

Abstract
Background and Objective
PL8177 is a selective melanocortin 1 receptor agonist in development for the treatment of various immunologic and inflammatory conditions. Here we describe the pharmacokinetics of PL8177 after subcutaneous (sc) delivery in animals and humans.
Methods
Mice, rats, and dogs were administered sc PL8177 at single doses of 1.0 and 3.0 mg/kg (mice); 1.0, 5.0, and 25.0 mg/kg/day (rats); or 1.5, 8.0, and 40.0 mg/day (dogs). Blood was collected over 24 h (mice) or 28 days (rats and dogs). Safety and pharmacokinetics of single and multiple sc doses were also examined in human volunteers. Two dose levels were tested in two dosing cohorts of 1.0 and 3.0 mg/day for 7 days. Blood samples were collected through Day 1 and on Days 2 to 6 at peak and trough times based on analysis of the first two single-dose cohorts.
Results
In mice, 3 mg/kg PL8177 resulted in an area under the plasma concentration–time curve from 0 to infinity (AUC∞) of 1727 ng·h/mL, a maximum plasma concentration (Cmax) of 2440 ng/mL, an elimination half-life (t½) of 0.5 h, and a time to maximum concentration (tmax) of 0.25 h. Results for the 1-mg/kg dose were generally proportional. In rats, mean tmax values were independent of dose and ranged from 0.25 to 1.0 h for single and multiple dosing. Cmax values ranged from 516 to 695 ng/mL (1-mg/kg dose) and from 666 to 1180 ng/mL (25-mg/kg dose). In dogs, mean tmax values ranged from 0.4 to 1.3 h for single and multiple dosing. Values for tmax decreased with increasing dose and mean plasma Cmax increased less than dose proportionally (96–129 ng·h/mL [1.5 mg], 275–615 ng·h/mL [8.0 mg], and 633–1280 ng·h/mL [40.0 mg]). In humans, PL8177 was observed in the plasma within 15 min after a single dose and persisted for up to 48 h at higher doses. The tmax was 30–45 min (single dose) and 15–45 min (multiple doses). In multiple-dose studies, maximum steady-state plasma concentration (Cmax,ss) and AUC∞ increased with dose. Geometric mean Cmax,ss values were 20.1 ng/mL (1.0 mg) and 57.2 ng/mL (3.0 mg). AUC∞ values were 54.3 ng·h/mL (1.0 mg) and 199 ng·h/mL (3.0 mg). Unchanged PL8177 excreted in the urine was ≤ 1%, and accumulation was minimal.
Conclusion
PL8177 administration resulted in a consistent pharmacokinetic profile. The measured exposure levels resulted in pharmacologically active PL8177 concentrations at the targeted MC1R. Rapid absorption was seen in healthy volunteers, and multiple-dose administration over 7 days resulted in pharmacokinetic characteristics similar to those observed after single-dose administration. Results support the continued development of PL8177 to treat immunologic and inflammatory conditions.
Key Points
| PL8177 is a melanocortin agonist being developed to treat inflammatory conditions. |
| PL8177 had consistent pharmacokinetics and low accumulation with repeated dosing, supporting further development. |
Introduction
PL8177 is currently being evaluated for a variety of immunologic and inflammatory indications. It is a synthetic, cyclic, 7-amino-acid analog of α-melanocyte-stimulating hormone (α-MSH), a peptide that modulates inflammation centrally and peripherally by binding to the melanocortin 1 receptor (MC1R) [1, 2]. Stimulation of this receptor results in an increase in cyclic adenosine monophosphate, which suppresses nuclear transcription factor NF-κB [3], leading to a decrease in the production of proinflammatory cytokines, reactive oxygen species, and chemokines [2, 4]. In addition to direct intervention in inflammatory signaling, melanocortin agonism is believed to induce the immune system to enter an inflammatory resolution phase [5].
Melanocortin agonists represent an innovative anti-inflammatory strategy based on augmenting natural processes that promote resolution of inflammation [5]. There are four melanocortin peptides: α-, β- and γ-MSH, and adrenocorticotropic hormone (ACTH); and five melanocortin receptors have been identified (MC1R–MC5R) [6, 7]. These receptors can be stimulated by all melanocortins [2] with the exception of MC2R, which is activated only by ACTH [8]. In addition to inflammation resolution, the melanocortins play a role in a multitude of physiological processes including body weight regulation [9], energy homeostasis [10], sexual function [11], adrenal gland development, steroidogenesis, and pigmentation [12, 13].
α-MSH produces anti-inflammatory effects both centrally and peripherally [14], and these effects have been demonstrated in experimental models in arthritis [15], systemic inflammation [16], cutaneous inflammation [17], brain inflammation [18], and lung inflammation [19]. Experimental studies have also shown that α-MSH ameliorates symptoms of inflammatory bowel disease (IBD) [20] and uveitis [21, 22].
Because MC1R is involved in α-MSH–mediated modulation of inflammation, MC1R agonists may be useful in a variety of inflammatory conditions. PL8177 is a potent and selective MC1R agonist with an in vitro affinity of 0.04 nM and a half maximal effective concentration (EC50) of 0.39 nM, compared with α-MSH values of 0.095 and 0.22 nM, respectively [23]. PL8177 has a carboxyamide C-terminus and an acetylated amino terminus. It consists of seven amino acids, including one D-amino acid (D-phenylalanine), and is cyclized by an amide bond between glutamic and diaminopropionic acid side chains. The selectivity of PL8177 for MC1R was demonstrated against 72 other potential molecular targets, and its ability to reduce markers of inflammation and autoimmune disease has been demonstrated in preclinical studies [23]. In human blood, PL8177 inhibited tumor necrosis factor-α, a primary mediator of the host inflammatory response, to a similar degree as α-MSH. In a rat model of IBD, PL8177 reduced bowel inflammation compared with controls; and in mice with experimental autoimmune uveitis, retinal inflammation was significantly reduced in animals treated with PL8177 compared with untreated animals [23].
PL8177 has been granted orphan drug status by the US Food and Drug Administration for the treatment of noninfectious intermediate, posterior, pan and chronic anterior uveitis, which cause symptoms such as impaired vision, ocular ache, and photophobia. Phase II trials are planned using a subcutaneous (sc) injection formulation. An oral formulation is also being developed for the local treatment of ulcerative colitis and other inflammatory bowel diseases. In this article, we describe the pharmacokinetic properties of PL8177 after sc delivery, based on animal and human studies.
Methods
Preclinical Studies
All preclinical studies complied with the US Department of Agriculture’s Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals. Formulations of PL8177 were prepared weekly at nominal concentrations of 0.075, 0.2, 0.4, 0.6, 1.0, 2.0, and 5.0 mg/mL and were stored refrigerated at 2–8 °C until dosing.
Mouse
Adult male C57BL/6 mice weighing 25–30 g were obtained from Hilltop Lab Animals, Inc., (Scottdale, PA, USA). Mice were housed two per cage with a divider in an environmentally controlled room.
PL8177 was administered sc to the mice at single doses of 1.0 and 3.0 mg/kg. Between 0.5 and 1.0 mL of blood was collected via cardiac puncture from four mice each at 5, 15, 30, and 45 min and at 1, 2, 4, and 24 h after administration for pharmacokinetic analysis. Blood was collected into tubes containing 20 µL/mL of 0.16 Na2-EDTA and were centrifuged on ice at 4 °C for 15 min. Plasma was collected in a clean tube of 8 µL/mL aprotinin (~ 6000 TIU/mL, Sigma A6279) and frozen at − 80 °C.
Plasma concentrations of PL8177 and its active metabolite PL8435 (the carboxylic acid resulting from hydrolysis of the C-terminal carboxamide) were analyzed via liquid chromatography–mass spectrometry under non-GLP conditions. Plasma was treated with MeOH/H2O, vortexed for 1–2 min, and centrifuged at 3600 rpm for 10 min. Supernatant was transferred to a clean tube for analysis. Evaluated pharmacokinetic parameters included area under the plasma concentration–time curve (AUC) from 0 to 4 h (AUC4), AUC from 0 to 2 h (AUC2), maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), and elimination half-life (t½).
Rat
Male and female CD® Sprague Dawley rats were obtained from Charles River Laboratories (Portage, MI, USA), weighing 233–264 g (male) and 172–201 g (female) at randomization. Animals were housed two to three per cage with nonaromatic bedding in an environmentally controlled room.
All animals were observed daily for 14 days with respect to general health and any signs of disease. A 28-day toxicology study that included a toxicokinetic analysis was undertaken in 70 male and 70 female rats using sc PL1877 at doses of 0 (control), 1.0, 5.0, and 25.0 mg/kg/day. Blood samples were collected via the sublingual vein from cohorts of three animals per sex, per dose, and at each time point. Samples were taken at predose and at approximately 0.25, 0.5, 1, 2, 4, 8, and 24 h post-dose on Days 1 and 28. The animals were not fasted before blood collection.
Incurred sample reanalysis was performed on ≥ 7% of the samples to assess reproducibility of bioanalytical data. Samples were considered below quantification level if the lower limit of quantification (LLOQ) was < 5.0 ng/mL. Pharmacokinetic parameters were determined from mean concentration–time data using a noncompartmental module and included AUC from dosing until the last measurable concentration (AUClast), Cmax, tmax, and t½.
Dog
Beagles aged approximately 5–6 months were obtained from Marshall BioResources (North Rose, NY, USA). The dogs were housed in same-sex pairs of two in double-sized stainless-steel cages with plastic-coated flooring. All animals were observed daily for 14 days with respect to general health and any signs of disease. A 28-day toxicology study was undertaken in 16 males and 16 females that included a pharmacokinetic analysis of PL8177 when administered as either a single 20-mL sc injection or as two 10-mL sc injections at different sites at doses of 0 (control), 1.5, 8.0, and 40.0 mg/day administered once a day for 28 days. Plasma was collected from all animals on Days 1, 14, and 28 at the following times: predose and at 0.25, 0.5, 1, 2, 4, 8, and 24 h post-dose. For control animals, only blood samples that were collected at 0.5, 1, and 2 h post-dose were analyzed.
Plasma concentration versus time data were assessed using Phoenix WinNonlin (Certara, Princeton, NJ, USA) noncompartmental analysis function (linear trapezoidal rule for AUC calculations). Nominal dose values and sampling times were used for calculations. Lambda z (r2 > 0.75), the elimination rate constant upon which t½ and AUC from time 0 to infinity (AUC∞) were based, was determined based on automatically selected time points. Because the lambda z range was generally 4–24 h, only t½ values < 8 h were considered reliable. Additionally, only AUC∞ values with ≤ 20% extrapolated area were considered reliable. Because the majority of dogs failed to meet these criteria, AUC∞ and t½ were not reported; reported pharmacokinetic parameters included AUClast, Cmax, and tmax. The LLOQ was < 5.0 ng/mL.
Healthy Volunteers
A double-blind, randomized, placebo-controlled study was undertaken to evaluate the safety and tolerability of single and multiple sc doses of PL8177 in healthy volunteers. Secondary objectives are reported here, which included determining the pharmacokinetics of sc PL8177 and the presence of any potential metabolites.
Subjects
Healthy men and women aged 18–55 years were enrolled. Additional inclusion criteria were the ability to complete all screening period evaluations, to stay in the clinical research facility for the duration of the study, and to return for the follow-up visit; a body mass index of 18–30 kg/m2 with body weight ≥ 50 kg; and absence of any clinically significant medical or psychiatric illness or disease as determined by medical and surgical history, physical examination, weight, 12-lead electrocardiogram, and clinical laboratory tests. Subjects were excluded from the study if they had a current or recent history of gastrointestinal disease, a history of drug or alcohol abuse, or previous exposure to PL8177.
Study Design
PL8177 or placebo was administered in a double-blind fashion via single or multiple abdominal sc injections. In the single-dose arm, five dose levels of PL8177 were tested in two dosing cohorts of six subjects each and in three dosing cohorts of eight subjects each: 0.1 mg (4 active, 2 placebo); 0.5 mg (4 active, 2 placebo); 1.0 mg (6 active, 2 placebo); 3.0 mg (6 active, 2 placebo); 5.0 mg (6 active, 2 placebo). Blood samples were collected on Day 1 at ≤ 30 min before dosing and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, and 16 h post-dose; on Day 2 at 24, 30, and 36 h post-dose; and on Day 3 at 48 h post-dose. In the multiple-dose arm, two dose levels were tested in two dosing cohorts of 10 subjects each: 1.0 mg (8 active, 2 placebo) and 3.0 mg (8 active, 2 placebo) administered once daily for 7 days. Blood samples were collected on Day 1 at ≤ 30 min before dosing and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16, and 24 h post-dose; on Days 2–6 at the anticipated peak and trough times based on the analysis of the first two single-dose cohorts (≤ 2 collections per day per subject); on Day 7 ≤ 30 min before dosing and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, and 16 h after dosing; on Day 8 at 24, 30, and 36 h after Day 7 dosing; and on Day 9 at 48 h after Day 7 dosing (total of 49 blood samples).
Analysis
Plasma samples were analyzed for PL8177 using a validated bioanalytical method, and appropriate pharmacokinetic parameters were calculated for each formulation using noncompartmental methods. In the single- and multiple-dose study arms, evaluated parameters on Day 1 included Cmax, tmax, t½, AUC, AUC∞, and total body clearance from plasma (CL/F). AUC is calculated using the linear trapezoidal method from the time of dosing to the last quantifiable concentration. Parameters from Day 7 of the multiple-dose arm are presented as steady-state (ss) levels and as AUC over the entire dosing interval (AUCτ). Accumulation ratio (R) was also accounted for in applicable studies.
Liquid Chromatography–Mass Spectrometry
Samples were prepared for analysis (30.0 μL aliquots) using protein precipitation. The samples were injected onto a Waters XBridge Amide XP, 2 × 100-mm, 2.5-μm column (Waters Corporation, Milford, MA, USA). The analyte and the internal standard in the HPLC effluent were monitored on an Applied Biosystems API-4000 mass spectrometric system (Applied Biosystems, Foster City, CA, USA) with turbo-ion spray ionization (electrospray) in positive ion mode and multiple reaction monitoring detection. Peak areas of the m/z 499.0–110.0 Da product ions for PL-8177 are measured against the m/z 513.3–435.8 Da product ions for the internal standard. Quantitation was performed using a weighted (1/×2) linear least squares regression analysis generated from the ratio of the analyte peak area to the IS peak area measured in fortified plasma calibration standards. The linear analytical range was 5.00–500 ng/mL.
Results
Preclinical Studies
Mouse
The PL8177 plasma concentration over time curve exhibited a steep initial rise followed by steady decline over 4 h. Subcutaneous administration of 3 mg/kg PL8177 resulted in an AUC∞ of 1727 ng·h/mL, a Cmax of 2440 ng/mL, a t½ of 0.5 h, and a tmax of 0.25 h (Fig. 1, Table 1A). The active metabolite PL8435 was also observed in the plasma with an AUC∞ and Cmax 6–7 times lower than PL8177, but with the same t½ and tmax. Pharmacokinetics of PL8177 after the 1-mg/kg sc dose administration were generally proportional with a t½ of 0.26 h, and a tmax of 0.083 h (Fig. 1B). Pharmacokinetics of PL8177 after the 1-mg/kg intravenous (iv) dose administration resulted in an AUC∞ of 753 ng·h/mL and a t½ of 0.2 h (Fig. 1C); Cmax and tmax data were unavailable. The bioavailability (F) of the 1-mg/kg dose is 0.63. Active metabolite PL8435 was identified in plasma, although PL8435 was not dosed separately to determine any PK parameters and did not undergo stability or potency testing in any of these preclinical and clinical studies.
Fig. 1.
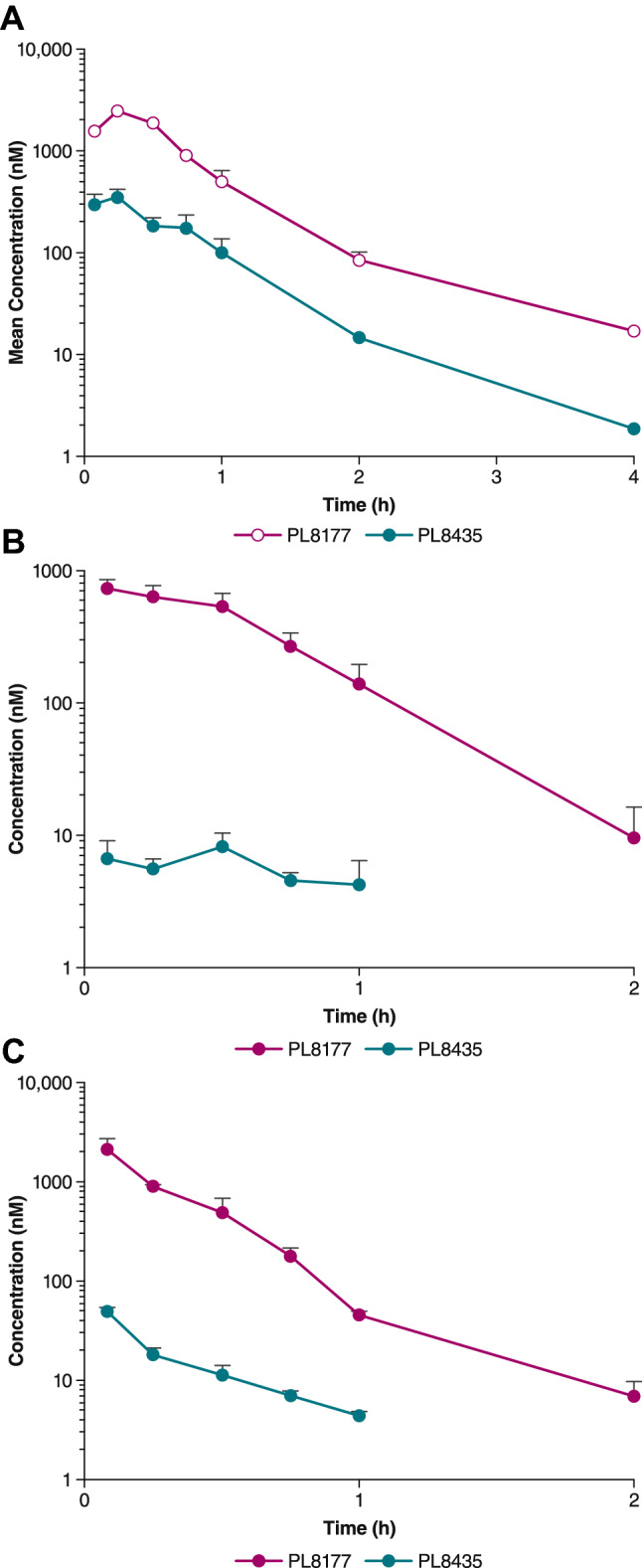
Plasma concentrations of PL8177 and PL8435 following administration of a single dose in mice of A 3 mg/kg sc, B 1 mg/kg sc, and C 1 mg/kg iv
Table 1.
Mean PL8177 pharmacokinetic parameters for mouse, rat, and beagle dogs
| Parameter | Dose | ||
|---|---|---|---|
| 3 mg/kg sc | 1 mg/kg sc | 1 mg/kg iv | |
| A. Mouse | |||
| AUC∞, ng·h/mL | 1727 | 476 | 753 |
| Cmax, ng/mL | 2440 | 703 | NC |
| CL/F, mL/min/kg | 22 | NC | NC |
| t½, h | 0.5 | 0.26 | 0.2 |
| tmax, h | 0.25 | 0.083 | NC |
| F | – | 0.63 | – |
| Dose mg/kg | Sex | tmax, h | Cmax (SE), ng/mL | AUClast (SE), ng·h/mL | t½, h | AUC∞, ng·h/mL | Vd/F, L/kg | CL/F, mL/min/kg |
|---|---|---|---|---|---|---|---|---|
| B. Rat PL8177 | ||||||||
| Day 1 | ||||||||
| 1.0 | F | 0.5 | 516 (41.7) | 880 (59.4) | 0.749 | 907 | 1.19 | 18.4 |
| M | 0.5 | 525 (86.7) | 1030 (78.4) | 0.868 | 1080 | 1.15 | 15.4 | |
| 5.0 | F | 0.5 | 1080 (36.8) | 4030 (122.0) | 5.91 | 4200 | 10.1 | 19.8 |
| M | 1.0 | 1180 (59.7) | 4300 (146.0) | 6.12 | 4510 | 9.78 | 18.5 | |
| 25.0 | F | 0.25 | 782 (60.8) | 4920 (181.0) | NC | NC | NC | NC |
| M | 0.5 | 666 (80.0) | 4930 (183.0) | NC | NC | NC | NC | |
| Day 14a | ||||||||
| 1.0 | F | 0.5 | 525 (5.82) | 712 (46.7) | NC | NC | NC | NC |
| M | 0.5 | 572 (65.4) | 708 (111.0) | NC | NC | NC | NC | |
| 5.0 | F | 1.0 | 1000 (91.6) | 1480 (71.8) | NC | NC | NC | NC |
| M | 1.0 | 1100 (28.05) | 1640 (59.6) | NC | NC | NC | NC | |
| 25.0 | F | 0.5 | 1190 (94.8) | 1710 (56.5) | NC | NC | NC | NC |
| M | 1.0 | 1140 (124.0) | 1860 (103.0) | NC | NC | NC | NC | |
| Day 28 | ||||||||
| 1.0 | F | 0.5 | 695 (138.0) | 818 (73.5) | 0.624 | 829 | 1.08 | 20.1 |
| M | 0.5 | 594 (64.6) | 933 (51.3) | 0.661 | 952 | 1.00 | 17.5 | |
| 5.0 | F | 0.25 | 1060 (158.0) | 3750 (247.0) | 9.29 | 4300 | 15.6 | 19.4 |
| M | 0.5 | 1080 (253.0) | 4550 (194.0) | 8.78 | 5160 | 12.3 | 16.2 | |
| 25.0 | F | 0.5 | 1180 (127.0) | 11,900 (406.0) | NC | NC | NC | NC |
| M | 0.25 | 1050 (71.6) | 12,600 (181.0) | NC | NC | NC | NC | |
| Dose, mg/kg | Sex | tmax (SD), h | Cmax (SD), ng/mL | Cmax/dose (SD) | AUClast (SD), ng·h/mL |
|---|---|---|---|---|---|
| C. Dog PL8177 | |||||
| Day 1 | |||||
| 1.5 | F | 1.3 (0.5) | 106 (43.5) | 70.5 (29.0) | 271 (93.1) |
| M | 1.1 (0.63) | 116 (30.2) | 77.4 (20.2) | 247 (16.2) | |
| 8.0 | F | 1.0 (0) | 615 (124) | 76.9 (15.5) | 1560 (141) |
| M | 0.75 (0.29) | 400 (117) | 50.0 (14.7) | 1280 (217) | |
| 40.0 | F | 0.56 (0.31) | 1010 (398) | 25.2 (9.94) | 3610 (792) |
| M | 0.88 (0.25) | 633 (257) | 15.8 (6.42) | 2640 (306) | |
| Day 14 | |||||
| 1.5 | F | 1.0 (0) | 128 (59.9) | 85.6 (39.9) | 258 (104) |
| M | 1.0 (0) | 129 (23.9) | 86.0 (15.9) | 239 (27.3) | |
| 8.0 | F | 0.63 (0.25) | 547 (52.4) | 68.4 (6.55) | 1650 (231) |
| M | 0.50 (0) | 359 (71.1) | 44.9 (8.89) | 1200 (108) | |
| 40.0 | F | 0.38 (0.14) | 1280 (305) | 31.9 (7.61) | 6220 (1250) |
| M | 0.50 (0) | 850 (196) | 21.2 (4.89) | 4760 (503) | |
| Day 28 | |||||
| 1.5 | F | 1.0 (0) | 96 (26.9) | 64.0 (17.9) | 210 (67.4) |
| M | 0.75 (0.29) | 98 (12.8) | 65.3 (8.51) | 194 (17.8) | |
| 8.0 | F | 0.63 (0.25) | 389 (114) | 48.6 (14.3) | 1250 (128) |
| M | 0.63 (0.25) | 275 (77.7) | 34.4 (9.72) | 1070 (159) | |
| 40.0 | F | 0.38 (0.14) | 1260 (407) | 31.5 (10.2) | 6620 (842) |
| M | 0.44 (0.13) | 860 (131) | 21.5 (13.28) | 4620 (250) | |
AUC area under the concentration–time curve, AUC0–t area under the concentration–time curve from time 0 to time of last measurable concentration, AUC2 area under the concentration–time curve from time 0 to 2 h, AUC4 area under the concentration–time curve from time 0 to 4 h, AUClast area under the concentration–time curve from time 0 to last measurable concentration, AUC∞ area under the concentration–time curve from time 0 to infinity, CL/F total clearance from plasma after administration, Cmax maximum plasma concentration, F bioavailability, iv intravenous, NC not calculated, sc subcutaneous, SD standard deviation, SE standard error of mean, t½ elimination half-life, tmax time to maximum plasma concentration, Vd/F apparent volume of distribution after extravascular administration
aDay 14 results are based on a truncated plasma concentration versus time curve (3 data points) and should be interpreted with caution
Rat
Plasma levels of PL8177 following 5-mg/kg, 1-mg/kg, and 25-mg/kg doses are shown in Figure 2. Evidence of systemic exposure was observed in all PL8177-treated rats following single- and multiple-dose sc administration of PL8177. Mean tmax values ranged from 0.25 to 1.0 h after a single dose, and from 0.25 to 0.50 h ours on Day 28 after multiple dosing (Table 1B). In general, tmax values appeared to be independent of dose. PL8177 plasma Cmax and AUClast increased in a less than dose-proportional manner over the full 25-fold dose range (1–25 mg/kg) on both Days 1 and 28. Mean t½ values ranged from 0.62 to 0.87 h at the 1-mg/kg dose and from 5.91 to 9.29 h at the 5-mg/kg dose on Days 1 and 28. Mean Cmax values on the 2 days ranged from 516 to 695 ng/mL for the 1-mg/kg dose, and from 666 to 1180 ng/mL for the 25-mg/kg dose. Mean CL/F values across all doses in both males and females ranged from 15.4 to 20.1 mL/min/kg and appeared to be dose independent throughout the study period.
Fig. 2.
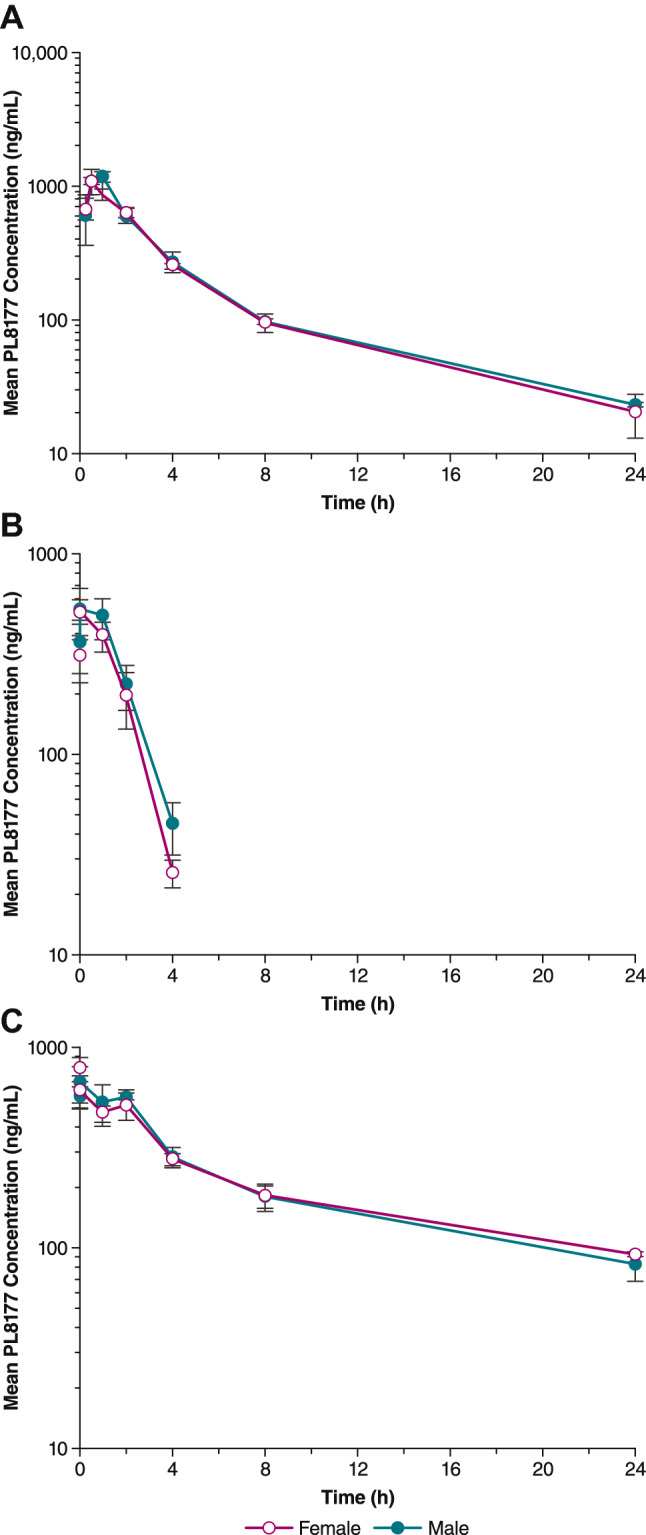
Mean PL8177 plasma concentration versus time following single subcutaneous administration in rats on Day 1 of A 5 mg/kg, B 1 mg/kg, and C 25 mg/kg
Dog
All dogs treated with PL8177 had evidence of systemic exposure following single- and multiple-dose sc administration. Plasma concentrations after 28 days of dosing with 8-mg PL8177 are shown in Fig. 3. After a single dose of PL8177, mean tmax values ranged from 0.56 to 1.3 h for all doses evaluated (Table 1C). Repeated dosing resulted in tmax values ranging from 0.38 to 1.0 h on Days 14 and 28. Values for tmax appeared to decrease with increasing dose. Mean plasma Cmax increased over the full dose range on all days, but in a manner that was less than dose proportional. Mean Cmax ranged from 96 to 129 ng·h/mL for the 1.5-mg dose, 275–615 ng·h/mL for the 8.0-mg dose, and 633.0–1280.0 ng·h/mL for the 40.0-mg dose for the 3 time points. Plasma AUClast values increased in a dose-proportional manner on Days 14 and 28 but not on Day 1. A < 2-fold difference was observed between mean Cmax and AUClast values with multiple-dose versus single-dose administration. There was no consistent evidence of sex differences. Accumulation ratios were almost identical for male and female dogs. For 1.5- and 9.0-mg doses, the accumulation ratio (Rac) as calculated by AUClast, day 28/AUClast, day 1 was 0.8, and for the 40-mg dose Rac was 1.8.
Fig. 3.
Mean PL8177 plasma concentration versus time following multiple subcutaneous administrations in dogs on Day 28 of A 8 mg, B 1.5 mg, and C 40 mg
Human Studies
In both the single-dose and multiple-dose study arms, the PL8177 concentration versus time curves showed distinct ascending and descending portions; distribution and elimination phases were evident, especially at higher doses, and ≤ 1% of intact drug was excreted in the urine. In both arms, Cmax was achieved by 45 min post-dose. Cmax was generally reached 30–45 min after dosing in the single-dose arm, and 15–45 min after dosing in the multiple-dose arm (Tables 2, 3).
Table 2.
Pharmacokinetic parameters for all single-dose human cohorts
| Dose, mg | Statistic | Cmax, ng/mL | AUC0–t, ng·h/mL | AUC∞, ng·h/mL | CL/F, L/h | t½, h | tmax, h | Vd/F, L | Cmax/dose, ng/mL/mg | AUC∞/dose, ng·h /mL/mg |
|---|---|---|---|---|---|---|---|---|---|---|
| 5.0 | n | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean (SD) | 87.8 (31.1) | 226 (36.0) | 240 (37.1) | 21.3 (3.6) | 16.3 (2.9) | 502 (133) | 17.6 (6.2) | 48.0 (7.4) | ||
| Median | 80.9 | 235 | 246 | 20.3 | 17.3 | 0.8 | 496 | 16.2 | 49.2 | |
| Geometric mean | 83.4 | 224 | 237 | 21.1 | 16.1 | 488 | 16.7 | 47.5 | ||
| 3.0 | n | 6 | 6 | 6 | 6 | 6 | 6 | 4 | 6 | 4 |
| Mean (SD) | 54.4 (11.3) | 138 (18.9) | 158 (19.4) | 19.3 (2.5) | 19.5 (17.5) | 375 (154) | 18.1 (3.8) | 51.3 (7.7) | ||
| Median | 49.1 | 138 | 163 | 18.4 | 12.3 | 0.6 | 394 | 16.4 | 51.1 | |
| Geometric mean | 53.5 | 137 | 157 | 19.1 | 15.2 | 348 | 17.8 | 50.9 | ||
| 1.0 | n | 6 | 6 | 6 | 6 | 6 | 6 | 3 | 6 | 3 |
| Mean (SD) | 17.4 (5.0) | 40.3 (9.4) | 46.2 (12) | 22.9 (5.9) | 6.7 (3.1) | 173 (35.1) | 17.4 (5.0) | 35.4 (1.8) | ||
| Median | 18.3 | 39.2 | 45.5 | 22.7 | 6 | 0.6 | 186 | 18.3 | 34.5 | |
| Geometric mean | 16.7 | 39.4 | 44.9 | 22.3 | 6.1 | 171 | 16.7 | 35.4 | ||
| 0.5 | n | 4 | 4 | 4 | 4 | 4 | 4 | 2 | 4 | 2 |
| Mean (SD) | 8.2 (3.7) | 19.9 (14.9) | 22.4 (15.8) | 28.6 (12.0) | 2.8 (1.2) | 74.4 (9.7) | 16.4 (7.5) | 59.9 (45.8) | ||
| Median | 7.2 | 13.3 | 15 | 33.6 | 2.6 | 0.5 | 74.4 | 14.3 | 59.9 | |
| Geometric mean | 7.6 | 16.8 | 19.4 | 25.8 | 2.6 | 74.1 | 15.3 | 50.4 | ||
| 0.1 | n | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Mean (SD) | 2.3 (1.0) | 2.7 (0.8) | 3.5 (0.8) | 29.8 (7.2) | 0.9 (0.3) | 36.1 (9.3) | 23.4 (10.1) | 34.9 (7.82) | ||
| Median | 1.9 | 2.6 | 3.5 | 28.5 | 0.8 | 0.6 | 36.6 | 18.9 | 35.3 | |
| Geometric mean | 2.2 | 2.6 | 3.4 | 29.2 | 0.8 | 35.1 | 22.1 | 34.2 |
AUC0–t area under the concentration–time curve from time 0 to time of last measurable concentration, AUC∞ area under the concentration–time curve from time 0 to infinity, CL/F total clearance from plasma after administration, Cmax maximum plasma concentration, SD standard deviation, t½ elimination half-life, tmax time to maximum plasma concentration, Vd/F apparent volume of distribution after extravascular administration
Table 3.
Pharmacokinetic parameters for human study (multiple-dose arm): Day 1
| Dose, mg | Statistic | Cmax, ng/mL | AUC0–t, ng·h/mL | AUC∞, ng·h/mL | CL/F, L/h | t½, h | tmax, h | Vd/F, L | Cmax/dose, ng/mL/mg | AUC∞/dose, ng·h/mL/mg |
|---|---|---|---|---|---|---|---|---|---|---|
| 1.0 | n | 8 | 8 | 6 | 6 | 6 | 8 | 6 | 8 | 6 |
| Mean (SD) | 23.0 (8.9) | 49.4 (15.9) | 52.3 (14.2) | 20.1 (4.6) | 4.3 (1.6) | 120 (44.4) | 23.0 (8.8) | 52.3 (14.2) | ||
| Median | 18.5 | 44.9 | 49.4 | 20.2 | 3.5 | 0.5 | 106 | 18.5 | 49.4 | |
| Geometric mean | 21.7 | 47.4 | 50.9 | 19.6 | 4.1 | 115 | 21.7 | 50.9 | ||
| 3.0 | n | 8 | 8 | 7 | 7 | 7 | 8 | 7 | 8 | 7 |
| Mean (SD) | 54.4 (9.6) | 154 (30.2) | 172 (30.6) | 17.9 (3.0) | 9.7 (2.1) | 257 (94.6) | 18.1 (3.2) | 57.2 (10.2) | ||
| Median | 50.0 | 145.0 | 159.0 | 18.9 | 9.6 | 0.6 | 261 | 16.7 | 53.0 | |
| Geometric mean | 53.7 | 152.0 | 169.0 | 17.7 | 9.5 | 242 | 17.9 | 56.5 |
AUC0–t area under the concentration–time curve from time 0 to time of last measurable concentration, AUC∞ area under the concentration–time curve from time 0 to infinity, CL/F total clearance from plasma after administration, Cmax maximum plasma concentration, SD standard deviation, t½ elimination half-life, tmax time to maximum plasma concentration, Vd/F apparent volume of distribution after extravascular administration on Day 1
In the single-dose arm, measurable amounts of PL8177 were detectable beginning 15 min after dosing and persisted for up to 48 h at higher doses (Fig. 4). Geometric mean Cmax values increased from 2.2 to 83.4 ng/mL as doses increased from 0.1 to 5.0 mg, and geometric mean AUC∞ values increased from 3.4 to 237 ng·h/mL (Table 2). The plasma t½ (mean ± standard deviation [SD]) ranged from 0.9 ± 0.3 h for the 0.1-mg dose to 16.3 ± 2.9 h for the 5-mg dose.
Fig. 4.
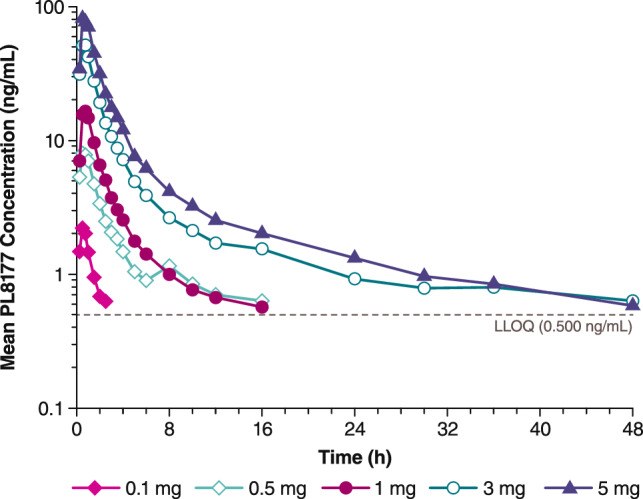
Mean plasma PL8177 concentration versus time following single-dose administration in healthy human volunteers. LLOQ lower limit of quantitation
Similar results were observed in the multiple-dose arm (Fig. 5). Following the first dose on Day 1, geometric mean Cmax values were 21.7 ng/mL after 1.0 mg PL8177 and 53.7 ng/mL after 3.0 mg PL8177 (Table 3). Geometric mean AUC∞ values after the first dose were 50.9 ng·h/mL for 1.0 mg compared with 169 ng·h/mL for 3.0 mg. The t½ (mean ± SD) on the first day of dosing was 4.3 ± 1.6 h and 9.7 ± 2.1 h with the 1.0- and 3.0-mg doses, respectively. Following the last dose on Day 7, steady-state parameters were assessed (Table 4). Geometric mean maximum steady-state plasma concentration (Cmax,ss) values were 20.1 ng/mL after the 1.0-mg dose and 57.2 ng/mL after the 3.0-mg dose. Geometric mean AUC∞ values after the last dose were 54.3 ng·h/mL for 1.0 mg and 199 ng·h/mL for 3.0 mg; t½ values (mean ± SD) for the 1.0- and 3.0-mg dosing groups were 7.1 ± 2.4 and 12.6 ± 3.8 h, respectively. Dose linearity was determined to be inconclusive for both single-dose (Fig. 6) and multiple-dose arms (Figure 7) for AUC∞ and Cmax as the 90% CI for the slope, although bracketing 1, extended beyond the critical upper or lower bound (Table 5). Less than a 36% increase in accumulation was observed after 7 days of dosing. The active metabolite PL8435 was identified; no other unique human metabolites were detected. PL8435 did not undergo stability or potency testing.
Fig. 5.
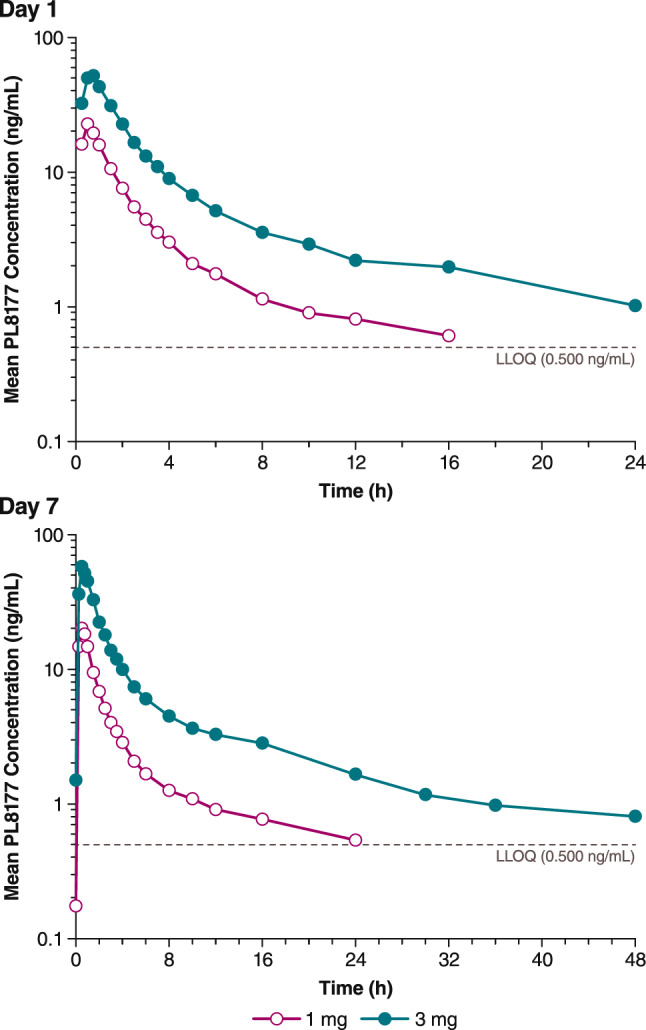
Mean plasma PL8177 concentration versus time following administration on Day 1 (top) and Day 7 (bottom) in healthy human volunteers. LLOQ lower limit of quantitation
Table 4.
Pharmacokinetic parameters for human study (multiple-dose arm): Day 7
| Dose, mg | Statistic | Cmax, ng/mL | AUCτ, ng·h/mL | AUC∞, ng·h/mL | t½, h | tmax, h | Vd/Fss, L | AI | Cmax/dose, ng/mL/mg | AUCτ/dose, ng·h/mL/mg |
|---|---|---|---|---|---|---|---|---|---|---|
| 1.0 | n | 8 | 8 | 5 | 5 | 8 | 5 | 5 | 8 | 8 |
| Mean (SD) | 20.9 (6.2) | 52.9 (15.0) | 57.2 (21.3) | 7.1 (2.4) | 200 (72.3) | 1.1 (0.1) | 20.9 (6.17) | 52.9 (15.0) | ||
| Median | 22.2 | 48.3 | 51.8 | 7.2 | 0.5 | 174 | 1.1 | 22.2 | 48.3 | |
| Geometric mean | 20.1 | 51.2 | 54.3 | 6.7 | 191 | 1.1 | 20.1 | 51.2 | ||
| 3.0 | n | 8 | 8 | 6 | 6 | 8 | 6 | 6 | 8 | 8 |
| Mean (SD) | 58.5 (13.6) | 177.0 (36.0) | 202.0 (37.5) | 12.6 (3.8) | 330 (111) | 1.4 (0.2) | 19.5 (4.52) | 59.1 (12.0) | ||
| Median | 52.7 | 171.0 | 200.0 | 11.4 | 0.5 | 288 | 1.3 | 17.6 | 57.1 | |
| Geometric mean | 57.2 | 174.0 | 199.0 | 12.3 | 316 | 1.4 | 19.1 | 58.1 |
AI accumulation index, AUCτ area under the concentration–time curve over dosing interval, AUC∞ area under the concentration–time curve from time 0 to infinity, Cmax maximum plasma concentration, SD standard deviation, t½ elimination half-life, tmax time to maximum plasma concentration, Vd/Fss apparent volume of distribution after extravascular administration on Day 7
Fig. 6.

Dose linearity for plasma PL8177 A Cmax and B AUC∞ in single-dose administration cohort. AUC∞ area under the plasma concentration–time curve from 0 to infinity, Cmax maximum plasma concentration
Fig. 7.

Dose linearity for plasma PL8177 A Cmax and B AUC∞ in multiple-dose administration cohort. AUC∞ area under the plasma concentration–time curve from 0 to infinity, Cmax maximum plasma concentration
Table 5.
Cmax and AUC∞ test for dose linearity
| Dosing day | Parameter | Slope (90% CI) | Critical lower/upper bound |
|---|---|---|---|
| A. Single-dose cohorts | |||
| AUC∞ | 1.08 (1.01–1.16) | 0.943, 1.06 | |
| Cmax | 0.953 (0.872–1.03) | ||
| B. Multiple-dose cohorts | |||
| 1 | Cmax | 0.824 (0.609–1.04) | 0.797, 1.2 |
| 7 | Cmax, ss | 0.950 (0.738–1.16) | |
| 1 | AUC∞ | 1.09 (0.903–1.29) | |
| 1 | AUCτ | 1.03 (0.833–1.23) | |
| 7 | AUCτ, ss | 1.12 (0.928–1.30) | |
AUC∞ area under the concentration–time curve from time 0 to infinity, AUCτ area under the concentration–time curve over dosing interval, AUCτ,ss steady-state area under the concentration–time curve over dosing interval, Cmax maximum plasma concentration, Cmax,ss maximum steady-state plasma concentration
The plasma concentration–time curves in human trials display a polyexponential decline that becomes obvious at higher doses. At lower doses, the plasma concentrations fall below the LLOQ by the time the terminal phase has occurred; therefore, the t½ for the lower doses is reflective of a distribution phenomenon (median times for last measurable concentrations are 2, 6, 16, 30, and 48 h for 0.1-, 0.5-, 1-, 3-, and 5-mg doses). Across both the single-dose and multiple-dose arms, there were no reports of adverse events (AEs) leading to study discontinuation, serious AEs, or severe AEs. Mild or moderate AEs occurred with similar frequencies across subjects who received treatment and placebo. In the single-dose arm, AEs occurred in nine subjects (90.0%) who received placebo and in two to six subjects (50.0–100%) across PL8177 dose levels; the most common AEs across all groups were injection-site reactions including injection-site pain, erythema, and induration. In the multiple-dose arm, AEs occurred in three subjects (75.0%) dosed with placebo and in 16 subjects (100%) who received any dose of PL8177. Similar to the single-dose arm, the most commonly reported AEs were injection-site pain, erythema, and induration; the second most commonly reported AE type was skin hyperpigmentation, experienced by nine subjects (45.0%) who received PL8177.
Discussion
The results of these studies suggest a promising pharmacokinetic profile for PL8177 when dosed subcutaneously. In humans, detectable plasma levels were seen within 15 min after a single dose and persisted for up to 48 h at higher doses. Dose-related increases in exposure were seen, although dose linearity could not be demonstrated. In multiple-dose studies, maximum plasma concentrations were reached 15–45 min after dosing. The amount of dose excreted unchanged in the urine was ≤ 1%, and accumulation was minimal. Because α-MSH has a very short t½ of ~ 20 min [24], an important focus of melanocortin peptide development has been to improve biological stability [25]. In multiple-dose studies, the t½ (mean ± SD) of PL8177 ranged from 9.7 ± 2.1 h after the first dose of 3.0 mg to 12.6 ± 3.8 h after 7 days of 3.0-mg dosing. An active metabolite, PL8435, was identified in rats, dogs, and humans, which may offer additional drug exposure to an active MC1R agonist. The t½ calculated in mice for different doses was similar for both PL8177 and PL8435, suggesting that the t½ for PL8435 is more likely a representation of the rate of formation due to the elimination of PL8177. Overall, PL8177 was well tolerated when administered as single and multiple ascending doses.
Published studies regarding other synthetic melanocortin agonists currently in development indicate a range of pharmacokinetic characteristics. Subcutaneous administration of the selective MC1R agonist BMS-470539 was associated with a Cmax of 11 µM and a short t½ of 1.7 h after a dose of 33 µmol/kg [26]. Subcutaneous administration of the pan-MCR agonist bremelanotide resulted in dose-related increases in plasma levels, with a tmax of 0.5–1.0 h and a t½ of 1.9–2.7 h [27].
Conclusion
The studies reported here demonstrate that sc administration of PL8177 results in a consistent pharmacokinetic profile, with levels of exposure that indicate pharmacologically active concentrations of PL8177 at the targeted MC1R. This is also consistent with in vitro cAMP functional studies of PL8177 on melanocortin receptor subtypes; PL8177 had the strongest potency relative to other receptor subtypes as determined by a half maximal effective concentration of 570 pM. The amount of dose excreted unchanged in the urine was ≤ 1%, and accumulation was minimal. Rapid absorption was seen in healthy volunteers, and multiple-dose administration over 7 days resulted in pharmacokinetic characteristics similar to those observed after single-dose administration. These results support the continued development of PL8177 for systemic administration to treat a variety of inflammatory conditions.
Acknowledgements
Editorial assistance was provided by The Curry Rockefeller Group, LLC, which was funded by Palatin Technologies, Inc.
Declarations
Funding
This work was funded by Palatin Technologies, Inc.
Conflict of interest
John Dodd, Carl Spana, Robert Jordan, Marie Makhlina, Wei Yang, and Keith Barnett are employees of Palatin Technologies, Inc. Luana Pesco Koplowitz and Barry Koplowitz are paid consultants for Palatin Technologies, Inc.
Availability of data and material
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics approval
The study was reviewed and approved by the IntegReview institutional review board and was conducted in accordance with the principles outlined in the Declaration of Helsinki and its amendments.
Consent to participate
All subjects provided written informed consent prior to study participation.
Author contributions
JD and RJ contributed to the design and oversight of the preclinical and clinical studies. MM contributed to the data handling, design, and oversight of the preclinical studies. LPK and BK provided the pharmacokinetic database, analysis, and report for the clinical study. KB was responsible for clinical operations oversight. WHY contributed to the conception and synthesis of PL8177. CS contributed to the design of preclinical and clinical studies. All authors contributed to manuscript development and reviewed and approved of the final draft.
References
- 1.Catania A, Lonati C, Sordi A, et al. The melanocortin system in control of inflammation. ScientificWorldJournal. 2010;10:1840–1853. doi: 10.1100/tsw.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catania A, Gatti S, Colombo G, et al. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol Rev. 2004;56(1):1–29. doi: 10.1124/pr.56.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Manna SK, Aggarwal BB. Alpha-melanocyte-stimulating hormone inhibits the nuclear transcription factor NF-kappa B activation induced by various inflammatory agents. J Immunol. 1998;161(6):2873–2880. [PubMed] [Google Scholar]
- 4.Luger TA, Scholzen TE, Brzoska T, et al. New insights into the functions of alpha-MSH and related peptides in the immune system. Ann N Y Acad Sci. 2003;994:133–140. doi: 10.1111/j.1749-6632.2003.tb03172.x. [DOI] [PubMed] [Google Scholar]
- 5.Perretti M, Leroy X, Bland EJ, et al. Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol Sci. 2015;36(11):737–755. doi: 10.1016/j.tips.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Cone RD, Mountjoy KG, Robbins LS, et al. Cloning and functional characterization of a family of receptors for the melanotropic peptides. Ann N Y Acad Sci. 1993;680:342–363. doi: 10.1111/j.1749-6632.1993.tb19694.x. [DOI] [PubMed] [Google Scholar]
- 7.Mountjoy KG, Robbins LS, Mortrud MT, et al. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257(5074):1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 8.Malik S, Dolan TM, Maben ZJ, et al. Adrenocorticotropic hormone (ACTH) responses require actions of the melanocortin-2 receptor accessory protein on the extracellular surface of the plasma membrane. J Biol Chem. 2015;290(46):27972–27985. doi: 10.1074/jbc.M115.668491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacNeil DJ, Howard AD, Guan X, et al. The role of melanocortins in body weight regulation: opportunities for the treatment of obesity. Eur J Pharmacol. 2002;440(2–3):141–157. doi: 10.1016/S0014-2999(02)01425-5. [DOI] [PubMed] [Google Scholar]
- 10.Hagan MM, Rushing PA, Schwartz MW, et al. Role of the CNS melanocortin system in the response to overfeeding. J Neurosci. 1999;19(6):2362–2367. doi: 10.1523/JNEUROSCI.19-06-02362.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van der Ploeg LH, Martin WJ, Howard AD, et al. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci USA. 2002;99(17):11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chida D, Nakagawa S, Nagai S, et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc Natl Acad Sci USA. 2007;104(46):18205–18210. doi: 10.1073/pnas.0706953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thody AJ. Alpha-MSH and the regulation of melanocyte function. Ann N Y Acad Sci. 1999;885:217–229. doi: 10.1111/j.1749-6632.1999.tb08679.x. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe T, Hiltz ME, Catania A, et al. Inhibition of IL-1 beta-induced peripheral inflammation by peripheral and central administration of analogs of the neuropeptide alpha-MSH. Brain Res Bull. 1993;32(3):311–314. doi: 10.1016/0361-9230(93)90192-E. [DOI] [PubMed] [Google Scholar]
- 15.Ceriani G, Diaz J, Murphree S, et al. The neuropeptide alpha-melanocyte-stimulating hormone inhibits experimental arthritis in rats. NeuroImmunoModulation. 1994;1(1):28–32. doi: 10.1159/000097087. [DOI] [PubMed] [Google Scholar]
- 16.Lipton JM, Ceriani G, Macaluso A, et al. Antiinflammatory effects of the neuropeptide alpha-MSH in acute, chronic, and systemic inflammation. Ann N Y Acad Sci. 1994;741:137–148. doi: 10.1111/j.1749-6632.1994.tb39654.x. [DOI] [PubMed] [Google Scholar]
- 17.Luger TA, Brzoska T, Scholzen TE, et al. The role of alpha-MSH as a modulator of cutaneous inflammation. Ann N Y Acad Sci. 2000;917:232–238. doi: 10.1111/j.1749-6632.2000.tb05388.x. [DOI] [PubMed] [Google Scholar]
- 18.Rajora N, Boccoli G, Burns D, et al. Alpha-MSH modulates local and circulating tumor necrosis factor-alpha in experimental brain inflammation. J Neurosci. 1997;17(6):2181–2186. doi: 10.1523/JNEUROSCI.17-06-02181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Getting SJ, Riffo-Vasquez Y, Pitchford S, et al. A role for MC3R in modulating lung inflammation. Pulm Pharmacol Ther. 2008;21(6):866–873. doi: 10.1016/j.pupt.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Rajora N, Boccoli G, Catania A, et al. Alpha-MSH modulates experimental inflammatory bowel disease. Peptides. 1997;18(3):381–385. doi: 10.1016/S0196-9781(96)00345-2. [DOI] [PubMed] [Google Scholar]
- 21.Lee DJ, Biros DJ, Taylor AW. Injection of an alpha-melanocyte stimulating hormone expression plasmid is effective in suppressing experimental autoimmune uveitis. Int Immunopharmacol. 2009;9(9):1079–1086. doi: 10.1016/j.intimp.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishida T, Miyata S, Itoh Y, et al. Anti-inflammatory effects of alpha-melanocyte-stimulating hormone against rat endotoxin-induced uveitis and the time course of inflammatory agents in aqueous humor. Int Immunopharmacol. 2004;4(8):1059–1066. doi: 10.1016/j.intimp.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Spana C, Taylor AW, Yee DG, et al. Probing the role of melanocortin type 1 receptor agonists in diverse immunological diseases. Front Pharmacol. 2019;9:1535. doi: 10.3389/fphar.2018.01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashton H, Millman JE, Telford R, et al. Psychopharmacological and endocrinological effects of melanocyte stimulating hormones in normal man. Psychopharmacology. 1977;55(2):165–172. doi: 10.1007/BF01457853. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed TJ, Montero-Melendez T, Perretti M, et al. Curbing inflammation through endogenous pathways: focus on melanocortin peptides. Int J Inflam. 2013;2013:985815. doi: 10.1155/2013/985815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang L, McIntyre KW, Gillooly KM, et al. A selective small molecule agonist of the melanocortin-1 receptor inhibits lipopolysaccharide-induced cytokine accumulation and leukocyte infiltration in mice. J Leukoc Biol. 2006;80(4):897–904. doi: 10.1189/jlb.1204748. [DOI] [PubMed] [Google Scholar]
- 27.Rosen RC, Diamond LE, Earle DC, et al. Evaluation of the safety, pharmacokinetics and pharmacodynamic effects of subcutaneously administered PT-141, a melanocortin receptor agonist, in healthy male subjects and in patients with an inadequate response to Viagra. Int J Impot Res. 2004;16(2):135–142. doi: 10.1038/sj.ijir.3901200. [DOI] [PubMed] [Google Scholar]