

Summary
Myeloid suppressor cells promote tumor growth by a variety of mechanisms which are not fully characterized. We identified myeloid cells (MCs) expressing the latency-associated peptide (LAP) of TGF-β on their surface and LAPHi MCs that stimulate Foxp3+ Tregs while inhibiting effector T cell proliferation and function. Blocking TGF-β inhibits the tolerogenic ability of LAPHi MCs. Furthermore, adoptive transfer of LAPHi MCs promotes Treg accumulation and tumor growth in vivo. Conversely, anti-LAP antibody, which reduces LAPHi MCs, slows cancer progression. Single-cell RNA-Seq analysis on tumor-derived immune cells revealed LAPHi dominated cell subsets with distinct immunosuppressive signatures, including those with high levels of MHCII and PD-L1 genes. Analogous to mice, LAP is expressed on myeloid suppressor cells in humans, and these cells are increased in glioma patients. Thus, our results identify a previously unknown function by which LAPHi MCs promote tumor growth and offer therapeutic intervention to target these cells in cancer.
Subject areas: Immunology, Cancer
Graphical abstract
Highlights
-
•
Several myeloid cell subsets express surface LAP
-
•
Myeloid cells that express surface LAP possess immunosuppressive properties
-
•
LAP expressing myeloid cells induce Tregs and inhibit effector T cell function
-
•
LAP expressing myeloid cells promote tumor growth
Immunology; Cancer
Introduction
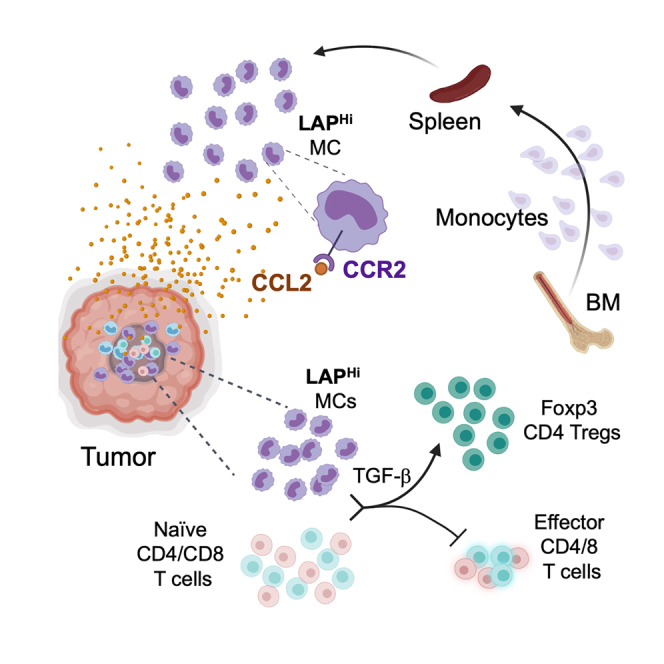
Myeloid cells (MCs) play a key role in priming and shaping the immune response and can either inhibit or promote tumor growth depending on their immunological status (Poh and Ernst, 2018; Sica and Massarotti, 2017). MCs represent a highly heterogeneous population of immune cells that includes myeloid suppressor cells (MSCs) or myeloid-derived suppressor cells (MDSCs) (Gabrilovich and Nagaraj, 2009; Marvel and Gabrilovich, 2015; Qian and Pollard, 2010). MSCs infiltrate tumor tissue and significantly contribute to tumor growth by attenuating anti-tumor immune responses (Gabrilovich et al., 2012; Tcyganov et al., 2018), but their diversity and mechanisms of function in the context of cancer remain largely unexplored. Two major populations of MDSCs, monocytic and granulocytic/polymorphonuclear MDCSs have been described in mice and humans (Marvel and Gabrilovich, 2015; Tcyganov et al., 2018). Murine monocytic MDSCs (mMDSCs) express high levels of Ly6C and CCR2 but are negative for Ly6G (CD11b+Ly6CHiLy6G−), whereas granulocytic MDCSs (gMDSCs) are CD11b+Ly6CLoLy6G+ (Peranzoni et al., 2010; Tcyganov et al., 2018). Of note, Ly6C and CCR2 are also present on inflammatory monocytes. The recruitment of myeloid cells expressing high levels of Ly6C to injured tissue is dependent on the CCR2-CCL2 signaling (Qian et al., 2011; Serbina and Pamer, 2006). Cancer cells, secreting CCL2, are known to attract CCR2 expressing myeloid cells to the tumor site, thereby promoting their accumulation in the tumor microenvironment (Chang et al., 2016; Qian et al., 2011; Zhao et al., 2013). The phenotypic markers that define human MDSCs are different from those in mice. Human mMDSCs are defined as CD11b+CD33+CD14+HLA-DRlo/- and gMDSCs are CD11b+CD33+CD14−CD15+ (Gabrilovich and Nagaraj, 2009; Peranzoni et al., 2010).
One of the mechanisms by which MSCs suppress the immune response is by secreting TGF-β, a highly immunosuppressive cytokine implicated in tumor growth (Batlle and Massague, 2019). TGF-β can be anchored on the surface of immune cells via its latency-associated peptide (LAP) bound to one of its trans-membrane receptors (Shi et al., 2011). Importantly, TGF-β and LAP are products of the same gene, associated as dimers in one tetrameric protein complex. Various immune cells, such as T cells, B cells, myeloid cells and platelets that express LAP on their surface membrane possess immunosuppressive properties in mice and humans (Gabriely et al., 2017; Gandhi et al., 2007; Scurr et al., 2014; Zhang et al., 2016). In fact, our group has shown that LAP+CD4+ T cells promote cancer progression in different mouse models (Gabriely et al., 2017). Although LAP-expressing myeloid cells have been identified in mice (Gabriely et al., 2017; Ma et al., 2019; Qin et al., 2018) and humans (Gandhi et al., 2007; Hanada et al., 2018; Oosterlynck et al., 1994; Slobodin et al., 2012, 2013; Tadmor et al., 2018), their role in cancer is not well understood.
In this study, we investigated the role that LAP-expressing myeloid cells play in cancer. For this, we employed mouse models of MC38 and CT26 colorectal cancer (CRC), and GL261 glioblastoma (GBM) and in vitro suppression assays to measure the effect of LAP-expressing MCs on T cell proliferation and stimulation of Tregs. In addition, we assessed the ability of LAP-expressing MCs to promote tumor growth and induce immune suppression in vivo. We also performed single-cell RNA sequencing (scRNA-Seq) on immune cells derived from CT26 tumor to characterize subtypes of LAP-expressing MCs in the tumor tissue. Finally, we analyzed circulating MDSCs that express surface LAP in glioma patients. We found that LAPHi MCs, via mature TGF-β, can orchestrate immunosuppression and cancer progression in the tumor micro-environment and macro-environment.
Results
LAPHi myeloid cells have a tolerogenic phenotype
To investigate the role of LAP-expressing myeloid cells in cancer, we first analyzed their phenotype in the spleen and tumor tissue of CT26 CRC and GL261 GBM mice. We found that LAP expression on CD11b+CD3− live cells in the tumor tissue ranges from high to negative without creating a distinctly separate population of LAP positive cells (Figure 1A). Therefore, we defined the cells gated above the fluorescence-minus one (FMO) control as LAPHi and the remaining myeloid cell population as LAPLo MCs. We found that LAPHi MCs express high levels of Ly6C but do not express Ly6G in the spleen (Figures 1A, 1B, and S1A), thereby sharing their markers with monocytic myeloid-derived suppressor cells (mMDSCs). We also found a similar population of mMDSCs that express LAP on their surface in the subcutaneous CT26 CRC and intracranial GL261 GBM tumors (Figure 1A). We then analyzed LAP expression on microglia, an important myeloid cell population known to express TGF-β and be associated with GBM (Butovsky et al., 2014; Geribaldi-Doldan et al., 2020; Wei et al., 2020). However, we did not find LAP expression on microglia (Figures 1A and S1B) and thus focused our investigation on the peripheral myeloid cells. Flow cytometry analysis revealed that LAPHi MCs express elevated levels of CD115/CSF-1R (M-CSF-R), which is associated with immunosuppression (Huang et al., 2006; Pyonteck et al., 2013), as compared to LAPLo MCs in the spleen of naive mice (Figures 1B and S1C) indicating that LAPHi MCs are more tolerogenic than LAPLo MCs. We also measured mRNA expression of various immunological markers in LAPHi and LAPLo MCs isolated from spleen, bone marrow (BM) and tumor tissue of CT26 tumor-bearing mice. We found that both Lrrc33/Nrros, a receptor that mediates LAP surface expression on myeloid cells (Qin et al., 2018), and Ccr2, a marker of Ly6CHi myeloid cells (Serbina and Pamer, 2006) were highly expressed in LAPHi MCs isolated from all these sites (Figure 1C). Interestingly, we found elevated levels of several tolerogenic markers on LAPHi MCs compared to LAPLo MCs. Although the LAPHi MCs isolated from the spleen and BM expressed higher levels of Msr1, within the tumor tissue, we found upregulation of several additional tolerogenic markers including Msr1, Mcr1, Stat3, and Il10 on LAPHi MCs (Figure 1C). On the other hand, markers that support the anti-tumor immunity were downregulated in LAPHi MCs as compared to LAPLo MCs. Specifically, we found that LAPHi MCs exhibited reduced levels of Ccl5 in the periphery and Ccl5, Ifng, and Il12 in the tumor (Figure 1C). We and others have shown that TGF-β can induce its own expression in an autocrine manner in myeloid cells (Garo et al., 2019; Kashiwagi et al., 2015). To assess whether TGF-β can regulate MCs in an autocrine manner, we measured the levels of Tgfb1 and its receptors, Tgfbr1 and Tgfbr2, in LAPHi MCs isolated from MC38 tumor. Although Tgfb1 expression was increased in LAPHi MCs, the levels of TGF-β receptors were comparable between LAPHi MCs and LAPLo MCs (Figure S1D). This increase in TGF-β expression in LAPHi MCs was associated with reduced inflammatory cytokines such as IFN-γ and IL-12 (Figure 1C), which are known to be suppressed by TGF-β suggesting that enhanced autocrine TGF-β signaling could be linked to the immunoregulatory phenotype of LAPHi MCs. Taken together, LAP expression is associated with the tolerogenic phenotype of LAPHi MCs, and these cells share markers with mMDSCs.
Figure 1.

LAP expressing myeloid cells have a tolerogenic phenotype
(A) LAP expression on MDSCs. Spleens of CT26 tumor-bearing mice and tumor tissues were dissociated, and cells were analyzed by flow cytometry. Representative FACS plots and calculations of frequencies of LAP expressing cells are presented (n = 5). MG, microglia.
(B) Expression of indicated immune markers on LAPHi and LAPLo MCs in the spleen of naive wild-type (WT) mice by flow cytometry (n = 5).
(C) Analysis of mRNA expression in LAPHi vs LAPLo MCs in the spleen of CT26 tumor-bearing mice and tumors by qPCR (n = 4).
(D) Modulation of LAPHi vs LAPLo MCs in the spleen and BM of tumor-bearing mice. Frequency (left panels) and mean fluorescent intensity (MFI, right panels) of LAPHi vs LAPLo MCs were measured in the spleen and BM of naive (n = 5) and MC38 tumor-bearing (n = 7) mice.
(E) Migration of LAPHi vs LAPLo MCs toward cancer cells. LAPHi vs LAPLo MCs were isolated from the spleen of tumor-bearing mice and migration toward CCL2 and tumor-conditioned media from MC38 and GL261 cancer cells measured in a migration assay described in the Materials and Methods section (n = 4). Data shown as mean ± SEM. Two-tailed t test (A, left and middle panels; (B–E) and one-way ANOVA (A, right panel) were used for p value calculations. ∗∗∗∗, p < 0.0001. See also Figure S1.
The tumor environment induces LAPHi myeloid cells
We then examined whether LAPHi MCs are induced during tumor development to support its growth. For this, we compared LAPHi MCs in the spleen and BM of naive and tumor-bearing mice. We found that LAP expression on myeloid cells (measured by mean fluorescent intensity, MFI) isolated from either the spleen or BM was higher in the tumor-bearing mice than in naive mice (Figures 1D and S1E). However, when we measured cell frequencies, we found decreased frequencies of LAPHi MCs in the BM but increased frequencies in the spleen from tumor-bearing mice compared to naive mice (Figures 1D and S1E). These observations suggest that LAP expression is induced on myeloid cells during tumor development and that LAPHi MCs may migrate from the BM to the tumor tissue and spleen. Because the chemokine receptor CCR2 is known to drive myeloid cells from the BM toward its ligand CCL2 produced by injured tissue (Swirski et al., 2009; Zhao et al., 2013), we investigated whether CCR2 is expressed on LAPHi MCs. We found that LAPHi MCs express higher levels of surface CCR2 than LAPLo MCs (Figure 1C). Consistent with this, LAPHi MCs migrated better toward CCL2 than LAPLo cells in an in vitro migration assay (Figure 1E). In addition, LAPHi MCs were strongly attracted by tumor-conditioned media from MC38 and GL261 cancer cells, which are known to secrete CCL2 (Figure 1E) (Zhao et al., 2013; Zhu et al., 2011). These findings suggest that tumor cells recruit LAPHi MCs via the CCL2-CCR2 axis to promote their growth.
LAPHi MCs suppress immune responses in vitro
To determine whether LAPHi MCs can suppress the anti-tumor T cell responses, we performed proliferation assays by culturing naive CD4+ T cells with either LAPHi or LAPLo MCs isolated from the tumor-bearing mice and measured T cell proliferation (Figures S1F and S2A). We found that LAPHi MCs isolated from either tumor or spleen of tumor-bearing mice (Figures 2A and 2B) and from spleen and BM of naive mice (Figure S2B) are more effective in preventing T cell proliferation compared to LAPLo MCs. Because previous studies have suggested that myeloid suppressor cells can induce regulatory T cells (Tregs) to promote tumor growth (Huang et al., 2006), we tested whether LAPHi MCs induce Tregs. We found that LAPHi MCs induced higher frequencies of Foxp3+ CD4+ Tregs than LAPLo MCs (Figures 2C, 2D, and S2C). As LAPHi MCs express TGF-β on their cell membrane, we investigated whether TGF-β is responsible for suppressing T cell proliferation and inducing Tregs. We found that treatment with either anti-TGF-β or anti-LAP antibodies abrogated the ability of LAPHi MCs to suppress T cell proliferation or induce Tregs (Figures 2E, 2F, and S2D), indicating that TGF-β plays a major role in the suppressive function of LAPHi MCs. In addition, we analyzed the ability of LAPHi MCs to modulate CD8+ T cell proliferation. Similar to our findings with CD4+ T cells, LAPHi MCs suppressed the proliferation of CD8+ T cells in a TGF-β-dependent manner (Figure S2E). Furthermore, we found that IFN-γ expression by CD4 T cells was diminished after exposure to LAPHi MCs (Figures 2G and 2H). In summary, our ex vivo studies revealed that LAPHi MCs can suppress the immune response by inhibiting T cell proliferation and inducing Tregs, and their function is mediated by TGF-β.
Figure 2.

LAPHi MCs have tolerogenic functions in vitro
(A and B) T cell proliferation assay of naive CD4+ T cells from OTII-Foxp3/GFP mice in the presence of LAPHi vs LAPLo MCs from MC38 tumors (A) and spleens of tumor-bearing mice (B). Fixed number of T cells was used in the culture. Myeloid:T cell ratio reflects relative numbers of each cell type. T cells were stimulated with OVA323-339 peptide. Percent of proliferation (left panels) and representative histograms (right panels) of responder CD4+ T cells are shown (n = 4).
(C and D) Foxp3+ Treg induction by LAPHi and LAPLo MCs isolated from MC38 tumor (C) or spleen (D) was measured in the assay described in (A). Quantification (left panels) and representative dot plots (right panels) are shown (n = 4).
(E) CD4 T cell proliferation assay in the presence of LAPHi vs LAPLo MCs isolated from spleen; non-treated (NT) or treated with anti-TGF-β, anti-LAP, or IgG1 control antibodies. Percent of proliferation is shown (n = 3).
(F) Foxp3+ Treg induction was measured in the assay described in (E). Percent of Foxp3+ T cells is shown (n = 3).
(G and H) IFN-γ was measured on CD4 T cells after co-culture with LAPHi or LAPLo MCs. Representative dot plots (G) and quantification (H) are shown (n = 4). Data shown as mean ± SEM. Two-tailed t test (A-D, H), two-way ANOVA (E) and one-way ANOVA (F) were used for p value calculations. ∗∗∗∗, p < 0.0001. See also Figure S2.
LAPHi MCs promote tumor growth in vivo
We then asked whether LAPHi MCs can facilitate tumor growth in mouse models. First, we tested whether the administration of anti-LAP antibodies, known to slow down tumor growth (Gabriely et al., 2017), also reduces LAPHi MCs in vivo in the subcutaneous models of MC38 or CT26 CRC. We found anti-LAP treatment slowed tumor growth, and the reduction in tumor growth was associated with decreased frequencies of LAPHi MCs in the spleen (Figures 3A–3D, S2F, and S2G). We then adoptively transferred LAPHi or LAPLo MCs to mice implanted with MC38 tumors and found that animals that received LAPHi MCs had increased tumor growth compared to mice transferred with LAPLo MCs (Figure 3E), indicating that LAPHi MCs can promote tumor growth. Importantly, we found higher frequencies of Foxp3+ Tregs in the spleens of animals that received LAPHi MCs compared to mice that received LAPLo MCs (Figure 3F). This is consistent with our in vitro findings that LAPHi MCs exhibit enhanced ability to promote Treg development. Moreover, CD103+Foxp3+ and LAP+ Tregs, both known for their superior regulatory function (Anz et al., 2011; Gabriely et al., 2017) were also increased after the LAPHi MCs transfer (Figures 3G and 3H), supporting the immunosuppressive role of LAPHi MCs in vivo. However, Foxp3+ Tregs were not significantly different in tumors of mice transferred with LAPHi MCs compared to mice that received LAPLo MCs suggesting that the main mechanism by which adoptively transferred LAPHi MCs function is through the induction of Tregs in the periphery thereby promoting systemic immunosuppression and tumor growth (Figure S2H). In summary, LAPHi MCs can facilitate tumor growth by promoting the generation of Tregs.
Figure 3.

LAPHi myeloid cells dampen tumor-specific immunity in vivo
(A) CT26 CRC tumor volume over time in WT mice treated with either anti-LAP or isotype control (IC) antibodies (n = 11).
(B) Effect of anti-LAP antibody treatment on myeloid cells (CT26 model). Mice were treated with anti-LAP or IC antibodies and frequencies of cells in the spleen measured by flow cytometry.
(C) MC38 CRC tumor volume over time in WT mice treated with either anti-LAP or IC antibodies (n = 5).
(D) Effect of anti-LAP antibody treatment on myeloid cells (MC38 model). Mice were treated with anti-LAP or IC antibodies and frequencies of cells in the spleen measured by flow cytometry.
(E) MC38 tumor volumes over time in WT mice adoptively transferred with LAPHi and LAPLo MCs (n = 5).
(F) Foxp3+ Tregs measured in the spleen of mice transferred with LAPLo or LAPHi MCs. Representative FACS plots (right panel) and quantification (left panel) are shown (n = 4).
(G) CD103+Foxp3+ Tregs measured in the spleen of mice adoptively transferred with LAPHi or LAPLo MCs. Representative FACS plots (right panel) and quantification (left panel) are presented (n = 4).
(H) LAP+ CD4 Tregs measured in the spleen of mice adoptively transferred with LAPHi or LAPLo MCs. Representative FACS plots (right panel) and quantification (left panel) are shown (n = 4). Data shown as mean ± SEM. Two-way ANOVA (A, C, and E) and two-tailed t test (B, D, and F–H) were used for p value calculations. ∗∗∗∗, p < 0.0001. See also Figure S2.
Single-cell analysis of LAPHi cells reveals distinct myeloid cell subsets in the CT26 tumor model
To better characterize LAP-expressing myeloid cell subtypes, we examined LAPHi immune cells that infiltrate CT26 tumors by single-cell RNA sequencing (scRNA-Seq). We isolated LAPHi and LAPLo CD45+ live cells from CT26 tumor tissue and performed scRNA-Seq using a Seq-Well approach (Gierahn et al., 2017) (Figure 4A). Combined gene expression analysis revealed upregulation of several tolerogenic myeloid cell genes, including Arg1/2, Csf1r (CD115), Mmp14 (MT1-MMP), Mrc1 (CD206), Msr1 (CD204), and Fn1 in LAPHi cells compared to LAPLo cells (Figure 4B). On the other hand, proinflammatory genes including Ccl5, Ccr7, Ifng, and Gzmb were downregulated in LAPHi cells. MHCII genes that mediate antigen presentation (e.g., H2-Aa, H2-Ab1, H2-Eb1, and CD74) were increased in LAPHi cells. In addition, we found that the number of cells that express markers of myeloid cells, Lyz2 (LysM) and Itgam (CD11b), was significantly higher among LAPHi cells (Figure 4C), indicating that LAPHi cells in the CT26 tumor are predominantly represented by myeloid cells.
Figure 4.

Single-cell RNA-seq reveals increased numbers of LAP-expressing myeloid cells within tolerogenic subsets in tumor
(A) Schematic workflow of the scRNA-Seq procedure. Created with BioRender.com.
(B) Fold change and adjusted p value plot demonstrating differential gene expression between LAPHi vs LAPLo cells.
(C) Number of Itgam+ (left panel) and Lyz2+ (right panel) cells in LAPHi and LAPLo samples.
(D) Graph showing unsupervised clustering of scRNA-Seq data using uniform manifold approximation and projection (UMAP). Each point represents a single cell. Clusters are marked by numbers and names of identified cell clusters are indicated.
(E) UMAP plots of representative LAPHi and LAPLo samples are shown. The area of myeloid cell clusters is marked by a dashed circle.
(F) Gene expression of key myeloid cells markers (Msr1 and Arg1) overlaid on the UMAP plots.
(G) Stacked bar chart shows percent of LAPHi and LAPLo cells in each cluster. Dashed line indicates the level where cell number in clusters reaches 50%. Data shown as mean ± SEM. Mann-Whitney test was used for p value calculations (C and G).
To identify cell subsets, we performed unsupervised clustering and dimension reduction using a uniform manifold approximation and projection (UMAP) based on cells from all samples (Figure 4D). This procedure grouped cells into distinct clusters that included populations of myeloid cells, T cells, B cells, and NK cells. Using the key myeloid cell marker, Itgam, we identified clusters enriched with myeloid cells (e.g., Clusters 2, 5, 15, 18, and 20). Analysis of LAPHi and LAPLo samples on the UMAP projection revealed that myeloid cell clusters were mostly represented by the LAPHi cells (Figure 4E) that express tolerogenic markers including Msr1 and Arg1 (Figure 4F). We then calculated the number of LAPHi and LAPLo cells in each cluster and found that LAPHi cells significantly outnumbered LAPLo cells in Clusters 2, 5, 15, and 18 (Figure 4G). Analysis of various tolerogenic markers helped define the identity and phenotype of the clusters (Figures 5A and S3). All four of these clusters expressed relatively high levels of Csf1r, Msr1, and Il1b, the myeloid cell genes which have been associated with cancer (Figure 5A) (McLoed et al., 2016; Miyasato et al., 2017; Pyonteck et al., 2013), indicating that these cell subpopulations acquire tumor supportive properties. Other pro-tumorigenic markers were predominantly expressed in specific clusters as follows. Cluster 2: Arg1, Cd274 (PD-L1), Mrc1, and Mmp14; Cluster 5: moderate expression of Arg1; Cluster 15: Chil3 (Ym1); Cluster 18: Arg1, Cd274, Mrc1, and Mmp12 (Figures 5A and S3). Of note, LAPHi cells were also prevalent in Clusters 20 and 21 indicated by borderline statistical significance (Figure 4G). Similar to Clusters 2, 5, 15 and 18, Cluster 20 expressed high levels of Csf1r, Msr1, and Il1b (Figure 5A). In addition, Cluster 20 expressed a unique signature of C1qa, C1qb, Mrc1, Arg1, and Mmp14 genes (Figures 5A and S3). Interestingly, the complement component C1q genes (C1qa and C1qb) have been previously linked to an anti-inflammatory macrophage phenotype, cancer metastasis and angiogenesis (Benoit et al., 2012; Bulla et al., 2016; Roumenina et al., 2019; Son et al., 2016), indicating that MCs from Cluster 20 may be involved in these processes. Conversely, Cluster 21, as we detail below, is mostly represented by CD11b negative tolerogenic dendritic cells (DCs) expressing high levels of H2-Ab1, Ccr7, Cd274, Flt3, and Itgax (Figures 5A and S3). Thus, we identified LAPHi MCs dominated clusters with different tolerogenic signatures suggesting that they contribute to cancer-promoting immune suppression.
Figure 5.

LAP-enriched myeloid cell subsets derived from tumor possess distinct tolerogenic signatures
(A) Violin plots showing expression of various markers in all clusters.
(B) MHCII/H2-Ab1 expressing cells marked in red overlaid on the UMAP plots. Localization of Cluster 21 is indicated by arrow.
(C) Analysis of MHCII mRNA expression in LAPHi vs LAPLo MCs in the tumor tissue in the CT26 model by qPCR (n = 4).
(D and E) Expression of MHCII in LAPHi and LAPLo MCs in the CT26 tumor tissue by flow cytometry (n = 5). Representative dot plots (D) and quantification (E) are shown.
(F) Foxp3+ Treg induction was measured after co-culture of naive CD4+ T cells from OTII-Foxp3/GFP mice with MHCII+LAPHi or MHCII+LAPLo MCs from the spleen of MC38 tumor-bearing mice. T cells were stimulated with OVA323-339. Quantification is shown (n = 4).
(G) T cell proliferation assay of naive CD4+ T cells from OTII-Foxp3/GFP mice in the presence of MHCII+LAPHi vs MHCII+LAPLo MCs from the spleen of MC38 tumor-bearing mice. T cells were stimulated with OVA323-339. Percent of proliferation of responder CD4+ T cells is shown (n = 4).
(H) Heatmap of the differential expression data for the key tolerogenic genes restricted to expressing cells in Cluster 2.
(I) Volcano plot demonstrating differential gene expression in LAPHi and LAPLo cells in Cluster 2.
(J) Percent of cells expressing genes which is significantly higher in LAPHi and LAPLo cells in Cluster 2. Key genes are highlighted. Data shown as mean ± SEM. Two-tailed t test (C, E, F, and G) was used for p value calculations. ∗∗∗∗, p < 0.0001. See also Figures S3 and S5.
Interestingly, Clusters 2, 18, and 20 expressed Arg1 and Mrc1 (Figures 5A and S3), genes associated with alternatively activated/M2-like macrophages or tumor associated macrophages (TAMs) (Gabrilovich et al., 2012; Murray et al., 2014; Schmieder et al., 2012), indicating that LAPHi MCs differentiate into TAMs in the tumor microenvironment. In addition, Clusters 2, 18, and 20 expressed matrix metalloproteinases (MMPs), including Mmp12 and Mmp14 (Figure S3), suggesting that they may be involved in the remodeling of the ECM to facilitate cancer cell tissue invasion and metastasis (Chou et al., 2016; Markovic et al., 2009). These clusters are presumably regulated by a positive feedback mechanism as MMPs can activate latent TGF-β, which in turn upregulates MMPs expression (Krstic and Santibanez, 2014). Tgfb1 was expressed in most clusters at moderate levels, with the highest expression in Cluster 15 (Figure S3), suggesting that TGF-β may be involved in the regulation of these clusters. Cancer is known for its ability to exploit different myeloid cell functions to promote its growth but how these multiple mechanisms are simultaneously regulated is not known. Our findings indicate that different LAPHi MCs predominated clusters contribute to cancer malignancy by involving unique mechanisms including cancer cell invasion, metastasis, angiogenesis and tumor-specific immunosuppression. The outcome of the combined actions of these cell subpopulations most likely leads to cancer progression, which is further promoted by the supply of mature TGF-β as a key component that maintains immunosuppression. Accordingly, we conclude that active TGF-β supplied by LAPHi MCs orchestrates the pro-tumorigenic mechanisms of MCs, suggesting that targeting these cells will be critical to restrict cancer progression.
Tumor-derived LAPHi MCs have tolerogenic and antigen presentation properties
Our scRNA-Seq analysis showed that H2-Ab1, the gene encoding MHCII, was not only upregulated in the clusters of DCs (Cluster 16) and B cells (Cluster 9) but also in the majority of the tolerogenic myeloid cell clusters including Clusters 2, 15, 20, 21 with the highest expression levels in Cluster 21 (Figures 5A and 5B). Cluster 21 also expressed markers of migratory DCs (such as Ccr7, Flt3, and Itgax) (Figure S3). Moreover, cells in this cluster are enriched with Cd274 (PD-L1) (Figure S3), a tolerogenic factor that contributes to tumor growth. Thus, Cluster 21 likely represents recently identified mregDCs (mature DCs enriched in immunoregulatory molecules) expressing Flt3, Ccr7, and Cd274 but negative for CD11b (Maier et al., 2020). We validated the increased expression of MHCII in LAPHi cells isolated from the CT26 tumor tissue by qPCR and flow cytometry (Figures 5C–5E). We also identified tumor-derived LAP-expressing myeloid cell subsets positive for MHCII and PD-L1 by flow cytometry (Figure S4). We then examined whether LAPHi MCs can induce tumor-specific tolerance by presenting tumor antigens to CD4 T cells and favoring their differentiation to Tregs. Accordingly, we found that MHCII+ LAPHi cells loaded with OVA323-339 peptide, which is presented by MHCII, effectively induce Foxp3+ Tregs compared to LAPLo cells (Figure 5F) while suppressing the proliferation of non-Treg CD4 T cells (Figure 5G).
To further understand differences in gene expression between LAPHi and LAPLo cells, we performed differential gene expression analysis within each cluster. For an unbiased analysis, we filtered out non-expressing cells and performed the conventional differential expression analysis on the expressing LAPHi vs LAPLo cells in each cluster. Consistent with our prior observations (Figures 5A and S3), we found a higher expression of tolerogenic markers in LAPHi than LAPLo cells across clusters (Figures 5H, 5I, and S5A). In addition, because certain genes were not detected in many cells, we analyzed the proportion of cells with detectable transcripts for each gene in each cluster using the binomial Generalized Linear Model test. With this approach, we found that the percentage of cells that expressed key tolerogenic markers was higher among LAPHi cells than LAPLo cells across all myeloid cell clusters (Figures 5J and S5B). Thus, this complementary analysis strongly supports the tolerogenic phenotype of LAPHi cells in each of the myeloid cell clusters. Finally, we found that the MHCII genes (e.g., H2-Aa, H2-Ab1, H2-Eb1, and CD74) were expressed at higher levels in LAPHi cells as compared to LAPLo cells in specific clusters, such as Cluster 2, 15, and 21 (Figures 5H–5J and S5). Taken together, our findings suggest that, while employing distinctive strategies to promote tumor growth, different subtypes of LAPHi MCs share common tolerogenic mechanisms sustained by active TGF-β.
LAPHi MCs are associated with cancer in humans
To explore the relevance of our findings in human cancer, we analyzed circulating MDSCs isolated from PBMCs of glioma patients. Analogous to mice, we found that LAP is expressed on mMDSCs (CD11b+CD33+CD14+HLA-DRLo/-), but not on gMDSCs (CD11b+CD33+CD15+) (Figures 6A and 6B). Interestingly, within the CD11b+CD33+CD14+ cell population, which partially identifies mMDSCs, MHCII+ (HLA-DR+) cells expressed LAP on their surface (Figures 6C, 6D, and S6A). Yet, most of circulating cells that express MHCII were LAP− (Figures 6C and 6D), indicating that not all human antigen-presenting cells express LAP. We found that LAP+ mMDSCs were increased in the blood of glioma patients as compared to healthy donors (Figure 6E, left panel), whereas LAP+HLA-DR+ myeloid cells were reduced (Figure 6E, right panel) suggesting that these cells may be recruited by the tumor tissue in the brain. To test this possibility, we measured CCR2 expression which enables migration of myeloid cells toward CCL2 secreted by gliomas (Chang et al., 2016) and found that LAP+HLA-DR+ MCs indeed express CCR2 (Figure 6F). Importantly, based on the TCGA data, the relatively high expression of human MHCII genes (e.g., HLA-DPB1, HLA-DMB, HLA-DRB1, HLA-DQB1) in the tumor tissue is strongly associated with poorer survival of low-grade glioma (LGG) patients (Figures 6G, left panel, and S6B, upper panels). We also found a positive correlation between the expression of MHCII genes (HLA-DPB1 and HLA-DMB) and TGFB1 (which encodes both LAP and TGF-β) in glioma samples (Figures 6H, upper panel, and S6C, left panel), and a combined high expression of the MHCII and LAP/TGF-β genes is associated with a worse prognosis for patients with LGG and GBM (Figures 6G, middle left panel, and S6B, lower left panels). Based on the OncoLnc algorithms (Anaya, 2016), the MHCII genes exhibit positive Cox coefficients, indicating a worse prognosis for patients in some types of cancer, including gliomas, and, to a lower extent, colon adenocarcinomas (COAD) (Figure S6D), thus supporting our findings that LAP-expressing antigen-presenting cells promote cancer. Furthermore, combined expression of either CD274 (PD-L1) and TGFB1; CD274 and HLA-DPB1 (Figure S6B, two lower right panels); or CD274, HLA-DPB1, and TGFB1 (Figure 6G, right panels) is associated with poor survival in glioma patients. Finally, the expression of CD274 positively correlates with HLA-DPB1 as well as TGFB1 expression in GBM and LGG patients (Figures 6H, lower panels and S6C, right panels). In summary, LAP is expressed on human mMDSCs and MHCII+ MCs which are associated with cancer in humans.
Figure 6.

LAP expressing myeloid cell subsets are modulated in glioma patients
(A and B) Expression of LAP on MDSCs derived from glioma patients (n = 6–18). Representative flow cytometry dot plots including fluorescence minus one (FMO-LAP) controls and gating strategy (A) and quantification of cell frequencies (B) are shown.
(C and D) LAP expression on MHCII+ cells in glioblastoma patients (GBM) and healthy donors (HC) (n = 18). Representative flow cytometry dot plots including FMO controls (C) and quantification of cell frequencies (D) are shown.
(E) Changes in MHCII− and MHCII+ MCs expressing LAP in the blood of GBM patients (n = 18). Frequencies of LAP expressing MHCII− and MHCII+ MCs in GBM patients and healthy controls are shown.
(F) CCR2 expression on LAP+MHCII+ MCs. Representative flow cytometry dot plots in two GBM patients and a healthy donor (HC), including FMO-LAP control are shown.
(G) Percent survival of patients with relatively high or low mRNA expression of indicated genes, including MHCII (HLA-DPB1), combined expression of MHCII and LAP genes (HLA-DPB1/TGFB1) and PD-L1, MHCII and LAP (CD274/HLA-DPB1/TGFB1) in the tumor tissue of glioma patients (low grade glioma, LGG and GBM). Graphs and p values were downloaded from the TCGA dataset via cBioPortal (https://www.cbioportal.org; (Cerami et al., 2012; Gao et al., 2013)).
(H) Correlation between TGFB1 and HLA-DPB1, TGFB1 and CD274, HLA-DPB1 and CD274 expression in the tumor tissue of GBM patients. Graphs and p values were downloaded from TCGA dataset via cBioPortal (Cerami et al., 2012; Gao et al., 2013). Data shown as mean ± SEM. Mann-Whitney test (B) and two-tailed t test (D and E) were used for p value calculations. ∗∗∗∗, p < 0.0001. See also Figure S6.
Discussion
Myeloid cells play a critical role in immune suppression in cancer, thus being a major obstacle for successful immunotherapy. Our study investigated LAPHi MCs producing a highly suppressive cytokine, TGF-β, which is primarily responsible for mediating their tolerogenic effects. We found LAP expression on different types of MCs including mMDSCs, TAMs and tolerogenic DCs in the context of cancer. We also identified subsets of LAPHi MCs with distinct genomic signatures that can contribute to the tumor promoting immune tolerance by different mechanisms. LAPHi MCs infiltrate the tumor tissue from the periphery, where they are likely involved in systemic tolerance. These cells can be targeted with anti-LAP antibodies resulting in reduced cancer-associated immune suppression. Some highly malignant and aggressive types of cancer, such as GBM, do not benefit from current immunotherapies aiming to restore the activity of exhausted T cells. This is likely because of cancer-established immune suppression largely driven by tolerogenic myeloid cells. Thus, inhibiting the function of LAPHi MCs in the context of cancer is of high importance.
Immune suppression within the tumor depends on the recruitment of tolerogenic cells including MCs to the tumor microenvironment. Our findings indicate that LAPHi MCs are recruited from the BM to the tumor tissue, which secretes CCL2 to attract leukocytes expressing CCR2, similar to what was previously reported (Swirski et al., 2009; Zhao et al., 2013). Within the tumor microenvironment, LAPHi MCs acquire tumor-promoting functions including the ability to suppress the proliferation of effector T cells and induce tumor antigen-specific Tregs, as we demonstrated by ex vivo assays. Both effects are likely to be mediated by mature TGF-β present on the cell surface of LAPHi MCs and thus bioavailable for a rapid response. Interestingly, it has been reported that normal monocytes acquire immunosuppressive properties after exposure to human glioma cells (Rodrigues et al., 2010), suggesting that LAPHi MCs are further modulated after recruitment to the brain tumor to promote cancer growth. In support of this idea, we found that LAPHi MCs isolated from the tumor tissue possess a stronger tolerogenic phenotype than their peripheral counterparts. Tumor-derived LAPHi MCs express high levels of Il10 and low levels of Ifng and Il12, further highlighting their tolerogenic properties. In addition, we found that LAPHi MCs express CSF-1R/CD115, which is consistent with a previously reported tolerogenic function of Gr-1+ CD115+ MSCs that share markers with LAPHi MCs, including CSF-1R (Huang et al., 2006). Inhibition of CSF-1R has anti-tumor effects in GBM models (Pyonteck et al., 2013), and CSF-1R is currently a target for clinical trials in glioma patients (Akkari et al., 2020). Thus, we propose that anti-LAP antibodies, which decrease LAPHi MCs in animal cancer models, will also be therapeutically beneficial for human cancer. Of note, it has been postulated that LAP is expressed on the surface of microglia, the brain’s resident myeloid cells (Qin et al., 2018). However, our results, based on well-established tools for detecting surface LAP by flow cytometry, do not support this hypothesis. We found LAP expression on peripherally derived MCs infiltrating intracranial GL261 GBM but not on microglia.
We found that LAPHi MCs are increased in the periphery of both tumor-bearing mice and GBM patients. The accumulation of LAPHi MCs may account for the lymphopenia reported in the blood of glioma patients (Chongsathidkiet et al., 2019; Dix et al., 1999; Roszman et al., 1985) and be associated with systemic immunosuppression (Brooks et al., 1972; Dix et al., 1999; Gustafson et al., 2010). LAP-expressing monocytes may have beneficial roles in autoimmune diseases, such as rheumatoid arthritis (Slobodin et al., 2013). Furthermore, changes in macrophages that express LAP were identified in the peritoneal fluid of patients with endometriosis (Hanada et al., 2018), and monocytes expressing LAP are associated with plasma cell dyscrasias (Tadmor et al., 2018). Finally, LAP positive DCs isolated from human blood inhibit T cell activation in an in vitro assay and this effect is mediated by TGF-β (Gandhi et al., 2007). Owing to the production of mature TGF-β, LAPHi MCs can potentially contribute to lung fibrosis, as it was reported for CCR2+ mMDSCs (Lebrun et al., 2017). Thus, LAPHi MCs may be important modulators of immunological responses not only in cancer but also in other disorders where immune tolerance may promote disease.
For detailed characterization of LAPHi MCs, we performed scRNA-Seq on immune cells isolated from the tumor tissue and identified tolerogenic myeloid cell clusters enriched with LAPHi MCs. Although these clusters share common tolerogenic markers, each of them possesses a unique signature, suggesting that these sub-populations of cells use different mechanisms to support tumor growth. This is consistent with the multiple mechanisms MDSCs known to use for immunosuppression (Gabrilovich et al., 2012; Veglia et al., 2021). Of note, LAPHi cells outnumber LAPLo cells in these clusters, demonstrating that the majority of tolerogenic myeloid cells in tumor express surface LAP. Myeloid cells are known to promote cancer by supporting multiple pro-tumorigenic pathways, including extracellular matrix (ECM) remodeling to facilitate metastasis (MMPs), recruiting immune cells to the tumor (CCR2/CCL2), depriving effector T cells of essential metabolites (Arg1), and immune suppression (TGF-β, IL-10, PD-L1) (Veglia et al., 2021). As mentioned earlier, unique tolerogenic signatures of the clusters dominated by LAPHi MCs suggest that each of the clusters employs a different mechanism to promote tumor growth. For example, the tolerogenic function of Clusters 2, 5 18, and 20 may be primarily driven by depriving effector T cells from arginine, as they express Arg1 (Gabrilovich et al., 2012). Clusters 2, 18 and 20 can also facilitate invasion and metastasis via MMPs, as they express Mmp12 and Mmp14 (Markovic et al., 2009). Cluster 20 has additional tools to drive metastasis and angiogenesis by complement activation (Benoit et al., 2012; Bulla et al., 2016; Roumenina et al., 2019; Son et al., 2016). Cluster 15 can contribute by involving Chil3/Ym1, a marker of alternatively activated macrophages promoting tissue remodeling (Gensel and Zhang, 2015; Murray et al., 2014). On the other hand, Cluster 21, most likely represented by mregDCs expressing Cd274 (PD-L1) and Ccr7 (Maier et al., 2020), can induce tumor-specific Tregs in the draining lymph nodes. We demonstrate that LAPHi MCs can play a key role in this process by supplying mature TGF-β. Thus, single cell analysis revealed unique tolerogenic features of LAP-associated myeloid sub-groups indicating that, through LAPHi MCs, cancer employs diverse immunity inhibition strategies.
Unexpectedly, we found that LAP is not only expressed on the conventionally defined human mMDSCs, which, by definition, do not include MHCII+ cells, but also on the MHCII+ MCs in both mice and humans. In fact, the Gabrilovich group demonstrated that MDSCs require MHCII to induce CD4 T cell-specific tolerance in the MC38 cancer model (Nagaraj et al., 2012), and, as mentioned earlier, intratumoral MHCII+ DCs can contribute to cancer in humans (Maier et al., 2020). The immunosuppressive function of LAPHi MHCII+ MCs is explained by the expression of mature TGF-β on the cell surface, readily available for activation. Indeed, our group has previously demonstrated that LAP+ DCs inhibit T cell proliferation in a TGF-β-dependent manner (Gandhi et al., 2007), and tolerogenic MHCII+ MCs have been described in humans (Costa et al., 2017). Interestingly, we observed lower frequencies of LAPHi MHCII+ MCs in GBM patients’ blood, which could be because of their migration to the brain tumor where they are presumably involved in the production of tumor-specific Tregs. As such, LAPHi MHCII− and LAPHi MHCII+ MCs likely play spatially distributed roles. Although circulating LAPHi MHCII−, representing classical mMDSCs, may suppress the immune response systemically by inhibiting the proliferation of T cells, LAPHi MHCII+ cells are recruited by the tumor to suppress the anti-tumor immune response in the brain via Treg stimulation. This hypothesis would have to be further validated by investigating the local functions of LAP expressing MCs. In summary, we identified a subset of MHCII+ MCs that express surface LAP/TGF-β and can contribute to immune suppression within the tumor microenvironment.
Notably, LAPHi MCs are not only found in tumor-bearing hosts but also in healthy human donors and naive mice, demonstrating that LAP-expressing myeloid cells are present in normal biological conditions. This suggests that LAPHi MCs play a role in supporting homeostasis in non-pathological settings. Cancer, however, utilizes the tolerogenic features of these cells to create an immunosuppressive environment for fostering tumor growth. Thus, our findings suggest that LAP can be a marker of pro-tumorigenic monocyte precursors, including MDSCs, TAMs, mregDCs, which have the potential to become immunosuppressive in the presence of tumor. In summary, we identified LAP-expressing myeloid cells that play a critical role in promoting tumor growth by favoring tolerance through multiple mechanisms involving TGF-β. Targeting these cells may constitute a potential therapy for cancer types in which LAPHi MCs play a major role.
Limitations of the study
Our study reveals a previously unknown function by which LAP expressing myeloid cells mediate immune tolerance in cancer. Conclusions of this study are based primarily on data obtained from subcutaneous mouse models of colorectal cancer. Follow up studies using additional cancer models are necessary to inform whether LAP expressing myeloid cells also play a similar tumor-promoting role in other cancer settings. Although we found LAP expressing myeloid cells are increased in the blood of GBM patients, the precise function of these cells is not explored in the current study. Further studies are required to investigate the immune suppressive role of LAP-expressing myeloid cells in patients with GBM and other forms of cancer.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-B220 (RA3-6B2); APC | eBioscience | Cat# 17-0452-82; RRID: AB_469395 |
| anti-CD103 (M290); PE-CF594 | BD Biosciences | Cat# 565849; RRID: AB_2739377 |
| anti-LAP (TW7-16B4); PE | Biolegend | Cat# 141404; RRID: AB_10720867 |
| anti-CD115 (AFS98); BrV421 | Biolegend | Cat# 135513; RRID: AB_2562667 |
| anti-CD11b (M1/70); BB515 | BD Biosciences | Cat# 564454; RRID: AB_2665392 |
| anti-CD11c (HL3); BrV711 | BD Biosciences | Cat# 563048; RRID: AB_2734778 |
| anti-CD19 (1D3); PE-Cy7 | eBioscience | Cat# 25-0193-82; RRID: AB_657663 |
| anti-CD3 (17A2); APC | Biolegend | Cat# 100236; RRID:AB_2561456 |
| anti-CD3e (145-2C11); BUV650 | BD Biosciences | Cat# 564378; RRID: AB_2738779 |
| anti-CD4 (GK1.5); BrV421 | Biolegend | Cat# 100437; RRID: AB_10900241 |
| anti-CD4 (GK1.5); BUV496 | BD Biosciences | Cat# 564667; RRID: AB_2722549 |
| anti-CD8a (53-6.7); BrV421 | Biolegend | Cat# 100737; RRID: AB_10897101 |
| anti-CD44 (IM7); PE | Biolegend | Cat# 103008; RRID: AB_312959 |
| anti-CD45 (30-F11); BUV661 | BD Biosciences | Cat# 565079; RRID: AB_2739057 |
| anti-IFN-γ (XMG1.2); eFluor660 | eBioscience | Cat# 50-7311-82; RRID: AB_11217680 |
| anti-FOXP3 (FJK-16s); FITC | eBioscience | Cat# 11-5773-82; RRID: AB_465243 |
| anti-PDL1 (MIH5); APC | eBioscience | Cat# 17-5982-80; RRID: AB_2688091 |
| anti-CD41 (MWReg30); PE-Cy7 | eBioscience | Cat# 25-0411-82; RRID: AB_1234970 |
| anti-Ly6A/E (Sca-1; D7); PE-Cy7 | eBioscience | Cat# 25-5981-82; RRID: AB_469669 |
| anti-TER-119 (TER-119); PE-Cy7 | eBioscience | Cat# 25-5921-82; RRID: AB_469661 |
| anti-MHC Class II (I-A/I-E); Brv605 | BD Biosciences | Cat# 563413; RRID: AB_2738190 |
| anti-CD49b (DX5); APC | eBioscience | Cat# 17-5971-82; RRID: AB_469485 |
| anti-CD62L (MEL-14); APC | Biolegend | Cat# 104412; RRID: AB_313099 |
| anti-CD8 (53-6.7); BUV805 | BD Biosciences | Cat# 564920; RRID: AB_2716856 |
| anti-Ly6C (HK1.4); APC-Fire750 | Biolegend | Cat# 128046; RRID: AB_2616731 |
| anti-Ly6G (1A8-Ly6g); APC | eBioscience | Cat# 17-0452-82; RRID: AB_2573307 |
| anti-Ly6G (1A8); BUV563 | BD Biosciences | Cat# 565707; RRID: AB_2739334 |
| anti-CCR2 (K036C2); APC | Biolegend | Cat# 357207; RRID: AB_2562238 |
| anti-CD103 (2E7); APC | eBioscience | Cat# 17-1031-82; RRID: AB_1106992 |
| anti-CD11b (M1/70); BUV650 | BD Biosciences | Cat# 563402; RRID: AB_2738184 |
| anti-CD14 (MφP9); BrV711 | BD Biosciences | Cat# 563373; RRID: AB_2744290 |
| anti-CD15 (W6D3); APC-Cy7 | Biolegend | Cat# 323048; RRID: AB_2750190 |
| anti-CD3 (SK7); BrV421 | BD Biosciences | Cat# 563798; RRID: AB_2744383 |
| anti-CD33 (WM-35); PE-Cy7 | eBioscience | Cat# 25-0338-42; RRID: AB_1907380 |
| anti-CD4 (SK3); BUV395 | BD Biosciences | Cat# 563552; RRID: AB_2738275 |
| anti-CD61 (VI-PL2); PerCP | Biolegend | Cat# 336409; RRID: AB_2561306 |
| anti-CD8 (SK1); BUV805 | BD Biosciences | Cat# 612890; RRID: AB_2870178 |
| anti-HLA-DR (G46-6); BrV785 | BD Biosciences | Cat# 564041; RRID: AB_2738559 |
| anti-LAP (TW7-28G11); PE | Purified by Cell Essentials | Custom conjugation to PE |
| Lineage Cocktail 1 (CD3/MφP9, CD14/NCAM16.2, CD16/3G8, CD19/SK7, CD20/L27, CD56/SJ25C1); FITC | BD Biosciences | Cat# 340546; RRID: AB_400053 |
| IC MPC-11 (IgG2b) | BioXCell | Cat# BE0086; RRID: AB_1107791 |
| IC MG1-45 (IgG1) | Biolegend | Cat# 401408; RRID: AB_11148942 |
| anti-CD3ε (KT3) | BioXCell | Cat# BE0261; RRID: AB_2687740 |
| anti-TGF-β (1D11.16.8) | BioXCell | Cat# BE0057; RRID: AB_1107757 |
| anti-LAP (TW7-16B4, IgG1) | Purified by Cell Essentials | Custom order purification |
| anti-LAP (TW7-28G11, IgG2b) | Purified by Cell Essentials | Custom order purification |
| Chemicals, peptides, and recombinant proteins | ||
| CellTrace Violet | Invitrogen | Cat# C34557 |
| RNase-Free DNase | Qiagen | Cat# 79254 |
| Fixable Viability Dye; e-Fluor506 | eBioscience | Cat# 65-0866-14 |
| Brilliant Stain Buffer | BD Biosciences | Cat# 566349; RRID: AB_2869750 |
| Percoll | Sigma | Cat# GE17-0891-02 |
| DAPI | Biolegend | Cat# 422801 |
| DNase I | Roche | Cat# 10104159001 |
| Collagenase D | Roche | Cat# 11088882001 |
| MCP-1, RECOMBINANT MOUSE | BD Biosciences | Cat# 554590 |
| OVA 323-339 | InvivoGen | Cat# vac-isq |
| Phorbol 12-myristate 13-acetate | Sigma | Cat# P8139 |
| Ionomycin | Sigma | Cat# I3909 |
| GolgiStop (v/v) containing monensin | BD Biosciences | Cat# 554724 |
| Bovine Albumin Fraction V Low Endotoxin | MP Biomedicals | Cat# IC810683 |
| TaqMan® Universal PCR Master Mix | ThermoFisher | Cat# 4304437 |
| Critical commercial assays | ||
| EasySep Mouse Monocyte Isolation Kit | STEMCELL Technologies | Cat# 19861 |
| EasySep Mouse Naïve CD4+ T Cell Isolation Kit | STEMCELL Technologies | Cat# 19765 |
| EasySep Mouse Naïve CD8+ T Cell Isolation Kit | STEMCELL Technologies | Cat# 19858 |
| High Capacity cDNA Reverse Transcription Kit | ThermoFisher | Cat# 4368814 |
| Foxp3 Fixation/Permeabilization kit | eBiosceince | Cat# 00-5523-00 |
| miRNeasy Mini Kit (50) | Qiagen | Cat# 217004 |
| Deposited data | ||
| Raw scRNA-seq data | This paper | GEO: GSE174077 |
| Experimental models: Cell lines | ||
| CT26 | ATCC | CRL-2638; RRID: CVCL_7256 |
| MC38 | Lab of Ana Anderson | RRID: CVCL_B288 |
| MC38-OVA | Lab of Ana Anderson | RRID: CVCL_XJ96 |
| GL261 | NIH NCI | Cat# Glioma 261; RRID: CVCL_Y003 |
| Experimental models: Organisms/strains | ||
| Mouse; B6.SJL-Ptprc<a> Pepc/BoyJ | Jackson Laboratories | Cat# JAX: 002014; RRID: IMSR_JAX:002014 |
| Mouse; Balb/cJ | Jackson Laboratories | Cat# JAX: 000651; RRID: IMSR_JAX:000651 |
| Mouse; C57BL/6J | Jackson Laboratories | Cat# JAX: 000664; RRID: IMSR_JAX:000664 |
| Mouse; GFP-FoxP3 knockin | Lab of Vijay Kuchroo | N/A |
| Mouse; OT-II/GFP-FoxP3 knockin | Bred in house | N/A |
| Oligonucleotides | ||
| See Table S1 for TaqMan qPCR Oligonucleotides | ||
| Software and algorithms | ||
| Prism 9.0 | GraphPad Software | https://www.graphpad.com |
| FlowJo v10 | Tree Star | https://www.flowjo.com |
| BD FACSDiva 8.0.1 | BD Biosciences | https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software#Overview |
| viSNE | Cytobank | https://cytobank.org/ |
| Drop-seq tools 1.12 | Dropseq STAR 2.5.3a |
http://mccarrolllab.com/dropseq |
| Seurat v3.0 | Seurat | https://www.cell.com/cell/fulltext/S0092-8674(19)30559-8 |
| R/3.5.1 | Binomial Generalized Linear Model | https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/glm |
| Other | ||
| ViiA 7 Real-Time PCR System | Applied Biosystems | N/A |
| FACSymphony | BD Biosciences | N/A |
| BD FACS AriaIIIu | BD Biosciences | N/A |
| UltraComp eBeads™ Compensation Beads | ThermoFisher | Cat# 01-2222-42 |
| FcR Blocking Reagent, human | Miltenyi Biotec | Cat# 130-059-901 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Howard Weiner (hweiner@rics.bwh.harvard.edu).
Materials availability
This study did not generate new unique reagents. Reagents generated in our laboratory in previous studies are commercially available.
Experimental model and subject details
Human studies
Human blood samples were obtained at BWH from 18 healthy donors (41–68 years of age) and 18 glioblastoma patients (47–85 years of age) of both genders prior to tumor resection. The study and procedures were approved by the institutional review board (IRB). Blood samples from healthy donors were obtained under the IRB Protocol 2017P000717 and from glioma patients under DF/HCC Protocol 10-417. All participants had given informed written consent for the use of their blood samples. Periphery blood mononuclear cells (PBMCs) were isolated by using Ficoll-Paque® density gradient centrifugation, frozen in freezing media (80% FBS; 20% DMSO) and stored in LN2. An equal number of thawed cells from healthy donors and glioma patients were analyzed by multicolor flow cytometry on two different days and data was combined for presentation.
Animal studies
C57BL/6J, B6.SJL, and Balb/cJ 6-8 weeks old male mice were purchased from the Jackson Laboratories. Foxp3-green fluorescent protein (GFP) and OTII-FoxP3-GFP reporter mice were housed in a conventional specific pathogen-free facility at the Building of Transformative Medicine. All experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital (BWH). Subcutaneous tumor models included MC38, MC38-OVA, and CT26. In preparation for subcutaneous implantation, cell lines were cultured in complete media until reached 80% confluence. MC38 tumors were implanted in WT male C57BL/6J mice at the age of 8-10 weeks and CT26 tumors were implanted in WT BALB/cJ male mice at the age of 8–10 weeks. Each mouse was implanted with 5 × 104 (MC38-OVA), 5 × 105 (MC38), or 2 × 105 (CT26) cells in 50 μl PBS by subcutaneous injections into the animal’s right flanks. Subcutaneous tumors were monitored every other day for total volume and integrity. Tumor volume was obtained through measurements in two dimensions (length, L and width, W) by caliper and calculated with the following formula: Tumor volume = 0.5 × (L × W2). For intracranial tumor implantation, mice were fixed on the stereotaxis frame under general anesthesia with 1–3% isoflurane, and 1 × 105 GL261 cells were implanted into the right striatum; coordinates were 1.8 mm lateral, 0.5 mm anterior to the bregma and 3.0 mm deep from the cortical surface of the brain.
Cell lines
CT26 cell line (RRID: CVCL_7256) was purchased from ATCC. GL261 (RRID: CVCL_Y003) cell line was purchased from NIH. MC38 (RRID: CVCL_B288) and MC38-OVA (RRID: CVCL_XJ96) cell lines were obtained from Dr. Ana Anderson. Cells were cultured in complete media as the following: DMEM (Gibco) supplemented with 10% FBS (Gibco), and 100 U/mL penicillin-streptomycin (Lonza) for culturing MC38, MC38-OVA, and GL261 cells; RPMI (Gibco) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate (Lonza), and 10 mM HEPES (Lonza) for culturing CT26 cells. In addition, for culturing MC38-OVA, 50 μg/mL geneticin selective antibiotic (Gibco) was added freshly into each culture. All cells were cultured at 37°C with 5% CO2.
Method details
Antibody treatments
Mice were treated with either anti-LAP (TW7-28G11) or IC (MPC-11) monoclonal antibodies diluted in PBS. Mouse anti-LAP antibodies were isolated from hybridoma generated in-house and purified by Cell Essentials. IC antibodies were purchased from BioXCell. As a standard treatment, antibodies were administered intraperitoneally (i.p.), 8 mg/kg on Mondays and Wednesdays and 10 mg/kg on Fridays. The initial dose of antibodies was administrated within seven days post tumor implantation.
FACS sorting of LAPHi and LAPLo MCs
For in vitro Proliferation Assay, Migration Assay, qRT-PCR and Adoptive Transfer, LAPHi and LAPLo MCs were isolated according to following protocols. For isolation of cells from spleen and BM, monocytes were enriched using EasySep Mouse Monocyte Isolation Kit (STEMCELL Technologies) according to the manufacturer’s instruction. For isolation of cells from tumor tissue, mononuclear immune cells were enriched using Percoll gradient, as described below. Following pre-enrichment, cells were stained with anti-LAP-PE, anti-CD11b-BB515, DUMP channel antibodies (anti-Ly6G, anti-CD49b, anti-B220, anti-CD3, conjugated to APC) and DAPI. Aggregated cells, dead cells, and DUMP+ cells were excluded by flow cytometry gating strategy (Figure S1F) and equal number of CD11b+ LAPHi and LAPLo MCs was sorted and used in corresponding assays. Cell sorting was performed on an BD FACS AriaIIIu (BD Biosciences) using BD FACSDiva 8.0.1. software (BD Biosciences).
Adoptive transfer
Wild type (WT) C57BL/6J mice were adoptively transferred with LAPHi and LAPLo MCs isolated from MC38-OVA tumor-bearing B6.SJL mice on days -1 and 8. LAPHi and LAPLo MCs were obtained through flow cytometry cell sorting after monocytes were isolated from lymphoid organs using EasySep Mouse Monocyte Isolation Kit (STEMCELL Technologies) according to the manufacturer’s instruction. A total of 3–5 × 105 cells were diluted in serum-free medium and injected intravenous (i.v.) through tail veins. One day after the first cell transfer, mice were implanted with MC38-OVA cells as described in the Animal studies section, and tumor growth was monitored by caliper measurements.
Isolation of cells from tumor
Subcutaneous CT26 and MC38 colorectal cancer model. Tumors were removed, minced and enzymatically dissociated with 2 mg/ml Collagenase D (Roche), 10% FBS and 100 μg/ml DNAse I (Roche) in HBSS by incubation with rotation for 20 min at 37°C. The digested tissue was triturated and passed through a 40-μm cell strainer. Cells were washed in M199 medium supplemented with 10% FBS, centrifuged and resuspended in 30% isotonic Percoll (Sigma) underlaid by 70% isotonic Percoll, and overlaid with HBSS. Then, cells were centrifuged at 700g at RT for 20 min and collected from the 70/30% Percoll interphase, washed and used for FACS analysis or sorting.
Intracranial GL261 GBM model. Brains were triturated in cold HBSS using a homogenizer and passed through a 70-μm cell strainer. Cells were centrifuged, resuspended in 30% isotonic Percoll (Sigma), underlaid by 70% isotonic Percoll, and centrifuged at 900g at RT for 25 min. Immune cells were collected from the 70/30% interphase, washed and analyzed by FACS.
In vitro proliferation assay
Naive CD4+ T cells (CD4+CD44loCD62LhiFoxp3-) were isolated from the spleen and lymph nodes of OT-II/GFP-Foxp3 mice, GFP-Foxp3 mice using EasySep Mouse Naive CD4+ T cell Isolation kit (STEMCELL Technologies) and sorted by flow cytometry. Naive CD8+ T cells (CD8+CD44loCD62Lhi) were isolated from the spleen and lymph nodes of WT mice using EasySep Mouse Naive CD8+ T cell Isolation kit (STEMCELL Technologies). Cells were labeled with CellTrace Violet according to the manufacturer’s instructions (CellTrace Violet proliferation kit, Invitrogen) and used as responder cells. The LAPHi and LAPLo MCs were isolated and sorted from spleens, bone marrows (BM) or tumors (as applicable) from naive or tumor-bearing C57BL/6J mice. From lymphoid organs, monocytes were enriched using EasySep Mouse Monocyte Isolation Kit (STEMCELL Technologies) according to the manufacturer’s instruction, stained and FACS-sorted, and LAPHi and LAPLo MCs were isolated as described above. From tumor, cells were enriched using Percoll gradient and LAPHi and LAPLo MCs were isolated as described above. The sorted myeloid cells and responder naive T cells were diluted and cultured in complete medium at different ratios, as indicated. For non-specific stimulation, anti-CD3 (0.5 μg/ml for CD4+ T cells or 0.25 μg/ml for CD8+ T cell) was added. For antigen-specific stimulation of CD4+ T cells isolated from OT-II/GFP-Foxp3 mice, myeloid cells were preloaded with OVA323-339 peptides (30 μg/mL, InvivoGen) for 3.5 hours, and unbound peptides were washed off before addition of responder cells. When indicated, blocking antibodies anti-TGF-β, anti-LAP TW7-16B4, anti-LAP TW7-28G11 or control antibodies (IC MPC-11, IC MG1-45) were added at 0.25 mg/mL. T cell proliferation was assessed after 48 hours (CD8+ T cells) or 72 hours (CD4+ T cells) by flow cytometry.
Migration assay
LAPHi and LAPLo MCs were isolated from spleens of MC38 tumor-bearing C57BL/6J mice as described above and 3 × 105 cells in100 μL complete IMDM medium seeded in the upper chambers of a 24-well tissue culture plates with 5 μm pore-size inserts (Corning). The lower chambers were loaded with 600 μL chemotaxis medium (IMDM medium containing 0.5% endotoxin-free BSA (MP Biomedicals), 10% FBS, 0.01M HEPES) with or without 0.1 μg/mL CCL2/MCP-1 (BD Biosciences). Cancer cell lines (MC38 or GL261) were cultured until they reached 80%-confluence and 600μl tumor-conditioned media transferred to the lower chambers of 24-well tissue culture plates. Migrating cells were collected from the lower chambers and the bottom of cell inserts after 3 hours incubation at 37°C and quantified by flow cytometry.
Flow cytometry staining and acquisition
Analysis of mouse cells: Immune cells were isolated from spleens, BMs, lymph nodes, and tumors were resuspended in FACS buffer (HBSS containing 2% FBS, 2 mM EDTA, and 25 mM HEPES) and blocked with anti-mouse CD16/CD32 before staining. Cells were stained with fluorochrome-labeled antibodies purchased from eBioscience, BioLegend, or BD Pharmingen in Brilliant Stain Buffer (BD Biosciences). For intracellular cytokine staining, cells were stimulated for 4 hours at 37°C with 500 ng/ml PMA (phorbol 12-myristate 13-acetate; Sigma), 0.67 μM ionomycin (Sigma), and 1.3% GolgiStop (v/v) containing monensin (BD Biosciences) diluted in complete IMDM (Gibco). Cells were fixed and permeabilized with Foxp3 Fixation/Permeabilization kit (eBiosceince) according to the manufacturer's instructions. The fixed cells were then stained for intracellular proteins. The following surface and intracellular antibodies were used: CD103-PE-CF594 (clone M290), CD115-BrV421 (clone AFS98), CD11b-BB515 (clone M1/70), CD11c-BrV711 (clone HL3), CD3e-BUV650 (clone 145-2C11), CD4-BUV496 (clone GK1.5), CD45-BUV661 (clone 30-F11), CD8-BUV805 (clone 53-6.7), Ly6C-APC-Fire750 (clone HK1.4), Ly6G-BUV563 (clone 1A8), MHC Class II-BrV605 (clone I-A/I-E), PDL1-APC (clone MIH5), CD41-PE-Cy7 (clone MWReg30), Ly6A/E-PE-Cy7 (Sca-1; clone D7), CD19-PE-Cy7 (clone 1D3), TER-119-PE-Cy7 (clone TER-119), LAP-PE (clone TW7-16B4), FOXP3-FITC (clone FJK-16s), IFN-γ-eFluor660 (clone XMG1.2). DUMP channel included: CD41-PE-Cy7, Ly6A/E-PE-Cy7, CD19-PE-Cy7, TER-119-PE-Cy7 antibodies. Fixable viability dye e-Fluor506 (eBioscience) was used to exclude dead cells. Compensation was performed by using UltraComp eBeads™ Compensation Beads (ThermoFisher). Data acquisition and analysis were performed on a FACSymphony (BD Biosciences) and FlowJo version 10 (Tree Star). For viSNE analysis, 100,000 cells were sub-sampled from three tumor-derived immune cells and analyzed using multicolor flow cytometry. Gating was performed manually to exclude platelets, erythrocytes, undifferentiated cells, B cells, and dead cells; and the viSNE algorithm was run on concatenated samples by Cytobank (Amir el et al., 2013; Kotecha et al., 2010).
Analysis of human PBMCs: A small amount of gMDCSs was detected from frozen samples due to cryopreservation that compromises the viability of gMDSCs (Trellakis et al., 2013). For sufficient presentation, we chose to show results for samples with a minimum of 200 detected gMDSCs which we found in a total of six GBM patients’ samples. PBMCs were washed in FACS buffer (HBSS containing 2% FBS, 2 mM EDTA, and 25 mM HEPES) and blocked with human FcR Blocking Reagent (Miltenyi Biotec) before staining. Cells were stained in Brilliant Stain Buffer (BD Biosciences). The following surface Abs were used: Lineage Cocktail 1-FITC (CD3, CD14, CD16, CD19, CD20, CD56), CD61-PerCP (clone VI-PL2), CD103-APC (clone 2E7), CD15-APC-Cy7 (clone W6D3), CD3-BV421 (clone SK7), CD11b-BV650 (clone M1/70), CD14-BV711 (clone MφP9), HLA-DR-BV785 (clone G46-6), CD4-UV395 (clone SK3), CD8-BUV805 (clone SK1), LAP-PE (clone 28G11), CD33-PE-Cy7 (clone WM-35). Samples were analyzed on the FACSymphony (BD Biosciences) and FlowJo version 10 (Tree Star). Fixable viability dye e-Fluor506 was used to exclude dead cells. Data acquisition and analysis were performed on a FACSymphony (BD Biosciences) and FlowJo version 10 (Tree Star).
qRT-PCR
LAPHi and LAPLo cells were isolated and sorted from spleens and bone marrows of naive and tumor-bearing mice or MC38-OVA and CT26 subcutaneous tumors, as described above. mRNA was isolated with miRNAeasy kit (Qiagen) according to manufacturer’s protocol, including an on-column DNase Digestion step (RNase-Free DNase, Qiagen). cDNA was generated using the Reverse-Transcription PCR Kit following the manufacturer's instructions (ThermoFisher). qPCR was performed with the TaqMan® Universal PCR Master Mix on the ViiA 7 Real-Time PCR System (ThermoFisher), and the results were normalized to Gapdh or B2m. All TaqMan primers were provided by ThermoFisher (Table S1).
Single cell RNA sequencing analysis
CT26 tumors were implanted in WT Balb/cJ male mice at the age of 8 weeks with 2 × 105 CT26 cells in 50 μL PBS by subcutaneous injections into the animal’s flanks. When tumor sizes reached approximately 10mm in diameter, tumor tissue was removed, immune cells isolated by Percoll gradient, as described in the isolation of cells from tumor section. Live LAPHi and LAPLo CD45+ cells were sorted by FACS and processed for single-cell sequencing analysis.
Quantification and statistical analysis
Single cell RNA sequencing data processing
Mapping, QC, and counting. Single-cell transcriptomic data was processed using Drop-seq tools 1.12 (http://mccarrolllab.com/dropseq) as previously described (Gierahn et al., 2017; Macosko et al., 2015). In summary, cell barcodes and molecular barcodes were tagged, and the 5′ primer sequence and the 3′ polyA sequence were trimmed. The transcriptome sequences were then mapped to the GRCm38 reference genome using STAR 2.5.3a. The alignment files were sorted in queryname order and merged with cell/molecular barcodes. Gene/exon and other annotation tags were added. Bead synthesis errors were detected, fixed, or filtered if no straightforward fixation solution. A matrix of sequencing counts per gene per cell was output with each gene per row and each cell per column. Total sequence count, mapped sequence count, and exon sequence count per cell were then reported.
Single-cell clustering and LAPHiversus LAPLocell comparison. Single-cell clustering and differential expression analyses were performed by Seurat v3.0. Cells with more than 200 sequencing reads were selected, and genes with more than two reads across all samples entered downstream analyses. Expression counts were normalized by the “LogNormalize” method, and the top 2000 most variable genes were selected. The data from different samples were integrated by common anchors selected by Seurat. A linear transformation step was then applied by scaling the expression per gene to be mean 0 and variance 1. PCA was performed on the variable genes using the RunPCA function in Seurat. UMAP was created by the top 30 principal components. The differentially expressed genes in each cluster were output by FindAllMarkers. The clusters were identified basing on cell-type-specific key signature genes. The differentially expressed genes in LAPHi and LAPLo sample comparison were output in each cluster basing on cells with expression by FindMarkers. In addition to comparing the gene expression levels, the proportion of cells with detectable transcripts for each gene was compared between LAPHi and LAPLo samples in each cluster using binomial Generalized Linear Model in R/3.5.1.
Statistical analysis
Prism 9.0 (GraphPad Software) was used for statistical analysis. For comparison of two groups, an unpaired, two-tailed Student's t-test was used. For comparison of three or more groups, one-way ANOVA or two-way ANOVA with pairwise group comparisons using Tukey’s approach were used. Two-way ANOVA was used to determine the significance of the differences between tumor growth curves. Statistical differences between survival curves were calculated by log-rank (Mantel-Cox) test. Values of P < 0.05 were considered statistically significant. ∗∗∗∗, P < 0.0001.
Acknowledgments
We thank members of Tilos Therapeutics and Dr. Davide Mangani for helpful discussions, Dvora Ghitza for her technical assistance. Funding: This work was supported by the Ann Romney Center for Neurologic Diseases. The research in Gopal laboratory is supported by grants from the National Institutes of Health (R01AI127853 and R01AI151953).
Author contributions
GG, GM, and HLW designed research. GG, SS, LS, NPS, HAH, and LRW carried out experiments. AP and RK assisted with the experiments. DM and SSL analyzed scRNA-Seq data. TM, APC, SSL, BCH, RMR, MF, and GM provided input for the manuscript. BCH consulted statistical analyses. GG, GM, and HLW supervised the experiments and wrote the manuscript.
Declaration of interests
A patent for the use of anti-LAP antibodies for cancer treatment has been issued.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103347.
Contributor Information
Galina Gabriely, Email: ggabriely@jouncetx.com.
Gopal Murugaiyan, Email: mgopal@rics.bwh.harvard.edu.
Howard L. Weiner, Email: hweiner@rics.bwh.harvard.edu.
Supplemental information
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is GEO: GSE174077.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Amir el A.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C., Shenfeld D.K., Krishnaswamy S., Nolan G.P., Pe'er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkari L., Bowman R.L., Tessier J., Klemm F., Handgraaf S.M., de Groot M., Quail D.F., Tillard L., Gadiot J., Huse J.T., et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020;12:aaw7843. doi: 10.1126/scitranslmed.aaw7843. [DOI] [PubMed] [Google Scholar]
- Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016;2:e67. [Google Scholar]
- Anz D., Mueller W., Golic M., Kunz W.G., Rapp M., Koelzer V.H., Ellermeier J., Ellwart J.W., Schnurr M., Bourquin C., et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int. J. Cancer. 2011;129:2417–2426. doi: 10.1002/ijc.25902. [DOI] [PubMed] [Google Scholar]
- Batlle E., Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50:924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M.E., Clarke E.V., Morgado P., Fraser D.A., Tenner A.J. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J. Immunol. 2012;188:5682–5693. doi: 10.4049/jimmunol.1103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks W.H., Netsky M.G., Normansell D.E., Horwitz D.A. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J. Exp. Med. 1972;136:1631–1647. doi: 10.1084/jem.136.6.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulla R., Tripodo C., Rami D., Ling G.S., Agostinis C., Guarnotta C., Zorzet S., Durigutto P., Botto M., Tedesco F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016;7:10346. doi: 10.1038/ncomms10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O., Jedrychowski M.P., Moore C.S., Cialic R., Lanser A.J., Gabriely G., Koeglsperger T., Dake B., Wu P.M., Doykan C.E., et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A.L., Miska J., Wainwright D.A., Dey M., Rivetta C.V., Yu D., Kanojia D., Pituch K.C., Qiao J., Pytel P., et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76:5671–5682. doi: 10.1158/0008-5472.CAN-16-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chongsathidkiet P., Jackson C., Koyama S., Loebel F., Cui X., Farber S.H., Woroniecka K., Elsamadicy A.A., Dechant C.A., Kemeny H.R., et al. Author Correction: Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2019;25:529. doi: 10.1038/s41591-019-0355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J., Chan M.F., Werb Z. Metalloproteinases: a functional pathway for myeloid cells. Microbiol. Spectr. 2016;4:649–658. doi: 10.1128/microbiolspec.MCHD-0002-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M.L., Robinette M.L., Bugatti M., Longtine M.S., Colvin B.N., Lantelme E., Vermi W., Colonna M., Nelson D.M., Cella M. Two distinct myeloid subsets at the term human fetal-maternal interface. Front. Immunol. 2017;8:1357. doi: 10.3389/fimmu.2017.01357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix A.R., Brooks W.H., Roszman T.L., Morford L.A. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999;100:216–232. doi: 10.1016/s0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- Gabriely G., da Cunha A.P., Rezende R.M., Kenyon B., Madi A., Vandeventer T., Skillin Nathaniel, Rubino Stephen, Garo Lucien, Mazzola Maria A., et al. Targeting latency-associated peptide promotes anti-tumor immunity. Sci. Immunol. 2017;2:1–12. doi: 10.1126/sciimmunol.aaj1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich D.I., Ostrand-Rosenberg S., Bronte V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R., Anderson D.E., Weiner H.L. Cutting edge: immature human dendritic cells express latency-associated peptide and inhibit T cell activation in a TGF-beta-dependent manner. J. Immunol. 2007;178:4017–4021. doi: 10.4049/jimmunol.178.7.4017. [DOI] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garo L.P., Ajay A.K., Fujiwara M., Beynon V., Kuhn C., Gabriely G., Sadhukan S., Raheja R., Rubino S., Weiner H.L., et al. Smad7 controls immunoregulatory PDL2/1-PD1 signaling in intestinal inflammation and autoimmunity. Cell Rep. 2019;28:3353–3366.e3355. doi: 10.1016/j.celrep.2019.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel J.C., Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015;1619:1–11. doi: 10.1016/j.brainres.2014.12.045. [DOI] [PubMed] [Google Scholar]
- Geribaldi-Doldan N., Fernandez-Ponce C., Quiroz R.N., Sanchez-Gomar I., Escorcia L.G., Velasquez E.P., Quiroz E.N. The role of microglia in glioblastoma. Front. Oncol. 2020;10:603495. doi: 10.3389/fonc.2020.603495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierahn T.M., Wadsworth M.H., 2nd, Hughes T.K., Bryson B.D., Butler A., Satija R., Fortune S., Love J.C., Shalek A.K. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods. 2017;14:395–398. doi: 10.1038/nmeth.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson M.P., Lin Y., New K.C., Bulur P.A., O'Neill B.P., Gastineau D.A., Dietz A.B. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12:631–644. doi: 10.1093/neuonc/noq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T., Tsuji S., Nakayama M., Wakinoue S., Kasahara K., Kimura F., Mori T., Ogasawara K., Murakami T. Suppressive regulatory T cells and latent transforming growth factor-beta-expressing macrophages are altered in the peritoneal fluid of patients with endometriosis. Reprod. Biol. Endocrinol. 2018;16:9. doi: 10.1186/s12958-018-0325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Pan P.Y., Li Q., Sato A.I., Levy D.E., Bromberg J., Divino C.M., Chen S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- Kashiwagi I., Morita R., Schichita T., Komai K., Saeki K., Matsumoto M., Takeda K., Nomura M., Hayashi A., Kanai T., et al. Smad2 and Smad3 inversely regulate TGF-beta autoinduction in Clostridium butyricum-activated dendritic cells. Immunity. 2015;43:65–79. doi: 10.1016/j.immuni.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;53:10–17. doi: 10.1002/0471142956.cy1017s53. Chapter 10, Unit10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic J., Santibanez J.F. Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. Sci. World J. 2014;2014:521754. doi: 10.1155/2014/521754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun A., Lo Re S., Chantry M., Izquierdo Carerra X., Uwambayinema F., Ricci D., Devosse R., Ibouraadaten S., Brombin L., Palmai-Pallag M., et al. CCR2(+) monocytic myeloid-derived suppressor cells (M-MDSCs) inhibit collagen degradation and promote lung fibrosis by producing transforming growth factor-beta1. J. Pathol. 2017;243:320–330. doi: 10.1002/path.4956. [DOI] [PubMed] [Google Scholar]
- Ma W., Qin Y., Chapuy B., Lu C. LRRC33 is a novel binding and potential regulating protein of TGF-beta1 function in human acute myeloid leukemia cells. PLoS One. 2019;14:e0213482. doi: 10.1371/journal.pone.0213482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B., Leader A.M., Chen S.T., Tung N., Chang C., LeBerichel J., Chudnovskiy A., Maskey S., Walker L., Finnigan J.P., et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. 2020;580:257–262. doi: 10.1038/s41586-020-2134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic D.S., Vinnakota K., Chirasani S., Synowitz M., Raguet H., Stock K., Sliwa M., Lehmann S., Kalin R., van Rooijen N., et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. U. S. A. 2009;106:12530–12535. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvel D., Gabrilovich D.I. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Invest. 2015;125:3356–3364. doi: 10.1172/JCI80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoed A.G., Sherrill T.P., Cheng D.S., Han W., Saxon J.A., Gleaves L.A., Wu P., Polosukhin V.V., Karin M., Yull F.E., et al. Neutrophil-derived IL-1beta impairs the efficacy of NF-kappaB inhibitors against lung cancer. Cell Rep. 2016;16:120–132. doi: 10.1016/j.celrep.2016.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasato Y., Shiota T., Ohnishi K., Pan C., Yano H., Horlad H., Yamamoto Y., Yamamoto-Ibusuki M., Iwase H., Takeya M., et al. High density of CD204-positive macrophages predicts worse clinical prognosis in patients with breast cancer. Cancer Sci. 2017;108:1693–1700. doi: 10.1111/cas.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T., et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj S., Nelson A., Youn J.I., Cheng P., Quiceno D., Gabrilovich D.I. Antigen-specific CD4(+) T cells regulate function of myeloid-derived suppressor cells in cancer via retrograde MHC class II signaling. Cancer Res. 2012;72:928–938. doi: 10.1158/0008-5472.CAN-11-2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterlynck D.J., Meuleman C., Waer M., Koninckx P.R. Transforming growth factor-beta activity is increased in peritoneal fluid from women with endometriosis. Obstet. Gynecol. 1994;83:287–292. [PubMed] [Google Scholar]
- Peranzoni E., Zilio S., Marigo I., Dolcetti L., Zanovello P., Mandruzzato S., Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Poh A.R., Ernst M. Targeting macrophages in cancer: from bench to bedside. Front. Oncol. 2018;8:49. doi: 10.3389/fonc.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck S.M., Akkari L., Schuhmacher A.J., Bowman R.L., Sevenich L., Quail D.F., Olson O.C., Quick M.L., Huse J.T., Teijeiro V., et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B.Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B.Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L.R., Kaiser E.A., Snyder L.A., Pollard J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y., Garrison B.S., Ma W., Wang R., Jiang A., Li J., Mistry M., Bronson R.T., Santoro D., Franco C., et al. A Milieu molecule for TGF-beta required for microglia function in the nervous system. Cell. 2018;174:156–171. doi: 10.1016/j.cell.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues J.C., Gonzalez G.C., Zhang L., Ibrahim G., Kelly J.J., Gustafson M.P., Lin Y., Dietz A.B., Forsyth P.A., Yong V.W., et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010;12:351–365. doi: 10.1093/neuonc/nop023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszman T.L., Brooks W.H., Steele C., Elliott L.H. Pokeweed mitogen-induced immunoglobulin secretion by peripheral blood lymphocytes from patients with primary intracranial tumors. Characterization of T helper and B cell function. J. Immunol. 1985;134:1545–1550. [PubMed] [Google Scholar]
- Roumenina L.T., Daugan M.V., Noe R., Petitprez F., Vano Y.A., Sanchez-Salas R., Becht E., Meilleroux J., Clec'h B.L., Giraldo N.A., et al. Tumor cells hijack macrophage-produced complement C1q to promote tumor growth. Cancer Immunol. Res. 2019;7:1091–1105. doi: 10.1158/2326-6066.CIR-18-0891. [DOI] [PubMed] [Google Scholar]
- Schmieder A., Michel J., Schonhaar K., Goerdt S., Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin. Cancer Biol. 2012;22:289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Scurr M., Ladell K., Besneux M., Christian A., Hockey T., Smart K., Bridgeman H., Hargest R., Phillips S., Davies M., et al. Highly prevalent colorectal cancer-infiltrating LAP(+) Foxp3(-) T cells exhibit more potent immunosuppressive activity than Foxp3(+) regulatory T cells. Mucosal Immunol. 2014;7:428–439. doi: 10.1038/mi.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina N.V., Pamer E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., Springer T.A. Latent TGF-beta structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A., Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J. Autoimmun. 2017;85:117–125. doi: 10.1016/j.jaut.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Slobodin G., Kessel A., Kuznets I., Peri R., Haj T., Rosner I., Toubi E., Odeh M. CD14brightLAP+ mononuclear cells in peripheral blood positively correlate with BASRI scores in patients with ankylosing spondylitis: a pilot study. Joint Bone Spine. 2012;79:633–634. doi: 10.1016/j.jbspin.2012.04.008. [DOI] [PubMed] [Google Scholar]
- Slobodin G., Kaly L., Peri R., Kessel A., Rosner I., Toubi E., Rimar D., Boulman N., Rozenbaum M., Odeh M. Higher expression of latency-associated peptide on the surface of peripheral blood monocytes in patients with rheumatoid arthritis may be protective against articular erosions. Inflammation. 2013;36:1075–1078. doi: 10.1007/s10753-013-9639-6. [DOI] [PubMed] [Google Scholar]
- Son M., Porat A., He M., Suurmond J., Santiago-Schwarz F., Andersson U., Coleman T.R., Volpe B.T., Tracey K.J., Al-Abed Y., et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood. 2016;128:2218–2228. doi: 10.1182/blood-2016-05-719757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski F.K., Nahrendorf M., Etzrodt M., Wildgruber M., Cortez-Retamozo V., Panizzi P., Figueiredo J.L., Kohler R.H., Chudnovskiy A., Waterman P., et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadmor T., Levy I., Vadasz Z. Hierarchical involvement of myeloid-derived suppressor cells and monocytes expressing latency-associated peptide in plasma cell dyscrasias. Turk J. Haematol. 2018;35:116–121. doi: 10.4274/tjh.2018.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcyganov E., Mastio J., Chen E., Gabrilovich D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018;51:76–82. doi: 10.1016/j.coi.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trellakis S., Bruderek K., Hutte J., Elian M., Hoffmann T.K., Lang S., Brandau S. Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immun. 2013;19:328–336. doi: 10.1177/1753425912463618. [DOI] [PubMed] [Google Scholar]
- Veglia F., Sanseviero E., Gabrilovich D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021;21:485–498. doi: 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Chen P., Gupta P., Ott M., Zamler D., Kassab C., Bhat K.P., Curran M.A., de Groot J.F., Heimberger A.B. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol. 2020;22:180–194. doi: 10.1093/neuonc/noz212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Morgan R., Chen C., Cai Y., Clark E., Khan W.N., Shin S.U., Cho H.M., Al Bayati A., Pimentel A., et al. Mammary-tumor-educated B cells acquire LAP/TGF-beta and PD-L1 expression and suppress anti-tumor immune responses. Int. Immunol. 2016;28:423–433. doi: 10.1093/intimm/dxw007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Lim S.Y., Gordon-Weeks A.N., Tapmeier T.T., Im J.H., Cao Y., Beech J., Allen D., Smart S., Muschel R.J. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology. 2013;57:829–839. doi: 10.1002/hep.26094. [DOI] [PubMed] [Google Scholar]
- Zhu X., Fujita M., Snyder L.A., Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011;104:83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is GEO: GSE174077.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.