

Abstract
Purpose of Review
Systemic lupus erythematosus is a complex disease with broad spectrum of clinical manifestations. In addition to abnormal B cell responsive leading to autoantibody production, various T cells also play different roles in promoting systemic autoimmunity and end organ damage. We aim to provide a review on recent developments in how abnormalities in different T cells subsets contribute to systemic lupus erythematosus pathogenesis and how they inform the consideration of new promising therapeutics.
Recent Findings
Distinct subsets of T cells known as T follicular helper cells enable the production of pathogenic autoantibodies. Detailed understanding of the B cell helping T cell subsets should improve the performance of clinical trials targeting the cognate T:B cell interaction. CD8+ T cells play a role in peripheral tolerance and reversal of its exhausted phenotype could potentially alleviate both systemic autoimmunity and the risk of infection. Research on the abnormal lupus T cell signaling also leads to putative therapeutic targets able to restore interleukin-2 production and suppress the production of the pathogenic IL-17 cytokine. Recently, several studies have focused on dissecting T cell populations located in the damaged organs, aiming to target the pathogenic processes specific to each organ.
Summary
Numerous T cell subsets play distinct roles in SLE pathogenesis and recent research in understanding abnormal signaling pathways, cellular metabolism, and environmental cues pave the way for the development of novel therapeutics.
Keywords: Lupus, SLE, T cells
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of multiple autoantibodies and diverse clinical presentations [1]. Chronic inflammation affects multiple organs, causing life threatening complications [2]. Genetic, epigenetic, hormonal and environmental factors alter the function of practically every cell of the adaptive and innate immune response to advance the autoimmune response and cause inflammation and eventually damage in multiple organs (reviewed in [3]). In addition to numerous B cells abnormalities contributing to lupus pathogenesis, T cells contribute in important ways by propagating the autoimmune response, by providing help to B cells [4], and by amplifying systemic inflammation through the production of inflammatory cytokines [5]. In this article, we discuss T cell abnormalities expressed by different cell subsets in the context of SLE pathogenesis, and review aberrantly activated pathways and altered metabolism in lupus T cells leading to disease pathology.
T Follicular Helper Cell
SLE is the prototype of systemic autoimmune disease, characterized by the production of autoantibodies targeting various components within the nucleus, including chromatin-associated antigens, spliceosomes, and other ribonucleoproteins. Double-strand DNA (dsDNA) antibodies, which represent the hallmark of the disease, acquire high-affinity toward dsDNA binding through somatic hypermutation [6, 7], indicating the need of T cell help in generating autoreactive B cell clones. The MHC class II restricted nature suggests the cognate and contact-dependent nature of the T-B collaboration [8] (Fig. 1). This T cell help is mainly provided by a specialized group of B cell lymphoma 6 (BCL6)-expressing T cells residing in the follicles and are known as T follicular helper (Tfh) cells, which promote B cell differentiation through IL-21 production [9], and numerous contact-dependent molecules, including signaling lymphocyte activation molecule-associated protein (SAP) [10], CD40L-CD40 [11], and inducible T cell costimulator (ICOS)- ICOSL signaling [12]. The critical role of cognate T-B interaction in driving systemic autoimmunity has been demonstrated in murine lupus models by targeting each of these molecules and proved them being critical for T cell help. Roquin, a member of RING-type ubiquitin ligase family, represses the expression of OX40 and ICOS, and its mutation drives autoantibodies production mimicking lupus phenotype by overexpressing these costimulatory molecules on Tfh cells [13]. In Roquinsan/san lupus mouse model, SAP deficiency significantly reduces the number of germinal center Tfh cells, and abrogates the development of autoantibodies and tissue damage [14]. The importance of contact-dependent costimulatory pathways in promoting autoantibody production in murine lupus models has also been demonstrated by using blocking antibodies against ICOSL [15] and CD40L [16]. In addition to contact-dependent costimulation, the generation of autoreactive B cell clones depends on IL-21R signaling as demonstrated in experiments which involved its genetic modification [17] or the use of blocking antibodies [18]. Taken together, cognate T-B collaboration plays a pivotal role in driving self-reactive B cell clones and their further differentiation into pathogenic autoantibody-producing plasmablasts. These studies provide the foundation for clinical trials targeting cognate T-B interaction in SLE patients.
Fig. 1.
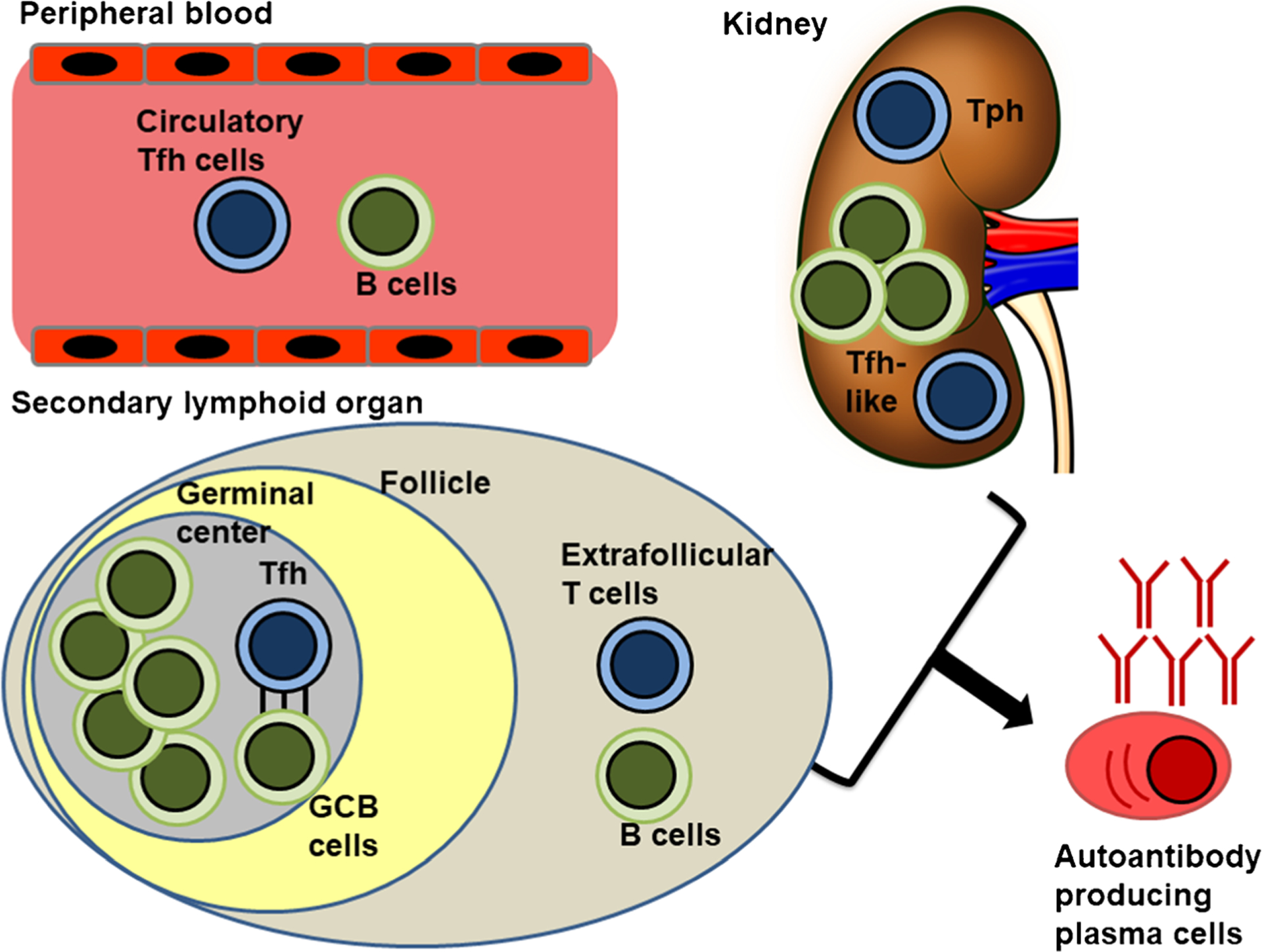
T cell subsets involved in promoting autoantibody-producing plasma cells through aberrant T-B interaction. T follicular helper (Tfh) promotes germinal center B cells (GCB) affinity maturation and class switch in a contact-dependent manner. Other non-canonical T cells subsets include extrafollicular T cells, circulatory Tfh cell, peripheral helper (Tph) cells, and Tfh-like cells residing in damaged organ
Extrafollicular and Peripheral T Helper Cell
In addition to the canonical follicular CD4+ T cells, there is evidence of pathogenic B cell maturation outside of germinal center. In the lupus-prone MRL.Faslpr mice, the site of proliferation and somatic hypermutation for autoreactive B cells occurs outside of germinal centers [19], and the extrafollicular CD4+ T cell help is also ICOS-dependent, indicating a similar T-B cognate interaction despite the non-classical location [20] (Fig. 1). Similar to germinal center Tfh cells, these extrafollicular CD4+ T cells express Bcl-6 and the extrafollicular antibody response depends also on IL-21 [21]. Beyond the secondary lymphoid organs, circulating Tfh-like cells—a population of peripheral blood T helper cells [22]—are increased proportionally to lupus disease activity in people with SLE [23]. These circulating CXCR5+ CD4+ T cells possess the same properties to promote differentiation of autoantibody-producing plasmablasts in a manner similar to that of germinal center Tfh cells, suggesting a role in the pathogenesis of disease [24]. Similar Bcl6+ IL-21+ Tfh-like cells have been documented in lupus nephritic kidneys where they are in close contact with B cells [25] (Fig. 1). These data suggest the presence of various Tfh-like populations, which not only phenotypically resemble follicular helper cells but they drive autoreactive B cell clones outside of germinal centers.
Analysis of damaged joint tissue in rheumatoid arthritis patients revealed another population of possible B cell-helper peripheral T helper cells (Tph) [26•] (Fig. 1). Despite being CXCR5− BLIMP1+, these CD4+ T cells express critical factors to enable B cell help, including ICOS expression and IL-21 production. This population is also found in the peripheral blood and kidney tissues of patients with lupus nephritis and their numbers correlate with the frequency of CD11c+ T-bet+ B cells as well as disease activity [27]. Single-cell RNA-sequencing analysis of human lupus nephritis samples also confirmed the presence of these Tfh-like and Tph populations [28•]. The analysis of the immune landscape of the same cohort also revealed signatures of autoantibody-producing T-bet+ antibody forming B cells among the locally activated B cells [28•]. In addition to the evidence suggesting a potential T:B collaboration, local anti-dsDNA producing plasma cells were found in both NZB/W lupus-prone mice and patients with lupus nephritis [29]. All these data suggest the possibility of autoantibody-producing B cell selection occurring in the peripheral blood and also in situ in the nephritic kidney, and also raise the question about the extent to which local autoantibody-producing cells contribute to the development and perpetuation the tissue injury.
Cytotoxic CD8+ T Cell—Friends or Foes
CD8+ T cells are cytotoxic immune cells that kill infected or damaged cells by releasing cytotoxins, such as granzymes and perforins. Peripheral blood CD8+ T cells from SLE patients generally have decreased production of granzyme B and perforin [30], and display impaired cytolytic function [31]. These functional defects in cytotoxicity likely contribute to the pathogenesis of autoimmunity. Accelerated autoimmunity in perforin-deficient lupus-prone mice suggests that cytotoxic function of CD8+ cells serves as a peripheral tolerance checkpoint charged to remove autoreactive B cells [32]. In a murine model of graft-vs-host disease presenting with lupus-like disease, co-transfer of donor cytotoxic CD8+ T cells eliminated host B cells and the subsequent development of lupus, confirming the role of cytotoxic T cells in inducing peripheral tolerance [33]. A reduction in cytolytic capacity also correlates with poor control of Epstein-Barr virus infection [34, 35] and predisposition to infection [36]. The risk of infections in SLE patients correlates with the reduction of signaling lymphocytic activation molecule family member 4 (SLAMF4) positive cytotoxic CD8+ T cells [37], and the expansion of peripheral blood CD38+ CD8+ T cell population, which presents features of exhaustion including decreased granzyme and perforin production [38•]. CD38, a cyclic ADP ribose hydrolase expressed on cell membrane and an exhaustion marker, degrades NAD+; limits chromatin accessibility of RUNX3, EOMES, and TBX21 loci through the action of the histone methyl-transferase EZH2; and suppresses the expression of cytotoxic genes [38•]. These epigenetic modifications explain the functional defect of the CD38+ CD8+ T cells and explain the higher risk of infection in lupus patients. Administration of the CD38 antibody daratumumab to two patients with SLE not only reduced autoantibody titers i by eliminating long-lived plasma cells, but also restored cytotoxicity of peripheral CD8+ T cells [39], suggesting its multifaceted role in targeting lupus pathogenesis.
In contrast to reduced cytolytic function in systemic circulating CD8+ T cells, CD8+ T cells extracted from inflammatory sites mostly indicated heightened effector functions, contributing to tissue damage (Fig. 2). However, the work by Tilstra et al. demonstrated exhausted phenotypes in kidney-infiltrating T cells, with their cytokine producing capability almost completely abolished [40]. In contrast with those findings, single-cell RNA-sequencing of biopsies from human lupus nephritis patients revealed clusters of NK and CD8+ T cells expressing GZMB and GZMK transcripts [28•]. These seemingly inconsistent data suggest heterogeneity in the nature of the tissue-infiltrating T cells, and possible discrepancies between human and murine lupus. Another intriguing paradox is the sharp contrast in cytotoxic effector functions between systemic exhausted and locally activated CD8+ T cells, and these differences are likely attributed to changes in the inflamed and damaged tissue microenvironment. These discrepancies in local and systemic CD8+ T cells function also pose great challenges in prescribing immunosuppressive drugs, which prevents disease flares and organ damage at the expense of imposing higher risk of infections [41]. Resolution of this therapeutic dilemma requires more detailed analysis of the pathophysiology of the disease and the design of targeted treatments specific to the local tissue-infiltrating T cells.
Fig. 2.
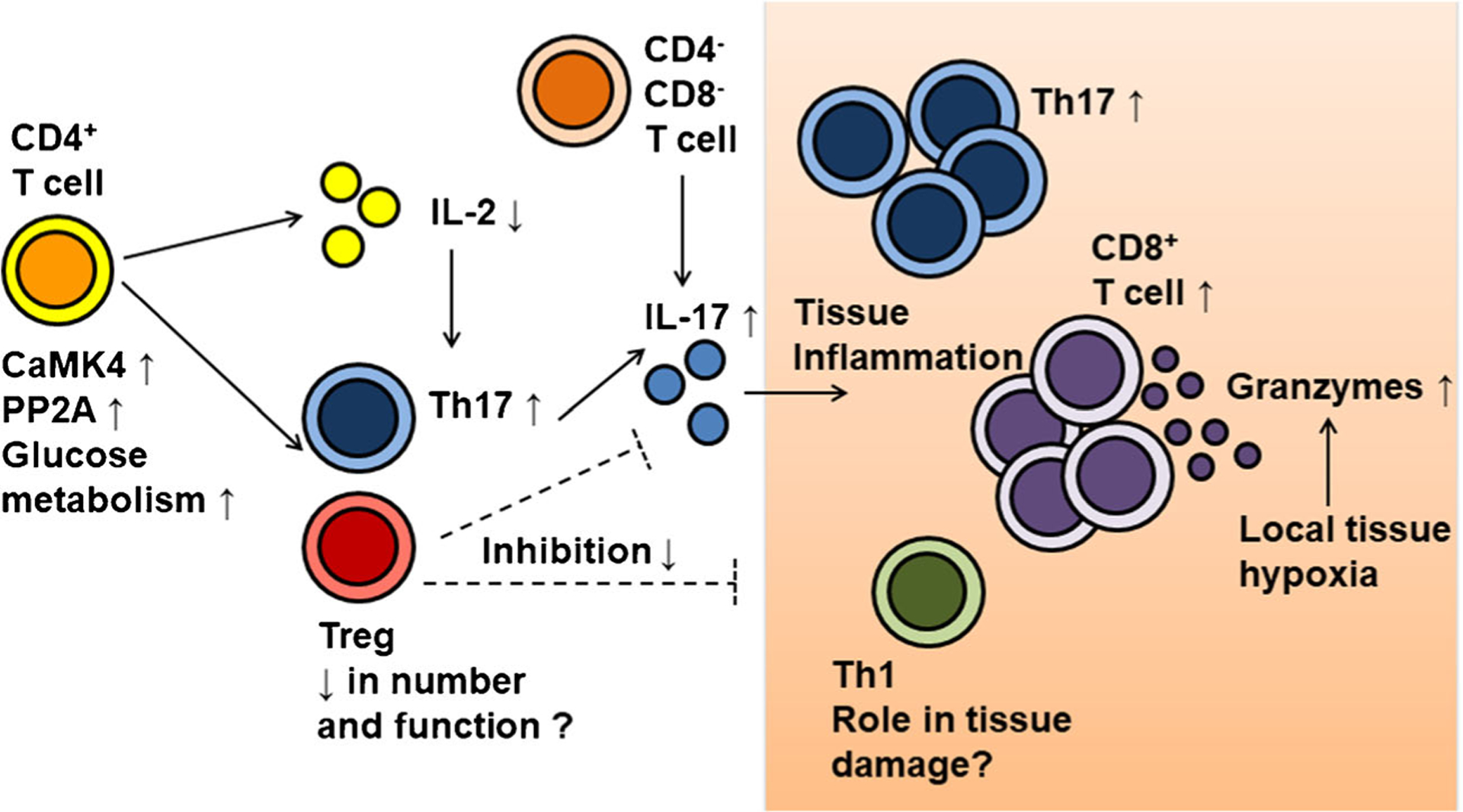
Dysregulated T cell subset distribution and function involved in driving lupus pathogenesis. Abnormality of CD4+ function and signaling, including increased calcium/calmodulin-dependent protein kinase IV (CaMK4), serine/threonine protein phosphatase 2A (PP2A), and glucose metabolism, cause reduction in IL-2 production and the imbalance of Th17/ regulatory T cells (Treg). Reduction of Treg numbers and function may partly play a role in lupus pathogenesis through inadequate suppression of systemic and tissue inflammation. Meanwhile, increased IL-17 production by Th17 and CD4− CD8− double-negative T cells contributes to tissue inflammation. Increased numbers of Th17 and CD8+ T cells indicate their pathogenic roles, and increased effector function of tissue-infiltrating T cells may be caused of local factors, such as hypoxia
Local tissue hypoxia and transcriptional activation of hypoxia-inducible factor-1 (HIF-1) are suggested as drivers of heightened effector function in tissue-infiltrating T cells [42•]. The pathogenic process in the lupus kidney is therefore seen as a chain reaction of tissue hypoxia, increased cytotoxic effector function of infiltrating T cells, and persistence of organ damage. Therapeutic HIF-1 blockade helps breakup this vicious cycle and shows promising effect in murine lupus nephritis. However, implementation of such treatment should also consider its effect on other local tissue-resident cell types. HIF stabilization in non-immune cells effectively reduces the degree of tissue injury, and this effect is most prominent in endothelial cells through the action of HIF-2α [43]. These HIF-dependent protective effects can be viewed as adaptive cellular changes to avoid hypoxic injury to the tissue. Considering HIF-2 mediated protective mechanisms of renal non-immune cells and also HIF-2-dependent erythropoietin production, a selective HIF-1α blockade may be used to ameliorate tissue damage incurred by infiltrating immune cells and minimize the adverse pharmacologic effects [42•]. Other approaches are also implemented to block expansion of these tissue-resident T cells.
In lupus nephritis biopsy samples, TCRβ sequencing shows persistence of oligoclonal effector CD8+ T cells in damaged tissues and the numbers of CD8+ T cells in diseased tissue correlate positively with lupus disease activity [44], suggesting features of tissue-resident memory T cells and the expansion of these culprit T cells locally. In lupus-porne mice, blocking JAK/STAT signaling by tofacitinib effectively blocks the expansion of tissue-resident memory CD8+ T cells and promotes their apoptosis [45]. These data provide promising approaches to develop novel therapeutics to target specifically the local tissue-resident memory CD8+ T cells involved in the promotion of tissue damage and could, potentially, minimize systemic adverse effect.
Pathogenic IL-17 Producing Cells
Two types of IL-17 family cytokines, IL-17A and IL-17F, promote local tissue inflammation by inducing chemokine production, such as monocyte chemoattractant protein-1 (MCP-1), to recruit monocytes and neutrophils to the site of inflammation [46]. Propagation of these proinflammatory Th17 cells in lupus patients is likely the result of multiple factors, including abnormal T cell signaling and environmental exposure of bacteria. In T cells from SLE patients and lupus-prone mice, T cells display increased IL-17 production as a result of activation of calcium/calmodulin-dependent protein kinase IV (CaMK4) [47] (Fig. 2). Inhibition of CaMK4 not only blocks Th17 cell expansion [48], but also restores IL-2 production and expands functional regulatory T cells in murine lupus [49]. Lupus CD4+ T cells also express higher TLR2, which upon activation increases histone acetylation and reduces methylation of the IL17A and IL17F promoter region [50]. Enhanced catalytic subunit of protein phosphatase 2A (PP2Ac) in SLE T cells also contributes to DNA hypomethylation, particularly of the Il17 locus, facilitating thus Th17 differentiation [51, 52]. Gut microbiota dysbiosis also account for the Th17/Treg imbalance seen in lupus patients and mice [53•], and the abnormal CD4+ T cells differentiation and activation likely stems from the molecular mimicry between bacterial epitopes and autoantigens.
IL-17 producing T cells are also expanded in the kidney and skin lesions of lupus patients, and have been proposed to play pathogenic roles [54]. Variants in CCR6—the protein product of which is the signature chemokine receptor expressed by Th17 cells—are associated with susceptibility to lupus nephritis [55], suggesting a pathogenic role of IL-17 producing cells in disease. In addition to the chemokine gradient, metalloprotease-cleaved CD95 ligand induces S1P receptor 3 in Th17 cells with transmigration into the inflamed kidney and the promotion of organ damage [56]. These tissue-resident Th17 cells likely accumulate in susceptible patients after exposure to numerous systemic pathogens. Study of kidney tissue samples from patients with antineutrophil cytoplasmic antibodies (ANCA)–associated glomerulonephritis revealed dominance of resident Th17 cells, and these are likely the result of exposure to pathogens derived from pathogens such as S. aureus [57].
In addition to the canonical Th17 cells, double-negative T cells (CD4− CD8−) represent another major source of IL-17 producing cells [58] (Fig. 2). These double-negative T cells present with mitochondrial hyperpolarization, increased mitochondrial mass, and mTOR activation prior disease flare [59]. They derive from CD8+ T cells through silencing of CD8A and CD8B by the cAMP-responsive element modulator α (CREMα) [60], and are reactive to self-antigens [61]. However, targeting IL-17 in lupus mice showed conflicting results. IL-17 deficiency (Il17−/−) prevents disease induction with reduction in the frequency of double-negative T cells in pristane-induced lupus mice [62]. By contrast, IL-17A-deficient lupus-prone MRL/lpr mice were not protected from renal damage, nor did IL-17A neutralization prevent nephritis in NZB/W F1 lupus-prone mice [63]. Taken together, these data suggest heterogeneity in lupus pathogenesis, and targeting only one inflammatory cytokine may be insufficient to completely reverse the disease. Certain patients may still benefit from IL-17 blockade, yet this may require detail characterization of the pathogenetic profile of each patient.
T Helper Type 1 Cells
Early studies of peripheral blood mononuclear cells from patients with proliferative lupus nephritis demonstrated the predominance of Th1 cells [64]. Two autoreactive T cell clones identified in murine lupus models were IFN-γ producing Th1 cells [65]. Meanwhile, IL-12 and IFN-γ signaling are necessary for the initiation and progression of kidney disease in murine lupus [66, 67]. Enrichment of CXCR3+ CD4+ T cells is also found in patients with lupus nephritis, and the number of Th1 cells in the urine correlates with disease activity [68]. Ifng−/− mice are resistant to pristane-induced lupus [69], and administration of anti-IFN-γ blocks the development of lupus nephritis in NZB/W F1 lupus-prone mice [63]. These data, along with the reduced renal damage in Cxcr3 knockout lupus mice [70], suggest that Th1 cells may be involved in promoting tissue injury. In addition to its role on organ damage, IFN-γ signaling also leads to Bcl-6 overexpression in Tfh cells, excessive germinal center response and autoantibody production, suggesting the potential benefits of blocking IFN-γ in SLE patients [71]. However, administering anti-interferon-γ monoclonal antibody to lupus patients failed to reduce disease activity, organ damage, or autoantibody titer [72], which casts doubts on the role of Th1 cells in lupus pathogenesis.
Regulatory T Cells
Regulatory T cells (Treg) are CD4+ T cells expressing Forkhead box P3 (FoxP3) with high levels of IL-2Rα (CD25) and are known to suppress the activation and proliferation of CD4+ and CD8+ T cells. Treg cells not only keep autoreactive T cell clones in check [73], but also inhibit the accumulation of autoreactive B cells [74]. Due to their role in maintaining peripheral immune tolerance [75], their functional or numeric defects are thought to contribute to SLE pathogenesis (Fig. 2). However, mixed results from SLE patients have yet to produce a clear consensus. In NZB × NZW F1 lupus mice, adoptive transfer of Treg cells leads to disease suppression and remission [76, 77]. In this lupus-prone strain, the imbalance of effector T cells and the reduced proliferation of Treg cells is due to IL-2 deprivation, which can be reversed by injecting recombinant IL-2 [78]. In another murine lupus MRL/lpr, Treg cells have reduced suppressive capacity against T cell proliferation [79, 80], which can also contribute to autoimmunity. These data in murine lupus suggest that either numeric or functional defects in Treg cells can cause of systemic autoimmunity. However, results from studies in SLE patients are mixed probably due to the heterogeneity of the studied cohorts and the use of different markers in defining Treg. While some studies revealed functional and quantitative deficiency of regulatory T cells in SLE patients [81, 82], others suggest the resistance of SLE effectors T cells against suppression rather than deficiencies in Treg cell function [83, 84]. In addition, educed IL-2 production in lupus patients is directly related to lower CD25 expression [85], making the definition of Treg by CD25 expression more problematic. Taking into account the IL-2 deficiency in lupus patients and its consequence of Th17/Treg balance, low-dose IL-2 treatment has shown promising results in inducing Treg, reducing Th17, as well as reducing disease activity [86].
Aberrant activation of the mechanistic target of rapamycin complex 1 (mTORC1) results in deficiency of Treg suppressive function and subsequent development of autoimmune disease [87]. Increased mTORC1 signaling and functional defects in Treg cells is directly related to the altered glucose metabolism, in which mTOC1-dependent Glut1 expression reduces both Foxp3 expression and the suppressive capacity of Treg cells [88]. Elevated glucose metabolism is also found in activated CD4+ T cells in both lupus-prone mice and SLE patients, and inhibition of both glycolysis and mitochondrial metabolism restores IL-2 production and ameliorates disease activity in lupus-prone mice [89]. In addition to these pharmacologic treatments, autologous Treg cell transfer has been tested in patients, and has shown promising results in accumulating Treg cells at the sites of inflammation [90]. In summary, despite the conflicting results in SLE patients, certain patients may still benefit from treatments that target Treg cells. More studies are needed to characterize patients who can benefit most from autologous Treg cell transfer and low-dose IL-2 administration.
Conclusions
T cells are involved in multiple aspects of SLE pathogenesis. Germinal center Tfh cells provide B cell help necessary for the development of autoreactive clones, which further leads to autoantibody production. T-B collaboration occurs also outside of germinal center through the action of extrafollicular Tfh cells, circulatory and tissue Tfh-like cells, and peripheral helper T cells. Exhausted phenotype of systemic CD8+ T cells likely also results in loss of peripheral tolerance and contributes to autoreactive B cells. Abnormal lupus T cells signaling pathways cause reduction in IL-2 production which drives the imbalanced Th17/Treg differentiation, and contributes to the acceleration of organ damage. Changes in the damaged organ microenvironment, such as hypoxia, also contribute to the altered T cells phenotypes and lead to further aggravation in organ damage. Studies in recent years have made significant breakthroughs in uncovering pathogenic T cell signaling pathways and provide the foundation for the development of novel therapies.
Footnotes
Conflict of Interest The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent This article does not involve human subjects or animal protocol necessary for ethics approval.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Olsen NJ, Karp DR. Autoantibodies and SLE: the threshold for disease. Nat Rev Rheumatol. 2014;10(3):181–6. 10.1038/nrrheum.2013.184. [DOI] [PubMed] [Google Scholar]
- 2.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–21. 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 3.Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol. 2020;21(6):605–14. 10.1038/s41590-020-0677-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seth A, Craft J. Spatial and functional heterogeneity of follicular helper T cells in autoimmunity. Curr Opin Immunol. 2019;61:1–9. 10.1016/j.coi.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Apostolidis SA, Lieberman LA, Kis-Toth K, Crispín JC, Tsokos GC. The dysregulation of cytokine networks in systemic lupus erythematosus. J Interf Cytokine Res. 2011;31(10):769–79. 10.1089/jir.2011.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellmann U, Letz M, Herrmann M, Angermüller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNA autoantibodies. Proc Natl Acad Sci U S A. 2005;102(26):9258–63. 10.1073/pnas.0500132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo W, Smith D, Aviszus K, Detanico T, Heiser RA, Wysocki LJ. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J Exp Med. 2010;207(10):2225–37. 10.1084/jem.20092712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobel ES, Kakkanaiah VN, Kakkanaiah M, Cheek RL, Cohen PL, Eisenberg RA. T-B collaboration for autoantibody production in lpr mice is cognate and MHC-restricted. J Immunol. 1994;152(12): 6011–6. [PubMed] [Google Scholar]
- 9.Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. 2010;207(2):365–78. 10.1084/jem.20091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal Centre formation. Nature. 2008;455(7214):764–9. 10.1038/nature07345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Randall TD, Heath AW, Santos-Argumedo L, Howard MC, Weissman IL, Lund FE. Arrest of B lymphocyte terminal differentiation by CD40 signaling: mechanism for lack of antibody-secreting cells in germinal centers. Immunity. 1998;8(6):733–42. 10.1016/s1074-7613(00)80578-6. [DOI] [PubMed] [Google Scholar]
- 12.Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature. 2015;517(7533):214–8. 10.1038/nature13803. [DOI] [PubMed] [Google Scholar]
- 13.Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435(7041):452–8. 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 14.Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206(3):561–76. 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwai H, Abe M, Hirose S, Tsushima F, Tezuka K, Akiba H, et al. Involvement of inducible costimulator-B7 homologous protein costimulatory pathway in murine lupus nephritis. J Immunol. 2003;171(6):2848–54. 10.4049/jimmunol.171.6.2848. [DOI] [PubMed] [Google Scholar]
- 16.Kalled SL, Cutler AH, Datta SK, Thomas DW. Anti-CD40 ligand antibody treatment of SNF1 mice with established nephritis: preservation of kidney function. J Immunol. 1998;160(5):2158–65. [PubMed] [Google Scholar]
- 17.Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC 3rd, et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A. 2009;106(5):1518–23. 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi JY, Seth A, Kashgarian M, Terrillon S, Fung E, Huang L, et al. Disruption of pathogenic cellular networks by IL-21 blockade leads to disease amelioration in murine lupus. J Immunol. 2017;198(7):2578–88. 10.4049/jimmunol.1601687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science (New York, NY). 2002;297(5589):2066–70. 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 20.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205(12):2873–86. 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. 2011;208(7):1377–88. 10.1084/jem.20102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1): 108–21. 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol (Hoboken, NJ). 2015;67(4):988–99. 10.1002/art.39020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Lindwall E, Gauthier C, Lyman J, Spencer N, Alarakhia A, et al. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus. 2015;24(9):909–17. 10.1177/0961203314567750. [DOI] [PubMed] [Google Scholar]
- 25.Liarski VM, Kaverina N, Chang A, Brandt D, Yanez D, Talasnik L, et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med. 2014;6(230): 230ra46. 10.1126/scitranslmed.3008146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.•.Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542(7639):110–4. 10.1038/nature20810 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first to demonstrate peripheral T helper, and its potential role in driving B cell maturation in the inflamed tissue.
- 27.Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, et al. PD-1hiCXCR5- T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI insight. 2019;4(20). 10.1172/jci.insight.130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.•.Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019;20(7):902–14. 10.1038/s41590-019-0398-x [DOI] [PMC free article] [PubMed] [Google Scholar]; A well-constructed cohort of single-cell RNA-seq of immune cells from patients with lupus nephritis.
- 29.Espeli M, Bokers S, Giannico G, Dickinson HA, Bardsley V, Fogo AB, et al. Local renal autoantibody production in lupus nephritis. J Am Soc Nephrol. 2011;22(2):296–305. 10.1681/asn.2010050515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comte D, Karampetsou MP, Yoshida N, Kis-Toth K, Kyttaris VC, Tsokos GC. Signaling lymphocytic activation molecule family member 7 engagement restores defective effector CD8+ T cell function in systemic lupus erythematosus. Arthritis Rheumatol (Hoboken, NJ). 2017;69(5):1035–44. 10.1002/art.40038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stohl W Impaired polyclonal T cell cytolytic activity. A possible risk factor for systemic lupus erythematosus. Arthritis Rheum. 1995;38(4):506–16. 10.1002/art.1780380408. [DOI] [PubMed] [Google Scholar]
- 32.Peng SL, Moslehi J, Robert ME, Craft J. Perforin protects against autoimmunity in lupus-prone mice. J Immunol. 1998;160(2):652–60. [PubMed] [Google Scholar]
- 33.Via CS, Sharrow SO, Shearer GM. Role of cytotoxic T lymphocytes in the prevention of lupus-like disease occurring in a murine model of graft-vs-host disease. J Immunol. 1987;139(6):1840–9. [PubMed] [Google Scholar]
- 34.Kang I, Quan T, Nolasco H, Park SH, Hong MS, Crouch J, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol. 2004;172(2):1287–94. 10.4049/jimmunol.172.2.1287. [DOI] [PubMed] [Google Scholar]
- 35.Larsen M, Sauce D, Deback C, Arnaud L, Mathian A, Miyara M, et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog. 2011;7(10):e1002328. 10.1371/journal.ppat.1002328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–30. 10.1038/nrrheum.2016.186. [DOI] [PubMed] [Google Scholar]
- 37.Kis-Toth K, Comte D, Karampetsou MP, Kyttaris VC, Kannan L, Terhorst C, et al. Selective loss of signaling lymphocytic activation molecule family member 4-positive CD8+ T cells contributes to the decreased cytotoxic cell activity in systemic lupus erythematosus. Arthritis Rheumatol (Hoboken, NJ). 2016;68(1):164–73. 10.1002/art.39410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.•.Katsuyama E, Suarez-Fueyo A, Bradley SJ, Mizui M, Marin AV, Mulki L, et al. The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. Cell Rep. 2020;30(1):112–23.e4. 10.1016/j.celrep.2019.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study nicely links CD38 with CD8+ T cell exhaustion and risk of infection in lupus patients.
- 39.Ostendorf L, Burns M, Durek P, Heinz GA, Heinrich F, Garantziotis P, et al. Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. N Engl J Med. 2020;383(12): 1149–55. 10.1056/NEJMoa2023325. [DOI] [PubMed] [Google Scholar]
- 40.Tilstra JS, Avery L, Menk AV, Gordon RA, Smita S, Kane LP, et al. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J Clin Invest. 2018;128(11): 4884–97. 10.1172/jci120859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houssiau FA, Vasconcelos C, D’Cruz D, Sebastiani GD, Garrido EER, Danieli MG, et al. Immunosuppressive therapy in lupus nephritis: the Euro-Lupus Nephritis Trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum. 2002;46(8):2121–31. 10.1002/art.10461. [DOI] [PubMed] [Google Scholar]
- 42.•.Chen PM, Wilson PC, Shyer JA, Veselits M, Steach HR, Cui C et al. Kidney tissue hypoxia dictates T cell-mediated injury in murine lupus nephritis. Sci Transl Med. 2020;12(538). doi: 10.1126/scitranslmed.aay1620. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first study to discuss how inflammatory microenvironment can affect T cell phenotype in promoting tissue damage, and this maladaptation can be intervened to reverse organ damage.
- 43.Kapitsinou PP, Sano H, Michael M, Kobayashi H, Davidoff O, Bian A, et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest. 2014;124(6):2396–409. 10.1172/jci69073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Couzi L, Merville P, Deminiere C, Moreau JF, Combe C, Pellegrin JL, et al. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. 2007;56(7):2362–70. 10.1002/art.22654. [DOI] [PubMed] [Google Scholar]
- 45.Zhou M, Guo C, Li X, Huang Y, Li M, Zhang T, et al. JAK/STAT signaling controls the fate of CD8(+)CD103(+) tissue-resident memory T cell in lupus nephritis. J Autoimmun. 2020;109: 102424. 10.1016/j.jaut.2020.102424. [DOI] [PubMed] [Google Scholar]
- 46.Woltman AM, de Haij S, Boonstra JG, Gobin SJ, Daha MR, van Kooten C. Interleukin-17 and CD40-ligand synergistically enhance cytokine and chemokine production by renal epithelial cells. J Am Soc Nephrol. 2000;11(11):2044–55. [DOI] [PubMed] [Google Scholar]
- 47.Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–45. 10.1172/jci73411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otomo K, Koga T, Mizui M, Yoshida N, Kriegel C, Bickerton S, et al. Cutting edge: nanogel-based delivery of an inhibitor of CaMK4 to CD4+ T cells suppresses experimental autoimmune encephalomyelitis and lupus-like disease in mice. J Immunol. 2015;195(12):5533–7. 10.4049/jimmunol.1501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koga T, Ichinose K, Mizui M, Crispín JC, Tsokos GC. Calcium/ calmodulin-dependent protein kinase IV suppresses IL-2 production and regulatory T cell activity in lupus. J Immunol. 2012;189(7):3490–6. 10.4049/jimmunol.1201785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Liao J, Zhao M, Wu H, Yung S, Chan TM, et al. Increased expression of TLR2 in CD4(+) T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur J Immunol. 2015;45(9):2683–93. 10.1002/eji.201445219. [DOI] [PubMed] [Google Scholar]
- 51.Apostolidis SA, Rauen T, Hedrich CM, Tsokos GC, Crispín JC. Protein phosphatase 2A enables expression of interleukin 17 (IL-17) through chromatin remodeling. J Biol Chem. 2013;288(37): 26775–84. 10.1074/jbc.M113.483743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunahori K, Nagpal K, Hedrich CM, Mizui M, Fitzgerald LM, Tsokos GC. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J Biol Chem. 2013;288(30):21936–44. 10.1074/jbc.M113.467266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.•.Choi SC, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci Transl Med. 2020;12(551). 10.1126/scitranslmed.aax2220 [DOI] [PMC free article] [PubMed] [Google Scholar]; A very nice illustration of how microbiota and altered metabolism contribute to lupus pathogenesis.
- 54.Crispin JC, Tsokos GC. Interleukin-17-producing T cells in lupus. Curr Opin Rheumatol. 2010;22(5):499–503. 10.1097/BOR.0b013e32833c62b0. [DOI] [PubMed] [Google Scholar]
- 55.Zhou XJ, Mu R, Li C, Nath SK, Zhang YM, Qi YY, et al. Association of variants in CCR6 with susceptibility to lupus nephritis in Chinese. Arthritis Rheumatol (Hoboken, NJ). 2015;67(11):3091–3. 10.1002/art.39268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poissonnier A, Sanseau D, Le Gallo M, Malleter M, Levoin N, Viel R, et al. CD95-mediated calcium signaling promotes T helper 17 trafficking to inflamed organs in lupus-prone mice. Immunity. 2016;45(1):209–23. 10.1016/j.immuni.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krebs CF, Reimers D, Zhao Y, Paust HJ, Bartsch P, Nuñez S, et al. Pathogen-induced tissue-resident memory T(H)17 (T(RM)17) cells amplify autoimmune kidney disease. Sci Immunol. 2020;5(50). 10.1126/sciimmunol.aba4163. [DOI] [PubMed] [Google Scholar]
- 58.Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181(12):8761–6. 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai ZW, Borsuk R, Shadakshari A, Yu J, Dawood M, Garcia R, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236–46. 10.4049/jimmunol.1301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hedrich CM, Rauen T, Crispin JC, Koga T, Ioannidis C, Zajdel M, et al. cAMP-responsive element modulator α (CREMα) trans-represses the transmembrane glycoprotein CD8 and contributes to the generation of CD3+CD4-CD8- T cells in health and disease. J Biol Chem. 2013;288(44):31880–7. 10.1074/jbc.M113.508655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodríguez-Rodríguez N, Apostolidis SA, Fitzgerald L, Meehan BS, Corbett AJ, Martín-Villa JM, et al. Pro-inflammatory self-reactive T cells are found within murine TCR-αβ(+) CD4(−) CD8(−) PD-1(+) cells. Eur J Immunol. 2016;46(6):1383–91. 10.1002/eji.201546056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amarilyo G, Lourenco EV, Shi FD, La Cava A. IL-17 promotes murine lupus. J Immunol. 2014;193(2):540–3. 10.4049/jimmunol.1400931. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol (Hoboken, NJ). 2015;67(2):475–87. 10.1002/art.38955. [DOI] [PubMed] [Google Scholar]
- 64.Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum. 1999;42(8):1644–8. 10.1002/1529-0131(199908)42:8<1644::aidanr12>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 65.Okamoto A, Fujio K, Tsuno NH, Takahashi K, Yamamoto K. Kidney-infiltrating CD4+ T-cell clones promote nephritis in lupus-prone mice. Kidney Int. 2012;82(9):969–79. 10.1038/ki.2012.242. [DOI] [PubMed] [Google Scholar]
- 66.Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR. IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1998;161(1):494–503. [PubMed] [Google Scholar]
- 67.Kikawada E, Lenda DM, Kelley VR. IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J Immunol. 2003;170(7): 3915–25. [DOI] [PubMed] [Google Scholar]
- 68.Enghard P, Humrich JY, Rudolph B, Rosenberger S, Biesen R, Kuhn A, et al. CXCR3+CD4+ T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients. Arthritis Rheum. 2009;60(1):199–206. 10.1002/art.24136. [DOI] [PubMed] [Google Scholar]
- 69.Richards HB, Satoh M, Jennette JC, Croker BP, Yoshida H, Reeves WH. Interferon-gamma is required for lupus nephritis in mice treated with the hydrocarbon oil pristane. Kidney Int. 2001;60(6):2173–80. 10.1046/j.1523-1755.2001.00045.x. [DOI] [PubMed] [Google Scholar]
- 70.Steinmetz OM, Turner JE, Paust HJ, Lindner M, Peters A, Heiss K, et al. CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis. J Immunol. 2009;183(7):4693–704. 10.4049/jimmunol.0802626. [DOI] [PubMed] [Google Scholar]
- 71.Lee SK, Silva DG, Martin JL, Pratama A, Hu X, Chang PP, et al. Interferon-γ excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity. 2012;37(5):880–92. 10.1016/j.immuni.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 72.Boedigheimer MJ, Martin DA, Amoura Z, Sánchez-Guerrero J, Romero-Diaz J, Kivitz A, et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci Med. 2017;4(1):e000226. 10.1136/lupus-2017-000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where CD4+ CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med. 2005;202(10):1387–97. 10.1084/jem.20051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood. 2013;121(9):1595–603. 10.1182/blood-2012-09-457465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–87. 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 76.Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol. 2006;177(3):1451–9. 10.4049/jimmunol.177.3.1451. [DOI] [PubMed] [Google Scholar]
- 77.Weigert O, von Spee C, Undeutsch R, Kloke L, Humrich JY, Riemekasten G. CD4+Foxp3+ regulatory T cells prolong drug-induced disease remission in (NZBxNZW) F1 lupus mice. Arthritis Res Therapy. 2013;15(1):R35. 10.1186/ar4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc Natl Acad Sci U S A. 2010;107(1):204–9. 10.1073/pnas.0903158107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Divekar AA, Dubey S, Gangalum PR, Singh RR. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. J Immunol. 2011;186(2):924–30. 10.4049/jimmunol.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parietti V, Monneaux F, Décossas M, Muller S. Function of CD4+, CD25+ Treg cells in MRL/lpr mice is compromised by intrinsic defects in antigen-presenting cells and effector T cells. Arthritis Rheum. 2008;58(6):1751–61. 10.1002/art.23464. [DOI] [PubMed] [Google Scholar]
- 81.Bonelli M, Savitskaya A, von Dalwigk K, Steiner CW, Aletaha D, Smolen JS, et al. Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int Immunol. 2008;20(7):861–8. 10.1093/intimm/dxn044. [DOI] [PubMed] [Google Scholar]
- 82.Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+ CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. 2007;178(4):2579–88. 10.4049/jimmunol.178.4.2579. [DOI] [PubMed] [Google Scholar]
- 83.Vargas-Rojas MI, Crispín JC, Richaud-Patin Y, Alcocer-Varela J. Quantitative and qualitative normal regulatory T cells are not capable of inducing suppression in SLE patients due to T-cell resistance. Lupus. 2008;17(4):289–94. 10.1177/0961203307088307. [DOI] [PubMed] [Google Scholar]
- 84.Venigalla RK, Tretter T, Krienke S, Max R, Eckstein V, Blank N, et al. Reduced CD4+,CD25- T cell sensitivity to the suppressive function of CD4+,CD25high,CD127 -/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum. 2008;58(7):2120–30. 10.1002/art.23556. [DOI] [PubMed] [Google Scholar]
- 85.Costa N, Marques O, Godinho SI, Carvalho C, Leal B, Figueiredo AM, et al. Two separate effects contribute to regulatory T cell defect in systemic lupus erythematosus patients and their unaffected relatives. Clin Exp Immunol. 2017;189(3):318–30. 10.1111/cei.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med. 2016;22(9):991–3. 10.1038/nm.4148. [DOI] [PubMed] [Google Scholar]
- 87.Apostolidis SA, Rodríguez-Rodríguez N, Suárez-Fueyo A, Dioufa N, Ozcan E, Crispín JC, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol. 2016;17(5):556–64. 10.1038/ni.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance T(reg) cell anabolic metabolism for suppression. Nat Immunol. 2016;17(12): 1459–66. 10.1038/ni.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dall’Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al. Adoptive Treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol (Hoboken, NJ). 2019;71(3):431–40. 10.1002/art.40737. [DOI] [PMC free article] [PubMed] [Google Scholar]