

Abstract
Fibrosis is the final common pathology of most chronic diseases as seen in the heart, liver, lung, kidney, and skin and contributes to nearly half of death in the developed countries. Fibrosis, or scarring, is mainly characterized by the transdifferentiation of fibroblasts into myofibroblasts and the excessive accumulation of extracellular matrix (ECM) secreted by myofibroblasts. Despite immense efforts made in the field of organ fibrosis over the past decades and considerable understanding of the occurrence and development of fibrosis gained, there is still lack of an effective treatment for fibrotic diseases. Therefore, identifying a new therapeutic strategy against organ fibrosis is an unmet clinical need. Naringenin, a flavonoid that occurs naturally in citrus fruits, has been found to confer a wide range of pharmacological effects including antioxidant, anti-inflammatory, and anticancer benefits and thus potentially exerting preventive and curative effects on numerous diseases. In addition, emerging evidence has revealed that naringenin can prevent the pathogenesis of fibrosis in vivo and in vitro via the regulation of various pathways that involved signaling molecules such as transforming growth factor-β1/small mother against decapentaplegic protein 3 (TGF-β1/Smad3), mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt), sirtuin1 (SIRT1), nuclear factor-kappa B (NF-κB), or reactive oxygen species (ROS). Targeting these profibrotic pathways by naringenin could potentially become a novel therapeutic approach for the management of fibrotic disorders. In this review, we present a comprehensive summary of the antifibrotic roles of naringenin in vivo and in vitro and their underlying mechanisms of action. As a food derived compound, naringenin may serve as a promising drug candidate for the treatment of fibrotic disorders.
1. Introduction
Currently, the incidence of fibrotic diseases is on the rise and presents a serious threat to global public health [1]. Nearly 45% of disease-related deaths in the developed countries are closely associated with fibrotic disorders, and the morbidity and mortality of these disorders are probably higher in the developing countries [2, 3]. Despite much progress made in uncovering the molecular mechanisms underlying the development and progression of fibrosis over the past decades, there is currently no effective antifibrotic treatment available for fibrotic diseases. Therefore, identification of new molecular mechanisms involved in the fibrotic process and development of novel therapeutic agents against fibrotic disorders are urgently needed.
Recently, increasing evidence has demonstrated that many natural products such as flavonoids have potent antifibrotic activities, and some of which have shown promise as emerging new antifibrotic agents [4]. Naringenin, a natural citrus flavonoid that possesses various biological properties, has been extensively reported to prevent the pathogenesis of fibrosis in several experimental studies [5–8]. The importance of naringenin in managing tissue fibrosis warrants a detailed review of the effects of naringenin on fibrosis and of its underlying mechanisms of action. Here, we will focus on the crucial role of naringenin in the suppression of tissue fibrosis and discuss its therapeutic potential as a promising agent for the treatment of fibrotic disorders.
2. The Cellular and Molecular Mechanisms of Fibrosis
Fibrosis often takes place in response to a trigger or tissue injury [9]. Initially, tissue injury is mild or transient, and the formation of fibrotic scars in organs is actually a normal tissue repair response and is beneficial for organisms. However, when the injury is severe or prolonged, sustained or uncontrolled fibrogenesis can result in adverse architectural remodeling, organ malfunction, and eventually organ failure. These defects contribute significantly to global morbidity and mortality [10].
Fibrosis is the final common pathology of many chronic inflammatory diseases as detected in the heart, kidney, liver, lung, and skin tissue [11]. The causative factors of fibrosis are diverse in the various organs (Figure 1), but the most common etiologies include inflammation, aging, and genetic alteration [4, 12]. Fibrosis is mainly defined as the activation and proliferation of fibroblasts, the production of inflammatory factors, and the massive deposition of extracellular matrix (ECM) proteins such as type I collagen (COL1), type III collagen (COL3), and fibronectin (FN) [13, 14]. Myofibroblasts, the activated form of fibroblasts, exhibit two unique characteristics: firstly, they are contractile due to the expression of α-smooth muscle actin (α-SMA), which results in the distortion of tissue cytoarchitecture. Secondly, they secrete ECM macromolecules, which lead to the replacement of normal tissue with a permanent fibrotic scar, thus causing an increase in tissue stiffness and the parenchymal destruction of organs [2, 15].
Figure 1.
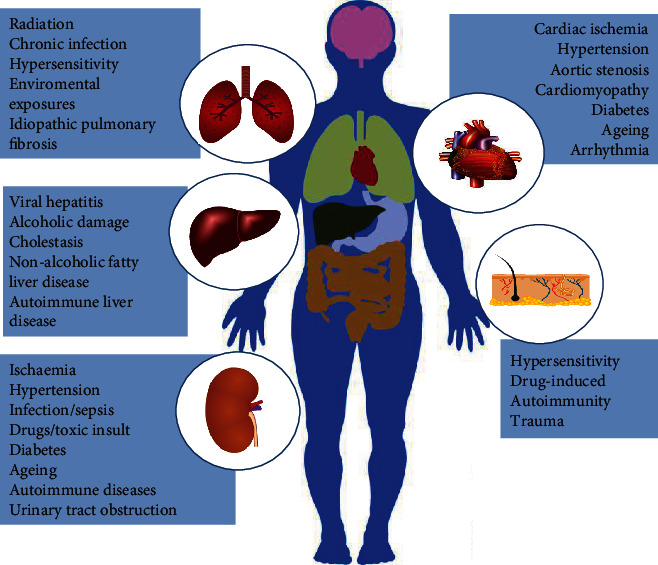
Major causes of organ fibrosis. In the different organs, a broad range of triggers and etiologies can result in occurrence and development of fibrosis. Fibrosis may lead to organ dysfunction or failure and accounts for substantial morbidity and mortality [adapted from ref. [10]].
The sources of myofibroblasts may vary across different tissues, depending on the injured organ and the specific fibrotic response [16–18]. Several potential sources and formation mechanisms of myofibroblasts are presented in Figure 2. Although epithelial/endothelial-to-mesenchymal transition (EMT/EndoMT), or pericyte to myofibroblast transition may play a role under special conditions, it is now widely accepted that the main source of myofibroblasts is the activation of tissue-resident fibroblasts [16, 18–20].
Figure 2.
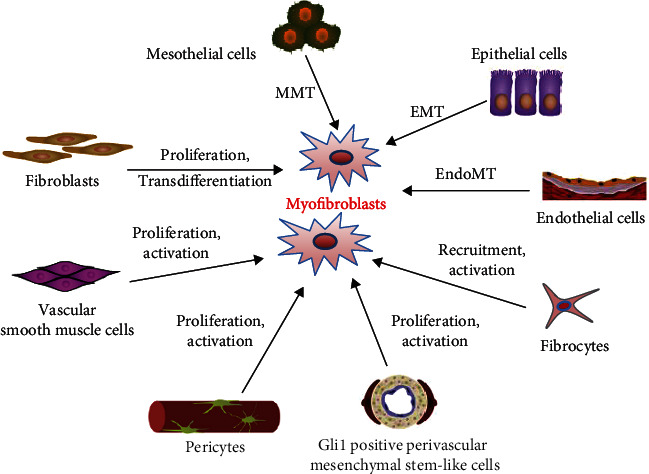
Potential sources and formation mechanisms of myofibroblasts. Activated myofibroblasts are central drivers for fibrosis and can secrete excess extracelluar matrix proteins. The cellular subsets may be originated from resident fibroblasts, epithelial cells, endothelial cells, circulating fibrocytes, mesothelial cells, vascular smooth muscle cells, pericytes, Gli1 positive perivascular mesenchymal stem-like cells, and others. Diverse mechanisms comprising cellular proliferation, activation, transdifferentiation, recruitment, mesothelial-to-mesenchymal transition (MMT), epithelial-to-mesenchymal transition (EMT), and endothelial-to-mesenchymal transition (EndoMT) can lead to myofibroblast formation [adapted from refs. [11, 15]].
Despite the highly complex mechanisms for fibrosis, the transdifferentiation of fibroblasts into myofibroblasts is a central driver for all forms of fibrosis [10, 15]. To date, a wide range of mediators have been found to activate fibroblasts and to promote the initiation and progression of fibrosis, including transforming growth factor-β1 (TGF-β1) [21, 22], angiotensin II (AngII) [23, 24], connective tissue growth factor (CTGF) [25, 26], platelet-derived growth factor (PDGF) [27, 28], interleukins (IL-6, IL-13, IL-33, IL-11, IL-17, etc.) [11, 12, 29], tumor necrosis factor-α (TNF-α) [30], endothelin-1(ET-1) [31, 32], reactive oxygen species (ROS) [33, 34], and hypoxia [35, 36]. Some of these mediators inducing fibrotic processes are illustrated in Figure 3. Among them, TGF-β1 is considered to be the most potent profibrotic factor and contributes to fibrosis primarily by activating its downstream canonical small mother against decapentaplegic (Smad) signaling pathway [37]. In addition, TGF-β1 has also been shown to act through several Smad-independent pathways (known as noncanonical signaling cascades) in the development of fibrosis, such as mitogen-activated protein kinase (MAPK) pathways mediated by extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK as well as phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) or Rho-like GTPases signaling pathways [38, 39]. Besides the TGF-β signaling pathway, there are numerous other signaling cascades that are also involved in the pathogenesis of fibrosis, such as nuclear receptors signaling (peroxisome proliferation-activated receptor-γ, PPAR-γ) [9], bone morphogenetic protein (BMP) signaling [40], Wnt/β-catenin signaling [41], Hedgehog signaling [42], Notch signaling [43], and epidermal growth factor receptor (EGFR) signaling [44]. Therefore, targeting these fibrotic mediators or signaling pathways could represent potential therapeutic strategies to combating fibrotic diseases.
Figure 3.
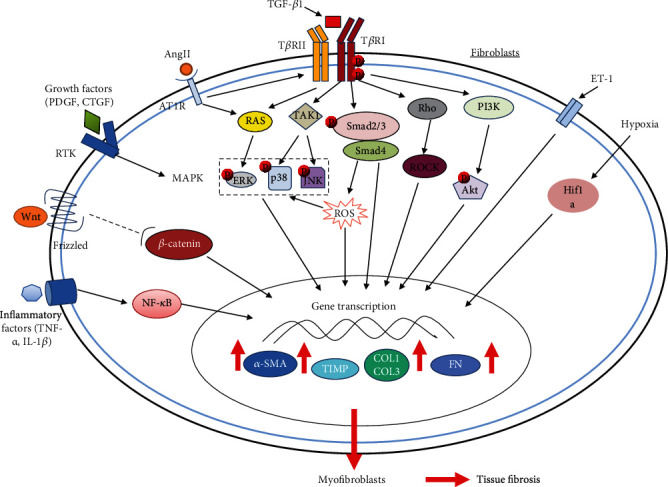
Molecular mechanisms in tissue fibrosis. The diagram shows the TGF-β1, AngII, ET-1, growth factors (PDGF, CTGF, etc.), inflammatory factors (TNF-α, IL-1β, etc.), Wnt, ROS, and hypoxia-inducible factor-1α (HIF-1α) pathways that may mediate tissue fibrotic responses. The central pathways for tissue fibrosis are TGF-β1 canonical (Smad-dependent) and noncanonical (Smad-independent) signaling pathways, among which the canonical TGF-β1/Smad pathway plays a major role in the development of fibrosis. Following TGF-β1 binding, type II TGF-β1 receptor (TβRII) recruits type I TGF-β1 receptor (TβRI) and activates it by phosphorylating it. The activated TβRI then specifically phosphorylates Smad2 and Smad3, which then bind to Smad4 to form a complex leading to their translocation to the nucleus and regulation of transcription of profibrotic genes. Apart from Smad-mediated signal transduction, TGF-β1 can also signal through several noncanonical signaling cascades such as PI3K, p38, ERK, JNK, and Rho-like GTPase pathways. Most of the other pathways have been indicated to regulate or to interact with the TGF-β1 signaling pathways. The final result of these signaling pathways activation is triggering a profibrotic gene transcriptional regulation program contributing to tissue fibrosis caused by the activation of myofibroblasts and their increased synthesis of various myofibroblast-specific and profibrotic proteins such as α-SMA, COL1, COL3, FN, and tissue inhibitors of metalloproteinase (TIMP) [Adapted from ref. [15]].
3. Naringenin
Naringenin, 5,7-Dihydroxy-2-(4-hydroxyphenyl) chroman-4-one, is one of the most important natural flavonoids and mostly exists in citrus fruits like grape fruits, orange and lemon [45, 46]. It has a molecular weight of 272.26 (C15H12O5) and exists predominantly in nature in two forms: the glycosylated form (naringin or naringenin-7-O-glucoside) and the aglycosylated form (naringenin) (Figure 4) [47]. Naringin can be hydrolyzed into naringenin by the liver enzyme naringinase [48], and naringin is responsible for the bitter taste of citrus fruits, whereas naringenin is flavorless.
Figure 4.
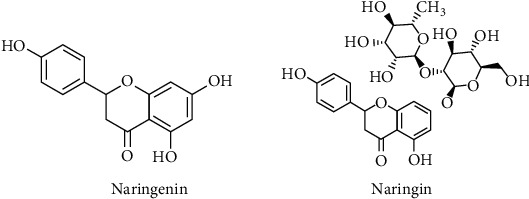
Chemical structures of naringenin and its glycosylated form naringin.
In nature, naringenin exists as a solid and is almost insoluble in water, but soluble in organic solvents such as dimethyl sulfoxide and ethanol. However, naringin can easily dissolve in water [49]. Although naringenin is quickly absorbed after its single oral administration in human subjects, it shows only 5.81% oral bioavailability due to its poor aqueous solubility, which compromises its clinical use [50]. Both active transport and passive diffusion aid its absorption into the gastrointestinal tract [45]. After absorption, naringenin is rapidly conjugated to form glucuronide or sulphoglucuronide and is bound to serum albumin and is rapidly transported to highly perfused organs like the kidney, heart, spleen, liver, and cerebrum [51, 52]. Before absorption, naringenin is hydrolyzed by β-glucosidase in the small intestine [53, 54]. Then, naringenin is further metabolized by the intestinal bacteria into p-hydroxyphenylpropionic acid, phenolic acids, and p-hydroxybenzoic acid which are mainly found in the plasm, urine, and bile [55, 56]. The excretion of naringenin occurs through two primary pathways: urinary and biliary pathways [57]. It is suggested that naringenin has low toxicity, and its LD50 is 5000 mg/kg [58]. Despite the fact that naringenin has relatively low bioavailability, there are currently some techniques and pharmacological formulations to improve its bioavailability, such as developing drug delivery systems by the use of liposomes, nanoparticles, nanosuspensions, solid dispersion, and inclusion complexation [45, 59].
In various in vitro and in vivo studies, naringenin has been demonstrated to exhibit extensive biological activities, including antioxidant [60, 61], anti-inflammatory [62, 63], antiviral [64, 65], antibacterial [66, 67], and anticancer actions [68, 69]. Owing to these pharmacological properties, naringenin has been reported to exert a strong protective role and has therapeutic potential against numerous diseases, like cardiovascular diseases [55, 70], liver diseases [71], lung diseases [72], diabetes [58, 73], neurodegenerative diseases [74, 75], and malignant tumors [76, 77]. In addition, recently, emerging evidence has suggested that naringenin is capable of inhibiting the progression of fibrosis in multiple organs and tissues, including the liver, heart, lung, kidney, and skin, through regulation of various signaling pathways (Table 1) and thus showing potential therapeutic effects on fibrotic disorders [8].
Table 1.
Summary of preclinical antifibrotic effects and underlying mechanisms of naringenin.
| Fibrotic disease | Models | In vitro/in vivo | Effects and related mechanisms | Reference |
|---|---|---|---|---|
| Liver fibrosis | DMN-induced liver damage in rats | In vivo | Reduced hepatic collagen accumulation via the inactivation of HSCs | [6] |
| TGF-β1-treated rat HSCs | In vitro | Suppression of ECM expression through inhibition of Smad3 signaling | [87] | |
| High cholesterol-induced NASH in rats | In vivo | Improvement of liver oxidative and inflammatory status and reduction of hepatic collagen deposition through the downregulation of NF-κB and MMP-2/9, respectively | [88] | |
| CCl4-induced fibrosis in rats | In vivo | Prevented CCl4-induced liver inflammation, necrosis, and fibrosis through suppression of oxidative stress, NF-κB, TGF-β/Smad3, and JNK/Smad3 pathways | [89, 90] | |
| Alcohol-induced hepatic damage in mice | In vivo | Attenuated liver inflammation, fibrosis, and hepatocyte apoptosis via decreasing the NF-κB, TGF-β1, and caspase-3 levels | [91] | |
| CCl4-induced fibrosis in mice, TGF-β1-treated rat HSCs | Both in vitro and vivo | Increased targeting of HSCs, ameliorated liver injury and fibrosis via SPARC-dependent pathways | [92] | |
| ApoE−/−-induced NASH in mice, mouse hepatocyte AML-12 | Both in vitro and vivo | Suppressed hepatic steatosis, oxidative stress, inflammation and fibrosis through modulating hepatic SIRT1-mediated signaling cascades | [93] | |
| CCl4-induced fibrosis in rats | In vivo | Reduced liver fibrosis and inflammation by the upregulation of MMP-2 activity and downregulation of proinflammatory cytokines levels | [94] | |
| Cardiac fibrosis | Pressure overload-induced cardiac remodeling in mice | In vivo | Attenuated cardiac hypertrophy and interstitial fibrosis via the inhibition of PI3K/Akt, ERK, and JNK signaling | [70] |
| TGF-β1-treated CFs | In vitro | Inhibited CF proliferation, differentiation, and collagen synthesis via G0/G1 arrest | [98] | |
| Hypertension-induced atrial fibrosis in rats, hydrostatic pressure-treated CFs | Both in vitro and vivo | Alleviated the atrial fibrosis in SHRs and inhibited CF proliferation and profibrotic marker expression by inactivating Smad3 signaling | [7] | |
| AngII-treated CFs | In vitro | Suppressed profibrotic genes expression via inactivating Smad3 signaling | [99] | |
| Lung fibrosis | Bleomycin-induced pulmonary fibrosis in mice | In vivo | Attenuated pulmonary fibrosis through inhibiting TGF-β1 secretion and decreasing regulatory T cells | [5] |
| Allergen-induced chronic asthma in mice | In vivo | Inhibited airway remodeling and peribronchial fibrosis probably through reducing Th2 cytokines levels and oxidative stress | [104] | |
| HDM-induced chronic asthma in mice | In vivo | Improved airway inflammation and fibrosis potentially through inhibiting the expression of proinflammatory cytokines and TGF-β | [105] | |
| MP-induced pneumonia in mice, MP-treated BEAS-2B cell line | Both in vitro and vivo | Suppressed lung inflammation and fibrosis by inhibition of autophagy activation after MP infection | [106] | |
| Radiation-induced lung injury in rodents | In vivo | Ameliorated the lung injury including lung fibrosis by lowering IL-1β level and maintaining the homeostasis of inflammatory factors | [107] | |
| Renal fibrosis | Daunorubicin-induced nephrotoxicity in rats | In vivo | Improved nephrotoxicity by reducing renal fibrosis, inflammation, and oxidative/ER stress through mitigating AT1R, ERK1/2-NF-κB p65 signaling pathways | [113] |
| A mouse model of UUO, TGF-β1-treated NRK52E cell line | Both in vitro and vivo | Relieved renal fibrosis in vitro and in vivo by blocking Smad3 signaling | [114] | |
| STZ-induced diabetic nephropathy in rats, high glucose-treated cell line | Both in vitro and vivo | Attenuated the deposition of ECM in vitro and in vivo and inhibited cell proliferation in vitro, through let-7a-mediated inhibition of TGF-β1/smad signaling | [115] | |
| A rat model of renovascular hypertension | In vivo | Ameliorated hypertensive renal damage, including interstitial fibrosis, by modulating the balance of components of the renin-angiotensin system | [116] | |
| A mouse model of lupus | In vivo | Reduced the autoimmunity and prevented kidney damage including fibrosis by modulating T-cell subsets and cytokine profile | [117] | |
| Skin fibrosis | Mechanical stretch-induced hypertrophic scars in mice | In vivo | Attenuated skin fibrosis and inhibited scar formation via the inhibition of dermal fibroblast activation and local inflammation | [124] |
Abbreviations used are DMN: dimethylnitrosamine; HSCs: hepatic stellate cells; ECM: extracellular matrix; NASH: nonalcoholic steatohepatitis; SPARC: secreted protein acidic and rich in cysteine, CFs: cardiac fibroblasts; HDM: house dust mite; MP: mycoplasma pneumonia; ER: endoplasmic reticulum; STZ: streptozotocin.
4. Naringenin and Organ Fibrosis
4.1. Naringenin and Liver Fibrosis
Liver fibrosis is a reversible wound-healing response to acute or chronic liver injury from various causative factors, like chronic viral infection, excess alcohol consumption, toxic exposure, cholestasis, autoimmune hepatitis, and nonalcoholic steatohepatitis (NASH) [78, 79]. Hepatic fibrosis, mainly characterized by the excessive accumulation of ECM protein and the formation of fibrous scar in injured liver [80], is the final common pathology of all chronic liver diseases. It can ultimately lead to irreversible liver cirrhosis, which is the end stage of liver disease and also one of the most common causes of morbidity and mortality worldwide [81, 82]. Thus, prevention and reversal of hepatic fibrosis are an effective strategy for treating various chronic liver diseases and combating cirrhosis. However, up to now, there is no effective therapeutic treatment for liver fibrosis except for the removal of the causative factor or liver transplantation [80]. Hepatic stellate cells (HSCs) are the central effectors in the development of liver fibrosis, which are the primary source of abnormal ECM constituents in the liver [83, 84]. Upon fibrogenic stimulation, such as exposure to injury or profibrotic factor, HSCs become activated and begin to over proliferate and transdifferentiate into myofibroblasts, which massively express α-SMA and ECM proteins, thereby leading to liver fibrogenesis [85, 86]. Therefore, targeting HSC activation and proliferation has been considered as a promising therapeutic strategy for the treatment of liver fibrosis.
There have been now many studies indicating the therapeutic roles of naringenin in preclinical models of liver fibrosis. Lee et al. first suggested some histological evidence that oral administration of naringenin could reduce hepatic collagen accumulation and exert potential antifibrotic effects in rats with liver damage induced by dimethylnitrosamine (DMN) via the inactivation of HSCs [6]. In an in vitro study, naringenin was for the first time demonstrated to be a Smad3 specific inhibitor and could suppress the TGF-β1-induced ECM protein expression in cultured rat HSCs by blocking the TGF-β1 signaling pathway via selectively inhibition of Smad3 activation [87]. In a rat model of high cholesterol-induced hepatic damage, naringenin supplementation alleviated hepatic oxidative stress and inflammatory response, as well as collagen deposition as indicated by Sirius Red staining of liver sections, by inhibiting NF-κB pathway and matrix metalloproteinases-2/9 (MMP-2/9) activities, respectively, ultimately attenuating fibrosis and the liver injury [88]. In addition, in another rat model of liver fibrosis, Hernández-Aquino et al. showed that naringenin was able to block carbon tetrachloride- (CCl4-) induced liver inflammation, necrosis, and fibrosis by reducing oxidative stress as well as by preventing NF-κB, TGF-β/Smad3, and JNK/Smad3 signaling pathways [89], which was in agreement with the findings reported by the same research team in another study [90]. In a mouse model of chronic alcohol-induced hepatic damage, Zhang et al. demonstrated that naringenin treatment could prevent hepatic inflammation, suppress liver fibrosis, and alleviate hepatocyte apoptosis, thus improving the liver function, through decreasing the levels of NF-κB, TGF-β1, and caspase-3, respectively [91]. In another report, to increase the bioavailability and HSCs-targeted property of naringenin, Wang et al. developed a novel activated HSCs-targeted drug delivery system, namely, naringenin-loaded albumin self-modified liposomes (NaAlLs), and demonstrated that NaAlLs significantly, and specifically, increased targeting of activated HSCs and ameliorated liver fibrosis in vitro and in vivo via the secreted protein acidic and rich in cysteine- (SPARC-) dependent pathway [92]. Moreover, in a mouse model of NASH, naringenin administration could suppress hepatic steatosis, reduced hepatic oxidative stress and inflammation, and prevented liver fibrosis, as evidenced by the decrease in hepatic collagen deposition and hydroxyproline content, as well as by the reduction of protein expression of TGF-β1 and α-SMA in the liver. This process was mediated by the activation of hepatic sirtuin1- (SIRT1-) mediated signaling cascades that led to the therapeutic effects of naringenin on NASH [93]. In a recent report, Yang et al. discovered that naringenin loaded nanoparticles, which could enhance the oral bioavailability of naringenin, markedly reduced CCl4-induced liver fibrosis and inflammation in rats, as assessed by liver histology and serum levels of inflammatory cytokines, via upregulating the activity of MMP-2 and decreasing the levels of proinflammatory cytokines [94].
4.2. Naringenin and Cardiac Fibrosis
Cardiac fibrosis is a final pathological outcome for multiple forms of cardiovascular diseases, including cardiomyopathy, hypertension, arrhythmias, and myocardial infarction [10]. It represents a substantial accumulation of ECM proteins in the interstitium of the heart and excessive cardiac scar formation, which causes electrical and mechanical dysfunction, thereby ultimately contributing to heart failure and death [95, 96]. The risk factors of myocardial fibrosis are diverse, and some of the common ones are described in Figure 1. Cardiac fibroblasts (CFs) are the predominant cell type within the myocardium that provides structural support [97]. Upon injury or stimuli, CFs can proliferate abnormally and transdifferentiate into activated myofibroblasts, which is the key event in cardiac fibrosis. Despite extensive research, the underlying mechanisms of cardiac fibrosis are not fully elucidated and currently, no evidence-based therapies show significant effectiveness on treating cardiac fibrosis.
The first direct evidence of an interaction between naringenin and cardiac fibrosis was that naringenin was shown to alleviate pressure overload-induced cardiac hypertrophy and interstitial fibrosis in mice, as assessed by histological analysis and quantitative PCR analysis of hypertrophy biomarkers and profibrotic genes [70]. The potential mechanisms of naringenin exerting its cardioprotective effect may be related to the suppression of ERK, JNK, and PI3K/Akt signaling pathways. An in vitro study by Liu et al. reported that naringenin was able to inhibit TGF-β1-induced proliferation, transformation, and collagen production of CFs, and the mechanism underlying the process may be in part due to the inhibition of DNA synthesis via G0/G1 arrest following treatment with naringenin, thus implying that naringenin may serve as a novel treatment strategy for cardiac fibrosis [98]. In addition, Wei et al. found that naringenin, as a Smad3-specific inhibitor, could attenuate hypertension-induced atrial fibrosis in spontaneously hypertensive rats (SHRs) and inhibit the proliferation and ECM protein expression of CFs induced by elevated hydrostatic pressure via the suppression of Smad3 signaling activation [7]. In a recent report, Liang et al. suggested that naringenin significantly inhibited the protein expression of profibrotic genes such as COL1, COL3, and ACTA2 (actin alpha 2, smooth muscle) through inactivating the Smad3 signaling pathway in AngII-stimulated mouse CFs [99], also revealing the potential of naringenin to treat cardiac fibrosis.
4.3. Naringenin and Lung Fibrosis
Pulmonary fibrosis refers to the end stage of various interstitial lung diseases, characterized by phenotypic alteration of both fibroblasts and alveolar epithelial cells, abnormal deposition of ECM, and the disruption of lung parenchyma, which results in impaired gas exchange, decreased lung function, and progressive respiratory failure [100]. So far, a variety of underlying etiologies have been identified to lead to lung fibrosis, such as ageing, environmental and occupational exposures, autoimmune diseases, and genetic disorders; yet, the most common form is idiopathic pulmonary fibrosis (IPF) [101]. IPF is a progressive and terminal lung disease with 3-5 years of median survival time after diagnosis. The incidence of this disease has risen. Currently, two small-molecule drugs, pirfenidone [102], and nintedanib [103], which have been demonstrated to slow disease progression, have been approved worldwide for the treatment of IPF; however, they have toxic side-effects and cannot reverse fibrosis [102, 103]. As such, for now, lung transplantation is the sole therapeutic strategy.
Studies have shown that naringenin exhibits antifibrotic effects, as a potential drug to treat pulmonary fibrosis that arises from various etiologies. In a mouse model of bleomycin-induced pulmonary fibrosis, Du et al. demonstrated that oral administration of naringenin attenuated bleomycin-induced pulmonary fibrosis, as shown by histological staining and quantification of collagen content in the lung, by inhibiting TGF-β1 secretion and decreasing regulatory T cells [5]. In a murine model of asthma, Shi et al. suggested that naringenin treatment inhibited allergen-induced airway remodeling and peribronchial fibrosis as evidenced by the decreases in peribronchial α-SMA areas, subepithelial collagen deposition, and hydroxyproline content in the lung, probably through reducing T-helper 2 (Th2) cytokine levels and oxidative stress [104]. Similarly, in another murine asthma model induced by house dust mite (HDM), Seyedrezazadeh et al. found that a combination of naringenin with other flavanone, hesperetin, could markedly alleviate HDM-induced airway inflammation and fibrosis, as assessed by histological analysis, potentially through interfering with the expression of proinflammatory cytokines and TGF-β [105]. In a study of mycoplasma pneumoniae (MP) pneumonia, Lin et al. identified that treatment with naringenin could suppress MP-induced lung inflammation and fibrosis in vivo and also suppressed MP-induced BEAS-2B cell injury in vitro, by inhibition of the autophagy pathway [106]. In addition, in rodent models of radiation-induced lung injury, Zhang et al. proved that naringenin treatment effectively ameliorated radiation-induced lung injury, including lung fibrosis as assessed by histological analysis, by lowering IL-1β and maintaining the homeostasis of inflammatory factors [107].
4.4. Naringenin and Renal Fibrosis
Chronic kidney diseases (CKD), with a high prevalence of morbidity and mortality, remain a major global public health problem imposing enormous economic burden on society [108, 109]. Renal fibrosis is the common final pathway of almost all progressive CKD with diverse etiologies (including ischaemia, infection, autoimmune disease, toxic/drug insult, diabetes, and genetic disorders), and it has been indicated to be the best predictor of CKD progression to end-stage renal disease, which requires dialysis or kidney transplantation [110, 111]. Renal fibrosis is typically marked by infiltration of inflammatory cells and activation and proliferation of myofibroblasts, which leads to excessive accumulation of ECM components in the glomeruli, interstitium, and vasculature. Currently, there are no specific antifibrotic drugs in use for kidney patients [112]. Therefore, development of effective therapeutic treatments to treat kidney fibrosis is of utmost importance.
A previous study showed that treatment with naringenin could improve daunorubicin-induced nephrotoxicity in rats by reducing renal fibrosis, inflammation, and oxidative/endoplasmic reticulum stress, which may be possibly through the mitigation of AngII type I receptor (AT1R), ERK1/2-NF-κB p65 signaling pathways [113]. In addition, Meng et al. demonstrated that only naringenin treatment markedly alleviated renal fibrosis in vitro and in a mouse model of unilateral ureteral obstruction (UUO) by blocking Smad3 signaling directly, and the combination of naringenin with asiatic acid, a triterpene from Centella Asiatica, demonstrated to be a Smad7 agonist, produced a better inhibitory effect on renal fibrosis by suppressing Smad3 while inducing Smad7 [114]. In a study of diabetic nephropathy (DN), Yan et al. suggested that naringenin could inhibit the expressions of ECM components in both kidney tissues of DN rats and glomerular mesangial cells treated by high glucose and also inhibited mesangial cell proliferation, by suppressing TGF-β1/Smad signaling pathway via the regulation of microRNA let-7a [115]. In an animal model of renovascular hypertension established by performing 2-kidney, 1-clip surgery in rats, Wang et al. observed that naringenin administration significantly ameliorated hypertensive renal damage in the nonclipped kidneys, including interstitial fibrosis as measured by histological analysis, by normalizing the imbalance of renin-angiotensin system [116]. Moreover, naringenin was identified to prevent autoimmune features and kidney injury, including renal fibrosis as evaluated by the decrease in collagen fibers, in lupus-prone mice, by modulating T-cell subsets and cytokines profile [117].
4.5. Naringenin and Skin Fibrosis
Skin fibrosis, as defined by excessive fibroblast proliferation and ECM protein deposition in the dermis, is the common pathological hallmark of multiple skin disorders such as systemic sclerosis, hypertrophic scars, keloids, restrictive dermopathy, and graft-versus-host disease [118]. Skin fibrosis affects over 100 million people per year in westernized countries and becomes a significant health problem worldwide [119, 120]. Cutaneous scars have a profound impact on patients' quality of life due to related pain and pruritus, functional impairment, and psychosocial distress [119, 121, 122]. Despite the socioeconomic burden, effective and durable scar treatment remains a major unmet need in clinical medicine [123].
In a mouse model of mechanical stretch-induced hypertrophic scars, topical application of naringenin could attenuate skin fibrosis and inhibit scar formation, as assessed by histological analysis, by the suppression of dermal fibroblast activation and local inflammatory response, thus implying that naringenin may serve as a novel agent for treating hypertrophic scars [124]. As there are few reports about the effect of naringenin on skin fibrosis, the antifibrotic role of naringenin in skin tissues remains to be further elucidated.
5. Conclusions and Perspectives
In this review, we summarize the recent advances of naringenin in fibrosis research and treatment. A growing body of evidence, both in vitro and in vivo, has indicated that naringenin exerts potential antifibrotic properties in multiple tissues and organs like the liver, heart, lung, kidney, and skin, and their mechanisms of action, which have been summarized in Figure 5, may involve mostly the regulation of TGF-β1/Smad3, MAPK, PI3K/Akt, SIRT1, and NF-κB signaling pathways, as well as oxidative stress. However, the antifibrotic effects of naringenin are mostly derived from animal studies and cellular models of fibrosis, and there is a lack of clinical trial evidence. In addition, the antifibrotic mechanisms of naringenin have not been fully delineated. The studies regarding the safety and efficacy of naringenin in humans are still lacking. Going forward, more mechanistic and clinical studies are needed to further support the utilization of this flavonoid in human diseases. Despite that naringenin has very low water solubility, which leads to its low bioavailability, there are some techniques and methods to solve this problem, such as designing an oral drug delivery system using liposomes, nanoparticles, or nanosuspensions [45, 59]. In summary, as a food-derived compound, naringenin may serve as a promising therapeutic agent for fibrotic disorders in the future.
Figure 5.
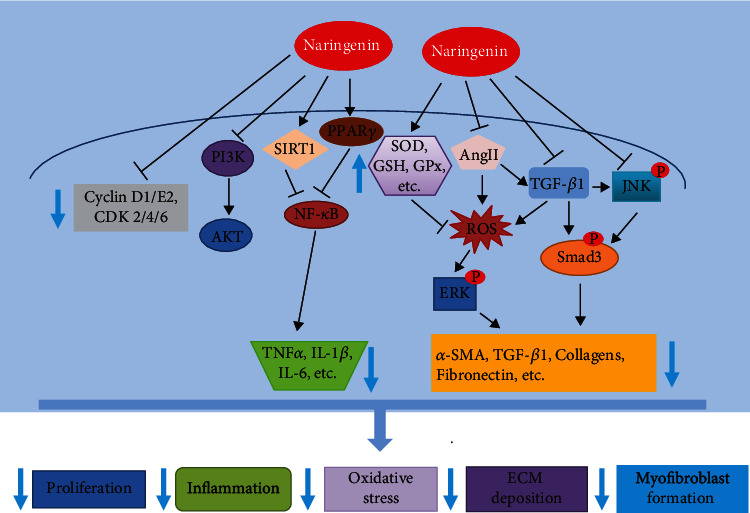
Antifibrotic mechanisms of naringenin: schematic representation of naringenin exerting its antifibrotic effects through affecting multiple signaling pathways related to fibrogenesis (↑: increase; ↓: decrease; SOD: superoxide dismutase; GSH: glutathione; GPx: glutathione peroxidase; CDK: cyclin-dependent kinases).
Acknowledgments
This work was supported by a grant from the Southwest Medical University (No. 2020ZRZD002).
Contributor Information
Guang Li, Email: liguang@swmu.edu.cn.
Qiang Ye, Email: yeqiangxnyd@126.com.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Authors' Contributions
Yanfei Du, Jun Ma, and Yu Fan contributed equally to this work.
References
- 1.Mehal W. Z., Iredale J., Friedman S. L. Scraping fibrosis: expressway to the core of fibrosis. Nature Medicine . 2011;17(5):552–553. doi: 10.1038/nm0511-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn T. A. Cellular and molecular mechanisms of fibrosis. The Journal of Pathology . 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenbloom J., Mendoza F. A., Jimenez S. A. Strategies for anti-fibrotic therapies. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease . 2013;1832(7):1088–1103. doi: 10.1016/j.bbadis.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Chen D. Q., Feng Y. L., Cao G., Zhao Y. Y. Natural products as a source for Antifibrosis therapy. Trends in Pharmacological Sciences . 2018;39(11):937–952. doi: 10.1016/j.tips.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Du G., Jin L., Han X., Song Z., Zhang H., Liang W. Naringenin: a potential immunomodulator for inhibiting lung fibrosis and metastasis. Cancer Research . 2009;69(7):3205–3212. doi: 10.1158/0008-5472.CAN-08-3393. [DOI] [PubMed] [Google Scholar]
- 6.Lee M. H., Yoon S., Moon J. O. The flavonoid naringenin inhibits dimethylnitrosamine-induced liver damage in rats. Biological & Pharmaceutical Bulletin . 2004;27(1):72–76. doi: 10.1248/bpb.27.72. [DOI] [PubMed] [Google Scholar]
- 7.Wei W., Rao F., Liu F., et al. Involvement of Smad3 pathway in atrial fibrosis induced by elevated hydrostatic pressure. Journal of Cellular Physiology . 2018;233(6):4981–4989. doi: 10.1002/jcp.26337. [DOI] [PubMed] [Google Scholar]
- 8.Zeng W., Jin L., Zhang F., Zhang C., Liang W. Naringenin as a potential immunomodulator in therapeutics. Pharmacological Research . 2018;135:122–126. doi: 10.1016/j.phrs.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Distler J. H. W., Györfi A. H., Ramanujam M., Whitfield M. L., Königshoff M., Lafyatis R. Shared and distinct mechanisms of fibrosis. Nature Reviews Rheumatology . 2019;15(12):705–730. doi: 10.1038/s41584-019-0322-7. [DOI] [PubMed] [Google Scholar]
- 10.Rockey D. C., Bell P. D., Hill J. A. Fibrosis--a common pathway to organ injury and failure. The New England Journal of Medicine . 2015;372(12):1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 11.Weiskirchen R., Weiskirchen S., Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Molecular Aspects of Medicine . 2019;65:2–15. doi: 10.1016/j.mam.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Henderson N. C., Rieder F., Wynn T. A. Fibrosis: from mechanisms to medicines. Nature . 2020;587(7835):555–566. doi: 10.1038/s41586-020-2938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wynn T. A., Ramalingam T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nature Medicine . 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh A. K., Quaggin S. E., Vaughan D. E. Molecular basis of organ fibrosis: potential therapeutic approaches. Experimental Biology and Medicine . 2013;238(5):461–481. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 15.Rosenbloom J., Macarak E., Piera-Velazquez S., Jimenez S. A. Human fibrotic diseases: current challenges in fibrosis research. Methods in Molecular Biology . 2017;1627:1–23. doi: 10.1007/978-1-4939-7113-8_1. [DOI] [PubMed] [Google Scholar]
- 16.McAnulty R. J. Fibroblasts and myofibroblasts: their source, function and role in disease. The International Journal of Biochemistry & Cell Biology . 2007;39(4):666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Hinz B., Phan S. H., Thannickal V. J., Galli A., Bochaton-Piallat M. L., Gabbiani G. The myofibroblast: one function, multiple origins. The American Journal of Pathology . 2007;170(6):1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Postlethwaite A. E., Shigemitsu H., Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Current Opinion in Rheumatology . 2004;16(6):733–738. doi: 10.1097/01.bor.0000139310.77347.9c. [DOI] [PubMed] [Google Scholar]
- 19.Piera-Velazquez S., Mendoza F. A., Jimenez S. A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. Journal of Clinical Medicine . 2016;5(4):p. 45. doi: 10.3390/jcm5040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphreys B. D., Lin S. L., Kobayashi A., et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. The American Journal of Pathology . 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng X. M., Nikolic-Paterson D. J., Lan H. Y. TGF-β: the master regulator of fibrosis. Nature Reviews Nephrology . 2016;12(6):325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 22.Akhurst R. J., Hata A. Targeting the TGFβ signalling pathway in disease. Nature Reviews Drug Discovery . 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy A. M., Wong A. L., Bezuhly M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis & Tissue Repair . 2015;8(1) doi: 10.1186/s13069-015-0023-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruiz-Ortega M., Rupérez M., Esteban V., et al. Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrology Dialysis Transplantation . 2006;21(1):16–20. doi: 10.1093/ndt/gfi265. [DOI] [PubMed] [Google Scholar]
- 25.Shi-Wen X., Leask A., Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine & Growth Factor Reviews . 2008;19(2):133–144. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Ponticos M., Holmes A. M., Shi-Wen X., et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis and Rheumatism . 2009;60(7):2142–2155. doi: 10.1002/art.24620. [DOI] [PubMed] [Google Scholar]
- 27.Bonner J. C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine & Growth Factor Reviews . 2004;15(4):255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Kazlauskas A. PDGFs and their receptors. Gene . 2017;614:1–7. doi: 10.1016/j.gene.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cook S. A., Schafer S. Hiding in plain sight: Interleukin-11 emerges as a master regulator of fibrosis, tissue integrity, and stromal inflammation. Annual Review of Medicine . 2020;71(1):263–276. doi: 10.1146/annurev-med-041818-011649. [DOI] [PubMed] [Google Scholar]
- 30.Bahcecioglu I. H., Koca S. S., Poyrazoglu O. K., et al. Hepatoprotective effect of infliximab, an Anti-TNF-α agent, on carbon tetrachloride-induced hepatic fibrosis. Inflammation . 2008;31(4):215–221. doi: 10.1007/s10753-008-9067-1. [DOI] [PubMed] [Google Scholar]
- 31.Hartopo A. B., Arfian N., Nakayama K., Suzuki Y., Yagi K., Emoto N. Endothelial-derived endothelin-1 promotes pulmonary vascular remodeling in bleomycin-induced pulmonary fibrosis. Physiological Research . 2018;67(Supplement 1):S185–S197. doi: 10.33549/physiolres.933812. [DOI] [PubMed] [Google Scholar]
- 32.Abraham D., Ponticos M., Nagase H. Connective tissue remodeling: cross-talk between endothelins and matrix metalloproteinases. Current Vascular Pharmacology . 2005;3(4):369–379. doi: 10.2174/157016105774329480. [DOI] [PubMed] [Google Scholar]
- 33.Siani A., Tirelli N. Myofibroblast differentiation: main features, biomedical relevance, and the role of reactive oxygen species. Antioxidants & Redox Signaling . 2014;21(5):768–785. doi: 10.1089/ars.2013.5724. [DOI] [PubMed] [Google Scholar]
- 34.Kliment C. R., Oury T. D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radical Biology & Medicine . 2010;49(5):707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 35.Lokmic Z., Musyoka J., Hewitson T. D., Darby I. A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. International Review of Cell and Molecular Biology . 2012;296:139–185. doi: 10.1016/B978-0-12-394307-1.00003-5. [DOI] [PubMed] [Google Scholar]
- 36.Haase V. H. Pathophysiological consequences of HIF Activation. Annals of the New York Academy of Sciences . 2009;1177(1):57–65. doi: 10.1111/j.1749-6632.2009.05030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu H. H., Chen D. Q., Wang Y. N., et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chemico-Biological Interactions . 2018;292:76–83. doi: 10.1016/j.cbi.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y. E. Non-Smad pathways in TGF-beta signaling. Cell Research . 2009;19(1):128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finnson K. W., Almadani Y., Philip A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: mechanisms and targets. Seminars in Cell & Developmental Biology . 2020;101:115–122. doi: 10.1016/j.semcdb.2019.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Bi J., Ge S. Potential roles of BMP9 in liver fibrosis. International Journal of Molecular Sciences . 2014;15(11):20656–20667. doi: 10.3390/ijms151120656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei J., Fang F., Lam A. P., et al. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis and Rheumatism . 2012;64(8):2734–2745. doi: 10.1002/art.34424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horn A., Palumbo K., Cordazzo C., et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis and Rheumatism . 2012;64(8):2724–2733. doi: 10.1002/art.34444. [DOI] [PubMed] [Google Scholar]
- 43.Kavian N., Servettaz A., Weill B., Batteux F. New insights into the mechanism of notch signalling in fibrosis. The Open Rheumatology Journal . 2012;6:96–102. doi: 10.2174/1874312901206010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhuang S., Liu N. EGFR signaling in renal fibrosis. Kidney International Supplements . 2014;4(1):70–74. doi: 10.1038/kisup.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joshi R., Kulkarni Y. A., Wairkar S. Pharmacokinetic, pharmacodynamic and formulations aspects of Naringenin: an update. Life Sciences . 2018;215:43–56. doi: 10.1016/j.lfs.2018.10.066. [DOI] [PubMed] [Google Scholar]
- 46.Gattuso G., Barreca D., Gargiulli C., Leuzzi U., Caristi C. Flavonoid composition of citrus juices. Molecules . 2007;12(8):1641–1673. doi: 10.3390/12081641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heidary Moghaddam R., Samimi Z., Moradi S. Z., Little P. J., Xu S., Farzaei M. H. Naringenin and naringin in cardiovascular disease prevention: a preclinical review. European Journal of Pharmacology . 2020;887 doi: 10.1016/j.ejphar.2020.173535. [DOI] [PubMed] [Google Scholar]
- 48.Ribeiro M. H. Naringinases: occurrence, characteristics, and applications. Applied Microbiology and Biotechnology . 2011;90(6):1883–1895. doi: 10.1007/s00253-011-3176-8. [DOI] [PubMed] [Google Scholar]
- 49.Orhan I. E., Nabavi S. F., Daglia M., Tenore G. C., Mansouri K., Nabavi S. M. Naringenin and atherosclerosis: a review of literature. Current Pharmaceutical Biotechnology . 2015;16(3):245–251. doi: 10.2174/1389201015666141202110216. [DOI] [PubMed] [Google Scholar]
- 50.Kanaze F. I., Bounartzi M. I., Georgarakis M., Niopas I. Pharmacokinetics of the citrus flavanone aglycones hesperetin and naringenin after single oral administration in human subjects. European Journal of Clinical Nutrition . 2007;61(4):472–477. doi: 10.1038/sj.ejcn.1602543. [DOI] [PubMed] [Google Scholar]
- 51.Tutunchi H., Naeini F., Ostadrahimi A., Hosseinzadeh-Attar M. J. Naringenin, a flavanone with antiviral and anti-inflammatory effects: a promising treatment strategy againstCOVID‐19. Phytotherapy Research . 2020;34(12):3137–3147. doi: 10.1002/ptr.6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orrego-Lagarón N., Martínez-Huélamo M., Vallverdú-Queralt A., Lamuela-Raventos R. M., Escribano-Ferrer E. High gastrointestinal permeability and local metabolism of naringenin: influence of antibiotic treatment on absorption and metabolism. The British Journal of Nutrition . 2015;114(2):169–180. doi: 10.1017/S0007114515001671. [DOI] [PubMed] [Google Scholar]
- 53.Felgines C., Texier O., Morand C., et al. Bioavailability of the flavanone naringenin and its glycosides in rats. American Journal of Physiology-Gastrointestinal And Liver Physiology . 2000;279(6):G1148–G1154. doi: 10.1152/ajpgi.2000.279.6.G1148. [DOI] [PubMed] [Google Scholar]
- 54.Hsiu S. L., Huang T. Y., Hou Y. C., Chin D. H., Chao P. D. Comparison of metabolic pharmacokinetics of naringin and naringenin in rabbits. Life Sciences . 2002;70(13):1481–1489. doi: 10.1016/S0024-3205(01)01491-6. [DOI] [PubMed] [Google Scholar]
- 55.Jeon S. M., Kim H. K., Kim H. J., et al. Hypocholesterolemic and antioxidative effects of naringenin and its two metabolites in high-cholesterol fed rats. Translational Research . 2007;149(1):15–21. doi: 10.1016/j.trsl.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Zaidun N. H., Thent Z. C., Latiff A. A. Combating oxidative stress disorders with citrus flavonoid: Naringenin. Life Sciences . 2018;208:111–122. doi: 10.1016/j.lfs.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 57.Justesen U., Knuthsen P., Leth T. Quantitative analysis of flavonols, flavones, and flavanones in fruits, vegetables and beverages by high-performance liquid chromatography with photo- diode array and mass spectrometric detection. Journal of Chromatography. A . 1998;799(1-2):101–110. doi: 10.1016/S0021-9673(97)01061-3. [DOI] [PubMed] [Google Scholar]
- 58.Ortiz-Andrade R. R., Sánchez-Salgado J. C., Navarrete-Vázquez G., et al. Antidiabetic and toxicological evaluations of naringenin in normoglycaemic and NIDDM rat models and its implications on extra-pancreatic glucose regulation. Diabetes, Obesity and Metabolism . 2008;10(11):1097–1104. doi: 10.1111/j.1463-1326.2008.00869.x. [DOI] [PubMed] [Google Scholar]
- 59.Rivoira M. A., Rodriguez V., Talamoni G., Tolosa de Talamoni N. New perspectives in the pharmacological potential of Naringin in medicine. Current Medicinal Chemistry . 2021;28(10):1987–2007. doi: 10.2174/0929867327666200604171351. [DOI] [PubMed] [Google Scholar]
- 60.Cavia-Saiz M., Busto M. D., Pilar-Izquierdo M. C., Ortega N., Perez-Mateos M., Muñiz P. Antioxidant properties, radical scavenging activity and biomolecule protection capacity of flavonoid naringenin and its glycoside naringin: a comparative study. Journal of the Science of Food and Agriculture . 2010;90(7):1238–1244. doi: 10.1002/jsfa.3959. [DOI] [PubMed] [Google Scholar]
- 61.Jayaraman J., Veerappan M., Namasivayam N. Potential beneficial effect of naringenin on lipid peroxidation and antioxidant status in rats with ethanol-induced hepatotoxicity. The Journal of Pharmacy and Pharmacology . 2009;61(10):1383–1390. doi: 10.1211/jpp/61.10.0016. [DOI] [PubMed] [Google Scholar]
- 62.Jayaraman J., Jesudoss V. A., Menon V. P., Namasivayam N. Anti-inflammatory role of naringenin in rats with ethanol induced liver injury. Toxicology Mechanisms and Methods . 2012;22(7):568–576. doi: 10.3109/15376516.2012.707255. [DOI] [PubMed] [Google Scholar]
- 63.Hämäläinen M., Nieminen R., Vuorela P., Heinonen M., Moilanen E. Anti-Inflammatory Effects of Flavonoids: Genistein, Kaempferol, Quercetin, and Daidzein Inhibit STAT-1 and NF-κB Activations, Whereas Flavone, Isorhamnetin, Naringenin, and Pelargonidin Inhibit only NF-κB Activation along with Their Inhibitory Effect on iNOS Expression and NO Production in Activated Macrophages. Mediators of Inflammation . 2007;2007:10. doi: 10.1155/2007/45673.45673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cataneo A. H. D., Kuczera D., Koishi A. C., et al. The citrus flavonoid naringenin impairs the in vitro infection of human cells by Zika virus. Scientific Reports . 2019;9(1) doi: 10.1038/s41598-019-52626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nahmias Y., Goldwasser J., Casali M., et al. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology . 2008;47(5):1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moon S. H., Lee J. H., Kim K. T., et al. Antimicrobial effect of 7-O-butylnaringenin, a novel flavonoid, and various natural flavonoids against helicobacter pylori strains. International Journal of Environmental Research and Public Health . 2013;10(11):5459–5469. doi: 10.3390/ijerph10115459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang S., Li D. D., Zeng F., et al. Efficient biosynthesis, analysis, solubility and anti-bacterial activities of succinylglycosylated naringenin. Natural Product Research . 2019;33(12):1756–1760. doi: 10.1080/14786419.2018.1431633. [DOI] [PubMed] [Google Scholar]
- 68.Bao L., Liu F., Guo H. B., et al. Naringenin inhibits proliferation, migration, and invasion as well as induces apoptosis of gastric cancer SGC7901 cell line by downregulation of AKT pathway. Tumour Biology . 2016;37(8):11365–11374. doi: 10.1007/s13277-016-5013-2. [DOI] [PubMed] [Google Scholar]
- 69.Leonardi T., Vanamala J., Taddeo S. S., et al. Apigenin and naringenin suppress colon carcinogenesis through the aberrant crypt stage in azoxymethane-treated rats. Experimental Biology and Medicine (Maywood, N.J.) . 2010;235(6):710–717. doi: 10.1258/ebm.2010.009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang N., Yang Z., Yuan Y., et al. Naringenin attenuates pressure overload-induced cardiac hypertrophy. Experimental and Therapeutic Medicine . 2015;10(6):2206–2212. doi: 10.3892/etm.2015.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Esmaeili M. A., Alilou M. Naringenin attenuates CCl4-induced hepatic inflammation by the activation of an Nrf2-mediated pathway in rats. Clinical and Experimental Pharmacology & Physiology . 2014;41(6):416–422. doi: 10.1111/1440-1681.12230. [DOI] [PubMed] [Google Scholar]
- 72.Shi Y., Dai J., Liu H., et al. Naringenin inhibits allergen-induced airway inflammation and airway responsiveness and inhibits NF-κB activity in a murine model of asthma. Canadian Journal of Physiology and Pharmacology . 2009;87(9):729–735. doi: 10.1139/Y09-065. [DOI] [PubMed] [Google Scholar]
- 73.Annadurai T., Muralidharan A. R., Joseph T., Hsu M. J., Thomas P. A., Geraldine P. Antihyperglycemic and antioxidant effects of a flavanone, naringenin, in streptozotocin-nicotinamide-induced experimental diabetic rats. Journal of Physiology and Biochemistry . 2012;68(3):307–318. doi: 10.1007/s13105-011-0142-y. [DOI] [PubMed] [Google Scholar]
- 74.Lou H., Jing X., Wei X., Shi H., Ren D., Zhang X. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology . 2014;79:380–388. doi: 10.1016/j.neuropharm.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 75.Ghofrani S., Joghataei M. T., Mohseni S., et al. Naringenin improves learning and memory in an Alzheimer's disease rat model: insights into the underlying mechanisms. European Journal of Pharmacology . 2015;764:195–201. doi: 10.1016/j.ejphar.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 76.Kapoor S. Tumor growth attenuating effects of naringenin. Pathology & Oncology Research . 2014;20(2):p. 483. doi: 10.1007/s12253-013-9702-5. [DOI] [PubMed] [Google Scholar]
- 77.Kanno S., Tomizawa A., Hiura T., et al. Inhibitory effects of naringenin on tumor growth in human cancer cell lines and sarcoma S-180-implanted mice. Biological & Pharmaceutical Bulletin . 2005;28(3):527–530. doi: 10.1248/bpb.28.527. [DOI] [PubMed] [Google Scholar]
- 78.Gressner A. M., Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-? as major players and therapeutic targets. Journal of Cellular and Molecular Medicine . 2006;10(1):76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bataller R., Brenner D. A. Liver fibrosis. The Journal of Clinical Investigation . 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun M., Kisseleva T. Reversibility of liver fibrosis. Clinics And Research in Hepatology And Gastroenterology . 2015;39:S60–S63. doi: 10.1016/j.clinre.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lim Y. S., Kim W. R. The global impact of hepatic fibrosis and end-stage liver disease. Clinics in Liver Disease . 2008;12(4):733–746. doi: 10.1016/j.cld.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 82.Bukong T. N., Maurice S. B., Chahal B., Schaeffer D. F., Winwood P. J. Versican: a novel modulator of hepatic fibrosis. Laboratory Investigation . 2016;96(3):361–374. doi: 10.1038/labinvest.2015.152. [DOI] [PubMed] [Google Scholar]
- 83.Kumar S., Wang J., Shanmukhappa S. K., Gandhi C. R. Toll-like receptor 4-independent carbon tetrachloride-induced fibrosis and lipopolysaccharide-induced acute liver injury in mice: role of hepatic stellate cells. The American Journal of Pathology . 2017;187(6):1356–1367. doi: 10.1016/j.ajpath.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elpek G. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World Journal of Gastroenterology . 2014;20(23):7260–7276. doi: 10.3748/wjg.v20.i23.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friedman S. L. Evolving challenges in hepatic fibrosis. Nature Reviews Gastroenterology & Hepatology . 2010;7(8):425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 86.Reynaert H., Thompson M. G., Thomas T., Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut . 2002;50(4):571–581. doi: 10.1136/gut.50.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu X., Wang W., Hu H., et al. Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-β1 in cultured rat hepatic stellate cells. Pharmaceutical Research . 2006;23(1):82–89. doi: 10.1007/s11095-005-9043-5. [DOI] [PubMed] [Google Scholar]
- 88.Chtourou Y., Fetoui H., Jemai R., Ben Slima A., Makni M., Gdoura R. Naringenin reduces cholesterol-induced hepatic inflammation in rats by modulating matrix metalloproteinases-2, 9 via inhibition of nuclear factor κB pathway. European Journal of Pharmacology . 2015;746:96–105. doi: 10.1016/j.ejphar.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 89.Hernández-Aquino E., Zarco N., Casas-Grajales S., et al. Naringenin prevents experimental liver fibrosis by blocking TGFβ-Smad3 and JNK-Smad3 pathways. World Journal of Gastroenterology . 2017;23(24):4354–4368. doi: 10.3748/wjg.v23.i24.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hernández-Aquino E., Quezada-Ramírez M. A., Silva-Olivares A., et al. Naringenin attenuates the progression of liver fibrosis via inactivation of hepatic stellate cells and profibrogenic pathways. European Journal of Pharmacology . 2019;865:p. 172730. doi: 10.1016/j.ejphar.2019.172730. [DOI] [PubMed] [Google Scholar]
- 91.Zhao L., Zhang N., Yang D., et al. Protective effects of five structurally diverse flavonoid subgroups against chronic alcohol-induced hepatic damage in a mouse model. Nutrients . 2018;10(11):p. 1754. doi: 10.3390/nu10111754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang J., Ding Y., Zhou W. Albumin self-modified liposomes for hepatic fibrosis therapy via SPARC- dependent pathways. International Journal of Pharmaceutics . 2020;574 doi: 10.1016/j.ijpharm.2019.118940. [DOI] [PubMed] [Google Scholar]
- 93.Hua Y. Q., Zeng Y., Xu J., Xu X. L. Naringenin alleviates nonalcoholic steatohepatitis in middle-aged Apoe−/−mice: role of SIRT1. Phytomedicine . 2021;81 doi: 10.1016/j.phymed.2020.153412. [DOI] [PubMed] [Google Scholar]
- 94.Yang F., Hu S., Sheng X., Liu Y. Naringenin loaded multifunctional nanoparticles to enhance the chemotherapeutic efficacy in hepatic fibrosis. Biomedical Microdevices . 2020;22(4) doi: 10.1007/s10544-020-00524-1. [DOI] [PubMed] [Google Scholar]
- 95.Talman V., Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell and Tissue Research . 2016;365(3):563–581. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burstein B., Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. Journal of the American College of Cardiology . 2008;51(8):802–809. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 97.Banerjee I., Yekkala K., Borg T. K., Baudino T. A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Annals of the New York Academy of Sciences . 2006;1080(1):76–84. doi: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- 98.Liu M., Xu X., Zhao J., Tang Y. Naringenin inhibits transforming growth factor-beta1-induced cardiac fibroblast proliferation and collagen synthesis via G0/G1 arrest. Experimental and Therapeutic Medicine . 2017;14(5):4425–4430. doi: 10.3892/etm.2017.5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liang J. N., Zou X., Fang X. H., et al. The Smad3-miR-29b/miR-29c axis mediates the protective effect of macrophage migration inhibitory factor against cardiac fibrosis. Biochimica et Biophysica Acta-Molecular Basis of Disease . 2019;1865(9):2441–2450. doi: 10.1016/j.bbadis.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 100.Wuyts W. A., Agostini C., Antoniou K. M., et al. The pathogenesis of pulmonary fibrosis: a moving target. The European Respiratory Journal . 2013;41(5):1207–1218. doi: 10.1183/09031936.00073012. [DOI] [PubMed] [Google Scholar]
- 101.Karampitsakos T., Woolard T., Bouros D., Tzouvelekis A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. European Journal of Pharmacology . 2017;808:35–43. doi: 10.1016/j.ejphar.2016.06.045. [DOI] [PubMed] [Google Scholar]
- 102.King T. E., Jr., Bradford W. Z., Castro-Bernardini S., et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. The New England Journal of Medicine . 2014;370(22):2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 103.Richeldi L., du Bois R. M., Raghu G., et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. The New England Journal of Medicine . 2014;370(22):2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 104.Shi Y., Tan Y., Mao S., Gu W. Naringenin inhibits allergen-induced airway remodeling in a murine model of asthma. Molecular Medicine Reports . 2014;9(4):1204–1208. doi: 10.3892/mmr.2014.1940. [DOI] [PubMed] [Google Scholar]
- 105.Seyedrezazadeh E., Kolahian S., Shahbazfar A. A., et al. Effects of the flavanone combination hesperetin-naringenin, and orange and grapefruit juices, on airway inflammation and remodeling in a murine asthma model. Phytotherapy Research . 2015;29(4):591–598. doi: 10.1002/ptr.5292. [DOI] [PubMed] [Google Scholar]
- 106.Lin Y., Tan D., Kan Q., Xiao Z., Jiang Z. The protective effect of Naringenin on airway remodeling after mycoplasma pneumoniae infection by inhibiting autophagy-mediated lung inflammation and fibrosis. Mediators of Inflammation . 2018;2018:10. doi: 10.1155/2018/8753894.8753894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang C., Zeng W., Yao Y., et al. Naringenin ameliorates radiation-induced lung injury by lowering IL-1βLevel. The Journal of Pharmacology and Experimental Therapeutics . 2018;366(2):341–348. doi: 10.1124/jpet.118.248807. [DOI] [PubMed] [Google Scholar]
- 108.Webster A. C., Nagler E. V., Morton R. L., Masson P. Chronic kidney disease. The Lancet . 2017;389(10075):1238–1252. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 109.Jager K. J., Fraser S. D. S. The ascending rank of chronic kidney disease in the global burden of disease study. Nephrology Dialysis Transplantation . 2017;32(supplement 2):ii121–ii128. doi: 10.1093/ndt/gfw330. [DOI] [PubMed] [Google Scholar]
- 110.Liu M., Ning X., Li R., et al. Signalling pathways involved in hypoxia-induced renal fibrosis. Journal of Cellular and Molecular Medicine . 2017;21(7):1248–1259. doi: 10.1111/jcmm.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Djudjaj S., Boor P. Cellular and molecular mechanisms of kidney fibrosis. Molecular Aspects of Medicine . 2019;65:16–36. doi: 10.1016/j.mam.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 112.Klinkhammer B. M., Goldschmeding R., Floege J., Boor P. Treatment of Renal Fibrosis--Turning Challenges into Opportunities. Advances in Chronic Kidney Disease . 2017;24(2):117–129. doi: 10.1053/j.ackd.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 113.Karuppagounder V., Arumugam S., Thandavarayan R. A., et al. Naringenin ameliorates daunorubicin induced nephrotoxicity by mitigating AT1R, ERK1/2-NFκB p65 mediated inflammation. International Immunopharmacology . 2015;28(1):154–159. doi: 10.1016/j.intimp.2015.05.050. [DOI] [PubMed] [Google Scholar]
- 114.Meng X. M., Zhang Y., Huang X. R., Ren G. L., Li J., Lan H. Y. Treatment of renal fibrosis by rebalancing TGF-β/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget . 2015;6(35):36984–36997. doi: 10.18632/oncotarget.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yan N., Wen L., Peng R., et al. Naringenin ameliorated kidney injury through let-7a/TGFBR1 signaling in diabetic nephropathy. Journal of Diabetes Research . 2016;2016:13. doi: 10.1155/2016/8738760.8738760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Z., Wang S., Zhao J., et al. Naringenin ameliorates Renovascular hypertensive renal damage by normalizing the balance of renin-angiotensin system components in rats. International Journal of Medical Sciences . 2019;16(5):644–653. doi: 10.7150/ijms.31075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Abrego-Peredo A., Romero-Ramírez H., Espinosa E., et al. Naringenin mitigates autoimmune features in lupus-prone mice by modulation of T-cell subsets and cytokines profile. PLoS One . 2020;15(5) doi: 10.1371/journal.pone.0233138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nguyen J. K., Austin E., Huang A., Mamalis A., Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Archives of Dermatological Research . 2020;312(2):81–92. doi: 10.1007/s00403-019-01972-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bayat A., McGrouther D. A., Ferguson M. W. Skin scarring. BMJ . 2003;326(7380):88–92. doi: 10.1136/bmj.326.7380.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bush J. A., McGrouther D. A., Young V. L., et al. Recommendations on clinical proof of efficacy for potential scar prevention and reduction therapies. Wound Repair and Regeneration . 2011;19(Suppl 1):S32–S37. doi: 10.1111/j.1524-475X.2010.00607.x. [DOI] [PubMed] [Google Scholar]
- 121.Bock O., Schmid-Ott G., Malewski P., Mrowietz U. Quality of life of patients with keloid and hypertrophic scarring. Archives of Dermatological Research . 2006;297(10):433–438. doi: 10.1007/s00403-006-0651-7. [DOI] [PubMed] [Google Scholar]
- 122.Brown B. C., McKenna S. P., Siddhi K., McGrouther D. A., Bayat A. The hidden cost of skin scars: quality of life after skin scarring. Journal of Plastic, Reconstructive & Aesthetic Surgery . 2008;61(9):1049–1058. doi: 10.1016/j.bjps.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 123.Tziotzios C., Profyris C., Sterling J. Cutaneous scarring: Pathophysiology, molecular mechanisms, and scar reduction therapeutics: Part II. Strategies to reduce scar formation after dermatologic procedures. Journal of the American Academy of Dermatology . 2012;66(1):13–24. doi: 10.1016/j.jaad.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 124.Shan S., Zhang Y., Wu M., Yi B., Wang J., Li Q. Naringenin attenuates fibroblast activation and inflammatory response in a mechanical stretch-induced hypertrophic scar mouse model. Molecular Medicine Reports . 2017;16(4):4643–4649. doi: 10.3892/mmr.2017.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]