

Abstract
FGF2 is a potent stimulator of vascular growth; however, even with a deficiency of FGF2 (Fgf2−/−), developmental vessel growth or ischemia-induced revascularization still transpires. It remains to be elucidated as to what function, if any, FGF2 has during ischemic injury. Wildtype (WT) or Fgf2−/− mice were subjected to hindlimb ischemia for up to 42-days. Limb function, vascular growth, inflammatory- and angiogenesis-related proteins, and inflammatory cell infiltration were assessed in sham and ischemic limbs at various timepoints. Recovery of ischemic limb function was delayed in Fgf2−/− mice. Yet, vascular growth response to ischemia was similar between WT and Fgf2−/− hindlimbs. Several angiogenesis- and inflammatory-related proteins (MCP-1, CXCL16, MMPs and PAI-1) were increased in Fgf2−/− ischemic muscle. Neutrophil or monocyte recruitment/infiltration was elevated in Fgf2−/− ischemic muscle. In summary, our study indicates that loss of FGF2 induces a pro-inflammatory microenvironment in skeletal muscle which exacerbates ischemic injury and delays functional limb use.
Keywords: fibroblast growth factor 2, hindlimb ischemia, arteriogenesis, angiogenesis, inflammation, micro-CT
INTRODUCTION
Diseases of vascular insufficiency are one of the leading causes of morbidity and mortality in the United States and worldwide. According to the American Heart Association (AHA), over 83 million people in the U.S are currently affected with one or more forms of cardiovascular disease (CVD). Approximately 18.2 and 8.5 million of these CVD patients are living with coronary artery disease (CAD) and peripheral artery disease (PAD), respectively (Benjamin et al. 2019). In addition to CAD and PAD, a significant subset of these patients is afflicted with other comorbidities that render them unsuitable for conventional revascularization procedures including percutaneous angioplasties and bypass grafts. Other CVD risk factors such as obesity, diabetes, or hypercholesteremia can also diminish the collateralization (called “arteriogenesis”; remodeling of preexisting arteriolar vessels) response to vascular occlusion (Lotfi et al. 2013; Laham and Baim 2007). A potential alternative treatment for these “no-option” patients is therapeutic neovascularization which aims to stimulate or enhance physiological angiogenesis (capillary vessel growth) and/or collateralization with pro-angiogenic genes, proteins or stem cells (Silvestre, Smadja, and Lévy 2013; Ahn et al. 2008). Evaluation of angiogenic factors and stem cells for efficacious use in CAD and PAD necessitated the development of in vivo pre-clinical models that approximated the human diseases of muscle perfusion loss. Additionally, it has become important to visualize, characterize, and analyze the post-ischemic vascular network that develops in the presence or absence of pro-/anti- angiogenesis agents of interest.
Proof-of-concept studies in small and large animal models of chronic ischemia have provided evidence for the therapeutic potential of exogenous FGF2 (Simons et al. 2002; Lederman et al. 2002). This promise, however, did not translate into successful clinical use. To date, the precise role of endogenous FGF2 in vascular biology is largely inconclusive since no effect on vascular development has been observed in mice with a targeted deletion of Fgf2. Several, independently developed lines of mice deficient in FGF2 gene expression have been produced to describe the roles of FGF2 in vascular development and physiology. Our laboratory has shown that normal growth of capillary and smooth muscle-containing vessels occurs in Fgf2 knockout hearts of mice bred on a 50:50 129/Sv and Black Swiss background (House et al. 2003; Liao et al. 2007). A similar result was observed in Fgf2−/− mice of a different strain, C57BL/6J (Virag et al. 2007). Mice bred on a C57BL/6J x 129/Sv mixed background, however, had impaired myocardial capillarogenesis when Fgf2 gene expression was partially (+/−) or completely (−/−) disrupted (Amann et al. 2006). Fgf2-deficient mice have normal retinal vascularization and neovascularization responses to retinal or choroid layer injury (Ozaki et al. 1998; Tobe et al. 1998). When subjected to chronic ischemia, Fgf2−/− hindlimb has similar capillary and arteriole densities as WT hindlimb (Sullivan, Doetschman, and Hoying 2002). Further complicating the study of the function of FGF2 in vascular growth is the inhibition of angiogenesis observed in studies employing antibody neutralization. Capillary density was significantly reduced in skeletal muscle denervation or ischemic injury models in the presence of FGF2 neutralizing antibodies (Walgenbach et al. 1995; Lefaucheur et al. 1996). While these contradictory results can be partially accounted when differences in animal strains or experimental tools are considered as well as the differences in necessity vs. sufficiency, the role of Fgf2 expression in neo/revascularization has yet to be clearly defined. The objective of this study was to determine the nature of endogenous FGF2-regulated vascular remodeling during chronic ischemia.
MATERIALS AND METHODS
Animals
Mice were housed in a pathogen-free facility and handled in accordance with standard use protocols, animal welfare regulations, and the NIH Guide for the Care and Use of Laboratory Animals. All protocols were approved by the University of Cincinnati Institutional Animal Care and Use Committee. Mice with a targeted ablation of the Fgf2 gene (Fgf2−/−) were generated in the laboratory of Dr. Thomas Doetschman as previously described (Zhou et al. 1998; Schultz et al. 1999). All mice were bred on a (50%) Black Swiss/ (50%) 129 mixed background.
Hindlimb Ischemia Model
Age- (10–12 weeks) and sex-matched wildtype (WT) or Fgf2−/− mice were anesthetized with 2.5% Avertin (2, 2, 2-Tribromoethanol and tertiary amyl alcohol in 0.9% saline) at a dose of 0.02ml/g, intraperitoneally (i.p.) and placed on a heating pad at 37°C. Unilateral hindlimb ischemia was induced surgically as previously described (T Couffinhal et al. 1998; Adeyemo, Zhang, and Schultz 2012; Sullivan, Doetschman, and Hoying 2002). After a longitudinal skin incision at the medial thigh of the left hindlimb, the femoral artery and vein were exposed and dissected free of the associated nerve. The vessel pair was ligated proximal to the external iliac artery and distally at the bifurcation into the saphenous and popliteal branches. The intervening vessel pair was then excised. The overlying skin was then closed with 5–0 silk sutures and tissue adhesive. A sham procedure was performed on the contralateral (right) hindlimb, wherein a longitudinal skin incision and sutures were placed without ligation or excision.
Assessment of limb function
The incidence of hindlimb tissue necrosis and limb function was assessed daily up to 14 days of ischemia and then weekly up to 42 days of ischemia. Functional use of the ischemic limb was measured on the following scale modified from Stabile and group (Stabile et al. 2003) (4 = dragging of foot, 3 = no dragging of foot but no plantar flexion, 2 = plantar flexion but no flexing of toes and 1= normal function, with flexing of toes to resist gentle traction on the tail).
Histological analysis of vasculature
Following 42 days of ischemia, skeletal muscles were harvested from the thigh (gracilis, semi-membranosus, and semi-tendinosus) and calf (gastrocnemius) regions of the sham or ischemic limbs and immersion-fixed overnight in 4% paraformaldehyde followed by immersion in 70% ethanol for processing. 5μm thick transverse paraffin-embedded sections were deparaffinized with xylenes and followed by rehydration in alcohol/water (100%, 95% and 70%) solutions. A 10-minute antigen retrieval step was performed at 100°C using a pH 6.0 citric acid solution. Endogenous peroxidase activity was blocked with H2O2 followed by treatment with 10% normal goat serum to reduce nonspecific binding. Serial sections were then incubated overnight at 4°C with biotinylated Griffonia simplicifolia (Bandeiraea) isolectin B4 (1:100, Vector Labs) or rabbit polyclonal antibody against α-SMA (1: 15,000; Abcam) to detect endothelial or vascular smooth muscle cells respectively. Immunoperoxidase staining was detected with 3, 3’-diaminobenzidine (Vector Labs) and sections were counterstained with Mayer’s Hematoxylin (Life Technologies) to detect nuclei. Capillary vessels were identified as vessels with an outer diameter ≤10µm and positive staining for endothelial cells. α-SMA-positive vessels with a 20μm or more outer diameter were identified as arterioles and venules. Venules were distinguished from arterioles by their collapsed luminal structure and thinner layers of smooth muscle. Images were captured under 400X (capillaries) or 200X (arterioles) magnification from each section (2 sections/limb, 20 fields/section). Vessel densities were estimated as the total number of vessels per myofiber number in each field.
Micro-computed tomography imaging of the hindlimb vasculature
1). Contrast agent perfusion
After 42 days, mice were anesthetized and overdosed with sodium pentobarbital at a dose of 80mg/kg, i.p. A horizontal midline incision was made right below the diaphragm to avoid pneumothorax and to ensure blood flow during catheter insertion. The abdominal aorta was cannulated, the catheter secured with 5–0 suture and a 6mL heparinized (100U/mL) saline solution containing a mixture of vasodilators (10µM acetylcholine, 10µM adenosine, 100µM papaverine and 200µM sodium nitroprusside; Sigma-Aldrich, St. Louis, MO) was infused at 1 mL/min with a 3mL plastic syringe (Becton Dickson, Franklin Lakes, NJ) for blood washout. The inferior vena cava was severed to allow for drainage of blood and the infusing solution. For the Microfil perfusion, a curing agent (5% v/v) was added to catalyze the mixture of the components (compound and diluent mixed in equal amounts, according to the manufacturer recommendations; MV122 Flow-Tech Inc Carver, MA) with a working time of 20 minutes before the start of polymerization. 0.5mL of the prepared compound was drawn in a 1mL syringe and injected at 1mL/h with care taken to remove any perfusate that contaminated the surrounding tissue, skin or fur. Perfused specimens were placed at room temperature for 30 minutes and then stored at 4°C overnight to allow for complete polymerization of the contrast agent.
2). Specimen scanning protocol
Sham or ischemic limbs were dissected at the head of the femur for individual scanning at high resolution using the micro-CT scanner in the Tri-modal Siemens Inveon (Siemens Healthcare, Hoffman Estates, IL) and the Inveon Acquisition Workplace (IAW) (version 1.5.28) software. Each ex vivo limb was placed on the imaging pallet with a field of view (FOV) of 18 x 25mm, positioned on the dorsal side, and scanned from thigh to ankle. CT scanning parameters were 80kVp and 300μA (voltage and current) with 0.5mm Al filtration. A 192° half scan was performed for 384 steps with an exposure time of 2100ms for each step. Center offset and dark/light calibrations were performed immediately prior to scanning for image optimization. The acquisition protocol included using a high system magnification with 2x2 binning to generate an isotropic voxel size of 17μm3. Raw data reconstruction was performed with a Shepp-Logan filter. Prior to image reconstruction, beam hardening correction was applied to ensure the quantitative accuracy of the Hounsfield Units (HUs) in the final images. Images are reconstructed with the standard Filtered Back-Projection (FBP) algorithm with no additional down-sampling of the raw data (binning of image voxels).
3). Micro-CT Data Volume Rendering, Segmentation and Quantification
For vessel morphology analysis, bones were digitally removed and the vasculature was segmented from the surrounding tissue (background) based on the density of the X-ray-attenuated voxels of the Microfil within the vessels. Segmentation is a technique to designate voxels within the CT images as defined objects; essentially, image points were defined as vessel, bone or tissue. Due to the irregular and non-uniform shapes of the hindlimbs, the segmentation was performed manually (Fig 3, A and B). Once defined, the same manual segmentation method was consistently applied to all the scanned volumes. This was done by manually drawing volumes of interest (VOIs) a few voxels away from the edge of the bone regions from each of the serial 2-D sections (~500 in the coronal plane) (Bouxsein et al. 2010). For each hindlimb, two volume “segments” were generated consisting of bone or tissue (and vessels). Thresholding is a segmentation technique that separates portions of an image based on pixel/voxel values (HUs values in this case). The relatively high density of the Microfil compounds allowed for partitioning of vascular structures from the tissue (background). In some cases, the X-ray attenuation of the Microfil was similar to bone and made it even more important for the bone to be segmented from the images before thresholding.
Figure 3:
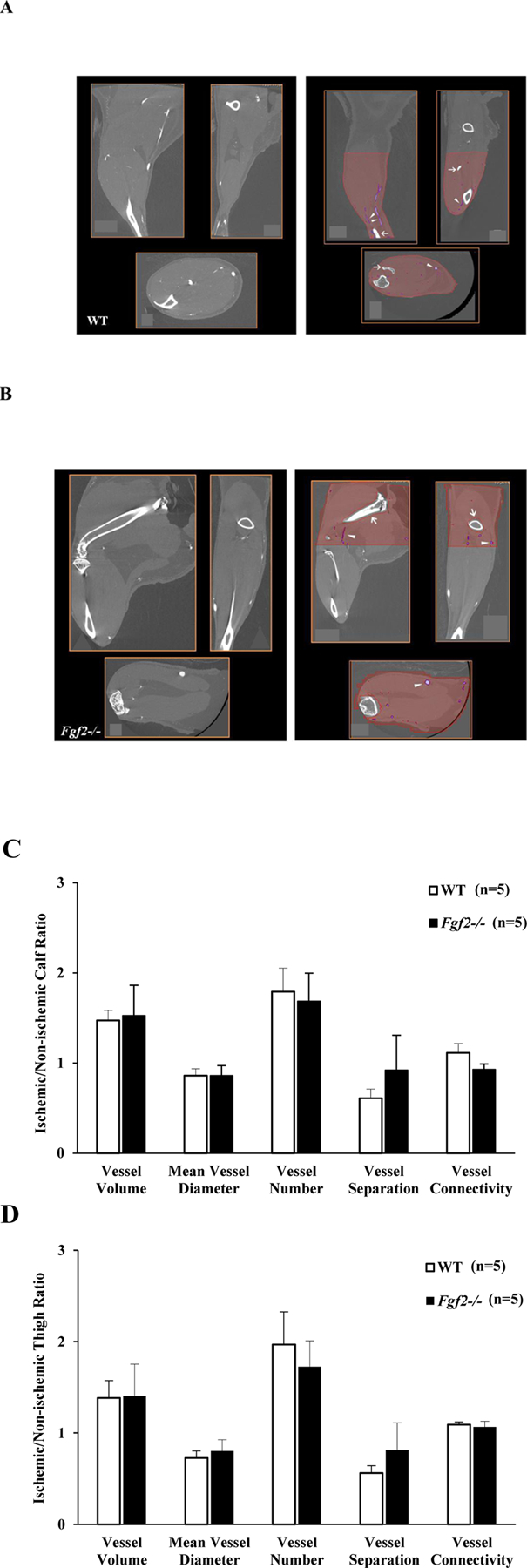
Representative 2-D cross-section coronal, sagittal, and axial views of non-ischemic WT (A) and Fgf2−/− (B) limbs before (left panel) and after (right panel) segmentation. The calf or thigh regions is highlighted in red with bones segments digitally deleted (white open arrows) and vessels segmented based on voxel intensity (white arrowheads). Quantification of vessel morphometry parameters in WT (white bars) and Fgf2−/− (black bars) expressed as a ratio between the ischemic and sham limbs. Calf parameters (C). Thigh WT parameters (D). Data are mean ± SEM, n=5.
Histomorphometric parameters (Parfitt et al. 1987; Hildebrand and Rüegsegger 1997; Bouxsein et al. 2010) including vessel volume (volume of segmented voxels normalized to tissue/limb volume), diameter (average local thickness of voxels), density (number of vascular structures intersected per mm) and separation (spacing i.e. average distance between vessels calculated from background voxels) were computed for each defined volume of interest (sham or ischemic calf or thigh) using the Siemens Inveon Research Workplace Bone Morphometry Tool. The values were reported as a ratio of ischemic to non-ischemic limbs for each animal to minimize any variations in the vascular morphology parameters that could be attributed to the Microfil perfusion protocol. For determination of vessel size (diameter) distribution, bone-deleted stacked 2-D image data were converted to DICOM and read into Fiji (NIH, Bethesda, MD). A plug-in of Fiji, developed for local thickness analysis, was used to assess the distribution of vessel diameters in the data set (Schindelin et al. 2012). An algorithm built into the Fiji plug-in first converts the gray scale images to binary ones using a thresholding procedure. The generated binary images will have non-zero signals in the segmented areas (Microfil-filled vessels). The non-zero voxels are treated as part of the object, and the space around the vessels (tissues) is regarded as background. A local thickness value is calculated for each non-zero voxel as the diameter of the largest sphere that fits inside the object and contains the voxel. The calculation relies on the 3-D spatial relationships to detect where the object/background interface lies and to determine the correct local diameter (Saito and Toriwaki 1994; Hildebrand and Rüegsegger 1997). Although the voxel size of the CT images in this study was 17µm3, the Fiji plug-in did not differentiate well between vessels with a diameter <17µm and those with a diameter ≥17µm and <34µm, because of the partial volume effect. For this reason, the smallest bin size of the diameter analysis has vessels between 0µm and 34µm. Histograms were generated representing the number of vascular segments over a range of diameters (34µm-442μm) in the sham or ischemic limbs.
Expression profile of angiogenesis-related proteins
Skeletal muscles were harvested from non-ischemic, sham or ischemic hindlimbs of WT and Fgf2−/− mice. Sham or ischemic thigh and calf muscles were dissected free of the femur and tibia/fibula, respectively and snap-frozen in liquid nitrogen at 7 days of ischemia. 100–120mg of skeletal tissues was pulverized and homogenized on ice in a glass dounce homogenizer in an extraction buffer containing 20mM Tris, 2mM EDTA, 2M NaCl, 1% NP40, EDTA-free protease inhibitor cocktail (Roche, 1 tablet/10mL), and 1mM PMSF. The homogenate was centrifuged at 15,000g for 15 minutes and the supernatant was collected. After protein concentration was determined using a Bio-Rad DC protein assay, 600µg of total protein in 1.5mL of array buffer from the proteome profiler mouse angiogenesis antibody array kit (R&D Systems) was mixed with 15µL of a detection antibody cocktail and incubated at 25°C for one hour. Each sample/antibody mixture was then added to a nitrocellulose membrane containing 52 different capture antibodies (listed in Table 2) dotted in duplicate and incubated at 4°C overnight. After washing, 2mL of buffered streptavidin-HRP was added to each membrane and allowed to incubate at 25°C for 30 minutes. Array membranes were developed using chemiluminescence with the provided Chem Reagent mix. The densitometry of the antibody/protein dots were quantified using a Fluorchem 8800 gel imager.
Table 2:
Vessel morphological parameters (volume, mean diameter, number, separation and connectivity) assessed from sham and ischemic WT or Fgf2−/− 3-D micro-CT angiograms after vascular segmentation. HLI (hindlimb ischemia)
| Volume (%) | P | Diameter (mm) | P | Number (1/mm) | P | Separation (mm) | P | Connectivity (1/mm) | P | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| WT Calf | Sham | 0.91 ± 0.05 | .02 | 0.08 ± 0.01 | .09 | 0.13 ± 0.02 | .03 | 8.31 ± 0.93 | .03 | 19.39 ± 0.93 | 0.28 |
| HLI | 1.34 ± 0.12* | 0.06 ± 0.01 | 0.22 ± 0.03* | 4.93 ± 0.77* | 20.62 ± 1.36 | ||||||
| Fgf2−/− Calf | Sham | 0.91 ± 0.13 | .03 | 0.07 ± 0.01 | .25 | 0.14 ± 0.02 | .01 | 7.87 ± 1.42 | .01 | 18.88 ± 1.58 | 0.04 |
| HLI | 1.44 ± 0.30* | 0.06 ± 0.003 | 0.22 ± 0.04* | 4.74 ± 0.89* | 20.59 ± 1.74* | ||||||
| WT Thigh | Sham | 0.54 ± 0.06 | .12 | 0.11 ± 0.01 | .04 | 0.05 ± 0.01 | .03 | 21.73 ± 3.44 | .02 | 26.19 ± 3.37 | 0.06 |
| HLI | 0.71 ± 0.10 | 0.08 ± 0.01* | 0.09 ± 0.01* | 11.68 ± 1.96* | 23.88 ± 2.41 | ||||||
| Fgf2−/− Thigh | Sham | 0.58 ± 0.09 | .09 | 0.09 ± 0.01 | .16 | 0.07 ± 0.02 | .01 | 16.73 ± 4.12 | .02 | 21.71 ± 0.61 | 0.46 |
| HLI | 0.88 ± 0.26 | 0.06 ± 0.01 | 0.13 ± 0.03* | 8.88 ± 1.70* | 23.90 ± 2.18 | ||||||
p < 0.05 vs Sham cohort
Immunohistochemical analysis detection of inflammatory cell infiltration/activation
5μm thick transverse paraffin-embedded sections were deparaffinized with xylenes and followed by rehydration in alcohol/water (100%, 95% and 70%) solutions. Endogenous peroxidase activity was blocked with H2O2 followed by treatment with 5% normal rabbit or goat serum to reduce nonspecific binding. Serial sections were then incubated overnight at 4°C with goat polyclonal antibody against myeloperoxidase (MPO 1:5000, R&D Systems), mouse monoclonal antibody against Mac-3 (CD107b 1:1000, BD Biosciences), or rabbit polyclonal antibody against CD206 (MMR 1:1000, Abcam). The MPO-positive population of inflammatory cell infiltrate were identified as neutrophils (Bradley et al. 1982; Oklu et al. 2013). Mac-3-positive cells were identified as macrophages (Koestler et al. 1984; Shireman et al. 2006) and CD206-positive cells were classified as M2a macrophages (Rőszer 2015; de Carvalho et al. 2013). Biotinylated secondary antibodies (1:500, Vector Labs) were then applied to the sections for 2 hours at room temperature; rabbit anti-goat for MPO, mouse absorbed rabbit anti-rat for Mac-3 or goat anti-rabbit for CD206. The antigen-antibody complexes were subsequently labeled utilizing the Vectastain ABC reagent kit (Vector Labs) and immunoperoxidase staining was detected with 3, 3’-diaminobenzidine (DAB, Vector Labs). Sections were counterstained with Mayer’s Hematoxylin (Life Technologies) to detect nuclei, dehydrated, and coverslipped with Permount mounting medium (Fisher Scientific). Images were captured at 400X magnification from each section (2 sections/limb, 40 fields/section). The number of MPO, Mac-3, or CD206-positive cells were counted in each image and presented as the density of cells per mm2 of muscle.
Statistical Analysis
Data are expressed as mean±S.E.M and analyzed using Graph Pad Prism software. Spearman correlation was used to compare the curves of limb function recovery over time. For micro-CT results, paired Student t-test was used to determine within group differences (i.e., sham vs. ischemic). Comparisons of ischemic/non-ischemic ratios were performed with unpaired Student t-test. Histological data were analyzed using two-way analyses of variance (ANOVA). When ANOVA evaluations produced significance (p≤0.05), Bonferroni post-hoc tests were carried out for pairwise comparisons. P<0.05 was considered as significant.
RESULTS
Delayed recovery of ischemic limb use/function in the absence of FGF2 expression
To address the role of the Fgf2 gene in the recovery of from ischemic limb injury, evaluation of the ischemic hindlimb use was performed at specific time points after induction of ischemia. A semi-quantitative scoring scale modified from Stabile and colleagues (Helisch et al. 2006; Stabile et al. 2003) was utilized to perform daily and weekly assessments of ischemic limb impairment or use (as described in Table 1). All WT and Fgf2−/− mice experienced impaired limb use immediately after the ischemia surgery which presented as dragging of the ischemic foot and this observation persisted for the next three days of ischemia (Fig. 1A), most likely due to ischemic pain and/or surgical procedure. Soon after, both groups of mice began to exhibit improved use of their ischemic limbs to varying degrees. At 7 days of ischemia, limb function improvement in WT mice began to diverge from the Fgf2−/− group. By 28 days, WTs had resumed normal hindlimb function and their ischemic limbs could no longer be differentiated from sham limbs. Conversely, Fgf2−/− mice had significant impairment of limb use and did not completely recover ischemic limb use by the end of the study at 42 days (p<0.05). These results provide evidence suggesting that in the absence of Fgf2 expression, there is an impaired functional recovery of hindlimb active use in response to ischemic injury.
Table 1:
Functional scoring for the assessment of hindlimb use during chronic ischemia
| Score | Hindlimb Use |
|---|---|
| 4 | Dragging of ischemic foot |
| 3 | No dragging of ischemic foot but no plantar flexion |
| 2 | Plantar flexion but no flexing of toes |
| 1 | Normal use of ischemic limb with flexing of toes to resist gentle traction on the tail |
Figure 1:
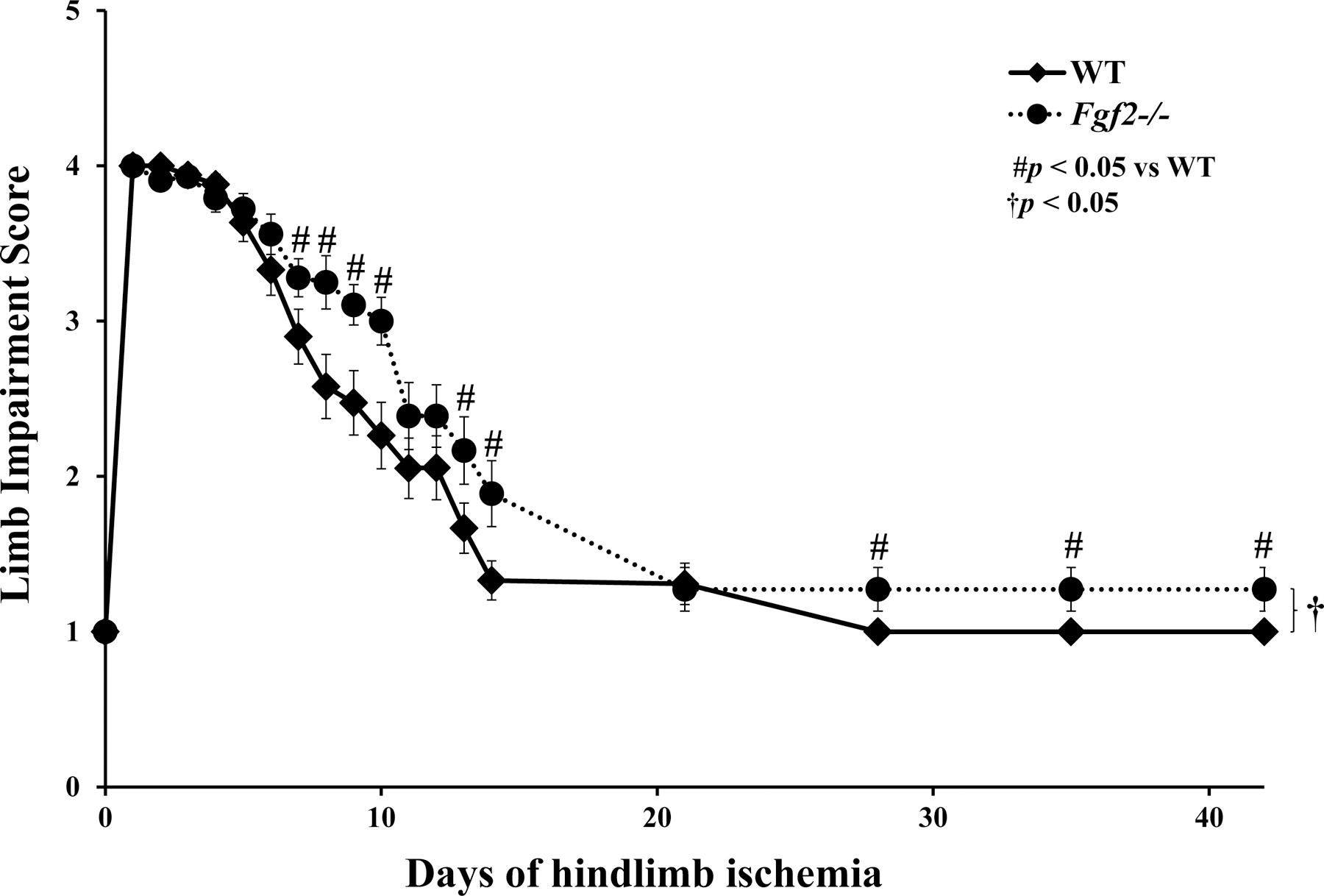
Average limb impairment scores of WT and Fgf2−/− ischemic limbs after induction of hindlimb ischemia. The scoring system listed in Table 1 was used to assign scores daily for up to 14 days of ischemia and then weekly for up to 42 days of ischemia. Data are presented as mean ± SEM, n=23–28. #p<0.05 vs. WT. †p<0.05 between WT and Fgf2−/− recovery curves.
Loss of Fgf2 expression does not lessen capillary or arteriole growth during chronic ischemia
To determine whether impaired function after limb ischemia in the Fgf2−/− mice was a function of altered angiogenesis or arteriogenesis, skeletal muscles from sham-operated and ischemic WT and Fgf2−/− mice were harvested and analyzed for vascular density. Muscle sections were collected 42 days after ischemia surgery and stained with GSI Lectin IB4, an endothelial cell marker to evaluate microvascular angiogenesis (Fig. 2A). No significant difference in capillary density was observed in non-ischemic (sham) muscles between WT and Fgf2−/− hindlimbs. Capillary density in WT or Fgf2−/− ischemic muscles was significantly increased relative to their respective sham limbs (p<0.05, Fig. 2B). However, the level of capillary vessel growth in Fgf2−/− ischemic limbs was not different from WT.
Figure 2:
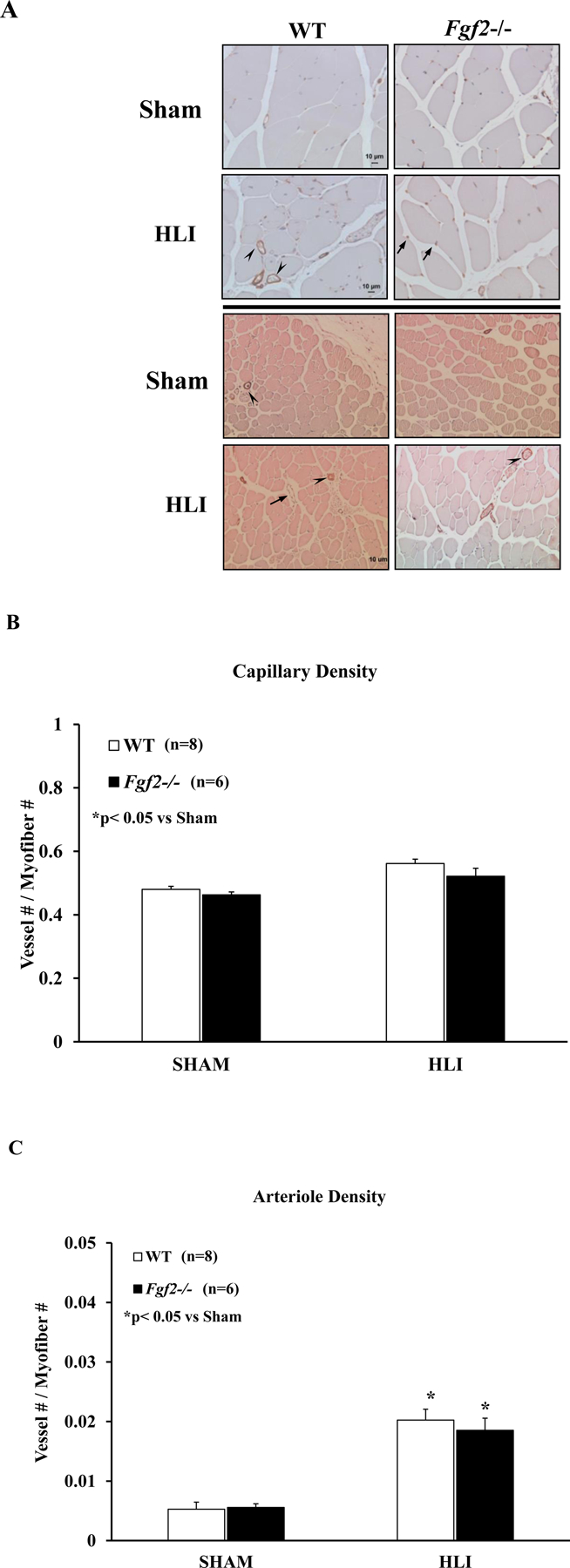
(A) Representative photomicrographs of GSI-Lectin+ (top panel) or α-SMA-stained vessels (bottom) in WT and Fgf2−/− sham and ischemic muscle sections harvested at 42 days of hindlimb ischemia. Capillary (B) and arteriole (C) density (expressed as vessel number/myofiber number) of sham and ischemic muscles after 42 days of ischemia in WT (white bars) and Fgf2−/− (black bars). Data are presented as mean ± SEM, n=6–8. *p<0.05 vs. SHAM cohort, #p<0.05 vs. WT HLI. Arrows (capillaries, venules), arrowheads (non-capillary GSI-Lectin-positive vessels, arterioles). Scale bar: 10μm
To determine the degree of ischemia-induced arteriogenesis, sham and ischemic muscle sections were immunostained with α-smooth muscle actin, a vascular smooth muscle cell marker to detect and quantify arteriolar vessels (Fig. 2A). Like the capillary density of the sham limbs, arteriole density was not different between WT and Fgf2−/− mice indicating that an underlying vascular defect is not the cause for the delayed recovery of limb function. Arteriole density was significantly increased in ischemic muscles of WT and Fgf2 mice compared to their respective sham muscles (Fig. 2C). No difference in arteriole density was detected between ischemic WT and Fgf2−/− limbs. Taken together, these findings suggest that the decreased functional capacity of Fgf2−/− limbs is likely not a result of differences in ischemia-induced vascular growth.
Post-ischemic network formation is similar between wildtype and Fgf2−/− mice
To further characterize the role of Fgf2 expression in ischemia-induced collateral growth and remodeling (arteriogenesis), contrast-enhanced micro-CT imaging was performed in addition to the histological staining. While immunological detection and 2-D counting of vessels is still the most-widely used method for quantifying post-ischemic vasculature (Madeddu et al. 2006; Waters et al. 2004; Thierry Couffinhal et al. 2009), it is possible that changes in arteriole vessel densities exist between WT and Fgf2−/− muscles that are not detectable by immunostaining due to differences in sensitivity. Furthermore, micro-CT analysis provides three-dimensional information on the stability of ischemic vascular network in the absence of Fgf2 expression. For 3-D quantification of the vasculature, 2 volumes of interest (VOIs) (calf or thigh) were defined for each limb using bone as anatomic guides. Serial z-slices of upper hindlimb volume extending from the top of the femur to the bottom of the patella were designated as the calf VOI, while the thigh volume included slices from the head of the fibula to the ankle joint (Fig. 3). Calf or thigh histomorphometric parameters, including vessel volume, mean diameter, density and separation, were evaluated from the 3-D volumes based on standardized methods using the IRW Bone Morphometry Tool. In this tool, the vessels are represented as “bone” and the calculation of these parameters requires a binarization of the VOI into a bone/mineralized “vessel” and a tissue/nonmineralized “non-vessel” segment and calculations are performed based on the parallel plate model of trabecular bone. Selection of the appropriate binarization threshold was very crucial for obtaining accurate results as high values will eliminate smaller vessels while a lower threshold can overestimate larger vessels. After visual inspection of 3-D volumes and 2-D cross-sections, comparisons between pre- and post-segmentation images of 4 non-ischemic limbs (n=2 from each group) were performed to determine the upper and lower limits of the vascular (‘bone”) density (Figs. 3 A and B). Based on this, a threshold of 1000–7000 HU was chosen and kept constant for all subsequent analyses of sham and ischemic limbs.
The volume parameter was determined from counting voxels in segmented volumes normalized to the total volume of interest. The mean vessel diameter was defined as the average thickness; density was defined as the number of vascular structures intersected per unit length (mm-1); while the separation represents the average distance between vascular structures within each VOI (mm)3 (Bouxsein et al. 2010; Parfitt et al. 1987). At 42 days of hindlimb surgery, the vascular volume fractions were significantly increased in the ischemic lower limbs (calf) WT and Fgf2−/− compared to the non-ischemic (sham) limbs (Table 2). Interestingly, the volumes were unchanged in response to ischemia in the upper limbs (thigh) in either group despite increased vessel numbers (density) in both the calf and thigh. The intervessel separation was also increased in the ischemic calves or thighs of WT or Fgf2−/− limbs. Mean vessel diameter was not altered except in the WT thigh where it was smaller. To minimize any variations attributable to the perfusion protocol, the quantitative parameters of each ischemic limb was also normalized to the sham (non-ischemic) limb. In the calf (Fig. 3C) or thigh (Fig. 3D), vessel volume and number ratios were greater than 1 while the mean diameter and separation ratios were less than 1. Ratios were similar between WT and Fgf2−/− limbs for all parameters.
To assess and compare the vessel size distribution present in the normal mouse hindlimb with the vasculature that develops in response to ischemia, additional quantitative analysis of the 3-D volumes was performed. The 3-D data were analyzed for local thickness and histograms were generated to display the volume and range of vessel sizes (diameter) across the entire hind limb (Fig. 4). Vessels of smaller diameter (≥34 µm≤204µm) were significantly increased in the ischemic WT limbs compared to sham (p<0.05, Fig. 4B). Fgf2−/− limbs also had higher volumes of smaller diameter vessels (p<0.05, Fig. 4C). Similar ratios of the size-lineated vessel volumes were present in WT and Fgf2−/− ischemic legs (Fig. 4D).
Figure 4:
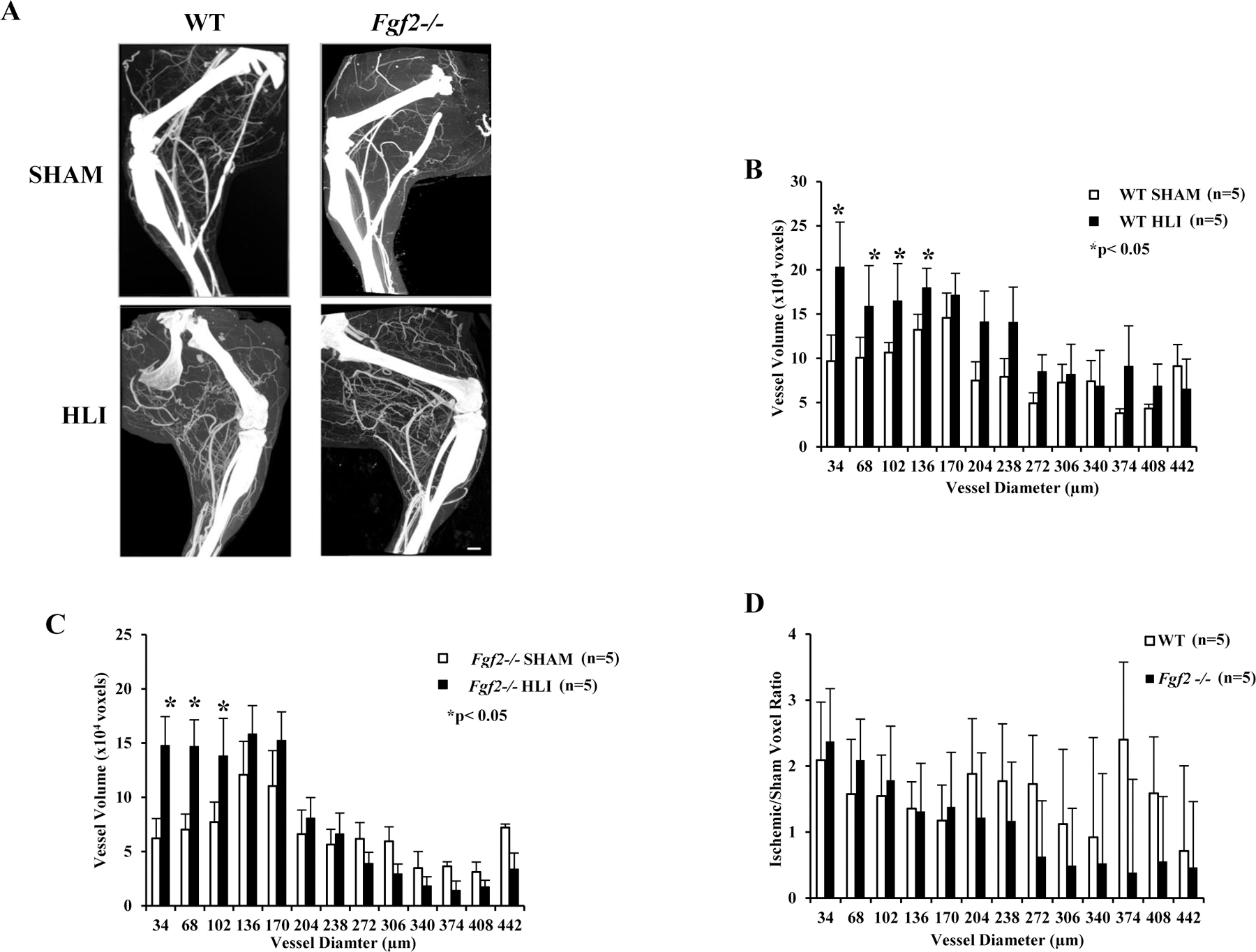
Representative 3-D angiograms (A) and quantitative analysis (B-D) of ischemic WT and Fgf2−/− hindlimbs scanned at 17µm resolution. Mean vessel thickness (diameter) distribution in WT (B) or Fgf2−/− (C) sham and ischemic limbs. The volumes of vessels were presented over a range of diameters (34µm - 442µm). (D) Vessel thickness of the ischemic limbs expressed as a ratio of the corresponding sham limb. Data are mean ± SEM, n=5. *p<0.05 vs. Sham cohort. Scale bar: 1mm.
Increased chemokine and ECM protein expression in ischemic Fgf2−/− muscles
To address the possibility that other pro-angiogenic proteins may compensate for the absence of Fgf2 gene expression in ischemic muscle and to identify potential mediators of the tissue response to ischemia, the expression profile of angiogenesis-related proteins at 7 days of ischemia was determined using a proteome antibody array. This timepoint was selected based on the observation of decreased function of the Fgf2−/− ischemic limbs between 3 and 10 days of ischemia. Relative expression of several growth factors including fibroblast growth factor 1 (FGF1), fibroblast growth factor 7 (FGF7), vascular endothelial growth factors (VEGFs) platelet-derived growth factors (PDGFs), and hepatocyte growth factor (HGF) in Fgf2-deficient ischemic muscles was not different from wildtype (see Supplemental Table). Several proteins were altered in expression relative to WT muscles. The subset of proteins that were changed due to loss of Fgf2 expression was chemokines, insulin growth factor binding proteins (IGFBP), proteases and a protease inhibitor (Fig. 5). The levels of the matrix metalloproteinases (MMP), MMP-8 and MMP-9 were significantly increased in the ischemic Fgf2−/− muscles (p<0.05 vs. WT). Fgf2−/− mice also had increased expression of the plasminogen activator inhibitor-1 (PAI-1). IGFBP-2 and IGFBP-3, which have activity in inflammation, proliferation, and apoptosis in injury and vascular alterations (Bach 2018), were significantly enhanced in the ischemic Fgf2−/− skeletal muscle (p<0.05 vs. WT). Lastly, monocyte chemoattractant protein-1 (MCP-1), CXCL12, and CXCL16, pro-inflammatory chemokines, were elevated in the absence of Fgf2 expression. Overall, the proteome profile in ischemic skeletal muscle suggests that complete loss of Fgf2 expression (in Fgf2−/− limbs) upregulates expression of proteins involved in ECM remodeling and inflammatory cell activation.
Figure 5:
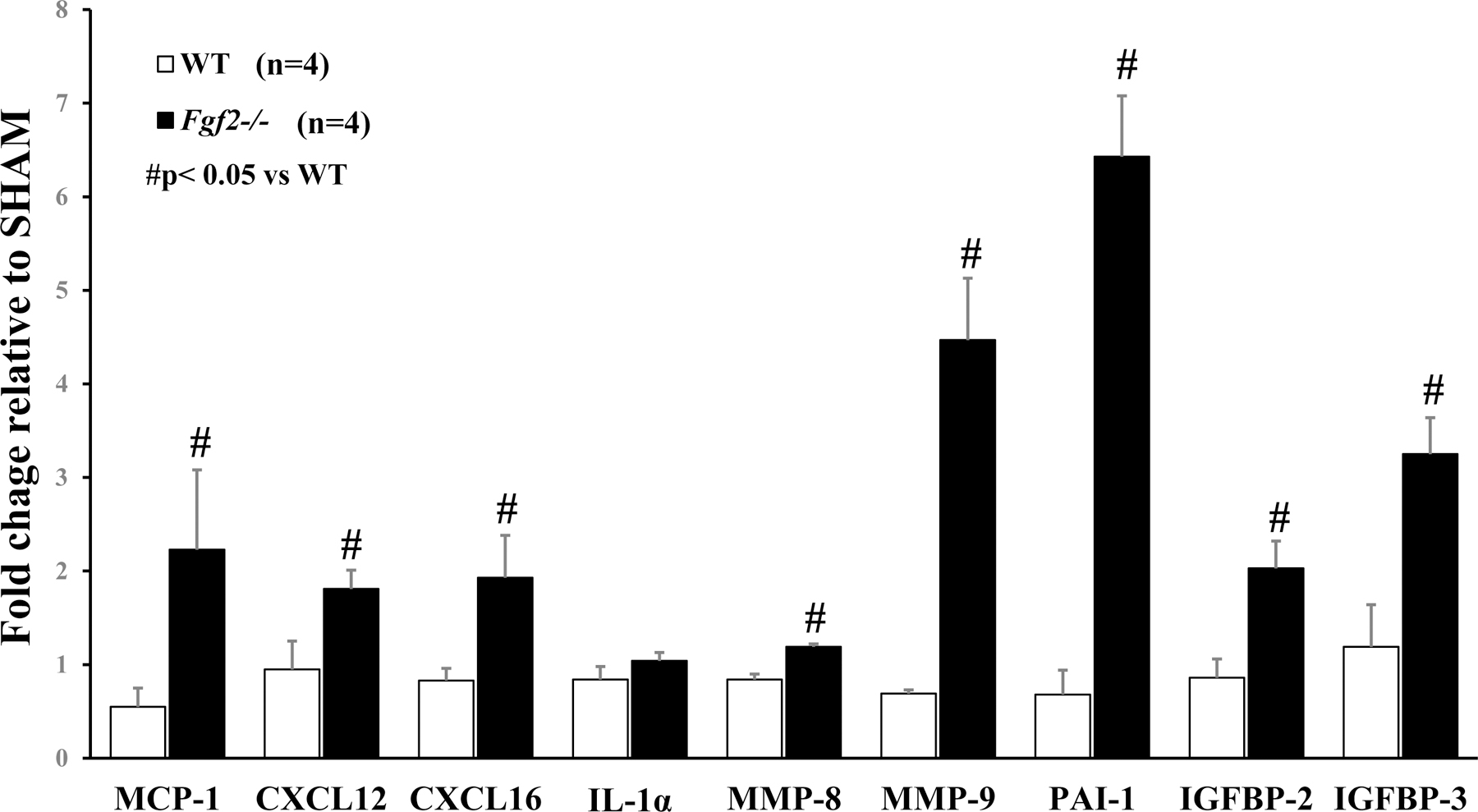
Expression level of angiogenic-related proteins in WT and Fgf2−/− muscles at 7 days of ischemia. For each limb, the fold change in ischemic muscle protein expression is presented as a ratio of the contralateral (sham) limb. Data are presented as mean ± SEM, n=4. #p<0.05 vs. WT.
Fgf2−/− mice display amplified inflammatory cell infiltration in response to ischemia
To identify the role of Fgf2 expression in the tissue inflammatory response to chronic hindlimb ischemia, neutrophil and monocyte/macrophage recruitment in non-ischemic, sham-operated or ischemic skeletal muscle were assessed. Calf (gastrocnemius) and thigh (gracilis, semi-membranosus and semi-tendinosus) muscles were harvested at baseline and at 3 and/or 7 days of ischemia from WT and Fgf2−/− mice. After immunodetection of a neutrophil marker, myeloperoxidase (MPO), no difference between the two groups in the number of neutrophils was present in non-ischemic muscles (Fig. 6A). Three days of ischemia showed significantly higher numbers of neutrophils in ischemic muscles from only Fgf2−/− mice relative to their respective sham muscles (p<0.05 vs. HLI). Furthermore, Fgf2−/− muscles had significantly increased neutrophil density relative to WT (p<0.05).
Figure 6:
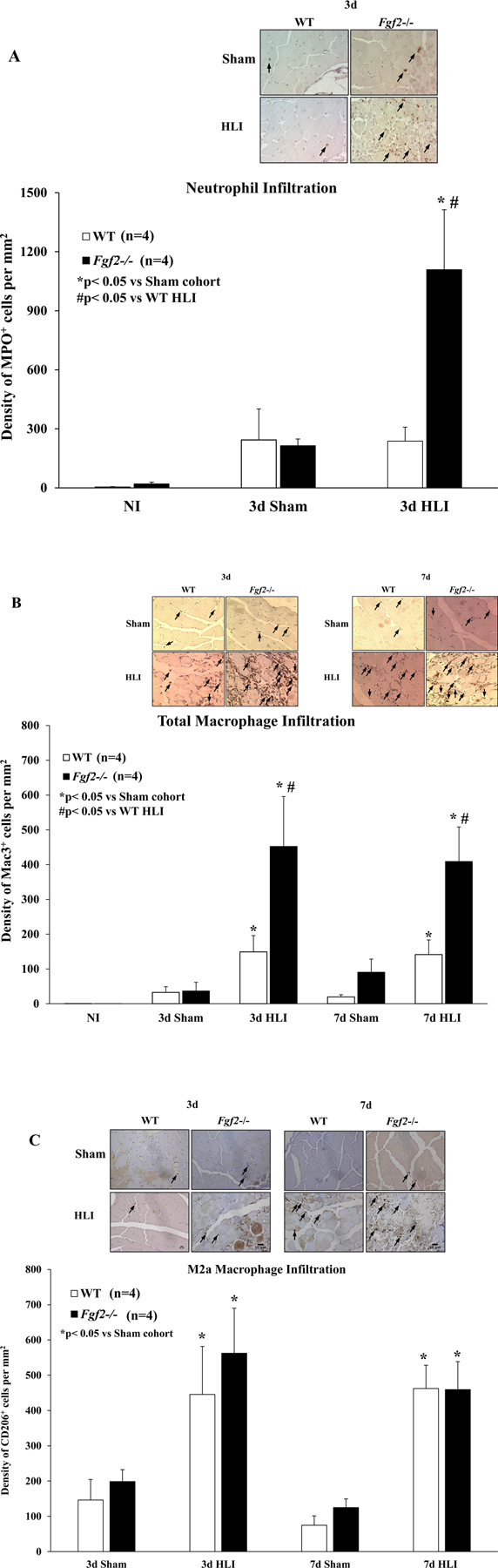
(A) Representative photomicrographs of neutrophils and neutrophil density expressed as the number of MPO-positive cells per mm2 of muscle in WT (white) and Fgf2−/− (black) sham and ischemic skeletal muscles after 3 days of ischemia. (B) Representative photomicrographs of of total macrophages (Mac-3 positive) and total macrophage density expressed as the number of Mac-3-positive cells per mm2 of muscle in WT (white) and Fgf2−/− (black) sham and ischemic skeletal muscles after 3 or 7 days of ischemia. (C) Representative photomicrographs of M2a (CD206-positive) macrophages and M2a macrophage density expressed as the number of CD206-positive cells per mm2 of muscle in WT (white) and Fgf2−/− (black) ischemic skeletal muscles after 3 or 7 days of ischemia. Data are presented as mean ± SEM, n=4. *p<0.05 vs. Sham cohort, #p<0.05 vs. WT. Arrowheads (immune-positive cells). Scale bar: 10μm
Quantitative analysis of immunostaining for Mac-3, a marker of macrophages (Tidball 2011) in non-ischemic skeletal muscle from WT and Fgf2−/− mice showed no difference in infiltrate density under baseline conditions (Fig. 6B). By 3 days of ischemia, there was significantly increased detection of macrophages in the ischemic muscles of both WT and Fgf2−/− mice compared to their respective sham muscles (p<0.05 vs. HLI, Figs. 6B, C). Similarly, levels of alternatively activated, anti-inflammatory M2a (CD206-positive) macrophages elevated after 3 days of ischemia in both WT and Fgf2−/− muscles (Fig. 6C). Total and M2a macrophages levels were also evaluated after 7 days of ischemia (Fig. 6B, C) and the degree of macrophage infiltration in the ischemic muscles were elevated in comparison to their respective sham cohorts (p<0.05 vs. HLI). Total macrophage numbers in the Fgf2−/− ischemic limbs was increased nearly 3-fold relative to ischemic WT muscles at 3 and 7 days after ischemia surgery. Interestingly, M2a macrophage density was not different between ischemic WT and Fgf2−/− mice at any time point of ischemia. Altogether, these results suggest that loss of Fgf2 expression produces an exaggerated and potentially harmful inflammatory response during hindlimb ischemia.
DISCUSSION
This study, for the first time, demonstrates that FGF2 is important for preserving ischemic limb functional capacity. Fgf2−/− mice had a delayed recovery of ischemic limb use compared to WT mice. These defects in the Fgf2−/− mice were independent of vascular growth as capillary and arteriole densities were similar to WT. These findings are important as they represent a function for Fgf2 in ischemic skeletal muscle recovery that is independent of vascular growth and suggests a role for inflammation in the FGF2-mediated response.
The functional impairment of the ischemic limb is a clinically appropriate endpoint for assessing the role of the Fgf2 expression in post-ischemic limb function. The observations in the mouse ischemic limb are similar to those detected in human patients of PAD whose symptoms present clinically as either intermittent claudication (IC) or critical limb ischemic (CLI) (Rutherford et al. 1997; Lotfi et al. 2013; Waters et al. 2004). This is relevant because one of the outcomes for IC or CLI patients enrolled in clinical trials of angiogenic/arteriogenic therapies included improvements in the Rutherford classification. The Rutherford scale is a clinical classification that ranks the severity of disease from mild claudication (Stage 1) to severe ischemic ulcers, gangrene, or tissue loss (Stage 6) (Annex 2013; Rutherford et al. 1997). As a measure of the Fgf2-dependent functional response to ischemia, mice were graded for the spontaneous or stimulated (tail traction) use of their ischemic limbs. This scoring method, a surrogate for the Rutherford scale, tracks the severity of and the recovery from ischemia in the mouse hindlimb. Movement of the ischemic limb was limited immediately following the surgical procedure and continued for the first three days following the ischemia surgery where mouse gait was altered and presented as dragging and no weight bearing of the ischemic foot. This observation was present in the ischemic foot of all WT and Fgf2−/− mice (Fig. 1). Loss of Fgf2 gene expression (Fgf2−/−) depressed the recovery of ischemic limb function. While WT mice resumed active foot and limb movement (plantar flexion with flexing of toes and full weight bearing) after 28 days, Fgf2−/− mice only recovered “normal” use of their ischemic limbs by the end of the study (42 days of ischemia). These results are reminiscent of the wound healing response that was observed in Fgf2−/− mice. Excisional dorsal skin wounds had a delay in complete healing rate relative to WT (Ortega et al. 1998). This response was confirmed to be Fgf2-specific because Fgf1/Fgf2 double knockouts have a similar delay as Fgf2−/− mice while Fgf1−/− wounds heal at a similar rate as WT (Miller et al. 2000).
In models of chronic ischemia, loss of or diminished tissue function is typically a consequence of impaired arteriogenesis, the outward remodeling of pre-existing collateral vessels or angiogenesis, the growth of the capillary network. When analyzed by immunohistochemistry, no alterations in angiogenesis or arteriogenesis were observed in ischemic Fgf2−/− limbs (Fig. 2). Ischemia induced a significant increase in Fgf2−/− capillary (Fig. 2B) or arteriole (Fig. 2C) densities. However, these vascular changes were similar to ischemic WT skeletal muscle. A complementary quantitative assessment of the effect of Fgf2 deletion on induced revascularization was performed with the use of micro-computed tomography (micro-CT). This imaging modality addresses the limitations inherent with the use of the histology for quantifying vascular growth including the subjectivity of the two-dimensional (2-D) measurements and small sampling. High resolution 3-D micro-CT imaging provides a powerful, objective and analytical tool for visualizing blood vessels, evaluating the volumetric architecture, and determining type and extent of the vascular network. Two distinct volumes of interest (VOI), calf and thigh, were defined in each of the binarized 3-D images for the computation of morphometric parameters including vascular volume, diameter, number and separation (Fig. 3). The vessel volume and number ratios (ischemic/sham) of the calf or thigh for all groups were all greater than 1 indicating an increase in vessel density as a response to ischemia (Table 2). Further analysis of the vessels generated in response to ischemia was performed by generating histograms of vessel size distribution across the entire hindlimb (Fig. 4). WT and Fgf2−/− ischemic limbs had increased volumes of smaller diameter vessels between 34 and 200 µm compared to their sham cohorts (Figs. 4A–B). Several studies employing the hindlimb ischemia model and micro-CT technology have also observed increases in volumes of small and/or medium-sized vessels ranging from 8 to 200 µm (Cristofaro et al. 2013; Duvall et al. 2004; W. Li et al. 2006). When compared to WT, Fgf2−/− limbs had similar vascular volume, number, diameter and separation ratios. Together, these findings parallel work by Sullivan and colleagues where ablation of Fgf2 gene expression did not reduce post-ischemic capillary or arteriole vessel densities (Sullivan, Doetschman, and Hoying 2002). Angiogenesis was also observed in the eye when Fgf2−/− mice were subjected to ischemic retinopathy or choroidal injury (Ozaki et al. 1998; Tobe et al. 1998). In the heart, however, permanent coronary artery occlusion resulted in reduced vascular density in Fgf2−/− hearts (Virag et al. 2007). Complete (Fgf2−/−) or partial (Fgf2+/−) disruption of Fgf2 gene expression decreased basal myocardial capillary density in adult hearts despite normal capillary supply at birth (Amann et al. 2006). The normal capillary growth at birth is consistent with studies from our laboratory and others in which no alterations to baseline capillary development in Fgf2−/− cardiac or skeletal muscles were detected (House et al. 2003; Sullivan, Doetschman, and Hoying 2002; Liao et al. 2007). The present study also did not uncover any changes to capillary vessel development in non-ischemic skeletal muscle (Figs. 2 & 3). The absence of any differences in vascular growth (angiogenesis or arteriogenesis) in normal or injured Fgf2-deficient tissues implies that Fgf2 does not play a role in revascularization. However, this conclusion might be too reductive when considering Fgf2 expression as a whole. FGF2 protein translation is a complex process that produces two classes of protein isoforms from a single transcript; one low molecular weight (LMW) and several high molecular weight (HMW) isoforms. There is increasing evidence that the LMW and HMW isoforms have disparate roles in cellular functions (Liao et al. 2009; Chlebova et al. 2009),(Liang et al. 2018). The availability of LMW or HMW FGF2 isoform specific knockout mouse models (Azhar et al. 2009; Liao et al. 2007; Adeyemo, Zhang, and Schultz 2012) does provide an avenue for the reconsideration of the role the FGF2 protein products in ischemia-induced revascularization. Ongoing studies in our laboratory are examining the vascular response to chronic ischemia in the presence of only LMW FGF2 or HMW FGF2 (Harris et al. 2016; Adeyemo, Zhang, and Schultz 2012).
Vascular growth is regulated by the coordinated balance between release and production of angiogenic and angiostatic modulators (Persson and Buschmann 2011). The angiogenesis-related proteins whose expression is altered during revascularization of the ischemic hindlimb include growth factors, pro-inflammatory and anti-inflammatory cytokines, cell-adhesion molecules, chemokines and their receptors. In stimulated vascular cells, FGF2 induces a distinct profile of these angiogenic and inflammatory cytokines and chemokines (Presta et al. 2008, 2009). Therefore, to determine if the diminished functional recovery in Fgf2−/− mice is a result of absent crosstalk between FGF2, other growth factor, cytokine, and chemokine expression in ischemic muscles, protein levels of these angiogenesis mediators were assessed in ischemic muscles after 7 days of ischemia. Several growth factors including FGF1, FGF7, VEGFs and platelet-derived growth factor (PDGFs) were similar in expression in Fgf2−/− muscles relative to WTs (see Supplemental Table). Insulin-like growth factor binding protein (IGFBP-3 and IGFP-2) expression was significantly increased in ischemic Fgf2−/− limbs (Fig. 5). Classically, IGFBPs are members of the endocrine IGF signaling pathway, where they serve as carriers of insulin growth factor (IGF) and regulate the turn-over of circulating IGFs (Yamada and Lee 2009; Baxter 2013). However, IGF-independent activities on cell growth, apoptosis, and anti-inflammation have also been attributed to these proteins. Expression of IGFBP-3, in particular, has an inhibitory effect on cell proliferation and tumor growth (Oh et al. 1995; Yamada and Lee 2009). Furthermore, microarray studies detected increased IGFBP3 gene expression within a day of hindlimb ischemia surgery and this change was associated with regeneration of the ischemic muscle (Paoni et al. 2003; Matsakas et al. 2012). Two other genes in the insulin-like growth factor (IGF) axis, IGFBP-2 and IGF2 were both increased two-fold during FGF2-induced cell proliferation in the AR4–2J cancer cell line (Hortala et al. 2005). IGFBP-2, synthesized by smooth muscle cells, acts in a ligand-dependent manner, interacting with insulin growth factor to inhibit proliferation and chemotaxis (Duan 2002). Yet, IGFBP-2 participates in proliferation and differentiation of myoblasts (Sharples et al. 2013) by regulating the transition of skeletal myoblasts from G0/G1 phase to S phase and suppression of myotubes via inhibition of MyoD and MyHC (Wang et al. 2019). Elevated IGFBP-2 may be linked to the limb impairment observed in ischemic Fgf2-deficient limbs. It is demonstrated that proinflammatory cytokines induce IGFBP-3 (Price, Moats-Staats, and Stiles 2002). Ligand-independent actions of IGFBP-3 are associated with anti-inflammatory effects via inhibiting NF-κB signaling (Lee et al. 2014).
Ischemia also increased expression of chemokines (MCP-1, CXCL12, and CXCL16) with the loss of Fgf2 gene expression (Fig. 5). MCP-1 (monocyte chemoattractant protein-1) and CXCL16 are upregulated by endothelial cells in response to FGF2 stimulation where they promote activation and recruitment of inflammatory cells, monocytes/macrophages by MCP-1 and T-lymphocytes by CXCL16 (Andrés et al. 2009; Fujii et al. 2006; Wempe, Lindner, and Augustin 1997). During femoral artery ligation, shear stress and circumferential wall stress mechanically activates the endothelium. FGF2 released from the activated endothelium and mesenchymal cells attracts and activates monocytes, which then in turn supply additional growth factors including FGF2 and other cytokines that promote continued inflammatory cell recruitment and collateral artery growth (Arras et al. 1998; Fujii et al. 2006). The activated and FGF2-stimulated endothelium also releases chemotactic factors like MCP-1 that recruit and prolong the survival of mononuclear inflammatory cells to amplify the inflammatory response (Ito et al. 1997; Arras et al. 1998; Presta et al. 2009). CXCL16 is expressed by endothelial cells and leukocytes (Shireman 2007). CXCL16/CXCR6 ligand/receptor binding has been associated with inflammatory diseases and may have some relevance in chronic wounds to promote angiogenesis (Bodnar 2015; Andrés et al. 2009). The CXCR4 receptor for CXCL12 is expressed on endothelial cells and this receptor expression is modulated by FGF2 activity (Salcedo and Oppenheim 2003). Furthermore, the CXCL12/CXCR4 axis is a key regulator in neutrophil responses in inflammation and disease (De Filippo and Rankin 2018). Like MCP-1, CXCL16 has also been shown to regulate inflammatory cell entry (both monocytes and neutrophils) into injured muscle (Zhang et al. 2009; Rigamonti et al. 2014). Similarly, CXCL12 signaling enhanced pro-inflammatory responses in vascular development (Salcedo and Oppenheim 2003; Gao, Yu, and Tang 2019; Chatterjee et al. 2015; Kim et al. 2014), vascular repair (Schober 2008) and angiogenic processes for tumor growth and metastasis (Liekens, Schols, and Hatse 2011; Salvatore et al. 2010).
To determine if the increased expression of MCP-1, CXCL12, and CXCL16 serves a functionally relevant role in ischemic Fgf2−/− muscles, the degree of inflammatory cell infiltration was determined. Neutrophil numbers were increased in Fgf2−/− limbs after 3 days of ischemia (Fig. 6A). Neutrophils are a significant part of in early inflammation associated with muscle injury. Their invasion begins within hours of muscle injury and can persist for several days (Tidball 2005; Tidball and Villalta 2010). Although there is evidence that neutrophils play a beneficial or neutral role in muscle ischemia, neutrophils can also exaggerate muscle damage increase via generation of reactive oxygen species (Tidball 2005). Monocyte/macrophage infiltration was also increased in the presence of ischemia in both WT and Fgf2−/− skeletal muscles. Like neutrophils, macrophages are also activated during ischemic injury and repair. The phenotypes of infiltrating cells during this process are of two types, classically activated, macrophages and alternatively activated anti-inflammatory macrophages known as M2 (MacLeod and Mansbridge 2016; Rőszer 2015). The degree of infiltration of macrophages at either 3 or 7 days of ischemia was enhanced by the loss of Fgf2 expression (Fig. 6B). Chronic (non-healing) ulcers and wounds have been shown to have a persistent inflammatory phase and do not have the ability to transition to the repair phase (Krishna, Moxon, and Golledge 2015; Ruiter et al. 2010). The enhanced infiltration of macrophages in Fgf2−/− ischemic muscle coupled with the poor limb functional recovery suggests a decreased wound repair response in these mice. This is supported by the delayed healing reported in excisional dorsal skin wounds of Fgf2−/− mice (Ortega et al. 1998; Miller et al. 2000). While repair-associated M2 macrophages were activated in ischemic Fgf2−/− muscles, their levels were similar to WT (see Figure 6C), which may be insufficient when compared to the degree of the neutrophil and total macrophage infiltration present in these muscles. This suggests that the delay in ischemic limb recovery of Fgf2−/− mice in the presence of increased vascular growth may be related to an imbalance in the types of infiltrating inflammatory cells present in ischemic muscles.
Extracellular matrix (ECM) remodeling proteins (MMP-8, MMP-9 and PAI-1) were also upregulated in ischemic Fgf2−/− skeletal muscle (Fig. 5). In physiological or pathophysiological conditions, matrix metalloproteinases (MMP-8 and MMP-9) are secreted by vascular and inflammatory cells during vascular growth where they degrade the ECM to facilitate recruitment, proliferation, migration and invasion of endothelial and vascular smooth muscle cells (Chen et al. 2013; Huang et al. 2009). Additionally, MMPs release and process non-matrix molecules including growth factors, integrins, and adhesion molecules which also impact vascular remodeling (Chen et al. 2013). Furthermore, the ECM undergoes dynamic changes that can lead to matrix fragments activating inflammatory responses (Dobaczewski, Gonzalez-Quesada, and Frangogiannis 2010);or that modulate chemokine synthesis via growth factor signaling (Frangogiannis 2012; Cavalera and Frangogiannis 2014) in striated muscle. Like the MMPs, PAI-1 is also involved in ECM remodeling where it is the main inhibitor of the urokinase-type plasminogen activator (uPA) system which cleaves adhesion molecules, growth factors and their receptors and degrades the ECM during endothelial and smooth muscle cell migration (Papetti and Herman 2002). Pharmacologic inhibition of PAI-1 ameliorated tissue necrosis and enhanced angiogenesis in mice with hindlimb ischemia (Tashiro et al. 2012). This improved angiogenesis was associated with increased FGF2 in the PAI-1 inhibitor-treated muscles. Similarly, inhibition of PAI-1 restored the regenerative ability of skeletal muscle in type 1 diabetic mice (Krause et al. 2011). Like the IGFBP-2 data, this suggests that the presence of elevated PAI-1 and MMP-9 may underlie the reduced muscle viability (measured as functional recovery) observed in Fgf2−/− ischemic limbs. Evidence for this exists in mice with chronic muscle injury (muscular dystrophy) where inhibition of MMP-9 enhances inflammation and reduces pathology of disease (H. Li et al. 2009). Furthermore, higher serum levels of MMP-9 and increased inflammatory cell infiltration in diabetic patients has been linked to diminished healing of foot ulcers (Dinh et al. 2012).
In conclusion, this study reveals that the absence of Fgf2 expression compromises skeletal muscle function at baseline and during recovery from chronic ischemic injury which occurred in spite of the presence of ischemia-induced revascularization of these muscles. This was accompanied with upregulated chemokine and cytokine expression during ischemia. This increased neutrophil and monocyte recruitment whose actions in the ischemic muscle environment are important for vascular repair response to chronically ischemic tissues may further exacerbate the underlying functional defect of impaired wound healing that occurs with the loss of Fgf2 expression. In parallel, these inflammatory cells secreted ECM remodeling proteins that may also be destructive to ischemic limb recovery. These results support a more nuanced role for Fgf2 expression in the ischemic mouse hindlimb beyond regulation of vascular growth.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the University of Cincinnati Heart, Lung and Vascular Institute’s Near Horizons grant and National Institutes of Health Grant R01-HL-075633 (to J. J. Schultz) and fellowship support from T32 HL-07382 (to A. Adeyemo).
Abbreviations used
- α-SMA
alpha smooth muscle actin
- CAD
coronary artery disease
- CXCL16
chemokine (C-X-C motif) ligand 16
- CVD
cardiovascular disease
- FGF
fibroblast growth factor
- HGF
hepatocyte growth factor
- HLI
hindlimb ischemia
- HU
hounsfield units
- IGFBP
insulin growth factor binding protein
- MIP
maximum intensity projection
- Micro-CT
micro-computed tomography
- MMP
metalloproteinases
- MCP-1
monocyte chemoattractant protein-1
- MPO
myeloperoxidase
- PAI-1
plasminogen activator inhibitor-1
- PDGF
platelet-derived growth factor
- 3-D
three-dimensional
- 2-D
two-dimensional
- VEGF
vascular endothelial growth factor
- VOI
volume of interest
- WT
wildtype
Footnotes
DISCLOSURE OF INTEREST
The authors report no conflict of interest.
REFERENCES
- Adeyemo A, Zhang Y, Schultz JJ. 2012. Abstract 15739: The high molecular weight isoforms of fibroblast growth factor 2 are necessary for postischemic hindlimb functional recovery and revascularization. Circulation 126 (21_MeetingAbstracts): A15739. [Google Scholar]
- Ahn A, Frishman WH, Gutwein A, Passeri J, Nelson M. 2008. Therapeutic angiogenesis: a new treatment approach for ischemic heart disease--part II. Cardiol Rev 16 (4): 219–229. [DOI] [PubMed] [Google Scholar]
- Amann K, Faulhaber J, Campean V, Balajew V, Dono R, Mall G, Ehmke H. 2006. Impaired myocardial capillarogenesis and increased adaptive capillary growth in FGF2-deficient mice. Lab Invest 86 (1): 45–53. [DOI] [PubMed] [Google Scholar]
- Andrés G, Leali D, Mitola S, Coltrini D, Camozzi M, Corsini M, Belleri M, Hirsch E, Schwendener RA, Christofori G, Alcami A, Presta M. 2009. A pro-inflammatory signature mediates FGF2-induced angiogenesis. J Cell Mol Med 13 (8 B): 2083–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annex BH. 2013. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol 10 (7): 387–396. [DOI] [PubMed] [Google Scholar]
- Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. 1998. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest 101 (1): 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azhar M, Yin M, Zhou M, Li H, Mustafa M, Nusayr E, Keenan JB, Chen H, Pawlosky S, Gard C, Grisham C, Sanford LP, Doetschman T. 2009. Gene targeted ablation of high molecular weight fibroblast growth factor-2. Dev Dyn 238 (2): 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach LA. 2018. 40 Years of IGF1: IGF-binding proteins. J Mol Endocrinol 61 (1): T11–T28. [DOI] [PubMed] [Google Scholar]
- Baxter RC. 2013. Insulin-like growth factor binding protein-3 (IGFBP-3): novel ligands mediate unexpected functions. J Cell Comm Signaling 7 (3): 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, et al. 2019. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 139 (10): e56–e66. [DOI] [PubMed] [Google Scholar]
- Bodnar RJ. 2015. Chemokine regulation of angiogenesis during wound healing. Adv Wound Care 4 (11): 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. 2010. Guidelines for assessment of bone microstructure in rodents using micro–computed tomography. J Bone Mineral Res 25 (7): 1468–1486. [DOI] [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD Rothstein G. 1982. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol 78 (3): 206–209. [DOI] [PubMed] [Google Scholar]
- Carvalho SC, Apolinário LM, Michelin-Matheus SM, Santo Neto H, Marques MJ. 2013. EPA protects against muscle damage in the mdx mouse model of duchenne muscular dystrophy by promoting a shift from the M1 to M2 macrophage phenotype. J Neuroimmunol 264 (1–2): 41–47. [DOI] [PubMed] [Google Scholar]
- Cavalera M, Frangogiannis N 2014. Targeting the chemokines in cardiac repair. Curr Pharm Design 20 (12): 1971–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee M, Von Ungern-Sternberg SNI, Seizer P, Schlegel F, Büttcher M, Sindhu NA, Müller S, Mack A, Gawaz M. 2015. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis 6 (11): e1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Jin M, Yang F, Zhu J, Xiao Q, Zhang L. 2013. Matrix metalloproteinases: inflammatory regulators of cell behaviors in vascular formation and remodeling. Med Inflamm 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebova K, Bryja V, Dvorak P, Kozubik A, Wilcox WR, Krejci P. 2009. High molecular weight FGF2: the biology of a nuclear growth factor. Cell Mol Life Sci 66 (2): 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, Isner JM. 1998. Mouse model of angiogenesis. Am J Pathol 152 (6): 1667–1679. [PMC free article] [PubMed] [Google Scholar]
- Couffinhal T, Dufourcq P, Barandon L, Leroux L, Duplaa C. 2009. Mouse models to study angiogenesis in the context of cardiovascular diseases. Front Biosci (Landmark Edition) 14: 3310–3325. [DOI] [PubMed] [Google Scholar]
- Cristofaro B, Shi Y, Faria M, Suchting S, Leroyer AS, Trindade A, Duarte A, Zovein AC, Iruela-Arispe ML, Nih LR, Kubis N, Henrion D, Loufrani L, Todiras M, Schleifenbaum J, Gollasch M, Zhuang Zw, Simons M, Eichmann A, le Noble F. 2013. “Dll4-notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development (Cambridge, England) 140 (8): 1720–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Filippo K, Rankin SM. 2018. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease.” Eur J Clin Invest 48 (Suppl 2): e12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh T, Tecilazich F, Kafanas A, Doupis J, Gnardellis C, Leal E, Tellechea A, Pradhan L, Lyons TE, Giurini JM, Veves A. 2012. Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes 61 (11): 2937–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. 2010. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48 (3): 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan C 2002. Specifying the cellular responses to IGF signals: roles of IGF-binding proteins. J Endocrinol 175 (1): 41–54. [DOI] [PubMed] [Google Scholar]
- Duvall CL, Taylor WR, Weiss D, Guldberg RE. 2004. Quantitative microcomputed tomography analysis of collateral vessel development after ischemic injury. Am J Physiol - Heart Circ Physiol 287 (1): H302–310. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG. 2012. Regulation of the inflammatory response in cardiac repair.” Circ Res 110 (1): 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Yonemitsu Y, Onimaru M, Tanii M, Nakano T, Egashira K, Takehara T, Inoue M, Hasegawa M, Kuwano H, Sueishi K. 2006. Nonendothelial mesenchymal cell-derived MCP-1 is required for FGF-2-mediated therapeutic neovascularization: critical role of the inflammatory/arteriogenic pathway. Arterioscler Thromb Vasc Biol 26 (11): 2483–2489. [DOI] [PubMed] [Google Scholar]
- Gao JH, Yu XH, Tang CK. 2019. CXC chemokine ligand 12 (CXCL12) in atherosclerosis: an underlying therapeutic target. Clinica Chimica Acta 495 (April): 538–544. [DOI] [PubMed] [Google Scholar]
- Harris R, Adeyemo A, Redfield J, Huang C, Jiang M, Ren X, Jones WK, Rubinstein, Schultz JJ. 2016. “Long-term biological functions of fibroblast growth factor-2 (FGF2) in mouse hearts following myocardial infarction. FASEB J 30 (1 Supplement). [Google Scholar]
- Helisch A, Wagner S, Khan N, Drinane M, Wolfram S, Heil M, Ziegelhoeffer T, Brandt U, Pearlman JD, Swartz HM, Schaper W. 2006. Impact of mouse strain differences in innate hindlimb collateral vasculature. Arterioscler Thromb Vasc Biol 26 (3): 520–526. [DOI] [PubMed] [Google Scholar]
- Hildebrand T, Rüegsegger P. 1997. A new method for the model-independent assessment of thickness in three-dimensional images. J Microscopy 185 (1): 67–75. [Google Scholar]
- Hortala M, Estival A, Pradayrol L, Susini C, Clemente F. 2005. Identification of c-Jun as a critical mediator for the intracrine 24 KDa FGF-2 isoform-induced cell proliferation. Int J Cancer 114 (6): 863–869. [DOI] [PubMed] [Google Scholar]
- House SL, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G, Schultz JEJ. 2003. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia.” Circulation 108 (25): 3140–3148. [DOI] [PubMed] [Google Scholar]
- Huang PH, Chen YH, Wang CH, Chen JS, Tsai HY, Lin FY, Lo WY, Wu TC, Sata M, Chen JW, Lin SJ. 2009. Matrix metalloproteinase-9 is essential for ischemia-induced neovascularization by modulating bone marrow-derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol 29 (8): 1179–1184. [DOI] [PubMed] [Google Scholar]
- Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W. 1997. Monocyte chemotactic Ppotein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res 80 (6): 829–837. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim J, Yoon JH, Ghim J, Yea K, Song P, Park S, Lee A, Hong CP, Jang MS, Kwon Y, Park S, Jang MH, Berggren PO, Suh PG, Ryu SH. 2014. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 57 (7): 1456–1465. [DOI] [PubMed] [Google Scholar]
- Koestler TP, Rieman D, Muirhead K, Greig RG, Poste G. 1984. Identification and characterization of a monoclonal antibody to an antigen expressed on activated macrophages. Proc Nat Acad Sci 81 (14 I): 4505–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause MP, Moradi J, Nissar AA, Riddell MC, Hawke TJ. 2011. Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated type 1 diabetic mice. Diabetes 60 (7): 1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna SM, Moxon JV, Golledge J. 2015. A review of the pathophysiology and potential biomarkers for peripheral artery disease. Int J Mol Sci 16 (5): 11294–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laham RJ, Baim DS. 2007. No-option patients. In Angiogenesis and Direct Myocardial Revascularization, 1–17.
- Lederman RJ, Mendelsohn FO, Anderson RD, Saucedo JF, Tenaglia AN, Hermiller JB, Hillegass WB, Rocha-Singh K, Moon TE, Whitehouse MJ, Annex BH, TRAFFIC Investigators. 2002. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): a randomised trial. Lancet 359 (9323): 2053–2058. [DOI] [PubMed] [Google Scholar]
- Lee HS, Woo SJ Koh HW, Ka SO, Zhou L Jang KY, Lim HS, Kim HO, Lee SI, Park BH. 2014. Regulation of apoptosis and inflammatory responses by insulin-like growth factor binding protein 3 in fibroblast-like synoviocytes and experimental animal models of rheumatoid arthritis. Arthritis Rheumatol 66 (4): 863–873. [DOI] [PubMed] [Google Scholar]
- Lefaucheur JP, Gjata B, Lafont H, Sebille A. 1996. Angiogenic and inflammatory responses following skeletal muscle injury are altered by immune neutralization of endogenous basic fibroblast growth factor, insulin-like growth factor-1 and transforming growth factor-b1. J Neuroimmunol 70 (1): 37–44. [DOI] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. 2009. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Human Mol Genetics 18 (14): 2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Shen W, Gill R, Corbly A, Jones B, Belagaje R, Zhang Y, Tang S, Chen Y, Zhai Y, Wang G, Wagle A, Hui K, Westmore M, Hanson J, Chen YF, Simons M, Singh J. 2006. High-resolution quantitative computed tomography demonstrating selective enhancement of medium-size collaterals by placental growth factor-1 in the mouse ischemic hindlimb. Circulation 113 (20): 2445–2453. [DOI] [PubMed] [Google Scholar]
- Liang W, Wang Q Ma H, Yan W, Yang J. 2018. Knockout of low molecular weight FGF2 attenuates atherosclerosis by reducing macrophage infiltration and oxidative stress in mice. Cell Physiol Biochem 45 (4): 1434–1443. [DOI] [PubMed] [Google Scholar]
- Liao S, Bodmer J, Pietras D, Azhar M, Doetschman T, Schultz JEJ. 2009. Biological functions of the low and high molecular weight protein isoforms of fibroblast growth factor-2 in cardiovascular development and disease.” Dev Dyn 238 (2): 249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Porter D, Scott A, Newman G, Doetschman T, Schultz JEJ. 2007. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: the role of JNK signaling. J Mol Cell Cardiol 42 (1): 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liekens S, Schols D, Hatse S. 2011. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr Pharm Design 16 (35): 3903–3920. [DOI] [PubMed] [Google Scholar]
- Lotfi S, Patel AS, Mattock K, Egginton S, Smith A, Modarai B. 2013. Towards a more relevant hind limb model of muscle ischaemia. Atherosclerosis 227 (1): 1–8. [DOI] [PubMed] [Google Scholar]
- MacLeod AS, Mansbridge JN. 2016. The innate immune system in acute and chronic wounds. Adv Wound Care 5 (2): 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeddu P, Emanueli C, Spillmann F, Meloni M, Bouby N, Richer C, Alhenc-Gelas F, Van Weel V, Eefting D, Quax PH, Hu Y, Xu Q, Hemdahl AL, van Golde J, Huijberts M, de Lussanet Q, Struijker-Boudier H, Couffinhal T, Duplaa C, Chimenti S, Staszewsky L, Latini R, Baumans V, Levy BI 2006. Murine models of myocardial and limb ischemia: diagnostic end-points and relevance to clinical problems. Vasc Pharmacol 45 (5): 281–301. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Yadav V, Lorca S, Evans RM, Narkar VA. 2012. Revascularization of ischemic skeletal muscle by estrogen-related receptor-γ. Circ Res 110 (8): 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. 2000. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol 20 (6): 2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Gucev Z, Ng L, Müller HL, Rosenfeld RG. 1995. Antiproliferative actions of insulin-like growth factor binding protein (IGFBP)-3 in human breast cancer cells. Prog Growth Factor Res 6 (2–4): 503–512. [DOI] [PubMed] [Google Scholar]
- Oklu R, Albadawi H, Jones JE, Yoo HJ, Watkins MT. 2013. Reduced hind limb ischemia-reperfusion injury in toll-like receptor-4 mutant mice is associated with decreased neutrophil extracellular traps. J Vasc Surg 58 (6): 1627–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. 1998. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci U S A 95 (10): 5672–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki H, Okamoto N, Ortega S, Chang M, Ozaki K, Sadda S, Vinores MA, Derevjanik N, Zack DJ, Basilico C, Campochiaro PA. 1998. Basic fibroblast growth factor is neither necessary nor sufficient for the development of retinal neovascularization. Am J Pathol 153 (3): 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoni NF, Peale F, Wang F, Errett-Baroncini C, Steinmetz H, Toy K, Bai W, Williams PM, Bunting S, Gerritsen ME, Powell-Braxton L. 2002. Time course of skeletal muscle repair and gene expression following acute hind limb ischemia in mice. Physiol Genomics 11: 263–272.. [DOI] [PubMed] [Google Scholar]
- Papetti M, Herman IM. 2002. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol - Cell Physiol 282 (5 5): C947–C970. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. 1987. Bone histomorphometry: standardization of nomenclature, symbols, and units. report of the ASBMR histomorphometry nomenclature committee. J Bone Miner Res 2 (6): 595–610. [DOI] [PubMed] [Google Scholar]
- Persson AB, Buschmann IR. 2011. Vascular growth in health and disease. Front Mol Neurosci 4 (August): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta M, Andrés G, Leali D, Dell’Era P, Ronca R. 2009. Inflammatory cells and chemokines sustain FGF2-induced angiogenesis. Eur Cytokine Network 20 (2): 39–50. [DOI] [PubMed] [Google Scholar]
- Presta M, Mitola S, Dell’Era P, Leali D, Nicoli S, Moroni E, Rusnati M. 2008. Fibroblast growth factor-2 in angiogenesis. Angiogenesis: An Integrative Approach From Science to Medicine, 77–88.
- Price WA, Moats-Staats BM, Stiles AD. 2002. Pro- and anti-inflammatory cytokines regulate insulin-like growth factor binding protein production by fetal rat lung fibroblasts. Am J Resp Cell Mol Biol 26 (3): 283–289. [DOI] [PubMed] [Google Scholar]
- Rigamonti E, Zordan P, Sciorati C, Rovere-Querini P, Brunelli S. 2014. Macrophage plasticity in skeletal muscle repair. BioMed Rese Int 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rőszer T 2015. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Med Inflamm 2015: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiter MS, Van Golde JM, Schaper NC, Stehouwer CD, Huijberts MS. 2010. Diabetes impairs arteriogenesis in the peripheral circulation: review of molecular mechanisms. Clin Sci 119 (6): 225–238. [DOI] [PubMed] [Google Scholar]
- Rutherford RB, Baker JD, Ernst C, Johnston KW, Porter JM, Ahn S, Jones DN. 1997. Recommended standards for reports dealing with lower extremity ischemia: revised version.” J Vasc Surg 26 (3): 517–538. [DOI] [PubMed] [Google Scholar]
- Saito T, Toriwaki J-I. 1994. New algorithms for euclidean distance transformation of an N-dimensional digitized picture with applications. Pattern Recog 27 (11): 1551–1565. [Google Scholar]
- Salcedo R, Oppenheim JJ. 2003. Role of chemokines in angiogenesis: CXCL12/SDF-1 and CXCR4 interaction, a key regulator of endothelial cell responses. Microcirculation 10 (3–4): 359–370. [DOI] [PubMed] [Google Scholar]
- Salvatore P, Pagliarulo C, Colicchio R, Napoli C. 2010. CXCR4-CXCL12-dependent inflammatory network and endothelial progenitors. Curr Med Chem 17 (27): 3019–3029. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomacak P, Cardona A. 2012. “Fiji: an open-source platform for biological-image analysis. Nature Methods 9 (7): 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober Andreas. 2008. “Chemokines in Vascular Dysfunction and Remodeling.” Arteriosclerosis, Thrombosis, and Vascular Biology 28 (11): 1950–59. 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- Schultz JEJ, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, Pawlowski SA, Lorenz JN, Kimball TR, Doetschman T. 1999. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest 104 (6). 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples AP, Al-Shanti N, Hughes DC, Lewis MP, Stewart CE. 2013. The role of insulin-like-growth factor binding protein 2 (IGFBP2) and phosphatase and tensin homologue (PTEN) in the regulation of myoblast differentiation and hypertrophy. Growth Hormone IGF Res 23 (3): 53–61. [DOI] [PubMed] [Google Scholar]
- Shireman PK, Contreras-Shannon V, Reyes-Reyna SM, Robinson SC, McManus LM. 2006. MCP-1 parallels inflammatory and regenerative responses in ischemic muscle. J Surg Res 134 (1): 145–157. [DOI] [PubMed] [Google Scholar]
- Shireman PK. 2007. The chemokine system in arteriogenesis and hind limb ischemia. J Vasc Surg 45 Suppl A (June): A48–A56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre J-S, Smadja DM, Lévy BI. 2013. “Postischemic revascularization: from cellular and molecular mechanisms to clinical applications. Physiol Rev 93 (4): 1743–1802. [DOI] [PubMed] [Google Scholar]
- Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, Udelson JE, Gervino EV, Pike M, Whitehouse MJ, Moon T Chronos NA. 2002. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation 105 (7): 788–793. [DOI] [PubMed] [Google Scholar]
- Stabile E, Burnett MS, Watkins C, Kinnaird T, Bachis A, La Sala A, Miller JM, Shou M, Epstein SE, Fuchs S. 2003. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout Mice. Circulation 108 (2): 205–210. [DOI] [PubMed] [Google Scholar]
- Sullivan CJ, Doetschman T, Hoying JB. 2002. Targeted disruption of the Fgf2 gene does not affect vascular growth in the mouse ischemic hindlimb. J Appl Physiol 93 (6): 2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro Y, Nishida C, Sato-Kusubata K, Ohki-Koizumi M, Ishihara M, Sato A, Gritli I, Komiyama H, Sato, Dan T, Miyata T, Okumura K, Tomiki Y, Sakamoto K, Nakauchi H, Heissing B, Hattori K. 2012. Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood 119 (26): 6382–6393. [DOI] [PubMed] [Google Scholar]
- Tidball JG. 2005. Inflammatory processes in muscle injury and repair. Am J Physiol. Reg, Integ Comp Physiol 288 (2): 345–353. [DOI] [PubMed] [Google Scholar]
- Tidball JG. 2011. Mechanisms of muscle injury, repair, and regeneration. Comp Physiol 1 (4): 2029–2062. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Villalta SA. 2010. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol. Reg, Integ Comp Physiol 298 (5): R1173–R1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, Vinores SA, Basilico C, Campochiaro PA. 1998. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol 153 (5): 1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag JAI, Rolle ML, Reece J, Hardouin S, Feigl EO, Murry CE. 2007. Fibroblast growth factor-2 regulates myocardial infarct repair: effects on cell proliferation, scar contraction, and ventricular function.” Am J Pathol 171 (5): 1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walgenbach Klaus J., Gratas Catherine, Shestak Kenneth C., and Becker Dorothea. 1995. “Ischaemia-induced expression of BFGF in normal skeletal muscle: a potential paracrine mechanism for mediating angiogenesis in ischaemic skeletal muscle.” Nature Med 1 (5): 453–459. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Li Z, Abdalla BA, Chen Y, Nie Q. 2019. MiR-34b-5p mediates the proliferation and differentiation of myoblasts by targeting IGFBP2.” Cells 8 (4): 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters RE, Terjung RL, Peters KG, Annex BG. 2004. Preclinical models of human peripheral arterial occlusive disease: implications for investigation of therapeutic agents. J Applied Physiol 97 (2): 773–780. [DOI] [PubMed] [Google Scholar]
- Wempe F, Lindner V, Augustin HG. 1997. Basic fibroblast growth factor (BFGF) regulates the expression of the CC chemokine monocyte chemoattractant protein-1 (MCP-1) in autocrine-activated endothelial cells.” Arterioscler Thromb Vasc Biol 17 (11): 2471–2478. [DOI] [PubMed] [Google Scholar]
- Yamada PM, Lee KW. 2009. Perspectives in mammalian IGFBP-3 biology: local vs. systemic action. Am J Physiol - Cell Physiol 296 (5): 954–976. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ran L, Garcia GE, Wang XH, Han S Du J, Mitch WE. 2009. Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle, promoting muscle regeneration.” Am J Pathol 175 (6): 2518–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin G, Duffy JJ, Pawlowski SA, Doetschman T. 1998. Fibroblast growth factor 2 control of vascular tone. Nature Med 4 (2): 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.