

SUMMARY
As pluripotent human embryonic stem cells progress toward one germ layer fate, they lose the ability to adopt alternative fates. Using a low-dimensional reaction coordinate to monitor progression toward ectoderm, we show that a differentiating stem cell’s probability of adopting a mesendodermal fate given appropriate signals falls sharply at a point along the ectoderm trajectory. We use this reaction coordinate to prospectively isolate and profile differentiating cells based on their mesendoderm competence and analyze their RNA sequencing (RNA-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq) profiles to identify transcription factors that control the cell’s mesendoderm competence. By modulating these key transcription factors, we can expand or contract the window of competence to adopt the mesendodermal fate along the ectodermal differentiation trajectory. The ability of the underlying gene regulatory network to modulate competence is essential for understanding human development and controlling the fate choices of stem cells in vitro.
Graphical Abstract
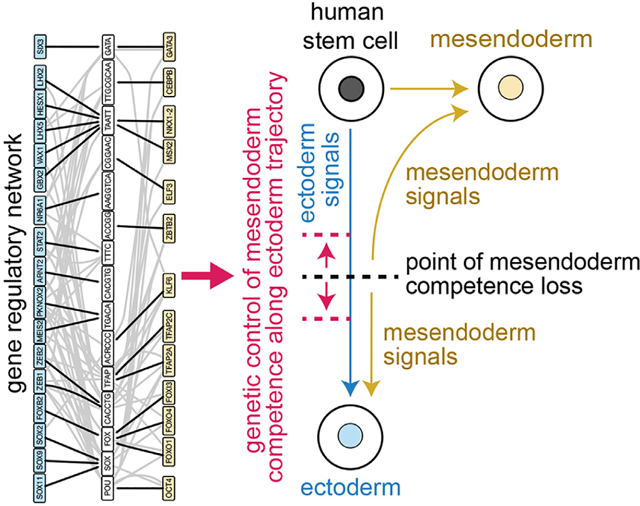
In brief
The competence of human embryonic stem cells to take on mesendoderm fates is lost at a particular point along the ectoderm trajectory. Valcourt et al. analyze transcriptomics and chromatin accessibility of cell populations before and after competence loss to reveal genes whose perturbation can expand or contract mesendoderm competence.
INTRODUCTION
Pluripotent cells have the ability to produce all the cell types of the adult body (Gilbert, 2000), but they lose this potential as they differentiate. During initial lineage specification, cells can change their fate choice upon exposure to signals that induce an alternative selection (Berg et al., 2011; Gilbert, 2000; Handyside, 1978; Pedersen et al., 1986), such as by transplantation to a different location in the embryo. In time, however, a cell’s fate becomes determined, and it is no longer competent to choose a different lineage in response to the same external signals (Oron and Ivanova, 2012; Rossant and Lis, 1979; Rossant and Vijh, 1980). This changing competence to adopt alternative fates has been pictured on a Waddington landscape (Waddington, 1957) as a pluripotent cell moving down into the valley corresponding to its chosen fate and being prevented from adopting the alternative fate by the barrier that rises between the valleys (Figure 1A). Thus, a cell’s ability to transition to the alternative fate depends both on its location along the developmental trajectory and on the position of the barrier. Although lineage specification is relatively well understood (Chng et al., 2010; Jang et al., 2016; Kiecker et al., 2016; Mullen et al., 2011; Patthey and Gunhaga, 2014; Sheng et al., 2003; Takaoka and Hamada, 2012; Tapscott, 2005; Trompouki et al., 2011; Zhang et al., 2010), whether and how competence for adopting alternative lineages can be tuned during differentiation is not. Determining how this competence is set and modulated is essential for understanding developmental patterning and plasticity. The first choice human embryonic stem cells have is between the ectodermal (neural and non-neural) and mesendodermal progenitor fates. Here, we studied how the competence to select the mesendodermal fate in response to mesendoderm-inducing signals changes during ectodermal differentiation of human embryonic stem cells (hESCs) and whether this competence can be modulated.
Figure 1. Stem cells lose competence to adopt mesendodermal fates upon BMP4 and Activin A signal exposure with increasing duration of activin/NODAL inhibition.
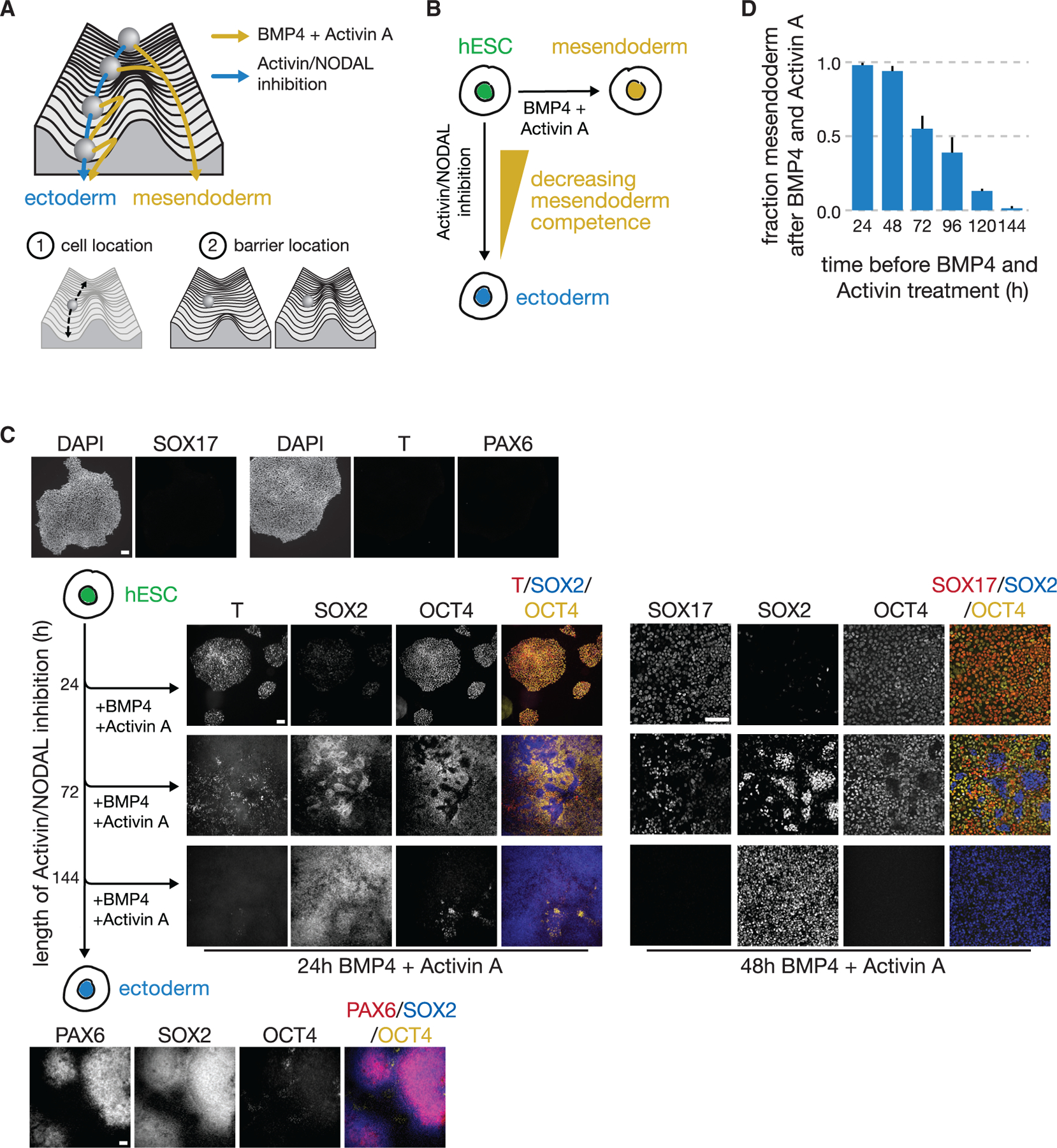
(A) Schematic of a Waddington landscape illustrating the ectoderm (blue) or mesendoderm (yellow) fate choice. In this picture, the competence of a cell to produce mesendoderm depends both on the cell’s location along the ectodermal developmental trajectory and on the position of the barrier between the two fates.
(B) hESCs choose the ectodermal lineage in response to activin/NODAL inhibition and mesendodermal lineages in response to BMP4 + Activin A.
(C) Fluorescence images of immunostained hESCs that were exposed to a pretreatment of activin/NODAL inhibition for 24, 72, or 144 h and then treated with 24 or 48 h of BMP4 + Activin A treatment. Increasing duration of activin/NODAL inhibition reduced the population’s competence to produce BRACHYURY(T)+ mesendoderm and SOX17+ endoderm and, more broadly, OCT4+ (yellow) SOX2− (blue) mesendoderm-derived cell types. The spatial structures seen here largely appear after BMP4 and Activin A signal induction and are likely due to a combination of local density impacts on ectoderm-directed differentiation rate and homophilic interactions between cells. After 144 h of activin/NODAL inhibition, cells become SOX2+, PAX6+ neuroectoderm (bottom). Cells in pluripotent state do not express T, SOX17, or PAX6. Scale bar represents 100 μm.
(D) Bar graph of the fraction of cells adopting mesendodermal fate in response to BMP4 + Activin A after 24–144 h of pretreatment with activin/NODAL inhibition (see STAR Methods). At the population level, the competence to choose mesendodermal fates in response to these signals decreases as the duration of prior ectodermal differentiation increases. Error bars: SD (n = 3).
There is a fundamental challenge in understanding the competence of a cell to choose a specific fate in response to a signal, because a cell’s fate choice is evident only after the expression of fate-specific marker genes. In mouse and humans, the fate markers for the germ layers are not expressed until at least 12 h after exposure to the appropriate signals (Li et al., 2015; Loh et al., 2016; Smith et al., 2008). Because the gene expression and chromatin accessibility state of the cell changes substantially during this period, determining how the molecular state of the cell at the time of signal exposure governs its competence to adopt alternative fates has been difficult.
To overcome this challenge, we identified a low-dimensional reaction coordinate to continuously monitor the progression of live single cells toward the ectodermal fate. Along this reaction coordinate, we measured the probability that a cell could transition to the mesendodermal fate in response to mesendoderm-inducing signals bone morphogenic protein 4 (BMP4) and Activin A. Using this probability distribution, we could prospectively sort and characterize cells based on their mesendoderm competence. Computational analysis of both the gene expression and chromatin accessibility profiles of these sorted cells allowed us to identify candidate genes that we predicted to control this competence. By perturbing the levels of these genes, we were able to change the mesendoderm competence along the ectodermal differentiation trajectory.
RESULTS
At the population level, hESCs lose mesendoderm competence along the ectodermal developmental trajectory
Given the appropriate signals, hESCs in vitro can adopt either mesendodermal or ectodermal progenitor fates: BMP4 and activin/NODAL signals induce mesendodermal fates, indicated by markers BRACHYURY (T) and SOX17 (Kanai-Azuma et al., 2002; Kavka and Green, 1997; Xu et al., 2011), and activin/NODAL inhibition promotes ectodermal fates (Smith et al., 2008; Figure 1B).
When we exposed hESCs to BMP4 and Activin A (see STAR Methods), the cells uniformly expressed BRACHYURY and SOX17 (Figures S1A and S1B). In contrast, inhibiting activin/NODAL signaling promoted ectoderm-derived fates, downregulating pluripotency factor OCT4 (POU5F1) and ultimately producing PAX6+ neurectoderm after 96–120 h (Figure S1C).
To study the ability of cells to adopt mesendoderm-derived fates in response to BMP4 and Activin A stimulation (hereinafter referred to as mesendoderm competence) along the ectoderm trajectory, we differentiated cells for increasing amounts of time toward the ectodermal fate with activin/NODAL inhibition and then treated with BMP4 + Activin A. We chose mTeSR as a base media and applied minimal, defined perturbations with signaling ligands or inhibitors. Our activin/NODAL inhibition robustly generated ectoderm (Figure 1C). We chose concentrations of BMP4 and Activin A to be consistent with literature reports of conditions that reliably and efficiently generate mesendoderm-derived fates in directed differentiation experiments (see STAR Methods; Loh et al., 2014). Consistent with previous data from mouse (Li et al., 2013, 2015), we demonstrated that competence to adopt mesendoderm-derived fates in response to BMP4 and Activin A decreases at the population level as cells differentiate toward the ectodermal fate. Increasing the duration of differentiation toward ectoderm reduced the fraction of cells that were mesendoderm competent, as shown by BRACHYURY (T) and SOX17 expression, which are not expressed by hESCs (Figures 1C, 1D, and S1D–S1F). The temporal decrease in the mesendoderm fraction occurred despite the cells’ continued ability to respond to BMP4 and Activin A signals throughout this period by phosphorylating SMAD proteins (Figures S1G and S1H). These findings suggested that the probability of cells transitioning to the mesendodermal fate in response to these signals decreases over the course of ectodermal differentiation. To understand how this reduction of probability occurs, we next studied the dynamics of fate choice at the single cell level.
Individual cells lose mesendodermal competence at a sharp point along the ectodermal trajectory
We sought to directly measure and predict the probability of individual cells along the ectodermal differentiation trajectory adopting a mesendodermal fate in response to BMP4 and Activin A. To do so, we first developed a measure of each live cell’s position along the developmental trajectory by choosing a low-dimensional coordinate—in this case, the expression levels of key genes—whose dynamics accurately report on the choice of the two germ layer lineages. Our recent computational work allows us to identify these key genes for a given lineage decision from single-cell gene expression data (Furchtgott et al., 2017; Jang et al., 2017; Melton and Ramanathan, 2021; Yao et al., 2017), and we have demonstrated the accuracy of this method for germ layer, cortical, and hematopoietic development in these works. For the decision between the two germ layer lineages, the genes that allow us to continuously monitor the progression of fate choice are the transcription factors (TFs) OCT4 and SOX2. Further, our previous work in mouse showed that the protein levels of Oct4 and Sox2 reflect the transitions of pluripotent cells to the mesendodermal or ectodermal fates (Furchtgott et al., 2017; Jang et al., 2017; Thomson et al., 2011). We validated that, in humans as in mouse, both OCT4 and SOX2 are symmetrically highly expressed in the pluripotent stem cell, but they are asymmetrically downregulated in the two lineages. OCT4 expression is maintained in the mesendoderm while SOX2 is downregulated; in contrast, ectoderm differentiation involves SOX2 maintenance and OCT4 downregulation (Thomson et al., 2011). Both TFs are also functionally important for these state transitions: OCT4 downregulation is necessary for neurectoderm induction (Greber et al., 2011; Thomson et al., 2011), and SOX2 downregulation is required for mesendoderm fate selection (Thomson et al., 2011). Furthermore, direct conversion to a neural fate silences OCT4 (Thomson et al., 2011), underscoring the fundamental nature of this reaction coordinate to the fate decision in question.
To monitor developmental trajectories in real time, we employed our validated hESC line in which one allele each of OCT4 and SOX2 had been tagged with red fluorescent protein (RFP) and yellow fluorescent protin (YFP), respectively, at the endogenous locus (Zhang et al., 2019; Figures S1I–S1K). Using flow cytometry, we followed the developmental trajectories of hESCs as they differentiated toward ectoderm under activin/NODAL inhibition. When we added BMP4 and Activin A signals at an intermediate stage of differentiation (Figure 2A), we could visualize a bifurcation of developmental trajectories: the mesendoderm-competent cells adopted OCT4+ SOX2− mesendoderm-derived fates, and the cells that were not mesendoderm competent proceeded toward OCT4− SOX2+ ectoderm-derived fates.
Figure 2. Dynamics of OCT4 and SOX2 accurately predict mesendoderm competence.
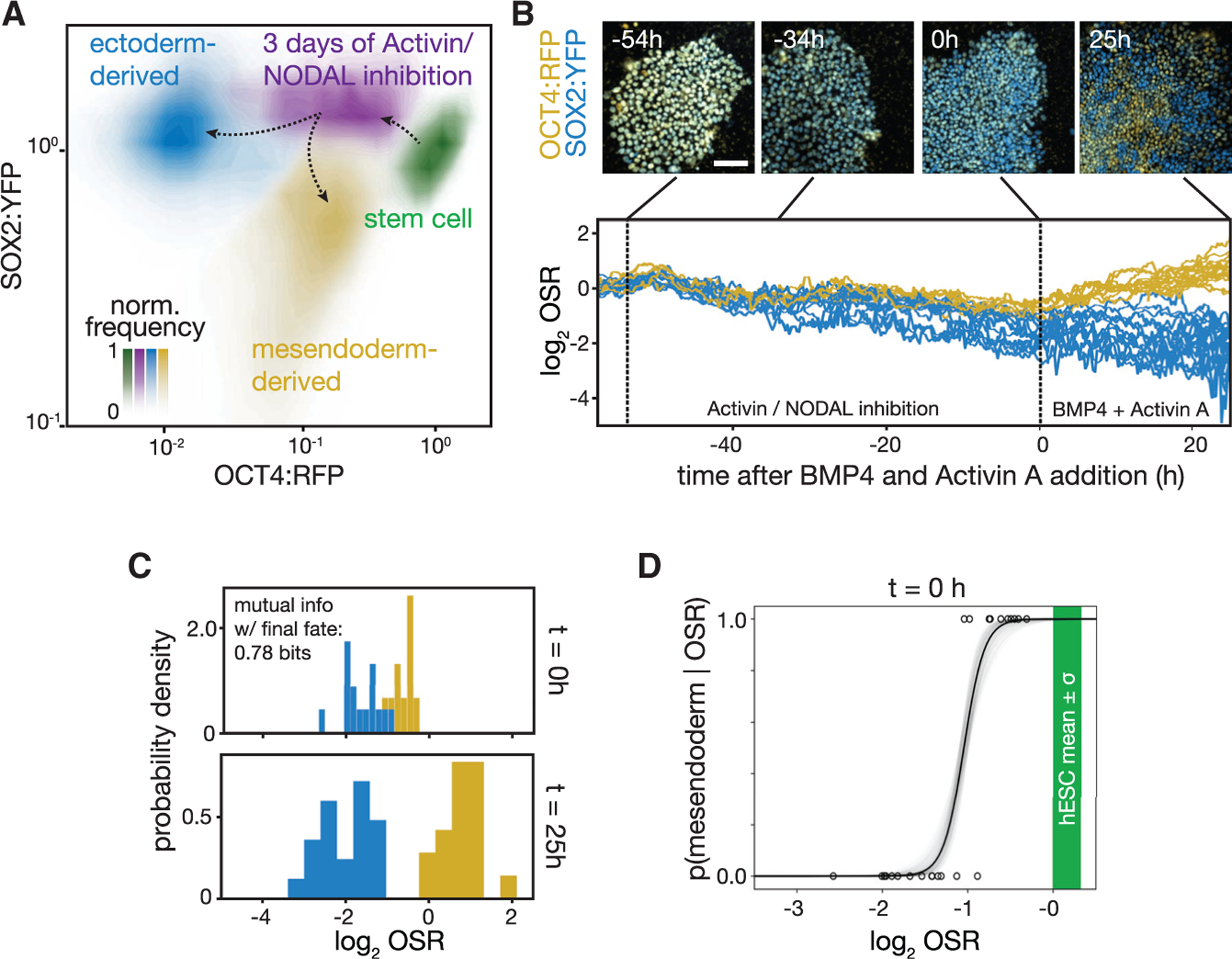
(A) Contour plots of flow cytometry data showing levels of OCT4:RFP and SOX2:YFP, each normalized to its mean level in the hESC population. hESCs (green) downregulate OCT4:RFP after 72 h of ectodermal differentiation (purple). After a subsequent 42 h of BMP4 + Activin A stimulation, the cells in this purple population bifurcate: the mesendoderm-competent fraction chooses a mesendodermal fate (yellow), while the remainder continues to become ectoderm (blue).
(B) Top: snapshots from a time-lapse microscopy experiment of a field of hESCs showing endogenous OCT4:RFP (yellow) and SOX2:YFP (blue). Cells started in pluripotency conditions are shown. Ectodermal differentiation began at t = −54 h. At t = 0 h, activin/Nodal inhibition was removed and BMP4 + Activin A signals were added (see STAR Methods); experiment ended at t = 25 h. Scale bar represents 100 μm. Bottom: plot of the log of the OCT4:RFP to SOX2:YFP fluorescence ratio (OSR) in individual cells through the time course above is shown. Time traces of cells are colored by their assigned fate at the end of the experiment: mesendoderm (ectoderm) in yellow (blue). Displayed: n = 40 cell tracks.
(C) Histogram of the log OSR from (B) shown at the time of signal addition (t = 0 h) and at the end of the experiment (t = 25 h). Histograms corresponding to cells adopting an eventual mesendodermal (ectodermal) fate at the end of the time course are in yellow (blue). The mutual information between the OSR at the moment of signal addition and the final fate choice is 0.78 bits.
(D) Plot of the probability of a single cell adopting a mesendoderm-derived fate given OSR, p(mesendoderm|OSR), calculated from time course in (B). Black line, mean; gray, 1 SD (see STAR Methods). Green bar, mean value ± SD of OSR in pluripotent stem cells. Each dot represents one cell.
Given the bimodal response of the population, we next sought to measure the probability of an individual cell adopting a mesendoderm-derived fate along the ectodermal differentiation trajectory. To do so, we performed a time-lapse experiment using the OCT4:RFP SOX2:YFP hESC line (Figure 2B). Because the cells in all of our experiments were grown on a membrane to allow the BMP4 and Activin A signals to access their basolateral receptors (see STAR Methods; Zhang et al., 2019), we developed a custom live-cell microscopy setup that was capable of imaging cells on the flexible membrane every 15 min for over 120 h (Figure S1L). Based on the timing of mesendoderm competence loss in our flow cytometry experiments, we first differentiated hESCs in this apparatus for 54 h in ectodermal differentiation conditions to obtain a heterogeneous population in which some cells had lost mesendoderm competence and others had not. We then added BMP4 and Activin A signals for 25 h, prompting mesendoderm-competent cells to adopt mesendodermal fates and non-mesendoderm-competent cells to adopt ectodermal fates.
Using our time-lapse data, we demonstrated that the OCT4:RFP to SOX2:YFP ratio (OSR) decreased with ectoderm differentiation, that OSR was predictive of a cell’s mesendoderm competence, and that this measure allowed such prediction days before the expression of classical master regulators. We tracked individual cells from pluripotency through ectoderm-directed differentiation and subsequent BMP4 + Activin A signal (Figures 2B and S1M). Pluripotent cells were initially tightly localized in OSR space but downregulated OSR at different rates upon ectoderm differentiation. Upon addition of BMP4 + Activin A, cells diverged sharply over the course of 25 h into two populations: OCT4+ SOX2− mesendoderm (yellow cells and tracks in Figure 2B) and OCT4− SOX2+ ectoderm (blue cells and tracks in Figure 2B; Figure S1N). Importantly, we found that, even more so than the levels of the individual proteins, OSR at the moment of BMP4 and Activin A signal addition was predictive of the ultimate fate of the cells with high accuracy. We binned cells by OSR at the time of signal addition and measured the fraction of cells that eventually adopted either fate (Figure 2C). Each cell carried 0.78 bits of mutual information (the maximum possible value being 1) about its mesendoderm competence state in its OSR at the moment of signal addition. Cells with a high OSR were competent to become mesendoderm in response to the BMP4 and Activin A signal, and cells with a low OSR were not. The final fates of only 4% of cells were wrongly predicted by OSR, while 16% and 8% were wrongly predicted when considering only OCT4:RFP or SOX2:YFP, respectively. We further investigated whether the cell cycle might affect mesendoderm competence (Pauklin and Vallier, 2013) and found that cells in our time-lapse did not show any fate bias based on their position in the cell cycle at the time of signal addition (p = 0.66; Figure S1O). Monitoring the dynamics of additional computationally identified genes (Furchtgott et al., 2017; Jang et al., 2017) along with OCT4 and SOX2 could decrease the prediction error below 4%, but given the high accuracy of the prediction given OSR and the potential adverse effect on the cells with increasing numbers of fluorescent tags, we proceeded with the double-tagged cell line.
From our direct measurements in Figure 2C, we computed the probability of a cell adopting a mesendodermal fate given its OSR (Figures 2D and S1P). This probability of adopting a mesendodermal fate, p(mesendoderm|OSR), had a sharp transition from 1 to 0 as OSR decreased, indicating that there was a defined point along the developmental trajectory at which cells lose their mesendoderm competence.
Prospective isolation of cell populations based on mesendoderm competence using the OSR
Having computed p(mesendoderm|OSR) for single cells in our time-lapse, we sought to predict the mesendoderm competence of cells in a heterogeneous differentiating population. Because cells in a population differentiate asynchronously and thus downregulate OSR at differing rates, at any given time, t, the cells have a distribution of OSR values defined by p(OSR|t). The probability of a given cell with a given OSR adopting a mesendodermal fate after BMP4 and Activin A stimulation is p(mesendoderm|OSR), as defined in the last section. At the population level, the predicted fraction of cells adopting a mesendodermal fate upon signal addition at a particular time is obtained by p(mesendoderm|OSR) multiplied by the fraction of cells in the population with that OSR value, p(OSR|t), and summed over the OSR of all the cells (Figure 3A):
| (Equation 1) |
We next reasoned that we should be able to use our ability to predict mesendoderm competence to prospectively isolate competent from non-competent cells from a single differentiating population. To this end, we measured p(OSR|t) of a population of differentiating hESCs using flow cytometry. Our analysis suggested that the cells in the high-OSR region where our computed p(mesendoderm|OSR)≈1 would be competent to differentiate into mesendoderm, while those in the low-OSR region where p(mesendoderm|OSR)≈0 would not (Figure 3B). We will hereafter refer to these populations as “pre-competence loss” and “post-competence loss,” respectively. To validate our predictions, we sorted cells based on their OSR using fluorescence-activated cell sorting (FACS) from a population that had been subjected to 72 h of ectodermal differentiation (Figure 3C). We then added BMP4 and Activin A to the sorted populations for 24 h to compare our predicted mesendoderm competence with the observed fate choice of these cells. From the post-competence-loss population, we obtained SOX2+ ectoderm-derived cells whose RNA sequencing results showed the exclusive expression of ectodermal genes (Figures 3D and S2G). In contrast, treatment of the pre-competence-loss population led to OCT4+ cells showing the expression of mesodermal and endodermal genes (Figures 3D and S2G). Given that pre-competence-loss cells will lose OCT4 expression and turn on PAX6 in response to extended activin/NODAL inhibition (Figures 1C and S1N), these cells are competent to form all three germ layers. Sorted populations were essentially pure, shown by counts of individual cells stained for OCT4 and SOX2 after treatment with mesendoderm signals (Figure 3E). Thus, we were indeed able to prospectively isolate individual cells at points before and after the loss of mesendoderm competence.
Figure 3. Prospective isolation of cell populations based on mesendoderm competence.
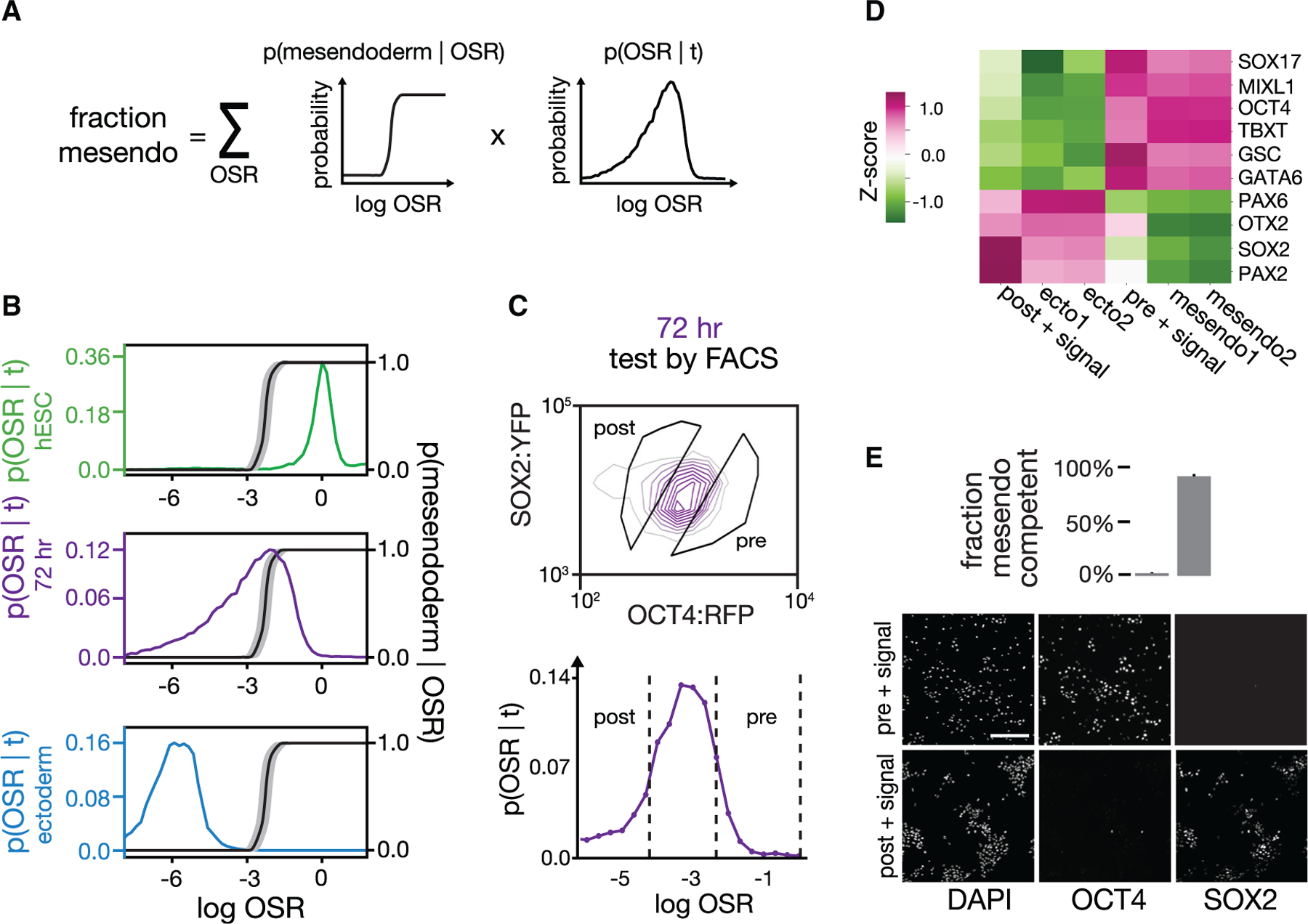
(A) The fraction of cells predicted to adopt mesendodermal fate in response to BMP4 + Activin A at given time t, p(mesendoderm|t), equals the sum over OSR of the probability of adopting mesendodermal fate given OSR, p(mesendoderm|OSR), times the probability distribution of OSR in the population at that given time, p(OSR|t).
(B) Plot of p(OSR|t) as a function of OSR for the populations in Figure 2A shown using the following colors: pluripotent stem cells (green); 72 h of ectodermal differentiation (purple); and ectodermal population (blue). p(mesendoderm|OSR) (black curve; see STAR Methods) is overlayed on the same plot (right y axis). p(OSR|t) moves leftward as cells differentiate. The cells in the purple population corresponding to the region with p(mesendoderm|OSR) = 1 are predicted to be mesendoderm competent, while the cells where p(mesendoderm|OSR) = 0 are predicted not to be.
(C) (Top) FACS density plot for a cell population at t = 72 h of activin/NODAL inhibition. Overlaid on the plot are the FACS gates used to sort subpopulation “post,” predicted to have lost mesendoderm competence, and subpopulation “pre,” predicted to retain mesendoderm competence. (Bottom) Histogram shows the same data in (top) with OCT4:RFP and SOX2:YFP collapsed into one vector, OSR.
(D) Competence testing of pre and post populations FACS sorted as in (C). Sorted populations were treated with 24 h of BMP4 + Activin A and analyzed with bulk RNA-seq. As expected, the post-competence-loss population formed ectodermal cell types that expressed SOX2, OTX2, and PAX6. The pre-competence-loss population formed mesendodermal cell types that expressed OCT4, SOX17, BRACHYURY/T, GSC, and GATA6. “mesendo” and “ecto” are reference bulk RNA-seq samples from ENCODE (see STAR Methods). Values shown are Z scores per gene across samples after normalization to units of transcripts per kilobase million (TPM).
(E) Fraction of cells in the post and pre populations sorted in (C) that adopt mesendodermal fate (top) after 36 h of BMP4 + Activin A treatment. Quantification was based on OCT4/SOX2 immunostaining (bottom, sample images). Error bars: SD (n = 3). The sorted populations uniformly adopt the predicted fate in response to mesendoderm signals.
Key TF families are remodeled upon loss of mesendoderm competence
We next sought to understand how mesendoderm competence is regulated by analyzing the gene expression and chromatin accessibility patterns of the pre- and post-competence-loss cell populations. To this end, we characterized both populations using RNA sequencing (RNA-seq) and assay for transposase-accessible chromatin using sequencing (ATAC-seq). We isolated populations of pre- and post-competence cells using FACS from a single heterogeneous population of stem cells that had been subjected to 72 h of ectoderm differentiation. As a control, we reserved a small fraction of sorted cells from each sample that we then treated with BMP4 and Activin A to confirm the competence of that sorted population. We also included a third, mesendoderm-derived population produced by subjecting pluripotent stem cells to BMP4 + Activin A for 42 h, which allowed us to distinguish lineage-specific changes in expression and chromatin accessibility from changes that are shared by cells entering either lineage (Figure S2A). These populations displayed significant and concerted changes in both their RNA-seq and ATAC-seq signatures upon the loss of mesendoderm competence (Figure 4A).
Figure 4. Key TF families show concordant changes in expression and motif accessibility upon mesendoderm competence loss, suggesting perturbation candidates.
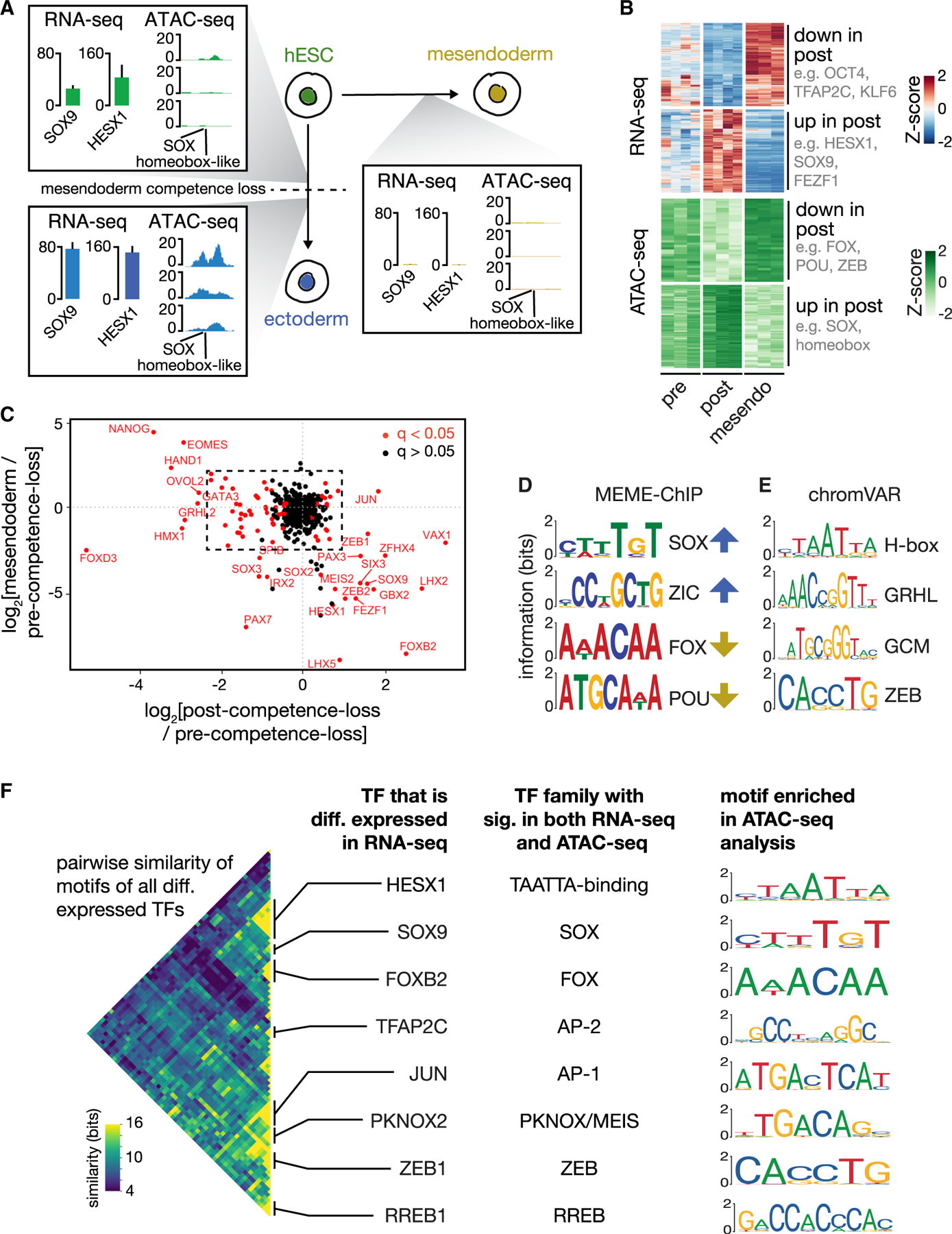
(A) Illustration showing the three assayed populations: pre- and post-competence loss (left) and mesendoderm (right). Inset boxes illustrate, with one example, combined analysis of gene expression and chromatin accessibility data from the three populations. Within each inset: (left) gene expression of SOX9 and HESX1 measured in TPM is shown; error bars: SD (n = 4; mesendoderm n = 3). (Right) ATAC-seq read depth for three biological replicates at a genomic locus that contains SOX and TAATTA homeobox-like motifs is shown, to which SOX9 and HESX1, respectively, are known to bind. SOX9 and HESX1 are upregulated in the post-competence-loss population in parallel with increased chromatin access to their binding site.
(B) (Top) Shown are all genes with significant lineage-specific differential expression level changes (heatmap with Z scores) between the pre-competence-loss and post-competence-loss populations (n = 4) as well as mesendoderm-derived outgroup (n = 3 biological replicates). Key TFs downregulated (upregulated) upon loss of mesendoderm competence include OCT4, TFAP2C, and KLF6 (HESX1, SOX9, and FEZF1). (Bottom) Heatmap shows row-normalized ATAC-seq read depth in all 250-bp peaks with a significant change in read depth between mesendoderm-competent and non-competent populations (n = 3). As with gene expression, these regions display clear, competence- and lineage-specific accessibility changes.
(C) Scatterplot showing log2 fold change in expression for all transcription factors between the (x axis) post- and pre-competence-loss populations and the (y axis) mesendoderm and pre-competence-loss populations. TFs that are significantly differentially expressed between the pre- and post-competence-loss populations (q < 0.05 as calculated by DESeq2) are shown in red. TFs in the second and fourth quadrants of this plot have lineage-specific expression patterns. See inset, Figure S2E.
(D) Selected motifs with significant enrichment in genomic regions that had a significant increase (blue arrows) or decrease (yellow arrows) from the pre-competence-loss to the post-competence-loss populations as discovered using MEME-ChIP. Motifs are labeled by the TF family or group to which they correspond. E-values: SOX, 3.7e-128; ZIC, 8.3e-033; FOX, 1.8e-02; POU, 2.9e-003.
(E) Top 4 nonredundant motifs that best explain the observed changes in ATAC-seq read depth between pre-competence-loss and post-competence-loss populations, as calculated using chromVAR. p values: homeobox (VAX2), 9.8E-05; GCM (GCM1), 3.2E-4; GRHL (GRHL1), 6.6E-4; ZEB (ZEB2), 6.9E-4.
(F) (Left) Heatmap showing an information-based measure of similarity (see STAR Methods) between the known DNA binding motifs of all pairs of differentially expressed TFs. Each row or column corresponds to the motif of one TF, and the matrix is arranged using hierarchical clustering. Only one half of the symmetric matrix is shown. TFs with similar preferences for DNA primary sequence cluster together, and notable families are labeled with the name of one cluster member in the column at center left. (Center right) The name of the TF family whose signatures are seen in both the RNA-seq analysis and ATAC-seq analysis is shown. (Right) The corresponding motif identified as significant in the ATAC-seq analysis for each labeled TF family is shown. These key TF families create concordant signatures in gene expression and chromatin accessibility data during mesendoderm competence loss.
Differential analysis of our RNA-seq data between the pre- and post-competence-loss populations using mesendoderm as an outgroup (see STAR Methods) showed that 544 genes were upregulated specifically in the post-competence-loss cells, 23 of which were TFs (Figures 4B and S2B). We also found 673 genes (32 TFs) that were specifically downregulated. In particular, we observed the differential expression of TFs, such as SOX9, HESX1, LHX2, FOXB2, TFAP2A, TFAP2C, PKNOX2, zinc finger E-box binding homeobox 1 (ZEB1), ZEB2, and GBX2, along with the expected expression pattern of OCT4 (Figure S2C). Consistent with our earlier observation (Figures S1G and S1H), the differentially expressed genes did not include signaling pathway components (Figure S2D). Our data further showed that competence loss occurred prior to the expression of master regulators, such as PAX6 and SOX1 (Figure S2C) and that these cells did not display signatures of extra-embryonic fates (Figure S2F). The expression pattern of all TFs is plotted in Figures 4C and S2E. We also validated our findings with complementary analysis of chromatin immunoprecipitation sequencing (ChIP-seq) target enrichment (Figure S2H).
From our ATAC-seq analysis, we observed DNA accessibility peaks that showed reproducible, clear changes between groups, alongside many peaks that were present in all samples (Figure S3A). Accessibility, as assayed by read depth, showed clear peaks at transcription start sites (Figure S3B). Differential analysis of our ATAC-seq data between the pre- and post-competence-loss populations using mesendoderm as an outgroup (see STAR Methods) showed thousands of regions that change accessibility between pre- and post-competence-loss populations. We found 2,071 regions that were more accessible after competence loss and 233 that were less accessible (false discovery rate [FDR] < 0.05; Figures 4B and S3C). We also confirmed that we could reproduce expected patterns in our ATAC-seq data (Figures S3D and S3E). Interestingly, we did not observe significant changes in accessibility at any of the Encyclopedia of DNA Elements (ENCODE)-annotated candidate regulatory elements of pluripotency genes, such as OCT4, SOX2, NANOG, KLF4, and MYC (Figure S3F).
To find TFs that potentially bind to the differentially accessible regions, we searched for sequence motifs that were enriched at these loci. We found more than 20 such motifs, many of which matched the known DNA-binding motifs of the differentially expressed TF families we had identified, including motifs that were similar to those bound by SOX, forkhead box (FOX), AP-2, AP-1, TAATTA-binding homeobox-like, PKNOX/MEIS, ZEB, and POU family TFs (Figure 4D), the latter of which includes OCT4 as a member. As a complementary analysis, we determined which known sequence motifs could best explain the changes in chromatin accessibility that we observed across the point of competence loss (Figure 4E).
We clustered the known binding motifs of the differentially expressed TFs by calculating a measure of similarity between each pair of motifs (see STAR Methods), and we found that many such TFs shared similar binding motifs (Figure 4F). These clusters correspond to major TF families—including the FOX, SOX, AP-2, PKNOX/MEIS, ZEB, and homeobox-like TAATTA-binding TF families—and multiple members of each family are differentially expressed. Each of these major TF family DNA-binding motifs was also enriched in the ATAC-seq analysis, indicating that the expression changes of these TFs have clear signatures in the chromatin accessibility data. Taken together, the gene expression and chromatin accessibility data revealed that a small, core set of TFs from key families are remodeled upon loss of mesendodermal competence (Figure S3G). Based on these results, we selected 36 genes, composed largely of the differentially expressed TFs in our core network plus their paralogs (Figures S3H and S3I; see STAR Methods), for genetic perturbation studies.
Computationally identified TFs modulate mesendoderm competence along the ectodermal developmental trajectory
We hypothesized that perturbation of the key TFs we identified from the data in the previous section could modulate mesendoderm competence at the population level, p(mesendoderm|t). Following Equation 1, such an effect could be the result of a change in ectodermal differentiation dynamics, measured by p(OSR|t); a change in mesendoderm competence along the ectodermal differentiation trajectory, measured by p(mesendoderm|OSR); or both. We therefore designed our analysis to measure the effects of perturbing the levels of these TFs both on p(OSR|t) and, more importantly, on p(mesendoderm|OSR). Thus, we were able to identify factors that expand or contract the window of mesendodermal competence without affecting the ectodermal differentiation dynamics in the absence of mesendoderm-inducing signals.
We first tested our hypothesis with FOXB2, a strong candidate from the RNA-seq and ATAC-seq data (Figures 4B–4D). Using a lentiviral delivery system, we transduced cells with a payload of mCerulean cyan fluorescent protein (CFP) separated at its C-terminal end from FOXB2 by a P2A self-cleaving peptide sequence, all under the control of an EF-1α promoter (Figure S4A). We performed epifluorescence time-lapse imaging experiments with overexpression of either CFP:P2A:FOXB2 or a CFP:P2A:CFP control. Cells were transduced with virus and then subjected to 72 h of ectodermal differentiation, followed by 24 h of BMP4 + Activin A treatment. We titrated viral concentration to achieve <50% transduction so that each sample also contained non-transduced (CFP−) cells to serve as an internal control population. By tracking individual cells as in Figure 2B, we showed that mutual information between OSR and final fate remains high upon FOXB2 perturbation (Figure 5A), thus ascertaining that OSR remains a good predictor of mesendoderm competence.
Figure 5. Perturbation of TF candidates from RNA-seq and ATAC-seq analyses tune mesendoderm competence along the ectodermal differentiation trajectory.
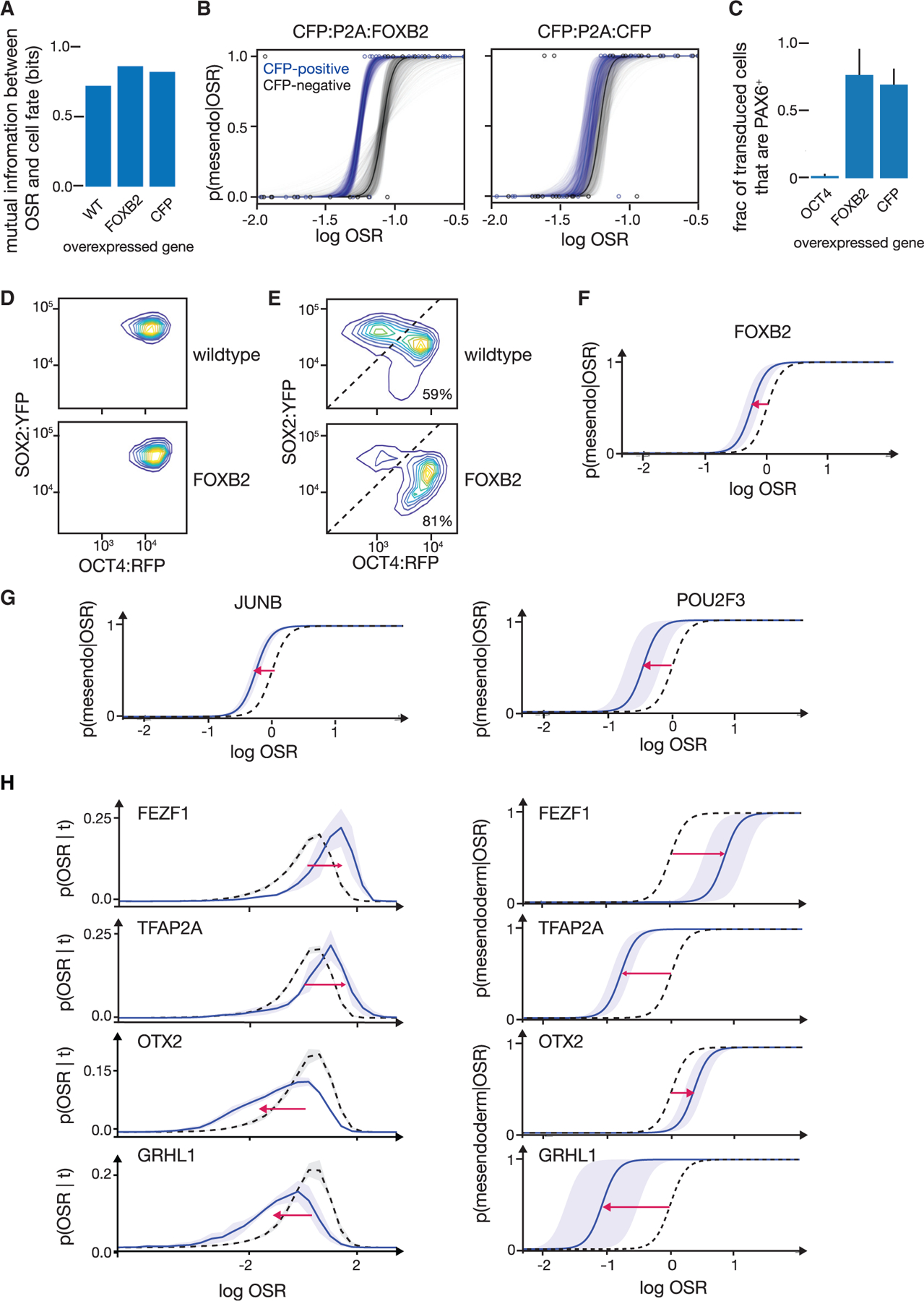
(A) Mutual information between final cell fate and OSR at the time of BMP4 + Activin A addition for wild-type, CFP:P2A:FOXB2 transduced, and CFP:P2A:CFP transduced cells. Mutual information was calculated from live, single-cell-tracked time lapse as in Figures 2B–2D. OSR at time of signal addition remains predictive of cell fate during overexpression of FOXB2 or a CFP control.
(B) Measured p(mesendoderm|OSR) for cells transduced with CFP:P2A:FOXB2 (left, blue) or CFP:P2A:CFP (right, blue) compared to the non-transduced cells in the same population (gray). Curves are measured from live-cell time lapses as in Figure 2D. Transparent curves show error as in Figure 2D. Curves are based on the following numbers of tracked cells: FOXB2 transduced, n = 40; FOXB2 non-transduced controls, n = 30; CFP transduced, n = 32; and CFP non-transduced controls, n = 44. FOXB2 overexpression (left) induces a shift that accords mesendoderm competence at lower OSR levels. This shift is not seen upon overexpression of a CFP:P2A:CFP control (right).
(C) Fraction of cells adopting neuroectoderm fate (PAX6+) after 144 h of neuroectodermal differentiation. Cells overexpressing CFP:P2A:FOXB2 or the control CFP:P2A:CFP show normal neuroectodermal differentiation. Overexpression of CFP:P2A:OCT4, a positive control for ectoderm disruption, precludes PAX6 expression. Error bars: SD (n = 3).
(D) (Top) FACS density plot of non-transduced control cells (CFP−) showing distribution ofOCT4:RFP and SOX2:YFP levels of cells after 72 h of activin/NODAL inhibition. (Bottom) CFP:P2A:FOXB2-overexpressing cells (CFP+) from the same population are shown. Overexpression of FOXB2 does not affect OCT4:RFP and SOX2:YFP dynamics during ectodermal differentiation.
(E) FACS density plots (OCT4:RFP versus SOX2:YFP) showing cell fates after 72 h of activin/NODAL inhibition followed by 42 h BMP4 + Activin A treatment. (Top) Non-transduced cells show two peaks corresponding to ectodermal lineage above diagonal (41% of cells) and mesendodermal lineage below diagonal (59% of cells). (Bottom) FOXB2-overexpressing cells (CFP+) from the same population show 81% of cells adopting mesendodermal fate. The ectodermal population shows over 2-fold higher modal SOX2-YFP expression compared to the mesendodermal population. Overexpression of FOXB2 increases the fraction of cells that adopt a mesendodermal fate.
(F) Inference of p(mesendoderm|OSR) from flow cytometry data, given the OSR distribution in (D) and the cell fate fractions in (E). Black dotted: CFP− internal controls; blue: CFP:P2A:FOXB-overexpressing cells, shaded: SD (n = 4). Consistent with microscopy data in (B), overexpression of FOXB2 shifts p(mesendoderm|OSR) to the left and thus accords mesendoderm competence at lower OSR. x axis values between (F) and (B) are not directly comparable as fluorescence values were measured by two different methods.
(G) Inferred p(mesendoderm|OSR) from flow cytometry measurements for cells overexpressing CFP:P2A:JUNB and CFP:P2A:POU2F3. Black dotted: CFP− internal controls; blue: transduced cells; shaded: SD (n = 3). Overexpression of JUNB and POU2F3, like FOXB2, shifts p(mesendoderm|OSR) to accord mesendoderm competence at lower OSR.
(H) Inferred p(mesendoderm|OSR) from flow cytometry measurements for cells overexpressing CFP:P2A:FEZF1, CFP:P2A:TFAP2A, CFP:P2A:OTX2, and CFP:P2A:GRHL1 (top to bottom). Black dotted: CFP− internal controls; blue: transduced cells, shaded: SD (n = 3). (Left) p(OSR|t). (Right) p(mesendoderm|OSR). Overexpression of candidates can tune p(OSR|t) and p(mesendoderm|OSR) independently.
Second, by analyzing the fate of each individual cell given its OSR at time of signal addition as in Figure 2D, we showed that FOXB2 overexpression significantly shifts the p(mesendoderm|OSR) of individual cells in such a way that mesendoderm competence is retained at lower OSR levels (and, therefore, further along the trajectory). This shift is significant in comparison to both the non-transduced internal-control cells and the same experiment performed with a CFP overexpression control (Figure 5B). We confirmed the identities of FOXB2-overexpessing populations with RNA-seq and immunofluorescence (Figures S5A and S5B). Thus, FOXB2 extends the mesendoderm competence window by increasing the probability that cells further along the ectodermal trajectory will transition to a mesendodermal fate if given the appropriate signals.
Third, despite changing the cells’ mesendoderm competence, FOXB2 overexpression did not prevent normal neuroectodermal differentiation as further assayed by N-cadherin and PAX6 expression in the absence of the BMP4 and Activin A signal (Figures 5C, S5B, and S5C). Thus, alternative fate competence can be modulated without preventing normal lineage progression in the absence of alternative-fate-inducing signals.
To further validate our results, we turned to flow cytometry to obtain measurements from a much larger number of cells at the cost of losing high time resolution tracking of single cells. We first validated our flow cytometry experimental design against the time-lapse microscopy results for FOXB2 perturbation. We seeded two parallel samples of pluripotent stem cell populations (Figure S4B). As with the microscopy experiment, both samples were transduced with the virus carrying CFP:P2A:FOXB2 at the onset of ectodermal differentiation. We again titrated viral concentration to achieve <50% transduction so that each sample had internal negative controls. After 72 h of ectodermal differentiation, we analyzed the first sample by flow cytometry to measure p(OSR|t) (Figure 5D). CFP:P2A:FOXB2 expression levels had no effect on OSR at this time point (Figure S5D), supporting our microscopy findings that FOXB2 does not alter normal ectodermal differentiation (Figure 5C). We switched the second sample to media containing BMP4 + Activin A for 42 h and assayed the final fate fractions by flow cytometry. We confirmed that our non-transduced control cells had produced about 50% OCT4:RFP+ SOX2:YFP− mesendoderm-derived cells and 50% OCT4:RFP− SOX2:YFP+ ectoderm-derived cells after signal addition. In contrast, a larger fraction of FOXB2-overexpressing cells than control cells took on a mesendoderm fate, as measured by OCT4+/SOX2− flow cytometry (Figure 5E). By using the measured mesendoderm fraction and p(OSR|t), we computed p(mesendoderm|OSR) using Equation 1. We assumed that the width parameter of the p (mesendoderm |OSR) sigmoid is similar to what we measured directly in our earlier time-lapse experiments both under perturbation and in wild type (Figures 2D and 5B). We confirmed that our results hold for wide ranges of this width parameter, so our conclusions would be robust even to substantial errors in this estimate (Figure S5F). The calculated p(mesendoderm|OSR) from this flow cytometry analysis confirmed that FOXB2 increases mesendoderm competence by shifting the OSR value at which p(mesendoderm|OSR) dropped to near zero (Figure 5F), consistent with the live time-lapse data (Figure 5B).
We similarly performed this lentiviral overexpression and flow cytometry experiment for all 36 candidate TFs to determine each candidate’s effect on p(OSR|t) and p(mesendoderm|OSR). Each perturbation was compared to its own internal control and to a control transduction of CFP:P2A:CFP (Figures 5B and S4C). We carried out flow cytometry analyses as described above, assuming for all perturbation conditions (as shown to be true for FOXB2) that (1) OSR remains a predictive coordinate of mesendoderm competence (Figures 2C and 5A), (2) OCT4 and SOX2 levels after 42 h of BMP4 + Activin A stimulation remain indicative of mesendoderm or ectoderm fates (Figures 1C, 3D, 5E, S1A–S1E, and S5A), and (3) single-parameter fitting of p(mesendoderm|OSR) is robust (Figures 2D, 5B, 5F, and S5F). For several key candidates, we confirmed cell fates with bulk RNA-seq and antibody staining (Figures S5A, S5B, and S5G) and ruled out differential cell death as a confounder (Figure S5H).
Overexpression of certain candidates principally affected p(mesendoderm|OSR), the point at which the cells’ competence for adopting the fate is lost. For example, overexpression of JUNB or POU2F3, like FOXB2, increased mesendoderm competence by shifting p(mesendoderm|OSR) (Figure 5G). In contrast, other candidates affected progression along the developmental trajectory, captured by p(OSR|t). For example, overexpression of SOX9 facilitated movement along the developmental trajectory, thereby shifting the distribution of p(OSR|t) toward the ectodermal fates (Figure S4D). In contrast, overexpression of TFAP2C hindered movement along the developmental trajectory, thereby shifting p(OSR|t) toward the pluripotent state (Figure S4D). We concluded that these candidates tuned fate competence by changing cellular position along the developmental trajectory.
Notably, several candidates in our network (Figure S3G) impacted competence by tuning p(mesendoderm|OSR) and p(OSR|t) independently of one another (Figures 5H and S4C). For example, although overexpression of FEZF1 and TFAP2A both shifted p(OSR|t) toward the pluripotent state, they shifted p(mesendoderm|OSR) in opposite directions. The outcomes seen upon overexpression of FEZF1, TFAP2A, OTX2, and GRHL1 together represent all four possible combinations of directional shifts in p(OSR|t) and p(mesendoderm|OSR). Thus, our candidates independently tuned p(OSR|t) and p(mesendoderm|OSR), thereby separately modulating ectoderm differentiation dynamics and mesendoderm competence.
DISCUSSION
Our findings show that genetic perturbations can directly modulate the competence for mesendoderm fate along the trajectory. Several candidates that confer mesendoderm competence (such as POU2F3, JUNB, TFAP2A, and TFAP2C) decrease in expression upon competence loss, and several candidates that promote competence loss (such as OTX2) show the opposite expression pattern. These patterns suggest that these candidates may have a role in the endogenous gene regulatory network (GRN) of mesendoderm competence. FOXB2 is one example of a candidate that confers mesendoderm competence but whose expression levels increase upon competence loss. How the effects of these endogenous expression dynamics contrast with overexpression prior to competence loss remains to be explored.
Our finding that competence for an alternative fate can be modulated suggests possible evolutionary and developmental consequences. During the patterning of the mammalian epiblast, for example, the progenitors are generated along the primitive streak as it extends anteriorly from the posterior end of the epiblast. We speculate that changing the dynamics of epiblast competence loss anterior to the primitive streak could be a possible mechanism for tuning the length and extent of the streak. Further investigation of the role of competence modulation during mammalian gastrulation could be an important element in a full description of this important process. More broadly, the regulation of competence could tune relative tissue sizes during any given cellular decision.
Our findings also highlight the importance of understanding how the competence is modulated to control the variability of cell fate decisions seen in vitro. Our approach of prospectively identifying pre- and post-competence-loss populations for molecular characterization was possible through monitoring the dynamics of reaction coordinates for the early germ layer fate decisions. Such an approach can be broadly applied to any fate decision by monitoring the dynamics of specific genes that serve as accurate reaction coordinates for that decision. We indeed have had success in identifying such genes for a wide range of lineage decisions (Furchtgott et al., 2017; Jang et al., 2017). In sum, understanding how competence is tuned will be crucial for elucidating the dynamics of mammalian embryonic patterning during development and the dynamics of fate decisions of multipotent cells.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sharad Ramanathan (sharad@cgr.harvard.edu).
Materials availability
Plasmids generated in this study will be shared by the lead contact upon request. No new cell lines were generated in this study.
Data and code availability
ATAC-seq and RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication underhttps://doi.org/10.5281/zenodo.5516285.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-OCT4 | Cell Signaling | CAT#C30A3; RRID AB_2167691 |
| Rat monoclonal anti-SOX2 | Thermo Fisher | CAT#14-9811-82; RRID AB_11219471 |
| Goat polyclonal anti-SOX17 | R&D Systems | CAT#AF1924; RRID AB_355060 |
| Rabbit monoclonal anti-pSMAD1/5/9 | Cell Signaling | CAT#12428; RRID: AB_2797908 |
| Rabbit monoclonal anti-pSMAD2 | Cell Signaling | CAT#18338; RRID: AB_2798798 |
| Mouse monoclonal anti-PAX6 | Developmental Studies Hybridoma Bank | CAT#PAX6; RRID: AB_528427 |
| Goat polyclonal anti-NANOG | R&D Systems | CAT#AF1997; RRID: AB_355097 |
| Rabbit monoclonal anti-CDH2 (NCAD) | Cell Signaling | CAT#13116; RRID: AB_2687616 |
| Bacterial and virus strains | ||
| NEB Stable competent E. coli | New England Biolabs | CAT#C3040 |
| Chemicals, peptides, and recombinant proteins | ||
| A 83-01 | R&D Systems | CAT#2939 |
| Human BMP4 | R&D Systems | CAT#314-BP |
| Human/Mouse/Rat Activin A Protein | R&D Systems | CAT#338-AC |
| γ-27632 | Stemgent | CAT#04-0012 |
| LDN-193189 | R&D Systems | CAT#6053 |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | CAT#74004 |
| KAPA mRNA Hyper Prep kit | Roche | CAT#07962363001 |
| Gibson Assembly Master Mix | New England Biolabs | CAT#E2611 |
| NEB Q5 High-Fidelity DNA Polymerase | NEB | CAT#M0491 |
| Deposited data | ||
| ATAC-seq | NCBI GEO | GSE149077 |
| RNA-seq | NCBI GEO | GSE148904 |
| Experimental models: cell lines | ||
| Human: H1 embryonic stem cells | WiCell | WA01, RRID CVCL_9771 |
| Human: H1 SOX2:YFP/OCT4:RFP embryonic stem cells | Zhang et al., 2019 | N/A |
| Oligonucleotides | ||
| Gibson homology arm to clone genes of interest into pWPXL-mCerulean-P2A, forward: gcaggtgacgtggaggagaatcccgggcct | This paper | N/A |
| Gibson homology arm to clone genes of interest into pWPXL-mCerulean-P2A, reverse: aatccagaggttgattatcatatga | This paper | N/A |
| Recombinant DNA | ||
| pWPXL | Addgene | CAT#12257; RRID: Addgene_12257 |
| pWPXL-mCerulean-P2A | This paper | N/A |
| pMD2.G | Addgene | CAT#12259; RRID: Addgene_12259 |
| psPAX2 | Addgene | CAT#12260; RRID: Addgene_12260 |
| Software and algorithms | ||
| Fiji | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| MATLAB 2019 | MathWorks | https://www.mathworks.com/products/matlab.html |
| MicroManager 2.0 beta | Edelstein et al., 2014 | https://micro-manager.org |
| CellProfiler 3.0 | McQuin et al., 2018 | https://cellprofiler.org/ |
| DESeq2 1.24.0 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| kallisto 0.45.1 | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| chromVAR 1.4.1 | Schep et al., 2017 | https://github.com/GreenleafLab/chromVAR |
| MEME-ChIP 5.0.3 | Machanick and Bailey, 2011 | https://meme-suite.org/meme/tools/meme-chip |
| PANTHER | Mi et al., 2019 | http://www.pantherdb.org |
| Enrichr | Kuleshov et al., 2016 | https://maayanlab.cloud/Enrichr/ |
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/ |
| Other | ||
| hESC-qualified Matrigel | Corning | CAT#354277 |
| Polyester membrane filters, 3 micron, 25mm | Sterlitech | CAT#PET3025100 |
| mTeSR 1 | STEMCELL | CAT#85850 |
| Accutase | Innovative Stem Cell Technologies | CAT#AT104-500 |
| jetPrime | Polyplus | CAT#114-07 |
| Lenti-X Concentrator | Clontech | CAT#631231 |
| ENCODE RNA-seq data | ENCODE | See method details |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
H1 human embryonic stem cells (WiCell ID WA01, male) and SOX2:YFP/OCT4:YFP double tagged stem cells were grown at 37°C in STEMCELL mTeSR 1 media. The SOX2:YFP/OCT4:YFP double tagged cells are of H1 background; pluripotency and karyotyping were performed in Zhang et al. (2019).
METHOD DETAILS
Cell lines
We conducted our experiments using WA01 (H1, WiCell) human embryonic stem cells. We also used an H1 cell line in which both OCT4 and SOX2 were tagged with fluorescent proteins as previously described (Zhang et al., 2019). In these cells, one endogenous copy of OCT4 was replaced with OCT4:tdTomato followed by an internal ribosomal entry site and a neomycin resistance gene to allow for selection, and one endogenous copy of SOX2 was replaced with SOX2:FLAG:Citrine:P2A:PuroR.
Cell culture
hESCs were cultured in 6-well tissue culture dishes treated with Matrigel (Corning 354277) and supplied with mTeSR media (STEMCELL Technologies 85850) according to the manufacturer’s specifications. For routine culture, we passaged by washing with phosphate buffered saline (PBS) followed by ReLeSR (Stem Cell) treatment according to the manufacturer’s instructions. Cells were passaged in clumps of 8–10 cells and seeded in mTeSR supplemented with the Rho-associated protein kinase inhibitor γ-27632 (STEMCELL Technologies 04-0012) at 10 μM for the first 24 hours to improve survival. All cell lines used were routinely tested for mycoplasma contamination.
For all experiments, we seeded cells using colony passage at a density of about 60,000 cells/cm2 on polyester membrane filters (Sterlitech PET3025100) with 3 μm pores that had been treated with Matrigel. We note that many cells seeded into the well attach to the plate rather than to the membrane, so the final effective density is somewhat less than the seeded cell count would otherwise indicate. We chose polyester membrane filters as a substrate to allow all cells to receive the BMP4 and Activin A signals we added to the media. TGF-β superfamily receptors, such as those for BMP4 and Activin A, are localized basolaterally in epithelial stem cell colonies and in vivo in the epiblast, so they are insulated from ligands in the apical media or luminal fluid (Etoc et al., 2016; Zhang et al., 2019). Typical tissue culture conditions allow for only the cells on the edge of the colony to receive signals, but growing cells on a membrane allows all cells in a colony access to the BMP4 and Activin A ligands.
For live cell imaging, membranes were glued to a custom 300 μm thick stainless-steel washer with Cytoseal 60 (Thermo Fisher), allowed to dry, sterilized with washes in 70% ethanol and with UV treatment, then treated with Matrigel for cell seeding.
Differentiation conditions
Differentiation toward the ectoderm lineage was effected using mTeSR supplemented with 0.5 μM A83-01 (R&D Systems 2939), a small molecule inhibitor of Activin and Nodal signaling. BMP4 + Activin A treatment was accomplished by treating cells with mTeSR supplemented with 3 ng/mL recombinant human BMP4 protein (R&D Systems 314-BP) and 100 ng/mL recombinant human Activin A protein (R&D Systems 338-AC). As endogenous concentrations of these molecules in the embryo are not known, concentrations were chosen to be consistent with literature reports of conditions that reliably and efficiently generate mesendoderm-derived fates (Loh et al., 2014). For neurectoderm-directed differentiation, we inhibited BMP signaling with 0.5 μM LDN-193189 (R&D Systems 6053) in addition to Activin/Nodal inhibition with 0.5 μM A83-01 for 144 hours.
Flow cytometry
Cells were washed with PBS (Lonza) and removed from membranes by treatment with Accutase (Innovative Cell Technologies AT104-500) until the cells were dissociated, about 20 minutes. Cells were analyzed on an LSRFortessa (BD Biosciences).
Fluorescence activated cell sorting
Accutase-dissociated cells were sorted using a BD Aria III (BD Biosciences) using a 100 μm nozzle. Cells were gated such that the pre-competence-loss population was taken as the cells with the top 10%–15% OCT4:RFP to SOX2:YFP ratio, and the post-competence-loss population was the bottom 10%–15% OCT4:RFP to SOX2:YFP ratio. We sorted around 250,000 cells per subpopulation in a typical experiment. Populations were sorted into 1.5 mL centrifuge tubes (Eppendorf) filled with 500 μL of mTeSR supplemented with 10 μM γ-27632; by the end of the sort, ~800 μL of sheath and sorted cells had been added to each tube. After the sort had completed, we pelleted the cells in a microcentrifuge at 250 ×g for 3 minutes, then resuspended in PBS.
For each sorted sample, about 10% of the sorted cells were reserved for competence testing to confirm the pre-/post-competence-loss status of the sorted population. These cells were seeded back into glass-bottom 24-well plates (Ibidi) treated with Matrigel and filled with 1 mL of mTeSR supplemented with γ-27632 and allowed to recover for 3 hours. The media was then changed to mTeSR supplemented with BMP4 and Activin A for 36 hours. Cells were fixed and stained for OCT4 and SOX2 according to the protocols described under “Immunofluorescence.”
RNA-seq
Total RNA was prepared from sorted or dissociated cells with an RNeasy Mini Kit (QIAGEN 74004) according to the manufacturer’s instructions. For the mesendoderm-derived outgroup samples, the input to the RNA extraction kit was a cell population directly after dissociation with Accutase; for FACS sorted populations, the input was sorted cells suspended in PBS. RNA integrity was quantified with a TapeStation 4200 (Agilent). All RINe scores were ≥ 9.9. Sequencing libraries were prepared by the Bauer Core at Harvard University using a Kapa mRNA Hyper Prep kit (Roche 07962363001) with Poly-A selection. Sequencing was performed on a NextSeq High output flow cell that generated paired-end 38bp reads. We obtained ≥ 42M reads per sample. Each group had n = 4 biological replicates, except for the mesendoderm group, which had n = 3, and the samples for confirming fate identity, which had n = 1.
ATAC-seq
ATAC-seq was performed as previously described (Buenrostro et al., 2015). Briefly, live cells were lysed and incubated with Tn5 transposase for 30 min at 37°C. After DNA purification, samples were amplified for the appropriate number of cycles as determined by qPCR to minimize PCR bias. Sequencing was performed by the sequencing core at Massachusetts General Hospital. We obtained ~100M mapped paired-end reads per sample. Each group had n = 3 biological replicates.
Plasmid construction
Overexpression targets were subcloned from plasmids available through the Harvard PlasmID database, where available. Other targets were cloned from complementary DNA (cDNA) libraries.
To prepare cDNA libraries for cloning, we differentiated human stem cells for 72 hours in mTeSR + 0.5 μM A8301 and extracted RNA with an RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. We then performed first strand cDNA synthesis using SuperScript II Reverse Transcriptase (Thermo Fisher). We amplified the relevant cDNAs using Phusion polymerase (NEB) or Kapa HiFi (Kapa Biosystems). The OCT4 DNA binding domain and the SOX2 DNA binding domain (OCT4DBD and SOX2DBD) were amplified from cDNA. The OCT4DBD consisted of amino acids 131–296 of OCT4A (NCBI reference sequence: NM_002701.5). The SOX2DBD consisted of amino acids 37–117 of SOX2 (NCBI reference sequence: NM_003106.3). All cDNA-amplified clones were fully sequence confirmed by Sanger sequencing (Genewiz).
We constructed a vector from the second-generation lentiviral transfer backbone pWPXL with an EF-1α promoter. pWPXL was a gift from Didier Trono (Addgene plasmid #12257; http://addgene.org/12257; RRID:Addgene_12257). We joined sequences for fluorescent protein mCerulean (CFP) and 2A peptide P2A (a ribosomal skip sequence) with Q5 (NEB M0491) fusion PCR and added them to the pWPXL vector with Gibson Assembly Master Mix (NEB E2611). We then constructed final transfer vectors by inserting target cDNA after the P2A using Gibson assembly. All constructed vectors were sequence confirmed at Gibson assembly junctions by Sanger sequencing (Genewiz) prior to use. Lentiviral transfer plasmids were grown and stored in NEB Stable E. coli (NEB C3040).
Lentiviral overexpression and flow cytometry analysis
To produce virus, we used jetPrime (Polyplus 114) according to the manufacturer’s instructions to transfect Lenti-X HEK293T cells (Takara) with lentiviral production plasmids pMD2.G and psPAX2 along with our individual transfer plasmids. pMD2.G and psPAX2 were gifts from Didier Trono (Addgene plasmid #12259; http://addgene.org/12259; RRID:Addgene_12259; Addgene plasmid #12260; http://addgene.org/12260; RRID:Addgene_12260). We collected viral media at 24 and 48 hours and concentrated using Lenti-X Concentrator (Clontech 631231) according to the manufacturer’s instructions.
We seeded human stem cells in mTeSR medium containing γ-27632 on Matrigel-treated membrane filters as described above. We treated cells with 1x and 3x viral titer at 24 hours and 48 hours post-seeding, respectively, in order to obtain transduction efficiency of ~10%. 1x viral treatment was performed simultaneously with the beginning of A83-01 treatment.
Two samples of each overexpression condition were performed in parallel. We harvested cells of one sample after 72 hours of treatment with A83-01 and the cells of the second sample after 72 hours of A83-01 followed by 42 hours of BMP4 + Activin A treatment. We analyzed each sample immediately after harvest using an LSRFortessa (BD Biosciences).
We analyzed differential OCT4:RFP to SOX2:YFP ratio distributions between CFP-positive and CFP-negative populations of each 72 hour sample by calculating the Kullback-Leibler divergence in MATLAB. To determine differences in proportions of end fates (OCT4:RFP+/SOX2:YFP− and OCT4:RFP−/SOX2:YFP+), we manually gated ectoderm and mesendoderm populations using a custom MATLAB script and used identical gates for both CFP-positive and CFP-negative populations. In some samples, the SOX2: YFP reporter was silenced in a small fraction of the cells, and we excluded such cells from our analysis. We performed at least 3 biological replicates for all candidates that showed an initial phenotype except for SOX2, AHR, ARNT2, and GBX2, each of which had two replicates. Significance compared to the CFP-overexpressing negative control was determined using a two-sided t test, and we controlled the FDR at 10% across the set of all candidates using the method of Benjamini and Hochberg (1995).
Immunofluorescence
Cells were fixed with 4% formaldehyde for 15 min at room temperature. Fixed cells were treated with blocking buffer (PBS + 5% normal donkey serum + 0.3% Triton X-100) for 1 h, then overnight at 4°C with primary antibody diluted in staining buffer (PBS + 1% BSA + 0.3% Triton X-100). The following primary antibodies were used: OCT4 (1:400, Cell Signaling C30A3); SOX2 (1:400, Thermo Fisher 14-9811-82); SOX17 (1:100, R&D Systems AF1924); phosphorylated SMAD1/5/9 (1:200, Cell Signaling 12428); phosphorylated-SMAD2 (1:200, Cell Signaling 18338); PAX6 (1:200, DSHB PAX6); and NANOG (1:500, R&D Systems AF1997), N-cadherin (1:200, Cell Signaling 13116). The PAX6 antibody was deposited to the DSHB by Kawakami, A. (DSHB Hybridoma Product PAX6). After overnight incubation, samples were washed three times with PBS, then secondary antibodies diluted in staining buffer were added. We used the following secondary antibodies all at a dilution of 1:1000: donkey anti-rabbit Alexa 568 (Thermo Fisher), donkey anti-rat Alexa 488 (Thermo Fisher), donkey anti-mouse Alexa 647 (Thermo Fisher), and donkey anti-goat Alexa 647 (Thermo Fisher). We incubated with a 300 nM DAPI (Thermo Fisher) solution in PBS for 5 minutes to visualize DNA. For analysis of the resulting images, we used CellProfiler 3.1.8 (McQuin et al., 2018) to segment well-separated nuclei for samples where automated segmentation performed well (Figure 2A). For more challenging images, we used Fiji to determine object centers and typical cell or nucleus size. We then used MATLAB (Mathworks) to integrate fluorescence over objects (Figures 1, 5, S1, and S5).
ATAC-seq analysis
Reads were trimmed using NGmerge 0.2_dev in adaptor removal mode with minimum overlap (-e flag) set to 20 to remove any remaining adaptor sequence. Reads were aligned to the hg38 build of the human genome using bowtie2 2.2.9 using the -very-sensitive preset and with a maximum fragment size of 2000, then collated with samtools 1.9. Duplicate fragments were removed with picard 2.8.0. Peaks were called with MACS2 2.1.1 in callpeak -f BAMPE mode. Differentially accessible peaks were identified using the Bio-conductor package DiffBind 2.12.0 in R 3.6.1. Peaks were annotated by genomic region type using ChIPSeeker 1.20.0.
For differential accessibility analysis with DiffBind and DESeq2, we used a design matrix with a “sample” column, which indicated the well from which the cells had been sorted (since each pair of pre- and post-competence-loss samples was derived from a single population sorted by FACS), and a “competenceloss” column, which was 1 for the post- competence-loss population and 0 for the pre-competence-loss and mesendoderm-derived populations. Thus, we identified regions that changed specifically with competence loss while controlling for original sample identity.
The primary DNA sequences of differentially accessible peaks were retrieved from Ensembl and examined for motifs using MEME-ChIP 5.0.3 (Machanick and Bailey, 2011). ATAC-seq read depth was modeled as a function of known motif presence using chromVAR 1.4.1 (Schep et al., 2017). Significant motif matches were identified with FIMO 5.0.3. For the gene regulatory network, possible associations between genomic regions and target genes were identified using CisMapper 5.0.5. The full list of human TFs and the motifs for each TF were extracted from the list in Lambert et al. (2018). Mutual information between pairs of motifs was calculated with a custom python script.
Enrichment of functions of genes near differentially accessible genomic regions was performed using the web interface of GREAT (McLean et al., 2010) in June 2019.
RNA-seq analysis
Reads were pseudoaligned using kallisto 0.45.1 (Bray et al., 2016) to transcripts from the human genome build hg38. Abundance estimates for each gene were output with sleuth 0.30.0. Differentially expressed genes were identified using DESeq2 1.24.0 (Love et al., 2014) on R 3.6.1. For analysis with DESeq2 when comparing pre- and post-competence-loss populations, we used a design matrix with a “sample” column, which indicated the well from which the cells had been sorted and a “population” column, which indicated the pre- or post-competence-loss state. For comparison of pre-competence-loss and mesendoderm-derived populations, our design matrix contained sequencing batch and pre-competence-loss or mesendoderm-derived population identity.
For clustering the motifs of differentially expressed transcription factors, similarity between each pair of motifs was quantified as the Kullback-Leibler divergence of the product of the two motifs from a reference distribution, which was the product of two uniform motifs (0.25 probability for each base at each position). Motif alignment was performed by calculating the aforementioned divergence at each possible offset and using the maximum value obtained at any offset. This calculation was performed using a custom python script. The linkage was computed using the scipy.cluster.hierarchy.linkage function from scipy 1.3.0 with the “average” clustering method and the “braycurtis” distance.
ChIP-seq target set enrichments were calculated using the Enrichr (Kuleshov et al., 2016) web interface in July 2019. GO term enrichment was calculated using the PANTHER (Mi et al., 2019) web interface in July 2019.
For bulk RNA-seq comparison with ENCODE samples, the Pearson correlation coefficient between samples was calculated with Pandas (1.1.0) and hierarchical clustering performed with seaborn (0.10.1). Correlations were calculated based on the expression levels in units of TPM of all human genes labeled with Gene Ontology term 0003700 (GO:0003700), “DNA-binding transcription factor activity.” The ENCODE accessions and short descriptions for the samples used for comparison were as follows: ENCFF034KRQ, ectoderm; ENCFF044YLS, mesendoderm; ENCFF081JBX, neural; ENCFF145AQN, neural progenitor; ENCFF183XSM, neuronal stem; ENCFF290ZZQ, neural crest; ENCFF342LYI, trophoblast; ENCFF419KMW, ectoderm; ENCFF425FGL, excitatory neuron; ENCFF466QUZ, mesendoderm; ENCFF483MRL, excitatory neuron; ENCFF567GQW, neural progenitor; ENCFF663ARH, neural progenitor; ENCFF672VVX, neural progenitor; ENCFF684BKA, neural crest; ENCFF699LBP, ectoderm; ENCFF760HDK, trophoblast; ENCFF789VZB, neuronal stem cell; ENCFF813LWT, neural cell. Figure 3D uses the mesendoderm samples ENCFF044YLS and ENCFF466QUZ and the ectoderm samples ENCFF538XVQ and ENCFF034KRQ.
Overexpression candidate selection
We selected TFs by incorporating information from both RNA-seq and ATAC-seq analyses. We began with all TFs that were differentially expressed between the pre- and post-competence-loss populations (q < 0.05 with DESeq2). A gene was considered to be a TF if it was so annotated in Lambert et al. (2018). We then limited this list only to those that were expressed in a lineage-specific pattern and had above-background expression levels in at least one of the three populations. We defined genes with a lineage-specific expression pattern as those genes that (1) were differentially expressed between pre- and post-competence-loss populations and (2) either were not differentially expressed from the pre-competence-loss to mesendoderm populations or were differentially expressed in the opposite direction (upregulated from pre- to post- and downregulated from pre- to mesendoderm, or vice versa). By these criteria, 23 TFs were specifically upregulated with competence loss and 32 were specifically downregulated with competence loss. We also added select paralogs of the TFs that passed our expression pattern cutoffs: POU6F1, GRHL1, POU2F3, and FOXJ2, along with the OCT4 and SOX2 DNA binding domains (OCT4DBD and SOX2DBD). We further restricted the list to those candidates that had a known, high-quality DNA binding motif that appeared in either the DiffBind/MEME-ChIP or chromVAR analyses of our data. We also added four TFs (OTX2, JUNB, ZSCAN23, and GSC) whose motifs appeared in our ATAC-seq analyses but did not pass our differential expression cutoffs. We excluded ZEB1 and ZEB2 as candidates because their size precluded delivery by lentivirus. We also eliminated 10 TFs for which a clone was not readily accessible to us, either from the Harvard PlasmID database, Addgene, or genes that had previously been cloned from cDNA in our lab. We note that one candidate that was tested before all RNA-seq analysis was complete, MBNL2, missed significance cutoffs in the final analysis but is nevertheless included for completeness. After adding three candidates based on the literature (NRF2, ZNF521, and ID2), we were left with 36 candidates in total.
Epifluorescence imaging of fixed samples
Samples were imaged on a Zeiss AxioObserver Z1 inverted microscope using Zeiss 10× and 20x plan apo objectives (NA 1.3) using the appropriate filter sets. Images were acquired using an Orca-Flash 4.0 CMOS camera (Hamamatsu). The 43 HE DsRed/46 HE YFP/47 HE CFP/49 DAPI/50 Cy5 filter sets from Zeiss were used. The microscope was controlled using the ZEN software.
Live cell time-lapse imaging
Samples were imaged on a Zeiss AxioObserver Z1 inverted microscope using a Zeiss 20x plan apo objective (NA 0.8) using the appropriate filter sets and a Hamamatsu ImagEM EMCCD camera. Cells were maintained in a 37 degree incubation chamber at 5% CO2. Cells were imaged every 15 minutes. Focus was maintained using a combination of Zeiss Definite Focus and, using a custom script in MicroManager 2.0 beta (Edelstein et al., 2014), software autofocus adjustments every hour to compensate for slight movement of the membrane. For maximum accuracy, cells in this time-lapse were tracked manually in Fiji (Schindelin et al., 2012) (Figure 2B, n = 40; Figure 5B, FOXB2 transduced, n = 40; Figure 5B FOXB2 nontransduced controls, n = 30; Figure 5B CFP transduced, n = 32; Figure 5B CFP nontransduced controls, n = 44), and the tracks were analyzed with a custom python script that performed illumination profile correction. All mitotic events were captured because we were imaging nuclear transcription factors. Occasionally, a cell track could not be resolved confidently from the beginning to the end of the time-lapse, and any such tracks were truncated to cover only the high-confidence portion of the track.
Confocal imaging
For Figures 1 and S1, cells were imaged on a Leica inverted microscope with a Zeiss 20x objective (NA 0.8) with the appropriate filter sets. Detection was performed with photomultiplier tubes (for detection of Alexa 488 and Alexa 647) and a Leica HyD Photon Counter (for Alexa 568). For Figure S5C, cells were imaged on a Zeiss LSM 880 with Airyscan using a Zeiss 20x objective (NA 0.8). Detection was performed with photomultiplier tubes (Alexa 568 and Alexa 647) and a GaAsP detector (CFP and Alexa 488).
p(mesendoderm|OSR) curve fitting and location inference
For the initial p(mesendoderm|OSR) curve fitting to the single cell data extracted from the time-lapse, we fit a two-parameter sigmoid function to the data using scipy.optimize.curve_fit to minimize the squared difference between data and prediction. We used the learned sigmoid shape parameter, a = 8.38, from Figure 2D for all p(mesendoderm|OSR) inference in Figures 5, S4, and S5. To infer p(mesendoderm|OSR) for a given population, we fit the location parameter, b, by minimizing the squared difference between the observed final mesendoderm fate proportion and the mesendoderm proportion predicted by p(mesendoderm|OSR) at varying locations, b, given the observed p(OSR|t).
Ethical compliance
We used hESCs in accordance with approvals by Harvard University IRB (protocol #IRB18- 0665) and Harvard University ESCRO (protocol E00065).
QUANTIFICATION AND STATISTICAL ANALYSIS
N-values in Figures 1D (n = 3), 3E (n = 3), 4A (n = 3, mesendoderm; n = 4, all other samples), 4B (n = 3, RNA mesendoderm outgroup; n = 4, all other RNA samples; n = 3, ATAC-seq samples), 5C (n = 3), 5F (n = 4), 5G (n = 3), 5H (n = 3), and S4C (n = 4) refer to biological replicates. Error bars represent standard deviation unless otherwise noted. N-values in figure captions 2B and 5B (n = 40, FOXB2-transduced cells; n = 30, FOXB2 internal control; n = 32, CFP transduced cells; n = 44, CFP non-transduced controls) refer to number of cells tracked in each time-lapse condition. Three biological replicates of up to 10,000 cells were performed for analysis of each genetic perturbation in Figures 5 and S4. Biological replicate numbers can be found in figure captions as well as RNA-seq Method details, ATAC-seq Method details, and Lentiviral overexpression Method details. We collected as many independent biological replicates and sample sizes as are consistent with standard practice in the field. No formal power calculation was performed and standard statistical tests were used through out. No data were excluded, except in Figure S4 where three candidate TFs that had the effect of making cells adopt a non-ectoderm, non-mesendoderm fate were excluded as described in the Figure S4 caption.
For p(mesendoderm|OSR) curve fitting to the single cell data extracted from the time-lapse in Figure 2D, we fit a two-parameter sigmoid function to the data using scipy.optimize.curve_fit to minimize the squared difference between data and prediction. We used the learned sigmoid shape parameter, a = 8.38, from Figure 2D for all p(mesendoderm|OSR) p(mesendoderm|OSR) inference in Figures 5, S4, and S5. To infer p(mesendoderm|OSR) for a given population, we fit the location parameter, b, by minimizing the squared difference between the observed final mesendoderm fate proportion and the mesendoderm proportion predicted by p(mesendoderm|OSR) at varying locations, b, given the observed p(OSR|t). In Figures 4B and S2B, we analyzed Z-scores in order to compare distance in RNaseq and ATACseq reads from the mean. Heatmaps in S2C and S2D are normalized by maximal expression observed in each row or 10 transcripts per kilobase million (TPM), whichever was greater. In Figures 4C and S2E, we analyzed differential expression of transcription factors using a significance cutoff of q < 0.05 as calculated by DESeq2. In Figure S2F, values are shown as Z-score after normalization to TPM. In S2F and S5A, matrices show the Pearson correlation between each pair of samples. Figures S3A, S3E, and S3F show normalized ATAC-seq read depth. In Figure S4C, divergence of distributions was calculated with Kull-back-Liebler divergence and significance was reported if false discovery rate <0.1. Details for statistical analysis can also be found in the corresponding figure captions and, when referenced, in Method details. The statistical tests used are standard in the field and consistent with a large body of literature both in biology, other sciences and statistics.
Supplementary Material
Highlights.
Mesendoderm competence of human embryonic stem cells is predicted by OCT4:SOX2 levels
Mesendoderm competence window closes sharply at a point along the ectoderm trajectory
Analysis of DNA accessibility and expression data reveals underlying gene network
Perturbation of predicted genetic factors changes the mesendoderm competence window
ACKNOWLEDGMENTS
We thank the staff of the Bauer Core at Harvard University for their work on the RNA sequencing used in this manuscript as well as for their expertise and assistance with flow cytometry and FACS. We would like to thank Ulandt Kim and the Massachusetts General Hospital Next-Generation Sequencing Core (supported by NIH P30 DK040561). We thank Manashree Damle for bioinformatics help, the Harvard Physics/SEAS Instructional Machine Shop for making the stainless-steel washers used in the live-cell imaging in this study, and the Harvard Center for Biological Imaging for the use of their equipment. We thank Andrew Murray, Sean Eddy, and all of the members of the Ramanathan Lab for their helpful comments. J.R.V. was funded by The Fannie and John Hertz Foundation, the National Science Foundation Graduate Research Fellowship Program (NSF GRFP), and the NSF-Simons Center for Mathematical and Statistical Analysis of Biology at Harvard, award no. 1764269. R.H. was funded by the NSF GRFP. Some confocal imaging was conducted on an instrument provided by the Harvard MRSEC (DMR-1420570). This work was supported in part by NIH R01GM131105-01.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109990.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300. [Google Scholar]
- Berg DK, Smith CS, Pearton DJ, Wells DN, Broadhurst R, Donnison M, and Pfeffer PL (2011). Trophectoderm lineage determination in cattle. Dev. Cell 20, 244–255. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 709,21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng Z, Teo A, Pedersen RA, and Vallier L (2010). SIP1 mediates cell-fate decisions between neuroectoderm and mesendoderm in human pluripotent stem cells. Cell Stem Cell 6, 59–70. [DOI] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etoc F, Metzger J, Ruzo A, Kirst C, Yoney A, Ozair MZ, Brivanlou AH, and Siggia ED (2016). A balance between secreted inhibitors and edge sensing controls gastruloid self-organization. Dev. Cell 39, 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchtgott LA, Melton S, Menon V, and Ramanathan S (2017). Discovering sparse transcription factor codes for cell states and state transitions during development. eLife 6, 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SF (2000). The developmental mechanics of cell specification. In Developmental Biology, Sixth Edition (Sinauer Associates; ). [Google Scholar]
- Greber B, Coulon P, Zhang M, Moritz S, Frank S, Müller-Molina AJ, Araúzo-Bravo MJ, Han DW, Pape H-C, and Schöler HR (2011). FGF signalling inhibits neural induction in human embryonic stem cells. EMBO J 30, 4874–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handyside AH (1978). Time of commitment of inside cells isolated from preimplantation mouse embryos. J. Embryol. Exp. Morphol 45, 37–53. [PubMed] [Google Scholar]
- Jang J, Wang Y, Lalli MA, Guzman E, Godshalk SE, Zhou H, and Kosik KS (2016). Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell 165, 410–420. [DOI] [PubMed] [Google Scholar]
- Jang S, Choubey S, Furchtgott L, Zou L-N, Doyle A, Menon V, Loew EB, Krostag A-R, Martinez RA, Madisen L, et al. (2017). Dynamics of embryonic stem cell differentiation inferred from single-cell transcriptomics show a series of transitions through discrete cell states. eLife 6, e20487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai-Azuma M, Kanai Y, Gad JM, Tajima Y, Taya C, Kurohmaru M, Sanai Y, Yonekawa H, Yazaki K, Tam PP, et al. (2002). Depletion of definitive gut endoderm in Sox17-null mutant mice. Development 129, 2367–2379. [DOI] [PubMed] [Google Scholar]
- Kavka AI, and Green JB (1997). Tales of tails: brachyury and the T-box genes. Biochim. Biophys. Acta 1333, F73–F84. [DOI] [PubMed] [Google Scholar]
- Kiecker C, Bates T, and Bell E (2016). Molecular specification of germ layers in vertebrate embryos. Cell. Mol. Life Sci 73, 923–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The human transcription factors. Cell 172, 650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu C, Biechele S, Zhu Q, Song L, Lanner F, Jing N, and Rossant J (2013). Location of transient ectodermal progenitor potential in mouse development. Development 140, 4533–4543. [DOI] [PubMed] [Google Scholar]
- Li L, Song L, Liu C, Chen J, Peng G, Wang R, Liu P, Tang K, Rossant J, and Jing N (2015). Ectodermal progenitors derived from epiblast stem cells by inhibition of Nodal signaling. J. Mol. Cell Biol 7, 455–465. [DOI] [PubMed] [Google Scholar]
- Loh KM, Ang LT, Zhang J, Kumar V, Ang J, Auyeong JQ, Lee KL, Choo SH, Lim CYY, Nichane M, et al. (2014). Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 14, 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, et al. (2016). Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166, 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: next-generation image processing for biology. PLoS Biol 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton S, and Ramanathan S (2021). Discovering a sparse set of pairwise discriminating features in high-dimensional data. Bioinformatics 37, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47 (D1), D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, and Young RA (2011). Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oron E, and Ivanova N (2012). Cell fate regulation in early mammalian development. Phys. Biol 9, 045002. [DOI] [PubMed] [Google Scholar]
- Patthey C, and Gunhaga L (2014). Signaling pathways regulating ectodermal cell fate choices. Exp. Cell Res 321, 11–16. [DOI] [PubMed] [Google Scholar]
- Pauklin S, and Vallier L (2013). The cell-cycle state of stem cells determines cell fate propensity. Cell 155, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen RA, Wu K, and Bałakier H (1986). Origin of the inner cell mass in mouse embryos: cell lineage analysis by microinjection. Dev. Biol 117, 581–595. [DOI] [PubMed] [Google Scholar]
- Rossant J, and Lis WT (1979). Potential of isolated mouse inner cell masses to form trophectoderm derivatives in vivo. Dev. Biol 70, 255–261. [DOI] [PubMed] [Google Scholar]
- Rossant J, and Vijh KM (1980). Ability of outside cells from preimplantation mouse embryos to form inner cell mass derivatives. Dev. Biol 76, 475–482. [DOI] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng G, dos Reis M, and Stern CD (2003). Churchill, a zinc finger transcriptional activator, regulates the transition between gastrulation and neurulation. Cell 115, 603–613. [DOI] [PubMed] [Google Scholar]
- Smith JR, Vallier L, Lupo G, Alexander M, Harris WA, and Pedersen RA (2008). Inhibition of activin/nodal signaling promotes specification of human embryonic stem cells into neuroectoderm. Dev. Biol 313, 107–117. [DOI] [PubMed] [Google Scholar]
- Takaoka K, and Hamada H (2012). Cell fate decisions and axis determination in the early mouse embryo. Development 139, 3–14. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ (2005). The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695. [DOI] [PubMed] [Google Scholar]
- Thomson M, Liu SJ, Zou L-N, Smith Z, Meissner A, and Ramanathan S (2011). Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 145, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu DC, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, et al. (2011). Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH (1957). The Strategy of the Genes (Routledge; ). [Google Scholar]
- Xu X, Browning VL, and Odorico JS (2011). Activin, BMP and FGF pathways cooperate to promote endoderm and pancreatic lineage cell differentiation from human embryonic stem cells. Mech. Dev 128, 412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Mich JK, Ku S, Menon V, Krostag A-R, Martinez RA, Furchtgott L, Mulholland H, Bort S, Fuqua MA, et al. (2017). A single-cell roadmap of lineage bifurcation in human ESC models of embryonic brain development. Cell Stem Cell 20, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang CT, Chen J, Pankratz MT, Xi J, Li J, Yang Y, Lavaute TM, Li XJ, Ayala M, et al. (2010). Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell 7, 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zwick S, Loew E, Grimley JS, and Ramanathan S (2019). Mouse embryo geometry drives formation of robust signaling gradients through receptor localization. Nat. Commun 10, 4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ATAC-seq and RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication underhttps://doi.org/10.5281/zenodo.5516285.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-OCT4 | Cell Signaling | CAT#C30A3; RRID AB_2167691 |
| Rat monoclonal anti-SOX2 | Thermo Fisher | CAT#14-9811-82; RRID AB_11219471 |
| Goat polyclonal anti-SOX17 | R&D Systems | CAT#AF1924; RRID AB_355060 |
| Rabbit monoclonal anti-pSMAD1/5/9 | Cell Signaling | CAT#12428; RRID: AB_2797908 |
| Rabbit monoclonal anti-pSMAD2 | Cell Signaling | CAT#18338; RRID: AB_2798798 |
| Mouse monoclonal anti-PAX6 | Developmental Studies Hybridoma Bank | CAT#PAX6; RRID: AB_528427 |
| Goat polyclonal anti-NANOG | R&D Systems | CAT#AF1997; RRID: AB_355097 |
| Rabbit monoclonal anti-CDH2 (NCAD) | Cell Signaling | CAT#13116; RRID: AB_2687616 |
| Bacterial and virus strains | ||
| NEB Stable competent E. coli | New England Biolabs | CAT#C3040 |
| Chemicals, peptides, and recombinant proteins | ||
| A 83-01 | R&D Systems | CAT#2939 |
| Human BMP4 | R&D Systems | CAT#314-BP |
| Human/Mouse/Rat Activin A Protein | R&D Systems | CAT#338-AC |
| γ-27632 | Stemgent | CAT#04-0012 |
| LDN-193189 | R&D Systems | CAT#6053 |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | CAT#74004 |
| KAPA mRNA Hyper Prep kit | Roche | CAT#07962363001 |
| Gibson Assembly Master Mix | New England Biolabs | CAT#E2611 |
| NEB Q5 High-Fidelity DNA Polymerase | NEB | CAT#M0491 |
| Deposited data | ||
| ATAC-seq | NCBI GEO | GSE149077 |
| RNA-seq | NCBI GEO | GSE148904 |
| Experimental models: cell lines | ||
| Human: H1 embryonic stem cells | WiCell | WA01, RRID CVCL_9771 |
| Human: H1 SOX2:YFP/OCT4:RFP embryonic stem cells | Zhang et al., 2019 | N/A |
| Oligonucleotides | ||
| Gibson homology arm to clone genes of interest into pWPXL-mCerulean-P2A, forward: gcaggtgacgtggaggagaatcccgggcct | This paper | N/A |
| Gibson homology arm to clone genes of interest into pWPXL-mCerulean-P2A, reverse: aatccagaggttgattatcatatga | This paper | N/A |
| Recombinant DNA | ||
| pWPXL | Addgene | CAT#12257; RRID: Addgene_12257 |
| pWPXL-mCerulean-P2A | This paper | N/A |
| pMD2.G | Addgene | CAT#12259; RRID: Addgene_12259 |
| psPAX2 | Addgene | CAT#12260; RRID: Addgene_12260 |
| Software and algorithms | ||
| Fiji | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| MATLAB 2019 | MathWorks | https://www.mathworks.com/products/matlab.html |
| MicroManager 2.0 beta | Edelstein et al., 2014 | https://micro-manager.org |
| CellProfiler 3.0 | McQuin et al., 2018 | https://cellprofiler.org/ |
| DESeq2 1.24.0 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| kallisto 0.45.1 | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| chromVAR 1.4.1 | Schep et al., 2017 | https://github.com/GreenleafLab/chromVAR |
| MEME-ChIP 5.0.3 | Machanick and Bailey, 2011 | https://meme-suite.org/meme/tools/meme-chip |
| PANTHER | Mi et al., 2019 | http://www.pantherdb.org |
| Enrichr | Kuleshov et al., 2016 | https://maayanlab.cloud/Enrichr/ |
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/ |
| Other | ||
| hESC-qualified Matrigel | Corning | CAT#354277 |
| Polyester membrane filters, 3 micron, 25mm | Sterlitech | CAT#PET3025100 |
| mTeSR 1 | STEMCELL | CAT#85850 |
| Accutase | Innovative Stem Cell Technologies | CAT#AT104-500 |
| jetPrime | Polyplus | CAT#114-07 |
| Lenti-X Concentrator | Clontech | CAT#631231 |
| ENCODE RNA-seq data | ENCODE | See method details |