochondrial metabolism (Roca-Agujetas et al., 2019). It also increases oxidative phosphorylation (OXPHOS) by enhancing the fatty acid availability to cells for OXPHOS (Bosc et al., 2020). Therefore, we asked whether β2-AR signaling in MDSCs increases autophagy and thus enhances the release of their immunosuppressive molecules associated with FAO, such as prostaglandin E2 (PGE2). To address this question, we used a Cyto-ID staining protocol to measure the presence of autophagic vesicles in MDSCs derived from WT and β2-AR−/− tumor-bearing mice. We found that WT MDSCs had higher levels of autophagy compared to β2-AR−/− MDSCs (Figures 4A and 4B). Furthermore, MDSCs were isolated from the bone marrow of tumor-bearing WT or β2-AR−/− mice, treated with ISO in vitro, and the levels of the protein Autophagy Related 5 (ATG-5) were elevated after 24 h in WT versus β2-AR−/− MDSCs (Figure 4C). In addition, WT MDSCs were treated with ISO or propranolol and ISO, and we found that ATG5 levels did not increase when ISO-mediated β-AR signaling was blocked with propranolol (Figure 4D).
Figure 4. β2-AR signaling in MDSCs increases autophagy.
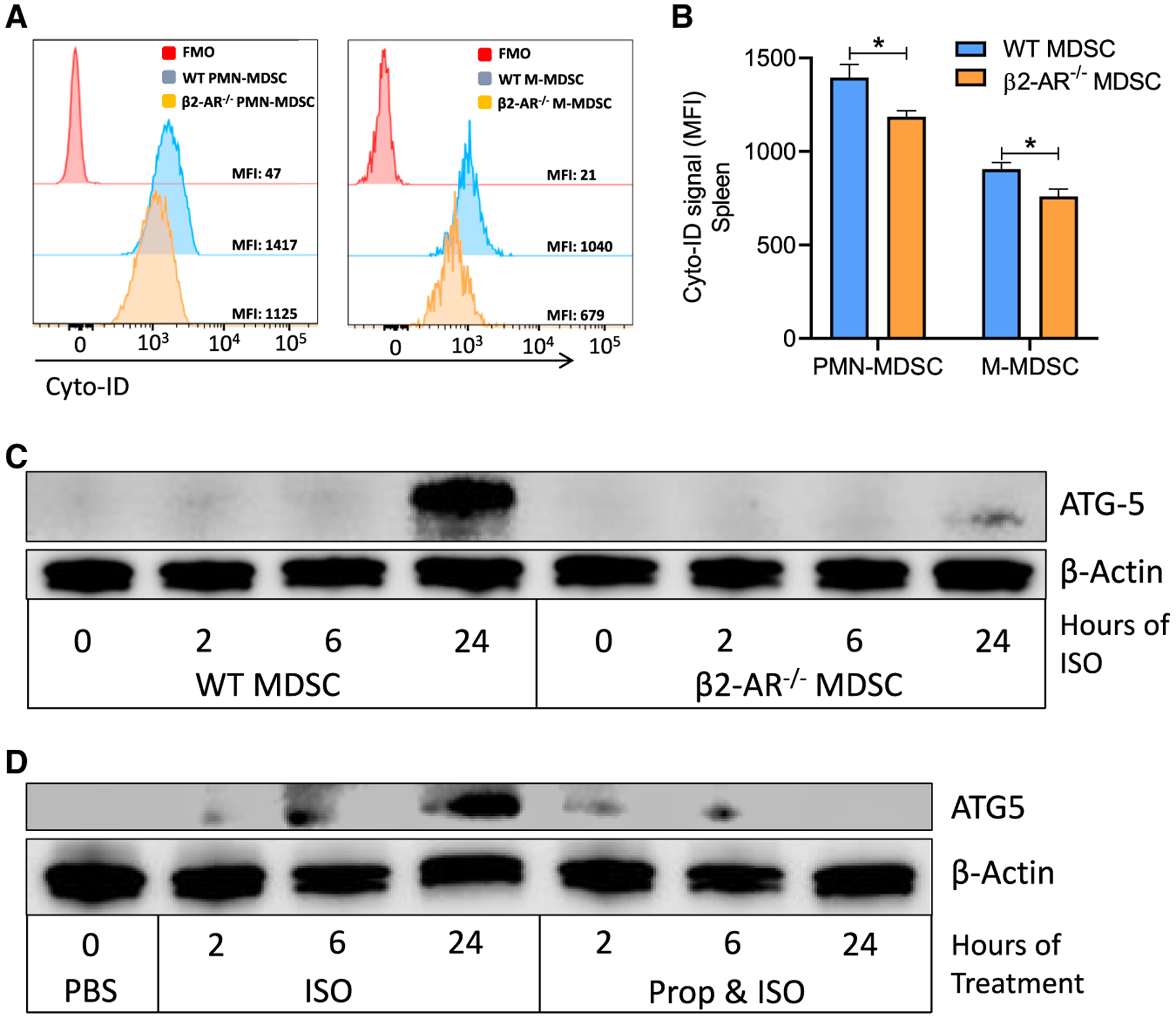
(A) Histogram presentation of Cyto-ID staining of autophagy vesicles in WT versus β2-AR−/− MDSC isolated from the spleens of WT and β2-AR−/− 4T1 tumor-bearing mice.
(B) Autophagy levels in splenic MDSCs isolated from WT or β2-AR−/− 4T1 tumor-bearing mice.
(C) PMN-MDSCs were sorted from WT and β2-AR−/− tumor-bearing mice. MDSCs were activated with or without ISO and ATG-5 expression was analyzed by using western blot.
(D) PMN- MDSCs were sorted from WT 4T1 tumor-bearing mice and ATG-5 expression was measured by western blot in MDSCs treated with ISO or propranolol plus ISO.
These data are presented as means ± SDs from 3 biological replicates in all of the graphs, and the Student’s t test was used to analyze statistical significance between 2 groups. In all of the panels, *p < 0.05, **p < 0.01, and ***p < 0.001. p < 0.05 was considered significant.
Higher FAO leads to increased levels of PGE2 production through cyclooxygenase 2 (COX2) overexpression
One of the critical immunosuppressive molecules closely related to fatty acid metabolism and autophagy is PGE2 (Koundouros and Poulogiannis, 2020). Veglia et al. (2019) reported that fatty acid transport 2 increases FAO and subsequently PGE2 production in PMN-MDSCs. Neural autophagy increases PGE2 production in response to neural injury (He et al., 2019), while the accumulation of lipids in monocytes enhances the metabolism of arachidonic acid (Hubler and Kennedy, 2016). PGE2, a product of the arachidonic acid pathway, is an important immunosuppressive mediator in the TME that can recruit MDSCs, suppress T cell function, and can increase the immunosuppressive function of MDSCs (Nakanishi and Rosenberg, 2013; Sinha et al., 2007; Tomić et al., 2019). PGE2 can also increase pro-survival signals in tumor cells and enhance angiogenesis (Wang and DuBois, 2018). It has been proposed that MDSCs may produce PGE2 (Serafini, 2010), but very little is known about the mechanistic details of PGE2 production by MDSCs.
We hypothesized that β2-AR signaling mediated metabolic reprogramming, and FAO in MDSCs leads to the production of higher levels of PGE2, resulting in increased immunosuppressive activity of MDSCs. During this process, fatty acids are metabolized by several enzymes, including COX2, to ultimately produce PGE2. When comparing NanoString RNA sequencing data from MDSCs isolated from WT or β2-AR−/− tumor-bearing mice, the gene for COX2 (ptgs2) is elevated in WT MDSCs (Figure S2). To investigate this process further, MDSCs isolated from the bone marrow of these mice were treated with ISO and protein levels of COX2 were analyzed by western blot. We found that COX2 levels in MDSCs were elevated in WT MDSCs compared to β2-AR−/− MDSCs (Figure 5A). To confirm that this increase in COX2 resulted in an increase in the effector molecule PGE2, we measured PGE2 levels by ELISA after MDSCs were cultured with various treatments in vitro. PMN-MDSCs were isolated from WT 4T1 tumor-bearing mice and cultured in media with either PBS, ISO, lipopolysaccharide (LPS) as a generic activator of myeloid cells, or ISO and LPS (Figure 5B). We found that ISO treatment significantly increased PGE2 production. In addition, we found that while LPS treatment alone did not increase PGE2 production, the combination of ISO and LPS did significantly increase PGE2 concentrations (Figure 4B), suggesting that β-AR signaling increases PGE2 production.
Figure 5. Higher FAO leads to increased levels of PGE2 production through COX2 overexpression.
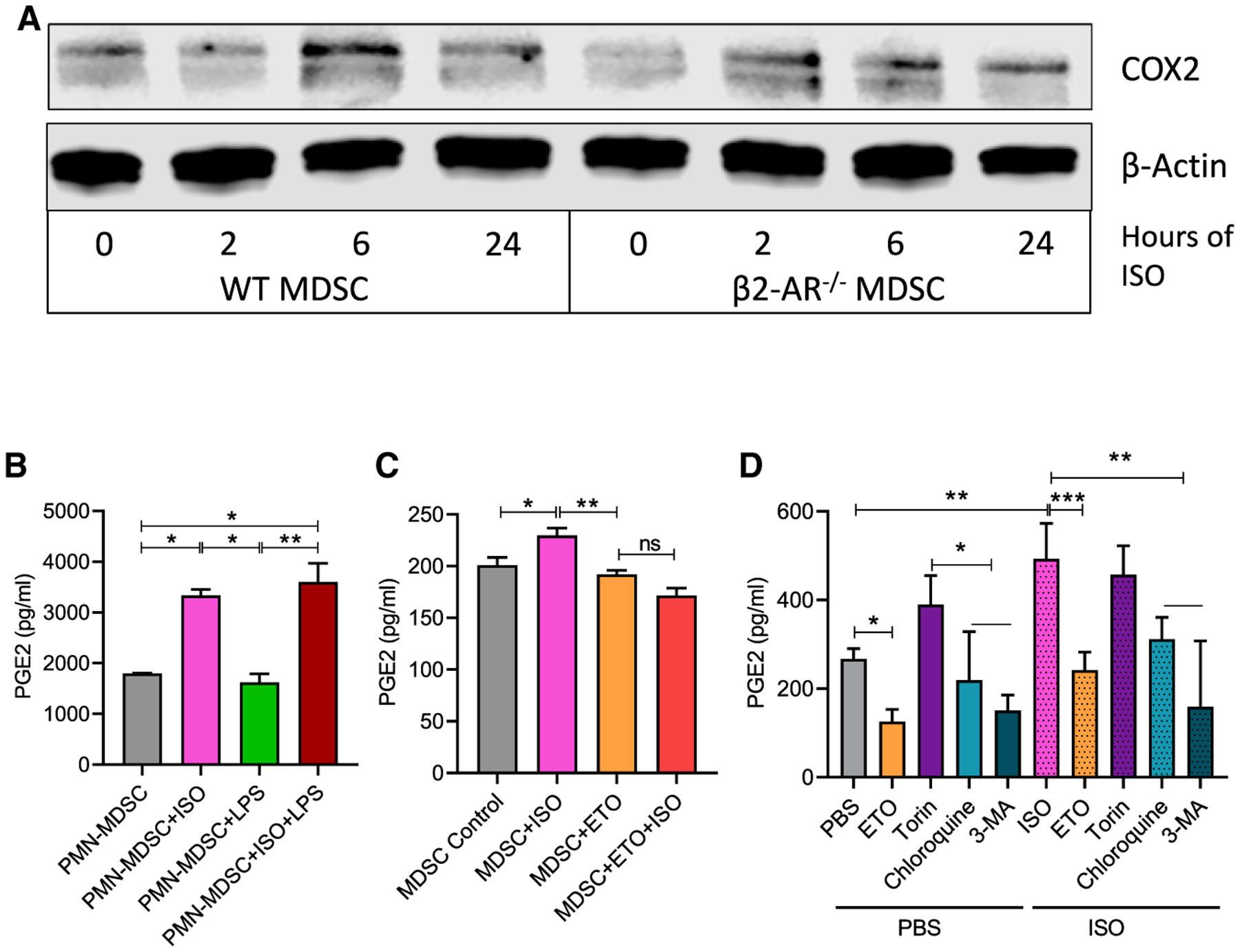
(A) PMN-MDSCs were sorted from WT and β2-AR−/− tumor-bearing mice. MDSCs were activated with or without ISO, and COX2 expression was analyzed by western blot.
(B) PMN-MDSCs were sorted from WT tumor-bearing mice. MDSCs were activated with ISO, LPS, or ISO plus LPS for 24 h, and PGE2 levels were measured by ELISA (n = 3).
(C) Human peripheral blood mononuclear cells (PBMCs) were cultured with IL-6 and GM-CSF for 7 days, treated with ISO, ETO, or ISO plus ETO, and at day 7, the level of PGE2 was measured by ELISA.
(D) PMN-MDSCs were sorted from bone marrow of WT EL-4 tumor-bearing mice. MDSCs were treated for 24 h with either PBS or ISO, and ETO, torin 1, chloroquine, or 3-methyladenine and PGE2 levels were measured by ELISA.
These data are presented as means ± SDs from 3 biological replicates in all of the graphs. One-way ANOVA was used to analyze the statistical significance between >2 groups, and the Student’s t test was used to analyze the statistical significance between 2 groups. In all of the panels, *p < 0.05, **p < 0.01, and ***p < 0.001. p < 0.05 was considered significant.
In addition, MDSCs were derived from human PBMCs using previously published methods in the presence or absence of ISO (Mohammadpour et al., 2019). Similar to observations in murine MDSCs, we found that ISO significantly increased PGE2 production in human MDSCs. In addition, when ETO was used to block CPT1A and inhibit fatty acid metabolism, levels of PGE2 were not increased by ISO treatment (Figure 5C). To determine how autophagy affects this process, several pharmacologic agents were used to induce or inhibit autophagy in MDSCs, in the presence or absence of ISO (Figure 5D). Torin 1 was used to induce autophagy, while chloroquine and 3-methyladenine (3-MA) were used to inhibit autophagy. ETO was also used to block CPT1A. We found that ISO was again able to significantly increase the production of PGE2 in all of the groups compared to those treated with PBS. An increase in autophagy from torin 1 treatment resulted in an increase in PGE2, while a decrease in autophagy from chloroquine or 3-MA treatment resulted in a decrease in PGE2 production (Figure 5D). These results suggest that FAO and autophagy induced by β2-AR signaling are key intermediate pathways leading to an increase in PGE2 production by MDSCs, and blocking either of them inhibits PGE2 production.
DISCUSSION
MDSCs are populations of hematopoietic cells that expand during cancer progression (Veglia et al., 2021), inhibit T cell proliferation, and constrain antitumor immune responses (Ostrand-Rosenberg and Fenselau, 2018). They also promote the growth of primary tumors, as well as metastasis to other organs (Wang et al., 2019). As a result, understanding the mechanisms responsible for driving their immunosuppressive function and clarifying their underlying metabolic regulation can reveal new avenues for improving cancer immunotherapies. We became very interested in studying the suppression of antitumor immunity we observed in response to adrenergic stress-induced signaling of β2-AR. We observed a significant increase in the immunosuppressive function of MDSCs in the TME (Mohammadpour et al., 2019) in mice experiencing chronic stress, or after exposure to β-AR agonists in vitro. A major question that emerged was whether stress signaling was influencing metabolic characteristics of MDSC. Answers to this question could reveal new therapeutic pathways by which MDSC-mediated immunosuppression may be reversed to improve immunotherapy. We not only found that β2-AR signaling increases oxidative phosphorylation and FAO in MDSCs but also that the expression of β2-AR on the cell surface of MDSCs increases during tumor progression. Thus, the ability of stress to undermine antitumor immunity may actually increase during tumor progression. Importantly, it has been shown that FAO is a key metabolic pathway in highly immunosuppressive MDSCs in the TME (Al-Khami et al., 2017; Hossain et al., 2015). We also showed that reducing β2-AR signaling by genetic or pharmacological manipulation suppresses FAO and oxidative phosphorylation. This is a crucial finding because there are other agents, such as ETO, that have been found to systemically block FAO. ETO, while reducing MDSC function, also targets other important antitumor immune cells such as memory T cells (Lochner et al., 2015; Raud et al., 2018), and thus may damage other arms of the immune system and cause major side effects, making it intolerable to many patients (Holubarsch et al., 2007). However, blocking adrenergic signaling by using well-studied, pharmacologic agents such as β-blockers provides a safe, targeted, and affordable opportunity to intervene in this axis and disrupt MDSC immunosuppression (Gosain et al., 2020). Importantly, while impairing MDSC function, β-blockers significantly improve the function of effector CD8 T cells (Qiao et al., 2019). We recently showed that using propranolol as a β-blocking agent significantly improves the metabolic fitness of T cells by increasing glycolysis and decreasing T cell exhaustion, indicating that β2-AR signaling has different metabolic effects on unique immune cell types (Qiao et al., 2021). In addition, MDSCs use different immunosuppressive tools, including the production of arginase-I, the release of nitric oxide (NO), PGE2, and peroxynitrite (PNT), and the expression of programmed cell death protein-ligand 1 (PD-L1), depending on the type of cancer (Yang et al., 2020). This suggests that the targeted inhibition of any one of these individual pathways may not result in a significant therapeutic effect. Stress, however, results in a pleiotropic and long- evolutionarily conserved set of pathways designed to help the organism survive and conserve energy. One can hypothesize that capitalizing on the ability of MDSCs to produce a wide range of immunosuppressive molecules would make them an excellent target for stress-mediated signaling.
Mechanistically, physiological and/or psychological stressors provide the stimuli to drive activation of a patient’s autonomic nervous system. This leads to an increased release of neuroendocrine mediators such as NE from sympathetic nerve endings that can be found both systemically and directly within the TME (Faulkner et al., 2019). Our work suggests that this elevated “adrenergic tone” is responsible for observed changes in MDSC metabolism, and overall promotes a pro-tumor milieu in the TME. Recent data reported by others show that tumors recruit and are innervated by sympathetic nervous system (SNS) fibers directly (Kamiya et al., 2019; Magnon et al., 2013), providing a conduit for chronic stress mediators such as NE to bathe cells within the TME. Here, our data reveal that MDSCs are highly sensitive to these signals and that the metabolic and secretory changes caused by adrenergic stress have the capacity to facilitate immunosuppression and tumor progression in response to even mild but chronic stress. Some of our data, while statistically significant, show a relatively modest impact of adrenergic receptor signaling. It is important to appreciate that chronic stress is a physiological perturbation that does not often result in outright pathology and therefore it is not likely to result in major immuno-logical changes. Nevertheless, repeated suboptimal immune control of tumor progression, occurring over long periods of chronic stress (occurring months to years, in many cases) could result in a significant difference in tumor progression and/or metastases. This point is reinforced by recent epidemiological data from several different cancer settings supporting the concept that coincidental daily use of β-blockers is associated with improved response to therapy and increased overall survival (Gandhi et al., 2021; Kokolus et al., 2017).
Previous studies have shown that the activation of myeloid cells in the TME leads to a switch toward oxidative phosphorylation and FAO induced by IL-4 derived from T helper 2 (Th2) cells and lactic acid (Biswas and Mantovani, 2010; Vats et al., 2006), and that this metabolic change from glycolysis toward oxidative phosphorylation intensifies with tumor growth (Biswas, 2015). We have shown that β2-AR expression on MDSCs increases in relation to tumor growth and is important for the metabolic switch of MDSCs within the TME. This indicates that chronic stress-mediated β2-AR signaling may play a key role in MDSC metabolic reprogramming alongside IL-4 and lactic acid. Our data showed that ETO decreased both tumor growth and the immunosuppressive function of MDSCs in tumor-bearing mice. One of the possible drivers of decreased immunosuppressive function of these MDSCs could be the indirect effect of tumor size on MDSC suppressive function, and further studies such as using CPT1a-deficient mice to examine the effect of β2-AR signaling-mediated FAO activation on MDSC-suppressive functions are needed. We also demonstrated that β2-AR signaling in MDSCs plays crucial roles in MDSC metabolic reprogramming, resulting in higher PGE2 production. Eruslanov et al. (2010) reported that tumor cells inhibit antigen-presenting cells in the TME through PGE2 released by myeloid cells, indicating that PGE2 plays a key role in tumor growth and suppression of antitumor immunity). Also, Veglia et al. (2019) showed that the fatty acid transporter protein 2, expressed by MDSCs, plays a key role in the immunosuppressive function of MDSC through increasing arachidonic acid uptake and PGE2 production. In addition, they showed that the immunosuppressive function of COX2-deficient MDSCs dramatically decreased, indicating the important role of PGE2 production in MDSC-suppressive function. Here, we showed that not only do MDSCs release PGE2 but also that β2-AR signaling significantly increases PGE2 production. Its production is mediated by FAO and autophagy, revealing a pathway through which chronic stress increases the immunosuppressive function of MDSCs within the TME. Further detailed studies are needed to clarify the connection between FAO and PGE2 production and the importance of PGE2 production in β2-AR-mediated immunosuppressive function in MDSCs.
In summary, our demonstration that β2-AR expression on MDSCs, a receptor for autonomic nerve-mediated stress signaling, increases with advancing tumor growth highlights an ominous hardwired mechanism for systemic nerves to maintain and increase local immunosuppression during tumor progression. Our work also determines that chronic stress, triggering activation of the β2-AR, significantly promotes FAO, autophagy, and PGE2 production in MDSCs. Therefore, this signaling pathway has the potential to significantly limit the efficacy of immunotherapies. Our data provide justification for further investigation of the therapeutic potential of blocking chronic stress-mediated β2-AR signaling in combination with other tumor therapy approaches, such as immunotherapy or chemo-radiation treatments.
Limitations of the study
Limiting β2-AR signaling by pharmacologic blockade can inhibit FAO and decrease the production of PGE2 by MDSCs. However, most β-AR agonists and antagonists lack complete specificity to one particular receptor; thus, additional research on pharmacologic agents that specifically block β2-ARs is needed to increase the precision of this therapy. Additional studies are also needed to investigate the contribution of other β-ARs, including β1 and β3-ARs, on MDSCs and their role in altering the immunosuppressive function and metabolism of MDSCs. Further research into understanding the mechanisms by which the expression of β2-AR can be modulated in MDSCs within the TME also warrants further investigation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Hemn Mohammadpour (hemn.mohammadpour@roswellpark.org).
Materials availability
This Study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. RNA sequencing data have been deposited at Figshare.com (https://figshare.com/articles/dataset/RNA_sequencing_data_from_MDSCs_sorted_from_4T1_tumor_bearing_WT_or_beta-2_adrenergic_receptor_knockout_mice_/16691767) and are publicly available as of the date of publication. This paper does not report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
BALB/c and C57BL/6 mice were purchased from Charles River. β2-AR knockout (β2-AR−/−) mice on the BALB/c background were a gift from David Farrar (UT Southwestern Medical Center). β2-AR−/− mice on the C57BL/6 background were developed at Roswell Park, and SCID mice were purchased from the Laboratory Animal Resource at Roswell Park. All mice were age maintained in specific-pathogen-free housing, and all experiments were performed in accordance with the animal care guidelines at Roswell Park Comprehensive Cancer Center. All mice were female ages 6–8 weeks. All mice were approximately 20 g, healthy, and drug/test naive prior to tumor implantation. Liter mates of the same sex we randomly assigned to experimental groups for both strains of β2-AR knockout mice bred at Roswell Park. Tumor growth was monitored in a blinded manner throughout experiments, and perpendicular diameters (width/length) were measured every 2–3 days. Tumor volume was calculated using the following equation: tumor volume = (W × W × L) / 2 mm3, where W is the small dimension and L is the large dimension. Generation of the mice and all mice studies were reviewed and approved by the Roswell Park Comprehensive Cancer Center IACUC (protocol numbers 757M and 1038M).
Humans
All demographic of patients and healthy volunteers related to Figure 1 can be found in Table S1. The Roswell Park Comprehensive Cancer Center IRB approved human subject studies (NHR 009510).
Cell lines
The female cell lines 4T1 (ATCC, catalog CRL-2539) and EL4 (ATCC, catalog TIB-39) tumor cell lines were purchased from ATCC and were confirmed to be mycoplasma-negative yearly using the Mycoplasma Plus PCR Primer Set (Agilent Technologies, catalog 302008). Both cell lines were cultured at 37°C and 5% CO2 in RPMI 1640 (Corning Cellgro) supplemented with 10% FBS, 1% l-glutamine, and 1% penicillin/streptomycin. Once thawed, cells were passed twice prior to use. 4T1 cells (1 × 105) were orthotopically injected into the fourth mammary fat-pad of female BALB/c mice in 100 μLs of PBS. EL4 cells (5 × 105) were subcutaneously injected into the flank of C57/BL6 mice in 100 μLs of PBS.
METHOD DETAILS
Reagents used
Mouse (Catalog I9646) and human (Catalog I1395) IL-6 were from MilliporeSigma. Mouse (Catalog 713704) and human (Catalog 713604) GM-CSF and mouse (Catalog 713502) and human (Catalog 713402) G-CSF were purchased from Biolegend. The T cell proliferation dye used was CellTrace Calcein Violet (ThermoFisher Scientific, Catalog C34858). Propranolol (Catalog P0884) and isoproterenol (Catalog I6504) were purchased from MilliporeSigma. In vitro, propranolol was used at a concentration of 1 μM and isoproterenol was used at a concentration of 10 μM. Etomoxir (Tocris, Catalog 4539) was used in vivo at a dose of 10mg/kg and in vitro at a concentration of 10 μM. To stain MDSCs for autophagic vesicles, the CYTO-ID® Autophagy detection kit 2.0 (Catalog ENZ-51031-0050) from Enzo Life Sciences was used. LPS (MilliporeSigma, Catalog L3024) was used at a concentration of 10ng/ml. Torin-1 (Cell Signaling, Catalog 14379S) was used at a concentration of 250 nM. 3-MA (Torics, Catalog 3977) was used at a concentration of 2 mM. Chloroquine (Enzo Life Sciences, Catalog 51031) was used at a concentration of 10 μM.
Generation of MDSCs from mouse bone marrow and human PBMCs
Mouse bone marrow cells were harvested from WT or β2-AR−/− mice by flushing femurs with sterile PBS. Human PBMCs were isolated from healthy volunteer donors by venipuncture and subsequent differential density gradient separation (Ficoll Hypaque, MilliporeSigma) was carried out. Bone marrow cells were then cultured in RPMI supplemented 10% FBS, 1% l-glutamine, 1% penicillin/streptomycin, IL-6 (20 ng/mL, Biolegend), and GM-CSF (20 ng/mL, Biolegend) for 7 days, in the presence or absence of ISO (10 μM). Media, cytokine and ISO were refreshed twice during generation, and adherent cells were removed using the Detachin Cell Detachment Solution (Genlantis, catalog T100100). Bone marrow derived murine MDSCs were characterized using CD11b, Ly6G, Ly6C and Gr-1 markers. Gr-1 positive cells were used for in vitro experiments. Human MDSC populations were characterized by flow cytometry using the markers CD14 and CD33, and CD33+ cells were isolated from each culture using EasySep HLA Chimerism CD33 Whole Blood Positive Selection Kit (STEMCELL Technologies) per manufacturer’s instructions. The purity of isolated cell populations was determined to be greater than 90% by flow cytometry.
Isolation of MDSCs and T cells
MDSCs were sorted from tumor–bearing mice using a mouse MDSC isolation kit (Stem Cell Technologies). CD4+ and CD8+ T cells were harvested from non-tumor bearing BALB/c mice using pan T Cell Isolation Kit II (Miltenyi Biotec).
Seahorse XF Analyzer
An XFe96 Extracellular Flux Analyzer (Seahorse Bioscience) was used to analyze the extracellular acidification rate (ECAR; mpH/min) and mitochondrial oxygen consumption rate (OCR; O2 mpH/min) in MDSCs isolated from bone marrow of WT and β2-AR−/− 4T1 tumor bearing mice or MDSCs derived from bone marrow in presence or absence of ISO. For ECAR analyses, MDSCs were isolated, washed, and resuspended in ECAR medium (DMEM base (no bicarbonate) with 2 mM L-glutamine, 143 mM NaCl, and 0.5% phenol red (pH 7.35)). The complete ECAR analysis consisted of four stages: basal (without pharmacologic agents), glycolysis induction (10 mM glucose), maximal glycolysis induction (2 mM oligomycin), and glycolysis inhibition (100 mM 2-Deoxy-glucose (2-DG)). For OCR analyses, MDSCs were washed and resuspended in OCR medium (DMEM base, 25 mM glucose, 1 mM pyruvate, 2 mM L-glutamine (pH 7.35)). Cells were plated in Cell-Tak coated 96-well flat-bottom plates and incubated in a non-CO2 incubator for 1 h at 37°C. A complete OCR study was performed with all groups simultaneously in four consecutive stages: basal respiration (without pharmacologic agents), mitochondrial complex V inhibition (2 mM oligomycin), maximal respiration induction (1 mM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP)), and electron transportation chain inhibition (1 mM rotenone and 1 mM antimycin A).
To assess fatty acid oxidation, bone marrow derived MDSCs were cultured with FAO assay media (KHB supplemented with saturating amounts of Palmitate-BSA (XF palmitate–BSA FAO substrate, Seahorse bioscience, Agilent Technology), 0.5 mM carnitine, and 5 mM HEPES and the pH was adjusted to 7.4). OCR was measured at baseline and after the injection two doses of etomoxir (100 μM) to obtain the maximal inhibition of exogenous Fatty acid oxidation. β-oxidation was evaluated as maximum OCR at baseline before etomoxir injection. In MDSCs isolated from WT or β2-AR−/− tumor bearing mice, OCR was measured at baseline and after injection of etomoxir, oligomycin, FCCP and rotenone and antimycin.
ELISA
The Prostaglandin E2 Parameter Assay kit was used to measure PGE2 levels (R&D Systems Catalog KGE004B).
Western blot
MDSCs were sorted from bone marrow of mice bearing 4T1 or EL4 tumors using a murine MDSC isolation kit as described above. After the indicated treatments, cells were washed with PBS and frozen as a pellet at −80°C. Lysis buffer composed of RIPA Buffer (Pierce, catalog 89900), protease and phosphatase inhibitor mini tablets (Pierce, catalog A32961), and 1 mM PMSF (ThermoFisher Scientific, catalog 36978) was used to lyse cells and extract protein from the MDSC samples. BCA assays were carried out using a clear, flat-bottom, 96-well plate (Costar, catalog 9018), and the BCA Protein Assay Kit (Pierce, catalog 23225) to determine the concentration of protein in each sample. A plate reader (Synergy H1) was used to detect the absorbance of each sample using. Protein resolution was achieved by SDS-PAGE, transferred to a polyvinylidene difluoride membrane (Millipore, catalog IPVH00010), and blocked with 5% nonfat milk or 5% BSA (ThermoFisher Scientific, catalog 10857) in Tris buffered saline (Bio-Rad, catalog 173–6435) with Tween 20 (Bio-Rad, catalog 170–6531) per primary antibody incubation specifications. Membranes were probed overnight at a concentration of 1:1000 for all primary antibodies. Horseradish peroxidase–conjugated anti-rabbit (Cell Signaling, catalog 7074) and anti-mouse (Cell Signaling, catalog 7076) secondary antibodies were used at a concentration of 1:3000. Membranes were developed using ECL-substrate (Bio-Rad, catalog 170–5060) and images were obtained on a LI-COR Odyssey Fc (catalog OFC-0756).
Flow cytometry
Single-cell suspensions were created by excising tumors and chopping them into 2 to 3 mm pieces. 4T1 and EL4 tumors were dissociated with collagenase/hyaluronidase (Catalog 07912, Stem Cell Technologies) following the manufacturer’s protocol prior to passage through a 70 μm nylon cell strainer (Corning). Spleens were mechanically disrupted and directly passed through a 70-μm nylon cell strainer (Corning). Red blood cells were lysed using ACK buffer (GIBCO). Cells were then washed with flow running buffer (0.1% BSA in PBS) and incubated with anti-CD16/32 (Fc receptors blocker, 1:200) at 4°C for 10 minutes. Cells were then stained with the antibodies listed above. Live/dead aqua (ThermoFisher Scientific) were used to gate out dead cells. All data were collected on an LSR Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo v7 software (Tree Star, Inc.). The absolute number of cells in both spleen and tumor tissues was calculated by multiplying the percentage of live CD45+ CD11b+ Ly6G+ (PMN-MDSC) and live CD45+ CD11b+ Ly6C+ (M-MDSC) by the cell numbers of the sample, divided by milligram weight. For staining with mitochondrial dye, cells were first stained with extracellular antibodies and live/dead fixable dye as previously described and then incubated in RPMI 1640 containing either 30 mM MitoTracker Green FM (mitochondrial mass) or MitoTracker Orange CMTMRos (mitochondrial membrane potential) at 37°C for 30 min. All mitochondria dyes were purchased from Thermo Fisher. To quantify the neutral lipid content, cells were first stained with surface markers and then with 250 ng/mL Bodipy 493/503 (Life Technologies) in PBS for 10 minutes at room temperature.
MDSC depletion
Anti–mouse Gr-1 antibody (clone RB6–8C5) and IgG2a isotype control antibody (clone LTF-2) were purchased from BioXCell. WT mice were randomized to receive treatment with either anti–Gr-1 antibody (200 μg) or an isotype antibody (200 μg). Mice received 5 injections spaced 4 days apart, and treatment began one day after tumors became detectable and mice were randomized.
Coculture of MDSCs and T cells
For coculture experiments, 2.5 × 105 pan T cells were cocultured with varying numbers of MDSCs in RPMI 1640 media supplemented with 1% l-glutamine, 1% penicillin/streptomycin, 10% heat-inactivated FBS, and CD3 and CD28 antibodies (T cell activation kit, miltenyi biotec) to activate mouse T cells. After 72 hours, cells were collected, and eFluor 670 dilutions were calculated by gating from live CD4+ or CD8+ T cells using flow cytometry.
RNA sequencing
WT and β2-AR−/− MDSCs were sorted from single cell suspensions of 4T1 tumors by FACS. RNA was isolated from cells using the RNeasy Plus Mini kit (QIAGEN). Analysis was performed with the NanoString Technologies nCounter Analysis System. The nCounter Mouse metabolic signaling Kit, which includes 235 metabolic signaling-related mouse genes, was used. The correlation between different genes and metabolic pathways we analyzed using Enricher (Kuleshov et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
All figure legends contain a description of the specific statistical analyses used to evaluate those data. The Student’s t test was used to compare data between 2 groups, 2-way ANOVA with Tukey’s post hoc analysis was used to generate tumor growth statistics using GraphPad Prism, and 1-way ANOVA with Tukey’s post hoc analysis was used to compare data between 3 groups or more using GraphPad Prism. All data are presented as mean ± SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti–mouse CD3 (clone 17A2) | BioLegend | RRID:AB_2715571 |
| anti–mouse CD4 (clone GK1.5) | eBioscience | RRID:AB_469533 |
| anti–mouse CD8 (clone 53–6.7) | eBioscience | RRID:AB_467086 |
| anti–mouse CD45 (clone 30F11) | BioLegend | Catalog 103114 |
| anti–mouse Gr-1 (clone RB6-8C5) | eBioscience | RRID:AB_1210822 |
| anti–mouse CD45 (clone 30-F11) | eBioscience | RRID:AB_469717 |
| anti–mouse CD11b (clone M1/70) | BioLegend | RRID:AB_2715571 |
| anti–mouse CD206 (clone C068C2) | BioLegend | Catalog 141729 |
| anti–mouse Ly6C (clone HK1.4) | BioLegend | Catalog 128004 |
| anti–mouse Ly6G (clone 1A8) | BioLegend | Catalog 127604 |
| anti–mouse CPT1A (clone ab171449) | Abcam | Catalog ab171449 |
| anti–mouse β2-AR (Catalog orb15065) | Biorbyt | Catalog orb15065 |
| anti-human CD33 (clone P67.6) | BioLegend | Catalog 366605 |
| anti-human CD33 (D3HL60.251) | Beckman Coulter | Catalog A54824 |
| anti-human HLA-DR (clone L243) | eBioscience | RRID:AB_1907430 |
| anti-human CD11b (clone D12) | BD Biosciences | Catalog 340937 |
| anti-human CD14 (clone M5E2) | BD Biosciences | RRID:AB_2033939 |
| anti-human CD14 (MϕP9) | BD Biosciences | Catalog 562691 |
| anti-human CD15 (clone HI98) | eBioscience | RRID:AB_464953 |
| anti-human CD4 (clone A161A1) | Biolegend | Catalog 357405 |
| anti-human CD8 (clone RPA-T6) | Biolegend | Catalog 301007 |
| AMPK | Cell signaling technologies | Catalog 5831 |
| phospho-AMPK | Cell signaling technologies | Catalog 2535 |
| ATG5 | Cell signaling technologies | Catalog 2630 |
| COX2 | Cell signaling technologies | Catalog 12282 |
| β-Actin | Cell signaling technologies | Catalog 3700 |
| Chemicals, peptides, and recombinant proteins | ||
| Mouse IL-6 | MilliporeSigma | Catalog I9646 |
| Human IL-6 | MilliporeSigma | Catalog I1395 |
| Mouse GM-CSF | Biolegend | Catalog 713704 |
| Human GM-CSF | Biolegend | Catalog 713604 |
| Mouse GM-CSF | Biolegend | Catalog 713502 |
| Human GM-CSF | Biolegend | Catalog 713402 |
| CellTrace Calcein Violet | ThermoFisher Scientific | Catalog C34858 |
| Propranolol | MilliporeSigma | Catalog P0884 |
| Isoproterenol | MilliporeSigma | Catalog I6504 |
| Etomoxir | Tocris | Catalog 4539 |
| LPS | MilliporeSigma | Catalog L3024 |
| Torin-1 | Cell Signaling | Catalog 14379S |
| 3-MA | Torics | Catalog 3977 |
| Chloroquine | Enzo Life Sciences | Catalog 51031 |
| Critical commercial assays | ||
| CYTO-ID® Autophagy detection kit 2.0 | Enzo Life Sciences | Catalog ENZ-51031-0050 |
| Cell Mito Stress Test Kit | Agilent | Part# 103010-100 |
| Palmitate Oxidation Stress Test Kit | Agilent | Part# 103693-100 |
| Long Chain Fatty Acid Oxidation Stress Test Kit | Agilent | Part# 103672-100 |
| Glycolysis Stress Test Kit | Agilent | Part# 103017-100 |
| Prostaglandin E2 Parameter Assay kit | R & D Systems | Catalog KGE004B |
| Deposited data | ||
| RNA Sequencing | FigShare | https://figshare.com/articles/dataset/RNA_sequencing_data_from_MDSCs_sorted_from_4T1_tumor_bearing_WT_or_beta-2_adrenergic_receptor_knockout_mice_/16691767 |
| Experimental models: cell lines | ||
| 4T1 | ATCC | Catalog CRL-2539 |
| EL4 | ATCC | Catalog TIB-39 |
| Mycoplasma Plus PCR Primer Set | Agilent Technologies | Catalog 302008 |
| Experimental models: organisms/strains | ||
| BALB/c | Charles River | Strain Code: 028 |
| C56BL/6 | Charles River | Strain Code: 027 |
| β2-AR knockout (BALB/c) | David Farrar (UT Southwestern Medical Center) | NA |
| β2-AR knockout (C57BL/6) | RPCCC | NA |
| SCID | RPCCC | NA |
| Software and algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/ |
Highlights.
Tumor progression increases the expression of β2-AR on MDSCs
β2-AR signaling increases fatty acid oxidation and oxidative phosphorylation in MDSCs
β2-AR signaling in MDSCs increases autophagy-mediated PGE2 production
ACKNOWLEDGMENTS
The authors thank Jeanne M. Prendergast, Samuel A. Ministero, Nishant Gandhi, and Claire Bleacher for technical assistance, Dr. David Farrar (University of Texas Southwestern Medical Center) for the generous gift of β2AR−/− mice (on BALB/c background), the Genomics Shared Resource (Roswell Park), Drs. Hans Minderman and Kieran O’Loughlin for technical help in human MDSC stating and data analysis, and the Roswell Park Flow Cytometry Core Facility for expertise and support. This project was supported by National Institutes of Health (NIH) grants R01 CA205246 and R01 CA099326 (to E.A.R.), F32 CA239356 and K99 HL155792 (to H.M.), T32 CA085183 and F30 CA265127 (to C.R.M.), the Roswell Park Alliance Foundation, and NCI grant P30 CA016056. S.I.A. was supported by NIH grant R01 CA172105.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109883.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abuatiq A, Brown R, Wolles B, and Randall R (2020). Perceptions of stress: patient and caregiver experiences with stressors during hospitalization. Clin. J. Oncol. Nurs 24, 51–57. [DOI] [PubMed] [Google Scholar]
- Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, Sanchez MD, Dean MJ, Rodriguez PC, and Ochoa AC (2017). Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. OncoImmunology 6, e1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, Polyzos A, Xilouri M, Wielockx B, Gogas H, and Verginis P (2018). Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Invest 128, 3840–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, Nee K, Hernandez G, Evans K, Torosian L, et al. (2020). Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol 5, eaay6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK (2015). Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449. [DOI] [PubMed] [Google Scholar]
- Biswas SK, and Mantovani A (2010). Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol 11, 889–896. [DOI] [PubMed] [Google Scholar]
- Bosc C, Broin N, Fanjul M, Saland E, Farge T, Courdy C, Batut A, Masoud R, Larrue C, Skuli S, et al. (2020). Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun 11, 4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, Liu H, Kokolus KM, Eng JW-L, Messmer MN, et al. (2017). β-adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8+ T cells and undermines checkpoint inhibitor therapy. Cancer Res. 77, 5639–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluxton D, Petrasca A, Moran B, and Fletcher JM (2019). Differential regulation of human Treg and Th17 cells by fatty acid synthesis and glycolysis. Front. Immunol 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eruslanov E, Daurkin I, Ortiz J, Vieweg J, and Kusmartsev S (2010). Pivotal advance: tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE2 catabolism in myeloid cells. J. Leukoc. Biol 88, 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner S, Jobling P, March B, Jiang CC, and Hondermarck H (2019). Tumor neurobiology and the war of nerves in cancer. Cancer Discov 9, 702–710. [DOI] [PubMed] [Google Scholar]
- Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, Utikal J, and Umansky V (2018). Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol 9, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Pandey MR, Attwood K, Ji W, Witkiewicz AK, Knudsen ES, Allen C, Tario JD, Wallace PK, Cedeno CD, et al. (2021). Phase I Clinical Trial of Combination Propranolol and Pembrolizumab in Locally Advanced and Metastatic Melanoma: Safety, Tolerability, and Preliminary Evidence of Antitumor Activity. Clin. Cancer Res 27, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosain R, Gage-Bouchard E, Ambrosone C, Repasky E, and Gandhi S (2020). Stress reduction strategies in breast cancer: review of pharmacologic and non-pharmacologic based strategies. Semin. Immunopathol 42, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, and Umansky V (2019). Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 120, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, and Jolicoeur M (2012). Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol 13, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H-Y, Ren L, Guo T, and Deng Y-H (2019). Neuronal autophagy aggravates microglial inflammatory injury by downregulating CX3CL1/fractalkine after ischemic stroke. Neural Regen. Res 14, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P, and Rhein S (2007). A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. (Lond.) 113, 205–212. [DOI] [PubMed] [Google Scholar]
- Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, et al. (2015). Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res 3, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Pang B, Lin G, Zhen Y, and Yi H (2020). Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br. J. Cancer 122, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubler MJ, and Kennedy AJ (2016). Role of lipids in the metabolism and activation of immune cells. J. Nutr. Biochem 34, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya A, Hayama Y, Kato S, Shimomura A, Shimomura T, Irie K, Kaneko R, Yanagawa Y, Kobayashi K, and Ochiya T (2019). Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci 22, 1289–1305. [DOI] [PubMed] [Google Scholar]
- Kokolus KM, Capitano ML, Lee C-T, Eng JW-L, Waight JD, Hylander BL, Sexton S, Hong C-C, Gordon CJ, Abrams SI, and Repasky EA (2013). Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc. Natl. Acad. Sci. USA 110, 20176–20181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokolus KM, Zhang Y, Sivik JM, Schmeck C, Zhu J, Repasky EA, Drabick JJ, and Schell TD (2017). Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. OncoImmunology 7, e1405205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koundouros N, and Poulogiannis G (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Patel S, Tcyganov E, and Gabrilovich DI (2016). The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 37, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, Wei S, Zhao L, Vatan L, and Wen B (2018). Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 28, 87–103.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner M, Berod L, and Sparwasser T (2015). Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 36, 81–91. [DOI] [PubMed] [Google Scholar]
- Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, and Frenette PS (2013). Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361. [DOI] [PubMed] [Google Scholar]
- Mohammadpour H, MacDonald CR, Qiao G, Chen M, Dong B, Hylander BL, McCarthy PL, Abrams SI, and Repasky EA (2019). β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J. Clin. Invest 129, 5537–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachmany I, Bogoch Y, Sivan A, Amar O, Bondar E, Zohar N, Yakubovsky O, Fainaru O, Klausner JM, and Pencovich N (2019). CD11b+Ly6G+ myeloid-derived suppressor cells promote liver regeneration in a murine model of major hepatectomy. FASEB J. 33, 5967–5978. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, and Rosenberg DW (2013). Multifaceted roles of PGE 2 in inflammation and cancer. Semin. Immunopathol 35, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, and Fenselau C (2018). Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol 200, 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou L, Shi Y, Dong W, Liu C, Schmidt TJ, Nagarkatti P, Nagarkatti M, Fan D, and Ai W (2015). Kruppel-like factor KLF4 facilitates cutaneous wound healing by promoting fibrocyte generation from myeloid-derived suppressor cells. J. Invest. Dermatol 135, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, Marasco D, Asm I, Chadli A, Hassan KA, et al. (2017). Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun 8, 14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KH, Horn LA, and Ostrand-Rosenberg S (2016). High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J. Leukoc. Biol 100, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelec G, Picard E, Bueno V, Verschoor CP, and Ostrand-Rosenberg S (2021). MDSCs, ageing and inflammageing. Cell. Immunol 362, 104297. [DOI] [PubMed] [Google Scholar]
- Qiao G, Bucsek MJ, Winder NM, Chen M, Giridharan T, Olejniczak SH, Hylander BL, and Repasky EA (2019). β-Adrenergic signaling blocks murine CD8+ T-cell metabolic reprogramming during activation: a mechanism for immunosuppression by adrenergic stress. Cancer Immunol. Immunother 68, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao G, Chen M, Mohammadpour H, MacDonald CR, Bucsek MJ, Hylander BL, Barbi JJ, and Repasky EA (2021). Chronic Adrenergic Stress Contributes to Metabolic Dysfunction and an Exhausted Phenotype in T Cells in the Tumor Microenvironment. Cancer Immunol. Res 9, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Q, Zeng F, Liu X, Wang QJ, and Deng F (2016). Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 7, e2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos BP, and Arnsten AF (2007). Adrenergic pharmacology and cognition: focus on the prefrontal cortex. Pharmacol. Ther 113, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raud B, McGuire PJ, Jones RG, Sparwasser T, and Berod L (2018). Fatty acid metabolism in CD8+ T cell memory: challenging current concepts. Immunol. Rev 283, 213–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca-Agujetas V, de Dios C, Lestón L, Marí M, Morales A, and Colell A (2019). Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid. Med. Cell. Longev 2019, 3809308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini P (2010). Editorial: PGE2-producing MDSC: a role in tumor progression? J. Leukoc. Biol 88, 827–829. [DOI] [PubMed] [Google Scholar]
- Sinha P, Clements VK, Fulton AM, and Ostrand-Rosenberg S (2007). Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 67, 4507–4513. [DOI] [PubMed] [Google Scholar]
- Tomić S, Joksimović B, Bekić M, Vasiljević M, Milanović M, Čolić M, and Vučević D (2019). Prostaglanin-E2 potentiates the suppressive functions of human mononuclear myeloid-derived suppressor cells and increases their capacity to expand IL-10-producing regulatory T cell subsets. Front. Immunol 10, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, and Chawla A (2006). Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Perego M, and Gabrilovich D (2018). Myeloid-derived suppressor cells coming of age. Nat. Immunol 19, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, et al. (2019). Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 569, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol 21, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistein R, and Puthenveedu MA (2013). Reprogramming of G protein-coupled receptor recycling and signaling by a kinase switch. Proc. Natl. Acad. Sci. USA 110, 15289–15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, and DuBois RN (2018). Role of prostanoids in gastrointestinal cancer. J. Clin. Invest 128, 2732–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ding Y, Guo N, and Wang S (2019). MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front. Immunol 10, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won W-J, Deshane JS, Leavenworth JW, Oliva CR, and Griguer CE (2019). Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 3, 47–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wen Y-A, Fairchild R, Zaytseva YY, Weiss HL, Evers BM, and Gao T (2020). Upregulation of CPT1A is essential for the tumor-promoting effect of adipocytes in colon cancer. Cell Death Dis. 11, 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, and Wan X (2019). Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front. Immunol 10, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li C, Liu T, Dai X, and Bazhin AV (2020). Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Micro-environmental Regulation. Front. Immunol 11, 1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. RNA sequencing data have been deposited at Figshare.com (https://figshare.com/articles/dataset/RNA_sequencing_data_from_MDSCs_sorted_from_4T1_tumor_bearing_WT_or_beta-2_adrenergic_receptor_knockout_mice_/16691767) and are publicly available as of the date of publication. This paper does not report original code.