

Abstract
Purpose
Histone deacetylases (HDACs) play a vital role in the epigenetic regulation of gene expression due to their overexpression in several cancer forms. Therefore, these enzymes are considered as a potential anticancer drug target. Different synthetic and natural structures have been studied as HDACs inhibitors; based on available structural design information, the capping group is important for the biological activity due to the different interactions in the active site entrance. The present study aimed to analyze high substituted pyridine as a capping group, which included carrying out the synthesis, antiproliferative activity analysis, and docking studies of these novel compounds.
Methods
To achieve the synthesis of these derivatives, four reaction steps were performed, generating desired products 15a-k. Their effects on cell proliferation and gene expression of p21, cyclin D1, and p53 were determined using the sulphorhodamine B (SRB) method and quantitative real-time polymerase chain reaction. The HDAC1, HDAC6, and HDAC8 isoforms were used for performing docking experiments with our 15a-k products.
Result
The products 15a-k were obtained in overall yields of 40–71%. Compounds 15j and 15k showed the highest antiproliferative activity in the breast (BT-474 and MDA-MB-231) and prostate (PC3) cancer cell lines at a concentration of 10 µM. These compounds increased p21 mRNA levels and decreased cyclin D1 and p53 gene expression. The docking study showed an increment in the strength, and in the number of interactions performed by the capping moiety of the tested molecules compared with SAHA; interactions displayed are mainly van der Waals, π-stacking, and hydrogen bond.
Conclusion
The synthesized compounds 2-thiophene (15j) and 2-furan (15k) pyridine displayed cell growth inhibition, regulation of genes related to cell cycle progression in highly metastatic cancer cell lines. The molecular coupling analysis performed with HDAC1, HDAC6 and HDAC8 showed an increment in the number of interactions performed by the capping moiety and consequently in the strength of the capping group interaction.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s40199-021-00406-8.
Keywords: Capping framework, High substituent pyridine, HDACs, Antiproliferative compounds, Docking, Gene expression
Introduction
Histone deacetylases (HDACs) are enzymes responsible for the regulation of gene expression [1, 2], by deacetylation of residues of lysine and arginine in histones and other proteins [3, 4]. Several studies showed an abnormal overexpression of HDACs, and such behavior is related to different types of cancer [5] and its different stages (initiation and progression) [6]. This allows us to visualize them as a therapeutic objective for treating different diseases, including cancer. The insight gained in diverse analyses reveal that abnormal expression of tumor suppressor genes (TSG) is modified in a cancer cell; nevertheless, it can be silenced using epigenetic drugs. The use of epigenetic modulators has been shown a change in the gene expression in cancer cells, resulting in apoptosis, cell differentiation, and recognition of cancer cells by the immune system [7, 8]. For example, those promoting the DNA hypermethylation catalyzed by DNA methyltransferases (DNMTs) and those allowing the expansion of chromatin, permitting the genetic transcription to take place catalyzed by HDACs [9]. The HDACs family has eleven zinc-dependent enzymes [10], the development of molecules to improve the biological activity of HDACs inhibitors has been widely studied. Among the strategies of such improvement are an efficient block of the metal and the strength of the displayed interactions of the capping group on the enzyme surface.
Synthetic HDACs inhibitors such as vorinostat (1) (SAHA), belinostat (2) (PXD 101), and panobinostat (3) (LDH589) have shown a broad spectrum of epigenetic activities, all these molecules characterized by containing a hydroxamic group as the chelating moiety. These drugs have been approved by the FDA for the treat different types of cancer [11–13]. Nowadays, there are interesting compounds in advanced preclinical phases, such as mocetinostat (4) [14], entinostat (5) [15], and chidamide (6) [16, 17], which draws the attention of these molecules is the benzamides as chelator and pyridine nuclei from nicotinic acid as capping group. This heterocycle is the subject of the present research, the principal difference with the molecules previously mentioned is the substitution in the position C2, which was considered as a potential metal coordination moiety, focusing on the two nitrogen atoms. Most of these inhibitors present a highly known pharmacophoric model consisting of three fragments: the capping framework, linker, and zinc-binding group (ZBG) (Fig. 1) [18, 19].
Fig. 1.

Compounds approved by the FDA and clinical candidates with inhibitor activity of HDACs
Several studies have shown the pharmacophoric models use to generate new compounds displaying HDAC inhibitory activity, some of which present interesting structural modifications in the capping group [20–24]. Some of such strategies are the increase of the hydrophobic region by including polycycles, or adding several substituents on the aromatic ring in different positions [23, 25], in addition to the inclusion of several heterocycles [26] as coumarin [27] and pyrrole [28] have been used. These nuclei have hydrophobic characteristics which can confer specific recognition of the HDAC at the edge of the active site cavity. These changes have generated compounds with similar or higher efficacy to SAHA. By considering all this research in the literature, it was planned to extend the knowledge of the relevant interactions between the biological target and the anticancer drugs, trying to develop new antiproliferative compounds. Here, we present the synthesis of novel compounds where the capping group shows a pyridine core highly substituted, which has not been investigated until now. The antiproliferative activity and their effects on genes implicated in the cell cycle were investigated in breast and prostate cancer cell lines. Besides, docking experiments were performed with HDAC1, HDAC6, and HDAC8 as biological targets.
Methods
Chemistry
Reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and used without prior purification. Melting points were determined on an Electrothermal digital 90,100 melting point apparatus and were uncorrected. The progress of reactions was monitored by thin-layer chromatography (TLC) using silica gel 60-F254 coated aluminum sheets in hexane/ethyl acetate (7:3) and visualized by a 254 nm UV lamp. The compounds were purified by column chromatography packed with silica gel of 230–400 mesh of Machery-Nagel Company using the system hexane/ethyl acetate. Pure compounds were characterized, 1H NMR and 13C NMR were recorded for solutions in CDCl3 with Me4Si as internal standard on Bruker UltraShield (500 MHz) instrument. High-resolution mass spectra were performed using a spectrometer Bruker ESI-QTOFMS maXis impact, and the samples were analyzed in combination with methyl stearate as an internal standard. CEM discovery SP microwave reactor was used for reactions. Chemical shifts are given in ppm (δ); multiplicities are indicated by s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), m (multiplet), or bs (broad singlet). X-ray data were collected on Oxford Diffraction Gemini “A” diffractometer with a CCD area detector.
General procedure for the synthesis ethyl 5-cyano-2-methyl-6-oxo-4-(aryl)-1,6-dihydropyridine-3-carboxylate (10a-k)
To obtain the compounds 10a-k, it was followed previously reported methodology, which does not deserve an in-depth analysis. The 4H-pyrans (8a-k) were obtained using different aldehydes (7a-k), ethyl acetoacetate, and malononitrile in high yield. Subsequently, the compounds 8a-k were transformed to tetrahidropyridin-2-one (9a-k) employing acid conditions. The latter compounds were oxidized to produce the 10a-k derivatives (Scheme 1) [29, 30].
Scheme 1.

Reagent and reaction conditions for 15a-k. i) POCl3, DMA, 106 °C, 7 d, 90–98%. ii) TEA, EtOH, MW, 120 °C, 3–4 h, 78–90%. iii) LiOH, THF/H2O, rt, 12 h, 78–97%. iv) NH2OH·HCl, DMAP, CDI, DCM, rt, 12 h, 75–84%
General procedure for the synthesis ethyl 4-(aryl)-6-chloro-5-cyano-2-methylnicotinate (11a-k)
A mixture of 2-pyridone derivative 10a-k (1 mmol), phosphorus oxychloride (42 mmol), and N,N-dimethylaniline (1.9 mmol) was heated under reflux for 6–7 days. After the reaction was finished monitored by TLC, the suspension was cooled down to room temperature, and the solution was quenched in a mixture of water/ice to remove the excess of POCl3; after quenching, the solution was kept under stirring, observing the precipitation of the product which was collected by vacuum filtration. The solid was dried at room temperature to afford a pure compound 11a-k.
Ethyl 6-chloro-5-cyano-2-methyl-4-phenylnicotinate (11a)
White solid; yield: 92%; mp:108–109 °C; 1H NMR (500 MHz, CDCl3) δ 7.75 – 7.42 (m, 3H), 7.44 – 7.21 (m, 2H), 4.05 (q, J = 7.1 Hz, 2H), 2.67 (s, 3H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.0, 160.2, 154.4, 153.0, 134.1, 130.3, 128.9, 128.9, 128.2, 114.0, 108.4, 62.3, 23.3, 13.6; HRMS (ESI + , [M + H]+) calculated for [C16H14ClN2O2]+: 301.0738, found 301.0741.
Ethyl 6-chloro-5-cyano-2-methyl-4-(4-nitrophenyl)nicotinate (11b)
White solid; yield: 96%; mp: 149–150 °C; 1H NMR (500 MHz, CDCl3) δ 8.38 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 2.71 (s, 3H), 1.01 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.2, 160.9, 153.2, 151.8, 148.9, 140.0, 129.5, 128.2, 124.0, 113.3, 108.0, 62.5, 23.5, 13.7; HRMS (ESI + , [M + H]+) calculated for [C16H13ClN3O4]+: 346.0589, found 346.0589.
Ethyl 6-chloro-5-cyano-2-methyl-4-(3-nitrophenyl)nicotinate (11c)
Gray solid; yield: 90%; mp: 129–130 °C; 1H NMR (500 MHz, CDCl3) δ 8.43–8.38 (m, 1H), 8.28 (s, 1H), 7.75 (d, J = 5.1 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 2.71 (s, 3H), 1.04 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.2, 160.9, 153.2, 151.4, 148.2, 135.3, 134.2, 130.2, 128.5, 125.0, 123.4, 113.4, 108.3, 62.6, 23.5, 13.7; HRMS (ESI + , [M + H]+) calculated for [C16H13ClN3O4]+: 346.0589, found 346.0591.
Ethyl 6-chloro-5-cyano-4-(4-fluorophenyl)-2-methylnicotinate (11d)
Gray solid; yield: 93%; mp: 98–99 °C; 1H NMR (500 MHz, CDCl3) δ 7.48 – 7.30 (m, 2H), 7.20 (t, J = 8.5 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 2.66 (s, 3H), 1.00 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.8, 164.8, 162.8, 160.2, 153.2 (d, J = 23.6 Hz), 139.8, 136.7, 130.3 (d, J = 8.7 Hz), 129.8 (d, J = 3.5 Hz), 128.8, 116.6, 116.2 (d, J = 22.1 Hz), 113.9, 111.8, 108.4, 62.3, 23.3, 13.6; HRMS (ESI + , [M + H]+) calculated for [C16H13ClFN2O2]+: 319.0644, found 319.0645.
Ethyl 4-(4-bromophenyl)-6-chloro-5-cyano-2-methylnicotinate (11e)
Green solid; yield: 87%; mp: 104–105 °C; 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 2.67 (s, 3H), 1.01 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.6, 160.3, 153.0, 153.0, 132.7, 132.2, 129.7, 128.5, 125.0, 113.7, 108.2, 62.3, 23.3, 13.6; HRMS (ESI + , [M + H]+) calculated for [C16H13BrClN2O2]+: 378.9843, found 378.9847.
Ethyl 6-chloro-4-(4-chlorophenyl)-5-cyano-2-methylnicotinate (11f)
Gray solid; yield: 91%; mp: 91–92°C; 1H NMR (500 MHz, CDCl3) δ 7.49 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.3 Hz, 2H), 4.10 (q, J = 7.1 Hz, 2H), 2.67 (s, 3H), 1.01 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.7, 160.3, 153.0, 152.9, 136.8, 132.2, 129.6, 129.2, 128.6, 113.8, 108.2, 62.3, 23.3, 13.6; HRMS (ESI + , [M + H]+) calculated for [C16H13Cl2N2O2]+: 335.0349, found 335.0348.
Ethyl 6-chloro-5-cyano-4-(4-methoxyphenyl)-2-methylnicotinate (11g)
Pale yellow; yield: 92%; mp: 83–84 °C; 1H NMR (500 MHz, CDCl3) δ 7.33 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 4.11 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 2.64 (s, 3H), 1.02 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.2, 161.2, 159.8, 154.0, 152.9, 129.8, 128.9, 126.0, 114.4, 114.2, 108.3, 62.1, 55.4, 23.2, 13.7; HRMS (ESI + , [M + H]+) calculated for [C17H16ClN2O3]+: 331.0844, found 331.0843.
Ethyl 6-chloro-4-(3-chlorophenyl)-5-cyano-2-methylnicotinate (11h)
Gray solid; yield: 94%; mp: 145–146 °C; 1H NMR (500 MHz, CDCl3) δ 7.50 (dt, J = 8.1, 1.8, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 1.7 Hz, 1H), 7.28 (dt, J = 4.9, 3.5 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.67 (s, 3H), 1.01 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.5, 160.4, 153.0, 152.6, 135.5, 135.0, 130.4, 130.2, 128.6, 128.9, 126.5, 113.6, 108.2, 77.3, 77.0, 76.8, 62.3, 23.3, 13.6; HRMS (ESI + , [M + H]+) calculated for [C16H13Cl2N2O2]+: 335.0349, found 335.0350.
Ethyl 6-chloro-5-cyano-4-(2,4-dichlorophenyl)-2-methylnicotinate (11i)
Green solid, yield: 81%; mp: 85–86°C; 1H NMR (500 MHz, CDCl3) δ 7.57 (d, J = 1.9 Hz, 1H), 7.39 (dd, J = 8.3, 1.9 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 4.10 (q, J = 7.1 Hz, 2H), 2.72 (s, 3H), 1.01 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 165.0, 161.5, 152.9, 151.3, 137.1, 133.6, 131.8, 130.5, 130.1, 128.4, 127.6, 113.2, 109.4, 62.4, 23.9, 13.7; HRMS (ESI + , [M + H]+) calculated for [C16H13Cl3N2O2]+: 368.9959, found 368.9966.
Ethyl 6-chloro-5-cyano-2-methyl-4-(thiophen-2-yl)nicotinate (11j)
Brown solid; yield: 93%; mp: 119–120 °C; 1H NMR (500 MHz, CDCl3) δ 7.59 (dd, J = 6.3, 5.7 Hz, 1H), 7.36 – 7.34 (m, 1H), 7.18 (dd, J = 4.9, 3.8 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 2.64 (s, 3H), 1.11 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.0, 160.1, 153.4, 147.0, 133.1, 130.8, 130.0, 129.2, 128.0, 114.2, 108.5, 62.6, 23.3, 13.8; HRMS (ESI + , [M + H]+) calculated for [C14H12ClN2O2S]+: 307.0303, found 307.0304.
Ethyl 6-chloro-5-cyano-4-(furan-2-yl)-2-methylnicotinate (11k)
Green solid; yield: 82%; mp: 78–79 °C; 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 1.4 Hz, 1H), 7.45 (d, J = 3.6 Hz, 1H), 6.65 (dd, J = 3.7, 1.8 Hz, 1H), 4.38 (q, J = 7.2 Hz, 2H), 2.62 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.6, 160.5, 153.9, 145.8, 145.6, 140.2, 125.3, 116.7, 114.9, 113.1, 103.4, 62.5, 23.2, 14.0; HRMS (ESI + , [M + H]+) calculated for [C14H12ClN2O3]+: 291.0531, found 291.0535.
General procedure for the synthesis Ethyl 4-(aryl)-5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13a-k)
In a pressure tube for microwave, the reaction was placed 11a-k (1 mmol), methyl 6-aminohexanoate hydrochloride 12 (1.1 mmol) triethylamine (2.2 mmol), and 3 mL of ethanol was added. The reaction mixture was microwave irradiated at 120 °C for 3–4 h. The end of the reaction was confirmed by TLC using Hex/Ethyl acetate (8:2). Once completed the reaction, the crude product was purified by column chromatography (Hex/Ethyl acetate 9:1) to obtain compounds 13a-k.
Ethyl 5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methyl-4-phenylnicotinate (13a)
White solid; yield: 90%; mp: 69–70 °C; 1H NMR (500 MHz, CDCl3) δ 7.44 – 7.42 (m, 3H), 7.33 – 7.31 (m, 2H), 5.43 (t, J = 5.4 Hz, 1H), 3.92 (q, J = 7.1 Hz, 2H), 3.68 (s, 1H), 3.58 (q, J = 6.9 Hz, 2H), 2.53 (s, 3H), 2.35 (t, J = 7.5 Hz, 2H), 1.75 – 1.62 (m, 4H), 1.46 – 1.40 (m, 2H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 167.7, 160.8, 158.0, 153.8, 136.4, 129.2, 128.5, 127.9, 118.2, 116.4, 88.9, 61.1, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 24.0, 13.5; HRMS (ESI + , [M + H]+) calculated for [C23H28N3O4]+: 410.2074, found 410.2073.
Ethyl 5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methyl-4-(4-nitrophenyl)nicotinate (13b)
Brown solid; yield: 84%; mp: 90–91 °C; 1H NMR (500 MHz, CDCl3) δ 8.32 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 5.53 (t, J = 5.4 Hz, 1H), 3.96 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.60 (q, J = 6.9 Hz, 2H), 2.58 (s, 3H), 2.35 (t, J = 7.5 Hz, 2H), 1.76 – 1.60 (m, 4H), 1.51 – 1.36 (m, 2H), 0.91 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 166.8, 162.0, 157.8, 151.7, 148.2, 143.1, 129.1, 123.7, 117.2, 115.7, 88.5, 61.3, 51.6, 41.2, 33.9, 29.0, 26.3, 24.5, 24.4, 13.6; HRMS (ESI + , [M + H]+) calculated for [C23H27N4O6]+: 455.1925, found 455.1927.
Ethyl 5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methyl-4-(3-nitrophenyl)nicotinate (13c)
White solid; yield: 91%; mp: 81–82 °C; 1H NMR (500 MHz, CDCl3) δ 8.41 – 8.25 (m, 1H), 8.21 (s, 1H), 7.70 – 7.60 (m, 2H), 5.46 (t, J = 5.3 Hz, 1H), 3.98 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.60 (q, J = 7,2 Hz, 2H), 2.57 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 1.74 – 1.65 (m, 4H), 1.47 – 1.41 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 166.8, 161.9, 157.9, 151.3, 148.1, 138.1, 134.0, 129.7, 124.0, 123.2, 117.6, 115.7, 88.8, 61.3, 51.6, 41.2, 33.9, 29.1, 26.3, 24.5, 24.5, 13.6; HRMS (ESI + , [M + H]+) calculated for [C23H27N4O6]+: 455.1925, found 455.1927.
Ethyl 5-cyano-4-(4-fluorophenyl)-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13d)
Brown solid; yield: 85%; mp: 88–89 °C; 1H NMR (500 MHz, CDCl3) δ 7.32 (dd, J = 7.6, 5.6 Hz, 2H), 7.14 (t, J = 8.4 Hz, 2H), 5.42 (bs, 1H), 3.96 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.58 (q, J = 6.5 Hz, 2H), 2.53 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 1.75 – 1.62 (m, 4H), 1.48 – 1.39 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 167.5, 163.2 (d, J = 249.4 Hz), 160.9, 158.0, 152.7, 132.3 (d, J = 3.5 Hz), 129.9 (d, J = 8.4 Hz), 118.3, 116.3, 115.7 (d, J = 21.9 Hz), 88.9, 61.2, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 24.1, 13.6; HRMS (ESI + , [M + H]+) calculated for [C23H27FN3O4]+: 428.1980, found 428.1986.
Ethyl 4-(4-bromophenyl)-5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13e)
White solid; yield: 88%; mp: 77–78 °C; 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.3 Hz, 2H), 5.40 (bs, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.58 (q, J = 6.7 Hz, 2H), 2.53 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 1.76 – 1.62 (m, 4H), 1.48 – 1.39 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 167.4, 161.2, 158.0, 152.6, 135.3, 131.8, 129.5, 123.7, 117.9, 116.2, 88.7, 61.2, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 24.1, 13.6; HRMS (ESI + , [M + H]+) calculated for [C23H27BrN3O4]+: 488.1179, found 488.1183.
Ethyl 4-(4-chlorophenyl)-5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13f)
White solid; yield: 87%; mp: 78–79°C; 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.4 Hz, 2H), 5.47 (bs, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.57 (q, J = 6.7 Hz, 2H), 2.53 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 1.74 – 1.62 (m, 4H), 1.49 – 1.38 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 167.4, 161.1, 158.0, 152.5, 135.4, 134.8, 129.3, 128.8, 118.0, 116.1, 88.7, 61.2, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 24.1, 13.6; HRMS (ESI + , [M + H]+) calculated for [C23H27ClN3O4]+: 444.1685, found 444.1685.
Ethyl 5-cyano-6-((6-methoxy-6-oxohexyl)amino)-4-(4-methoxyphenyl)-2-methylnicotinate (13g)
Pale yellow; yield: 85%; mp: 96–97 °C; 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.4 Hz, 2H), 5.43 (bs, 1H), 3.98 (q, J = 7.0 Hz, 2H), 3.83 (s, 3H), 3.67 (s,3H), 3.57 (q, J = 6.4 Hz, 2H), 2.51 (s, 3H), 2.34 (t, J = 7.4 Hz, 2H), 1.76 – 1.61 (m, 4H), 1.46 – 1.41 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 168.0, 160.4, 160.3, 158.1, 153.4, 129.3, 128.5, 118.5, 116.7, 113.9, 88.9, 61.1, 55.3, 51.5, 41.1, 33.9, 29.1, 26.3, 24.5, 23.9, 13.7; HRMS (ESI + , [M + H]+) calculated for [C24H30N3O5]+: 440.2180, found 440.2186.
Ethyl 4-(3-chlorophenyl)-5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13h)
White solid; yield: 91%; mp: 86–87 °C; 1H NMR (500 MHz, CDCl3) δ 7.43 – 7.37 (m, 2H), 7.31 (s, 1H), 7.22 (d, J = 7.4 Hz, 1H), 5.47 (t, J = 5.4 Hz, 1H), 3.98 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.58 (q, J = 6.8 Hz, 2H), 2.54 (s, 3H), 2.35 (t, J = 7.5 Hz, 2H), 1.73 – 1.64(m, 4H), 1.47 – 1.39 (m, 2H), 0.91 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 167.2, 161.3, 157.9, 152.2, 138.1, 134.5, 129.8, 129.3, 128.0, 126.2, 117.9, 116.0, 88.7, 61.2, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 24.2, 13.5; HRMS (ESI + , [M + H]+) calculated for [C23H27ClN3O4]+: 444.1685, found 444.1683.
Ethyl 5-cyano-4-(2,4-dichlorophenyl)-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13i)
White solid; yield: 78%; mp: 79–80 °C; 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.9 Hz, 1H), 7.32 (dd, J = 8.2, 2.0 Hz, 1H), 7.13 (d, J = 8.2 Hz, 1H), 5.43 (t, J = 5.3 Hz, 1H), 4.03 – 3.93 (m, 2H), 3.68 (s, 3H), 3.66 – 3.54 (m, 2H), 2.61 (s, 3H), 2.35 (t, J = 7.4 Hz, 2H), 1.76 – 1.62 (m, 4H), 1.49 – 1.38 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 166.3, 162.8, 157.7, 151.1, 135.6, 134.5, 133.3, 130.3, 129.6, 127.2, 117.1, 115.4, 89.8, 61.1, 51.6, 41.2, 33.9, 29.1, 26.3, 24.9, 24.5, 13.5; HRMS (ESI + , [M + H]+) calculated for [C23H26Cl2N3O4]+: 478.1295, found 478.1292.
Ethyl 5-cyano-6-((6-methoxy-6-oxohexyl)amino)-2-methyl-4-(thiophen-2-yl)nicotinate (13j)
White solid; yield: 83%; mp: 73–74 °C; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 4.9 Hz, 1H), 7.22 (d, J = 3.0 Hz, 1H), 7.12 – 7.09 (m, 1H), 5.44 (bs, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.57 (q, J = 6.7 Hz, 2H), 2.50 (s, 3H), 2.34 (t, J = 7.5 Hz, 2H), 1.75 – 1.62 (m, 4H), 1.48 – 1.39 (m, 2H), 1.03 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 167.7, 160.5, 158.1, 145.9, 135.8, 128.9, 128.1, 127.4, 118.9, 116.4, 89.0, 61.4, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 23.9, 13.7; HRMS (ESI + , [M + H]+) calculated for [C21H26N3O4S]+: 416.1639, found 416.1642.
Ethyl 5-cyano-4-(furan-2-yl)-6-((6-methoxy-6-oxohexyl)amino)-2-methylnicotinate (13k)
White solid; yield: 87%; mp: 105–106 °C; 1H NMR (500 MHz, CDCl3) δ 7.54 (s, 1H), 7.11 (d, J = 3.4 Hz, 1H), 6.56 (dd, J = 3.2, 1.6 Hz, 1H), 5.43 (t, J = 5.1 Hz, 1H), 4.24 (q, J = 7.1 Hz, 2H), 3.68 (s, 3H), 3.56 (q, J = 6.8 Hz, 2H), 2.49 (s, 3H), 2.34 (t, J = 7.5 Hz, 2H), 1.74 – 1.61 (m,4H), 1.47 – 1.38 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 174.0, 168.2, 160.6, 158.4, 147.5, 144.2, 139.9, 116.9, 115.8, 113.7, 112.2, 84.8, 61.5, 51.5, 41.2, 33.9, 29.1, 26.3, 24.5, 23.8, 14.0; HRMS (ESI + , [M + 1]+) calculated for [C21H26N3O5]+: 400.1867, found 400.1875.
General procedure for the synthesis 6-((4-(aryl)-3-cyano-5-(ethoxycarbonyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14a-k)
A mixture of 13a-k derivative (1 mmol), lithium hydroxide (6 mmol), and 3 mL of mixture Water/THF (2:1) was added. The solution was stirring at room temperature for 12 h. After the reaction was finished monitored by TLC. The mixture was acidified with 2 M HCl, subsequently diluted with distilled water, and extracted with CH2Cl2 (3 × 10 mL). All the organic layers were combined, dried (anhydrous Na2SO4), and the solvent evaporated to give the product pure 14a-k.
6-((3-cyano-5-(ethoxycarbonyl)-6-methyl-4-phenylpyridin-2-yl)amino)hexanoic acid (14a)
White solid; yield: 92%; mp: 128–129 °C; 1H NMR (500 MHz, CDCl3) δ 7.44 – 7.42 (m, 3H), 7.33 – 7.31 (m, 2H), 5.49 (bs, 1H), 3.92 (q, J = 7.1 Hz, 2H), 3.58 (q, J = 6.5 Hz, 2H), 2.53 (s, 3H), 2.39 (t, J = 7.3 Hz, 2H), 1.77 – 1.63 (m, 4H), 1.51 – 1.41 (m, 2H), 0.88 – 0.83 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.3, 167.7, 160.8, 158.0, 153.9, 136.4, 129.2, 128.5, 127.9, 118.2, 116.4, 88.9, 61.1, 41.2, 33.8, 29.1, 26.2, 24.3, 24.0, 13.5; HRMS (ESI + , [M + H]+) calculated for [C22H26N3O4]+: 396.1918, found 396.1899.
6-((3-cyano-5-(ethoxycarbonyl)-6-methyl-4-(4-nitrophenyl)pyridin-2-yl)amino)hexanoic acid (14b)
Brown solid; yield: 89%; mp: 135–136 °C; 1H NMR (500 MHz, CDCl3) δ 8.32 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 8.6 Hz, 2H), 5.52 (t, J = 5.1 Hz, 1H), 3.96 (q, J = 7.1 Hz, 2H), 3.61 (q, J = 6.7 Hz, 2H), 2.57 (s, 3H), 2.40 (t, J = 7.3 Hz, 2H), 2.17 (s, 1H), 1.75 – 1.66 (m, 4H), 1.50 – 1.44 (m, 2H), 0.91 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.1, 166.8, 162.0, 157.8, 151.7, 148.2, 143.0, 129.2, 123.7, 117.3, 115.7, 88.5, 61.3, 41.2, 33.7, 29.0, 26.2, 24.4, 24.2, 13.6; HRMS (ESI + , [M + H]+) calculated for [C22H25N4O6]+: 441.1769, found 441.1700.
6-((3-cyano-5-(ethoxycarbonyl)-6-methyl-4-(3-nitrophenyl)pyridin-2-yl)amino)hexanoic acid (14c)
Pale yellow solid; yield: 97%; mp: 120–121 °C; 1H NMR (500 MHz, CDCl3) δ 8.33 – 8.31 (m, 1H), 8.21 (s, 1H), 7.70 – 7.63 (m, 2H), 5.55 (t, J = 5.4 Hz, 1H), 3.98 (q, J = 7.1 Hz, 2H), 3.61 (q, J = 6.7 Hz, 2H), 2.57 (s, 3H), 2.40 (t, J = 7.4 Hz, 2H), 2.18 (s, 1H), 1.76 – 1.65 (m, 4H), 1.52 – 1.42 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.0, 166.8, 162.0, 157.9, 151.4, 148.1, 138.1, 134.0, 129.7, 124.0, 123.2, 117.6, 115.7, 88.7, 61.3, 41.2, 33.7, 29.0, 26.2, 24.5, 24.2, 13.6; HRMS (ESI + , [M + H]+) calculated for [C22H25N4O6]+: 441.1769, found 441.1704.
6-((3-cyano-5-(ethoxycarbonyl)-4-(4-fluorophenyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14d)
Brown solid; yield: 93%; mp: 119–120 °C; 1H NMR (500 MHz, CDCl3) δ 7.37 – 7.28 (m, 2H), 7.14 (t, J = 8.5 Hz, 2H), 5.48 (bs, 1H), 3.96 (q, J = 7.1 Hz, 2H), 3.58 (q, J = 6.6 Hz, 2H), 2.53 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.76 – 1.62 (m, 4H), 1.49 – 1.43 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.1, 167.6, 163.3 (d, J = 249.4 Hz), 160.9, 158.0, 152.8, 132.3 (d, J = 3.4 Hz), 129.9 (d, J = 8.4 Hz), 118.3, 116.3, 115.7 (d, J = 21.9 Hz), 88.9, 61.2, 41.2, 33.8, 29.1, 26.2, 24.3, 24.0, 13.6; HRMS (ESI + , [M + H]+) calculated for [C22H25FN3O4]+: 414.1824, found 414.1766.
6-((4-(4-bromophenyl)-3-cyano-5-(ethoxycarbonyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14e)
White solid; yield: 90%; mp: 127–128 °C; 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 5.48 (t, J = 5.4 Hz, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.58 (q, J = 6.6 Hz, 2H), 2.53 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.75 – 1.63 (m, 4H), 1.51 – 1.41 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.3, 167.4, 161.2, 158.0, 152.6, 135.3, 131.8, 129.6, 123.7, 117.9, 116.2, 88.7, 61.3, 41.1, 33.8, 29.1, 26.2, 24.2, 24.1, 13.6; HRMS (ESI + , [M + H]+) calculated for [C22H25BrN3O4]+: 474.1023, found 474.0955.
6-((4-(4-chlorophenyl)-3-cyano-5-(ethoxycarbonyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14f)
White solid; yield: 88%; mp: 124–125 °C; 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 5.49 (t, J = 5.1 Hz, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.58 (q, J = 6.6 Hz, 1H), 2.53 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.86 – 1.59 (m, 4H), 1.59 – 1.36 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.4, 167.4, 161.1, 158.0, 152.6, 135.5, 134.8, 129.3, 128.8, 118.0, 116.2, 88.7, 61.2, 41.2, 33.8, 29.1, 26.2, 24.3, 24.1, 13.6; HRMS (ESI + , [M + H]+) calculated for [C22H25ClN3O4]+: 430.1528, found 430.1467.
6-((3-cyano-5-(ethoxycarbonyl)-4-(4-methoxyphenyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14g)
White solid; yield: 84%; mp: 125–126 °C; 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.6 Hz, 2H), 5.43 (s, 1H), 3.98 (q, J = 7.1 Hz, 2H), 3.84 (s, 3H), 3.57 (q, J = 6.6 Hz, 2H), 2.51 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.79 – 1.60 (m, 4H), 1.52 – 1.40 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.1, 168.0, 160.4, 160.4, 158.1, 153.4, 129.4, 128.5, 118.5, 116.7, 114.0, 88.9, 61.1, 55.3, 41.1, 33.8, 29.1, 26.2, 24.3, 24.0, 13.7; HRMS (ESI + , [M + H]+) calculated for [C23H28N3O5]+: 426.2023, found 426.1963.
6-((4-(3-chlorophenyl)-3-cyano-5-(ethoxycarbonyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14h)
White solid; yield: 96%; mp: 128–129 °C; 1H NMR (500 MHz, CDCl3) δ 7.44 – 7.35 (m, 2H), 7.31 (s, 1H), 7.22 (d, J = 7.4 Hz, 1H), 5.50 (t, J = 5.4 Hz, 1H), 3.98 (q, J = 7.1 Hz, 2H), 3.59 (q, J = 6.7 Hz, 2H), 2.54 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.77 – 1.62 (m, 4H), 1.52 – 1.40 (m, 2H), 0.91 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.1, 167.3, 161.3, 157.9, 152.2, 138.1, 134.5, 129.9, 129.3, 128.0, 126.2, 117.9, 116.0, 88.7, 61.2, 41.1, 33.8, 29.1, 26.2, 24.2, 24.2, 13.5; HRMS (ESI + , [M + H]+) calculated for [C22H25ClN3O4]+: 430.1528, found 430.1473.
6-((3-cyano-4-(2,4-dichlorophenyl)-5-(ethoxycarbonyl)-6-methylpyridin-2-yl)amino)hexanoic acid (14i)
White solid; yield: 86%; mp: 124–125 °C; 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.6 Hz, 1H), 7.32 (dd, J = 8.2, 1.6 Hz, 1H), 7.13 (d, J = 8.2 Hz, 1H), 5.51 (t, J = 5.4 Hz, 1H), 4.03 – 3.95 (m, 2H), 3.72 – 3.48 (m, 2H), 2.61 (s, 3H), 2.40 (t, J = 7.4 Hz, 2H), 1.75 – 1.65 (m, 4H), 1.56 – 1.39 (m, 3H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.4, 166.3, 162.8, 157.7, 151.1, 135.6, 134.5, 133.3, 130.3, 129.6, 127.2, 117.1, 115.5, 89.8, 61.1, 41.2, 33.8, 29.1, 26.2, 24.9, 24.2, 13.5; HRMS (ESI + , [M + H]+) calculated for [C22H24Cl2N3O4]+: 464.1138, found 464.1072.
6-((3-cyano-5-(ethoxycarbonyl)-6-methyl-4-(thiophen-2-yl)pyridin-2-yl)amino)hexanoic acid (14j)
White solid; yield: 90%; mp: 117–118 °C; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 5.0 Hz, 1H), 7.22 (d, J = 3.5 Hz, 1H), 7.11 (t, J = 4.3 Hz, 1H), 5.46 (t, J = 5.3 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.57 (q, J = 6.7 Hz, 2H), 2.50 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.77 – 1.61 (m, 4H), 1.55 – 1.37 (m, 2H), 1.03 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.0, 167.7, 160.5, 158.2, 146.0, 135.7, 128.9, 128.1, 127.4, 118.9, 116.4, 89.0, 61.5, 41.1, 33.7, 29.1, 26.2, 24.3, 23.9, 13.7; HRMS (ESI + , [M + H]+) calculated for [C20H24N3O4S]+: 402.1482, found 402.1426.
6-((3-cyano-5-(ethoxycarbonyl)-4-(furan-2-yl)-6-methylpyridin-2-yl)amino)hexanoic acid (14k)
Brown solid; yield: 95%; mp: 111–112 °C; 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 1.1 Hz, 1H), 7.11 (d, J = 3.5 Hz, 1H), 6.56 (dd, J = 3.5, 1.8 Hz, 1H), 5.48 (t, J = 5.5 Hz, 1H), 4.24 (q, J = 7.1 Hz, 2H), 3.56 (q, J = 6.9 Hz, 2H), 2.49 (s, 3H), 2.39 (t, J = 7.4 Hz, 2H), 1.79 – 1.61 (m, 4H), 1.56 – 1.37 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 179.0, 168.2, 160.7, 158.5, 147.5, 144.2, 140.0, 117.0, 115.8, 113.7, 112.2, 84.7, 61.5, 41.2, 33.8, 29.1, 26.2, 24.3, 23.8, 14.0; HRMS (ESI + , [M + H]+) calculated for [C20H24N3O5]+: 386.1710, found 386.1661.
General procedure for the synthesis Ethyl 4-(aryl)-5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate 15a-k
A mixture of 14a-k derivative (1 mmol), 1.1´-carbonyldiimidazole (1.5 mmol), N,N-dimethylpyridin-4-amine (0.1 mmol), hydroxylamine hydrochloride (1.5 mmol) and dichloromethane 3 mL was added. The solution was stirring at room temperature for 12 h. After the reaction was finished monitored by TLC. The mixture was acidified with 2 M HCl, diluted with distilled water, and extracted with CH2Cl2 (3 × 10 mL). All the organic layers were combined, dried (anhydrous Na2SO4), and the solvent evaporated to give the product pure 15a-k.
Ethyl 5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methyl-4-phenylnicotinate (15a)
White solid; yield: 84%; mp: 116–117 °C; 1H NMR (500 MHz, CDCl3) δ 8.81 (bs, 1H), 7.51 – 7.38 (m, 3H), 7.36 – 7.27 (m, 2H), 5.55 (s, 1H), 3.92 (q, J = 7.1 Hz, 2H), 3.56 (m, 2H), 2.52 (s, 3H), 2.15 (d, J = 6.1 Hz, 2H), 1.74 – 1.59 (m, 4H), 1.47 – 1.36 (m, 2H), 1.26 (s, 1H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.5, 167.9, 161.0, 158.2, 154.0, 136.5, 129.3, 128.6, 128.0, 118.3, 116.6, 88.9, 61.2, 41.2, 32.8, 29.0, 26.2, 24.9, 24.2, 13.6; HRMS (ESI + , [M + Na]+) calculated for [C22H26N4O4Na]+: 433.1846, found 433.1852.
Ethyl 5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methyl-4-(4-nitrophenyl)nicotinate (15b)
Pale yellow solid; yield: 79%; mp: 134–135 °C; 1H NMR (500 MHz, CDCl3) δ 8.23 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 5.66 (s, 1H), 3.88 (q, J = 7.1 Hz, 2H), 3.49 (m, J = 5.4 Hz, 2H), 2.48 (s, 3H), 2.08 (s, 1H), 1.67 – 1.52 (m, 4H), 1.32 (s, 2H), 1.18 (s, 1H), 0.82 (t, J = 8.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.5, 166.9, 162.2, 158.0, 152.0, 148.3, 143.1, 129.3, 123.8, 117.3, 116.0, 88.4, 61.5, 41.2, 32.7, 28.9, 26.2, 24.9, 24.6, 13.7; HRMS (ESI + , [M + Na]+) calculated for [C22H25N5O6Na]+: 478.1697, found 478.1700.
Ethyl 5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methyl-4-(3-nitrophenyl)nicotinate (15c)
White solid; yield: 83%; mp: 92–93 °C; 1H NMR (500 MHz, CDCl3) δ 8.32 (dd, J = 7.0, 2.2 Hz, 1H), 8.21 (s, 1H), 7.70 – 7.63 (m, 2H), 5.57 (s, 1H), 3.98 (q, J = 7.1 Hz, 2H), 3.61 (q, J = 6.5 Hz, 2H), 2.57 (s, 3H), 2.20 (t, J = 7.0 Hz, 2H), 1.79 – 1.64 (m, 4H), 1.60 (s, 2H), 1.47 – 1.42 (m, 2H), 0.93 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.7, 167.0, 162.1, 158.0, 151.5, 148.1, 138.1, 134.2, 129.8, 124.1, 123.2, 117.5, 116.0, 88.6, 61.4, 41.2, 32.7, 28.8, 26.2, 24.9, 24.5, 13.7; HRMS (ESI + , [M + Na]+) calculated for [C22H25N5O6Na]+: 478.1697, found 478.1706.
Ethyl 5-cyano-4-(4-fluorophenyl)-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate (15d)
White solid; yield: 80%; mp: 95–96 °C; 1H NMR (500 MHz, CDCl3) δ 8.77 (bs, 1H), 7.34 – 7.28 (m, 2H), 7.13 (t, J = 8.4 Hz, 2H), 5.53 (s, 1H), 3.96 (q, J = 7.1 Hz, 2H), 3.56 (s, 2H), 2.52 (s, 3H), 2.16 (s, 2H), 1.70 – 1.65 (m, 4H), 1.41 (s, 2H), 1.26 (s, 1H), 0.91 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4, 167.6, 163.3 (d, J = 249.4 Hz), 161.0, 158.0, 152.8, 132.3 (d, J = 3.5 Hz), 129.9 (d, J = 8.4 Hz), 118.3, 116.4, 115.7 (d, J = 21.9 Hz), 88.8, 61.2, 41.0, 32.7, 28.9, 26.1, 24.8, 24.1, 13.6; HRMS (ESI + , [M + Na]+) calculated for [C22H25FN4O4Na]+: 451.1752, found 451.1752.
Ethyl 4-(4-bromophenyl)-5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate (15e)
White solid; yield: 81%; mp: 137–138 °C; 1H NMR (500 MHz, CDCl3) δ 8.37 (bs, 2H), 7.58 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 5.50 (s, 1H), 3.97 (q, J = 7.1 Hz, 2H), 3.59 – 3.58 (m, 2H), 2.53 (s, 3H), 2.17 (d, J = 6.4 Hz, 2H), 1.84 – 1.52 (m, 5H), 1.44 – 1.41 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 170.5, 166.4, 160.2, 157.0, 151.7, 134.2, 130.7, 128.5, 122.7, 116.8, 115.3, 87.5, 60.3, 40.1, 31.6, 27.9, 25.1, 23.8, 23.1, 12.5; HRMS (ESI + , [M + Na]+) calculated for [C22H25BrN4O4Na]+: 511.0951, found 511.0965.
Ethyl 4-(4-chlorophenyl)-5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate (15f)
White solid; yield: 78%; mp: 126–127 °C; 1H NMR (500 MHz, CDCl3) δ 8.31 (bs, 2H), 7.43 (t, J = 8.5 Hz, 2H), 7.29 – 7.26 (m, 2H), 5.50 (s, 1H), 3.97 (q, J = 7.2 Hz, 2H), 3.58 (s, 2H), 2.53 (s, 3H), 2.19 (s, 2H), 1.82 – 1.51 (m, 5H), 1.44 – 1.42 (m, 2H), 0.93 (t, J = 7.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.7, 170.2, 167.6, 163.9, 161.3, 159.8, 158.1, 154.7, 152.8, 135.6, 134.9, 129.4, 128.9, 118.0, 116.4, 88.7, 61.4, 41.2, 32.8, 29.0, 26.2, 25.0, 24.2, 13.7; HRMS (ESI + , [M + Na]+) calculated for [C22H25ClN4O4Na]+: 467.1457, found 467.1468.
Ethyl 5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-4-(4-methoxyphenyl)-2-methylnicotinate (15g)
White solid; yield: 76%; mp: 90–91 °C; 1H NMR (500 MHz, CDCl3) δ 8.69 (bs, 1H), 7.27 (d, J = 7.7 Hz, 2H), 6.95 (d, J = 7.8 Hz, 2H), 5.48 (s, 1H), 3.98 (q, J = 6.8 Hz, 2H), 3.84 (s, 3H), 3.56 (d, J = 4.2 Hz, 2H), 2.50 (s, 3H), 2.16 (s, 2H), 1.70 – 1.64 (m, 5H), 1.41 (s, 2H), 1.26 (s, 1H), 0.93 (t, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4, 168.2, 160.6, 160.6, 158.3, 153.6, 129.5, 128.6, 118.6, 117.0, 114.1, 88.9, 61.3, 55.5, 41.1, 32.8, 29.1, 26.2, 24.9, 24.1, 13.8; HRMS (ESI + , [M + Na]+) calculated for [C23H27N4O5Na]+: 463.1952, found 463.1966.
Ethyl 4-(3-chlorophenyl)-5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate (15h)
White solid; yield: 84%; mp: 111–112 °C; 1H NMR (500 MHz, CDCl3) δ 8.37 (bs, 1H), 7.36 – 7.28 (m, 2H), 7.23 (t, J = 1.7 Hz, 1H), 7.14 (dt, J = 7.3, 1.3 Hz, 1H), 5.54 (t, J = 5.2 Hz, 1H), 3.90 (q, J = 7.1 Hz, 3H), 3.48 (q, J = 6.5 Hz, 2H), 2.46 (s, 3H), 2.17 – 2.03 (m, 2H), 1.64 – 1.54 (m, 4H), 1.33 (d, J = 6.8 Hz, 2H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.5, 167.4, 161.5, 158.1, 152.4, 138.2, 134.6, 130.0, 129.4, 128.1, 126.3, 117.9, 116.3, 88.7, 61.4, 41.2, 32.8, 31.1, 29.0, 26.2, 25.0, 24.3, 13.7; HRMS (ESI + , [M + Na]+) calculated for [C22H25ClN4O4Na]+: 467.1457, found 467.1473.
Ethyl 5-cyano-4-(2,4-dichlorophenyl)-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate (15i)
White solid, yield: 80%; mp: 109–110 °C; 1H NMR (500 MHz, CDCl3) δ 8.80 (bs, 1H), 7.40 (s, 1H), 7.22 (d, J = 8.2 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 5.41 (d, J = 4.7 Hz, 1H), 3.90 – 3.86 (m, 2H), 3.52 – 3.46 (m, 2H), 2.58 (s, 3H), 2.30 (t, J = 7.3 Hz, 2H), 1.63 – 1.55 (m, 4H), 1.39 1.35 (m, 2H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.5, 165.3, 161.8, 156.7, 150.1, 134.6, 133.5, 132.3, 129.3, 128.6, 126.8, 116.1, 114.5, 88.8, 60.3, 40.2, 32.6, 28.0, 25.2, 23.9, 23.2, 12.5; HRMS (ESI + , [M + H]+) calculated for [C22H25Cl2N4O4]+: 479.1247, found 479.1182.
Ethyl 5-cyano-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methyl-4-(thiophen-2-yl)nicotinate (15j)
White solid; yield: 79%; mp: 95–96 °C; 1H NMR (500 MHz, CDCl3) δ 8.80 (bs, 1H), 7.47 (d, J = 4.8 Hz, 1H), 7.22 (d, J = 2.8 Hz, 1H), 7.16 – 7.06 (m, 1H), 5.56 (s, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.55 (d, J = 5.5 Hz, 2H), 2.49 (s, 3H), 2.16 (s, 2H), 1.81 – 1.52 (m, 4H), 1.40 (s, 2H), 1.26 (s, 1H), 1.02 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4, 167.8, 160.6, 158.2, 146.0, 135.7, 128.9, 128.2, 127.5, 118.8, 116.5, 88.9, 61.5, 41.1, 32.7, 28.9, 26.1, 24.8, 24.0, 13.7; HRMS (ESI + , [M + Na]+) calculated for [C20H24N4O4SNa]+: 439.1410, found 439.1431.
Ethyl 5-cyano-4-(furan-2-yl)-6-((6-(hydroxyamino)-6-oxohexyl)amino)-2-methylnicotinate (15k)
Brown solid; yield: 75%; mp: 123–124 °C; 1H NMR (500 MHz, CDCl3) δ 8.52 (bs, 1H), 7.46 (d, J = 1.4 Hz, 1H), 7.03 (d, J = 3.5 Hz, 1H), 6.49 (dd, J = 3.5, 1.8 Hz, 1H), 5.45 (s, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.48 (q, J = 6.5 Hz, 2H), 2.41 (s, 3H), 2.10 (s, 1H), 1.72 – 1.51 (m, 5H), 1.35 – 1.33 (m, 2H), 1.19 (s, 1H), 1.13 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4, 168.4, 160.9, 158.6, 147.6, 144.4, 140.1, 117.2, 116.0, 113.9, 112.3, 84.8, 61.7, 41.2, 32.8, 29.1, 26.2, 24.9, 23.9, 14.2; HRMS (ESI + , [M + Na]+) calculated for [C20H24N4O5Na]+: 423.1639, found 423.1639.
Cell culture
Breast (BT-474, MDA-MB-231) and prostate (PC3) cancer cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured and maintained according to the indications of the supplier. Cell cultures were kept and maintained in a humidified atmosphere with 5% CO2 at 37 °C.
Proliferation studies
The different compounds ability to inhibit cell proliferation was studied by the sulphorhodamine (SRB) assay. The cells were seeded in 96-well culture plates at a density of 3000 cells/well and treated in the present of DMSO, at different concentrations (0.1–10 µM) of compounds 15a–k or SAHA. The treated cells were incubated for 72–144 h (depending on the cell line) at 37 °C in a humid environment with 95% air and 5% CO2. Cell proliferation was determined by using the SRB assay [31]. Briefly, the cells were fixed with 10% trichloroacetic acid at 4 °C for 1 h. The plates were then washed with tap water and air-dried. The cells were stained for 1 h with 0.4% SRB dissolved in 1% acetic acid. To remove the unbound stain, plates were washed four times with 1% acetic acid and air-dried. The bound protein stain was solubilized with 10 mM of unbuffered tris base [tris(hydroxymethyl) aminomethane]. Absorbance was determined at 492 nm in a microplate reader (BioTek, Winooski, VT, USA). All biological assays were performed in triplicate. The concentrations that caused 50% cell growth inhibition (IC50) were obtained by non-linear regression analysis using sigmoidal fitting with a dose–response curve, with initial and final values fixed through a scientific graphing software (OriginPro 8, OriginLab Corporation, Northampton MA).
Real time RT-PCR
For gene expression analysis, the cells were incubated in the absence or presence of 10 µM of 15a, 15c, 15j, 15k, and SAHA for 24 h. After treatment, the cells were harvested, and total RNA was extracted using Trizol® reagent (Life Technologies). For the cDNA synthesis, a commercial kit was used (Transcriptor First-Strand cDNA Synthesis, Roche). RT-PCR was carried out on a LightCycler 2.0 instrument from Roche (Roche Diagnostics, Mannheim, Germany) according to the following protocol: activation of Taq DNA polymerase and DNA denaturing at 95 °C for 10 min, then 45 amplification cycles consisting of 10 s at 95 °C, 30 s at 60 °C, and 1 s at 72 °C. The oligonucleotides employed were cyclin D1 (CCND1)-F, GAAGATCGTCGCCACCTG; CCND1-R, GACCTCCTCCTCGCACTTCT; p21 (CDKN1A)-F, TCACTGTCTTGTACCCTTGTGC; CDKN1A -R, GGCGTTTGGAGTGGTAGAAA; p53 (TP53)-F, GTCCCAAGCAATGGATGATT; and TP53-R, TCTGGACCTGGGTCTTCAGT. The gene expression of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), was used as an internal control. It was AGCCACATCGCTGAGACAC for GAPDH-F and GCCCAATACGACCAAATCC for GAPDH-R.
Statistical analysis
Data are expressed as mean ± standard deviation (S.D.). Statistical analysis was done using one-way ANOVA, and all pairwise multiple comparisons were determined by the Holm-Sidak method on a specialized software package (SigmaStat Version 3.5, Jandel Scientific Systat Software, Inc., Richmond, VA, USA).
Computational methodology
The set of ligands (tested molecules: 15a, 15c, 15 g, 15j, and 15 k) were previously built and optimized with the semiempirical PM6 method using Gaussian 09 [32]. The optimized geometries were exported to pdb format files to be used with the docking software. All flexible bonds were set free to proceed with the flexible computational experiments.
The crystal structures of human HDAC1, 6, and 8 were obtained from the protein data bank (PDB codes 6Z2J, 5EDU, and 1T69, respectively) [33–35]. The former was acquired co-crystallized with hexakisphosphate, HDAC6 was obtained in a complex with Trichostatin A (TSA), and HDAC8 was obtained co-crystallized with suberoylanilide hydroxamic acid (SAHA). Hexakisphosphate, TSA, and SAHA were used to validate the parameters of the Docking experiments. With this regard, molecular docking was performed of each HDACs with the respective co-crystalized ligands. The root-mean-square deviations (RMSD) found were: 2.00 Å for inositol hexakisphosphate on HDAC1, 1.16 Å for TSA on HDAC6, and 1.51 Å for SAHA on HDAC8. Molecular docking was carried out on the AUTODOCK 4.2 software suite. For all calculations, we have used the Lamarckian Genetic Algorithm search function with 10 runs, 2,500,000 number of evaluations, a maximum number of generations of 27,000, a maximum population size of 10 individuals, a maximum initial energy for pose generation of 0.010000 kcal/mol, and a neighbor distance factor of 1.00 Å.
Result and Discussion
Chemistry
As a strategy to analyze the behavior of substituents into pyridine moiety, we envisioned using the same linker and ZBG groups because previously, the effect of these moieties was reported [27, 28]. The synthetic procedures adopted to obtain the target compounds are depicted in Scheme 1. Using previously reported methodologies 2-pyridones derivatives 10a-k were obtained by MCR procedure in good yields (87–92%) [29, 30]. Thus, latter compounds (10a-k) underwent to chlorination process through excess phosphorus oxychloride and N,N-dimethylaniline as a base to produce the compounds (11a-k) [36], the structures were confirmed by spectroscopy analysis and X-ray studies (Fig. 2). The structure for 11i is shown; the ORTEP diagram analysis was carried out where the dihedral angle between the pyridine nucleus and the aromatic ring at position 4 is 62.02°, while the length of C–Cl bond is 1.73 Å. The next step was to analyze the reactivity between 11a-k and amine 12; previous reports show that to afford this reaction, it is necessary to use transition metals [37]. However, this reaction was performed without the use of metals, and the best reaction conditions were microwave-heating at 140 °C in TEA and ethanol as solvent to afford 13a-k by nucleophilic aromatic substitution in good yields (78–90%). The structures were elucidated by NMR and HRMS; in all NMR spectra, it was possible to observe a triplet signal between 5.3 and 5.5 ppm showing a corresponding hydrogen NH bond; all compounds showed characteristic signals according to their identity. As a later stride, hydrolysis of the methyl ester group in the compounds 13a-k was performed using saponification reaction conditions, through lithium hydroxide in a mixture THF/water and stirring 12 h in room temperature to give the corresponding acids 14a-k in good yields (78–97%) [38], the structures were confirmed by spectroscopy analysis and X-ray studies of 14f (Fig. 2). Finally, the conversion of carboxylic acids 14a-k to hydroxamic derivatives 15a-k was achieved through reaction with hydroxylamine hydrochloride, 4-N,N-dimethylaminopyridine (DMAP), and 1,1´-carbonyldiimidazole in dichloromethane as a solvent, yielding the desired products 15a-k [39] in good yields (75–84%), all compounds were elucidated by NMR and HRMS.
Fig. 2.

The ORTED diagram for the X-ray resolved structure of 11i and 14f [40]
Biological activity
Effects of compounds 15a-k on cell proliferation
HDAC inhibitors include diverse compounds, which vary in structure, biological activity, and specificity. They affect many biological processes, including cell cycle, DNA repair, apoptosis, and gene expression alterations [41]. Consequently, HDAC inhibitors, such as SAHA, have been emerging as drugs with potential cancer treatment applications [42].
SAHA has been considering as a potential therapeutic agent for human breast cancer treatment. In fact, this inhibitor suppresses cell proliferation of various cancer cell lines, including human breast and prostate cancer cells [15, 43–45]. Therefore, the latter two cancer cell lines were employed to determine the possible antiproliferative activity of compounds 15a-k. Breast (BT-474 and MDA-MB-231) and prostate (PC3) cancer cells were incubated in the presence of different concentrations of the new synthesized compounds or SAHA as a reference control. Most compounds inhibited cancer cell proliferation in a dose-dependent manner (Fig. 3). Based on the calculated IC50 values (Table 1), the sensitivity of the cells to the compounds varied depending on the chemical structure of the compounds; although in general, for most the IC50 value is in the range of 1–3 µM, except for 15i, which did not modify cell growth and 15d that showed few activities at 10 µM concentration in BT-474 and PC3 cells, whence its IC50 value cannot obtain. Even SAHA holds the smallest value of the series, the displayed IC50 values of the proposed molecules reach the µM scale, which represents an excellent goal of an anticancer drug.
Fig. 3.

Antiproliferative effect of new synthesized compounds on cancer cells. A BT-474, B MDA-MB-231, and C PC3 cancer cell lines were incubated in the presence of different concentrations of compounds 15a-k or SAHA for 3–6 days. Cell proliferation was evaluated by the SRB assay. Results are shown as the mean ± S.D. of triplicate determinations of three independent experiments. Data from vehicle-treated cells (0) were normalized to 100%
Table 1.
IC50 values of new compounds on breast and prostate cancer cell proliferation

| ||||
|---|---|---|---|---|
| Compound | R | BT-474 (µM) | MDA-MB-231 (µM) | PC3 (µM) |
| SAHA | 0.55 ± 0.01 | 0.55 ± 0.12 | 0.88 ± 0.09 | |
| 15a | C6H5- | 2.93 ± 0.54 | 1.85 ± 0.46 | 2.20 ± 0.49 |
| 15b | 4-NO2C6H4- | 2.49 ± 0.46 | 1.74 ± 0.37 | 2.03 ± 0.44 |
| 15c | 3-NO2C6H4 | 2.06 ± 0.37 | 1.90 ± 0.28 | 1.93 ± 0.37 |
| 15d | 4-FC6H4 | - | 2.07 ± 0.77 | - |
| 15e | 4-BrC6H4 | 3.35 ± 0.47 | 2.52 ± 0.19 | 1.84 ± 1.22 |
| 15f | 4-ClC6H4 | 2.46 ±0.47 | 1.89 ± 0.32 | 2.08 ± 1.29 |
| 15g | 4-OCH3C6H4 | 2.90 ± 0.30 | 1.75 ± 0.82 | 2.29 ± 0.69 |
| 15h | 3-ClC6H4 | 2.87 ± 0.44 | 2.76 ± 0.33 | 3.11 ± 0.78 |
| 15i | 2,4-Cl2C6H3 | - | - | - |
| 15j | 2-C5H3S | 3.11 ± 1.44 | 1.39 ± 0.89 | 1.64 ± 0.06 |
| 15k | 2-C5H3O | 2.92 ± 1.46 | 1.78 ± 0.36 | 2.29 ± 0.33 |
Results are expressed as the mean ± S.D
Concerning growth inhibitory effects exerted by compounds in cancer cells at 10 µM concentration, all compounds significantly diminished cell proliferation compared with non-treated cells, with different potencies among the cell lines tested. The compounds 15a, 15c, 15j, and 15k, as well as 15 g, 15j, and 15k, showed similar growth inhibitor effects that SAHA on BT-474 and MDA-MB231 cells, respectively. Notably, these compounds obtained lesser IC50 values in each cell line. In the case of PC3 cells, the 15k compound exhibited an inhibitory effect comparable to SAHA. Notably, the 15k compound exerted the greatest antiproliferative activity in all cancer cell lines (Table 2). Another compound with an important growth inhibitory effect was 15j, being that it inhibits the proliferation of both breast cancer cell lines similar to the reference control.
Table 2.
Growth inhibitory effects (GI%) exerted by compounds 15a-k on breast and prostate cancer cells
| Compounda | R | BT-474 | MDA-MB-231 | PC3 |
|---|---|---|---|---|
| SAHA | 98.22 ± 1.82b | 97.54 ± 1.59b | 87.86 ± 7.85b | |
| 15a | C6H5 | 82.32 ± 3.40b | 89.60 ± 2.35b,c | 63.62 ± 13.55b,c |
| 15b | 4-NO2C6H4 | 65.82 ± 20.06b,c | 86.71 ± 3.65b,c | 64.32 ± 9.72b,c |
| 15c | 3-NO2C6H4 | 84.14 ± 11.11b | 86.79 ± 3.96b,c | 74.20 ± 7.61b,c |
| 15d | 4-FC6H4 | 27.68 ± 18.59b,c | 74.71 ± 3.43b,c | 36.73 ± 18.26b,c |
| 15e | 4-BrC6H4 | 74.75 ± 17.70b,c | 84.68 ± 4.36b,c | 66.14 ± 5.81b,c |
| 15f | 4-ClC6H4 | 56.81 ± 23.23b,c | 80.15 ± 3.75b,c | 54.59 ± 10.20b,c |
| 15g | 4-OCH3C6H4 | 76.07 ± 22.49b,c | 93.75 ± 3.20b | 58.98 ± 11.75b,c |
| 15h | 3-ClC6H4 | 54.85 ± 23.48b,c | 79.50 ± 5.89b,c | 57.71 ± 6.44b,c |
| 15i | 2,4-Cl2C6H3 | 1.60 ± 6.90c | 0.43 ± 6.28c | 0.00 ± 9.34b,c |
| 15j | 2-C5H3S | 82.32 ± 11.41b | 93.31 ± 1.86b | 68.46 ± 7.04 b,c |
| 15k | 2-C5H3O | 96.18 ± 2.63b | 98.42 ± 1.09b | 81.31 ± 15.35b |
Results are expressed as the mean ± S.D. percent growth inhibition of triplicate determinations and represent at least three different experiments. a10μM, bP < 0.05 vs. vehicle, cP < 0.001 vs. SAHA
The structure–activity relationship was important in the activity of the compounds. In this sense, in compounds 15k and 15j, the addition of the furan and thiophene substituents at C4 of pyridine conferred relevant antiproliferative properties since 15k showed the greatest antiproliferative effect in the three cancer cell lines used in this study and 15j in the breast cancer lines. It is important since both compounds can affect cell proliferation of cancer cell lines with an aggressive phenotype [46, 47]. The C6H5, 3-NO2C6H4, 4-OCH3C6H4 substituents at C4 of pyridine also confer antiproliferative activity to compounds 15a, 15c, and 15g, respectively, which was comparable to SAHA in breast cancer cells. In contrast, the addition of 2,4-Cl2C6H4 substituent to compound 15i implied the total loss of the antiproliferative effect in the three cancer cell lines.
Effects of compounds 15a, 15c, 15j, and 15 k on the expression of cell cycle regulatory genes
Considering that compounds 15a, 15c, 15j, and 15k have antiproliferative properties like SAHA in BT-474 cells, we evaluated their effects on mRNA expression of three genes related to the cell cycle progression in this cell line. For these experiments, the cells were cultured in the absence or presence of the compounds or SAHA. The results demonstrated that the compounds significantly upregulated CDKN1A (p21) and downregulated CCND1 (cyclin D1) and TP53 (p53) gene expression when compared to vehicle-treated cells. Compound 15c did not modify mRNA expression levels of p21 (Fig. 4).
Fig. 4.
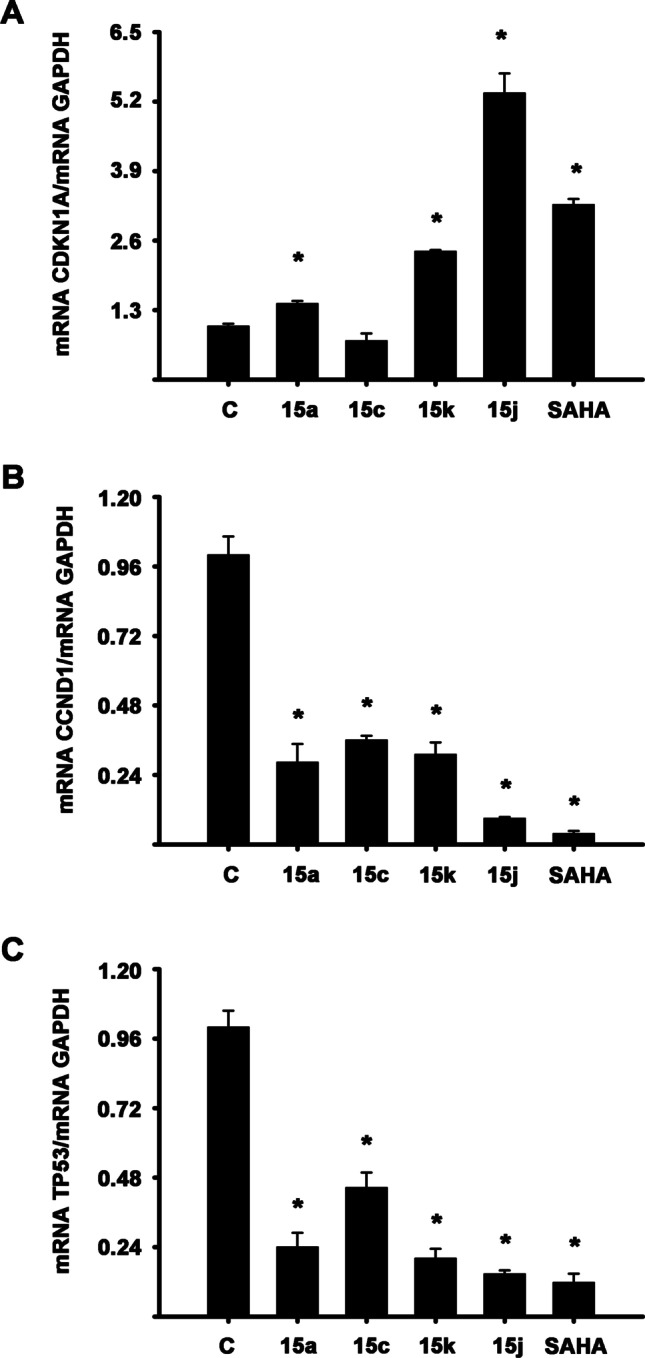
Effects of the compounds 15a, 15c, 15j, and 15 k on the expression of genes implicated in the cell cycle. BT-474 cells were incubated in the absence (C) and presence of 10 µM of compounds or SAHA for 24 h. Results shown are the mean ± S.D. of genes/GAPDH mRNA normalized ratio of three independent experiments per triplicate. The value of vehicle-treated cells (C) was set to one. *P ≤ 0.05 vs. C
Overexpression and aberrant activity of HDACs are critical in tumorigenesis by regulating the transcription of genes essential for controlling normal cell growth and differentiation. This has led to the development of many small molecules with the capacity to interfere with HDAC activity, and therefore with potential application to cancer treatment. SAHA has been the first HDAC inhibitor approved by the FDA to treat cutaneous T-cell lymphoma (CTCL), and several HDAC inhibitors are currently in clinical trials [48, 49]. SAHA inhibits growth and induces cell cycle arrest through p21, p53, and cyclin D1 regulation in cancer cells [50–52]. In particular, in the MDA-MB-231 breast cancer cells (p53 mutated), SAHA treatment down-regulated p53 and induced p21; and in MCF7 breast cancer cells, the mRNA encoding cyclin D1 was diminished by treatment with SAHA [44, 52]. Similar to these results, BT-474 cells (p53 mutated) treated with compounds 15a, 15c, 15j, and 15k reduced p53 and cyclin D1 mRNA levels and increased p21 gene expression. Except for 15c, which did not modify p21 mRNA expression.
Interestingly, compound 15c was able to suppress cell growth and diminish cyclin D1 and p53 gene expression, but it did not alter p21 mRNA levels in BT-474 cells. Maybe, 3-NO2C6H4 substituent to compound 15c can confer antiproliferative activity but hinder some level of transcriptional induction of p21, but it deserves further investigations. Like these results, RG7388, a small molecule that induces cell cycle arrest and triggers cell death, did not elevate p21; but it significantly increased p53 levels in MCF-7 cells (p53 wild-type) [50].
Concerning p21, it has been demonstrated that SAHA increases the accumulation of acetylated histone H3 and H4 in the p21 promoter, which was associated with the increase of its gene expression [53]. Therefore, future research is needed to directly analyze the compounds' effects in histone acetylation and resulting changes in chromatin structure to provide further insight into the mechanism action of new compounds.
These results indicate that the new compounds derived from pyridine with substituents 2-furan (15k) and 2-thiophene (15j) could be proposed as good candidates for continuing with future preclinical assays due to their inhibition of cell growth and regulation of genes related to cell cycle progression in highly metastatic cancer cells.
Docking studies
The HDACs are a group of corepressors of transcriptional activators, and their levels of expression are potentially dysregulated in cancer [54]. We investigated the antiproliferative effect of compounds with a high substituted pyridine as capping in breast and prostate cancer cell lines, which overexpress certain HDAC isoforms. The overexpression of HDAC1 is present in the BT-474 [55] and PC3 cell lines [43, 54]. In the MDA-MB-231 cell line, HDAC6 and HDAC8 play a critical role in proliferation and metastasis [56, 57]. Hence, such isoforms are ideal biological targets for docking experiments. Compounds 15a, 15c, 15 g, 15j, and 15k were docked into the active site of HDAC1, HDAC6, and HDAC8 to gain insight into the ligand-receptor binding mode. The negative interaction free energies and ligand efficiencies, shown in Tables 3 and 4. Binding modes of SAHA, 15a, 15c, 15g, 15j, and 15k with HDAC1, HDAC6, and HDAC8 are displayed in Figs. 5, 6, and 7, respectively.
Table 3.
Interaction energies of molecules docked into the active site of isoforms HDAC
| Ligand | HDAC1* | HDAC6* | HDAC8* |
|---|---|---|---|
| SAHA | -8.05 | -9.79 | -8.86 |
| 15a | -7.97 | -9.27 | -9.65 |
| 15c | -10.01 | -11.42 | -10.57 |
| 15g | -8.32 | -10.17 | -8.26 |
| 15j | -9.43 | -10.32 | -9.97 |
| 15k | -8.19 | -9.49 | -9.64 |
*Kcal/mol
Table 4.
Ligand efficient of molecules docked into the active site of isoforms HDAC
| Ligand | HDAC1* | HDAC6* | HDAC8* |
|---|---|---|---|
| SAHA | -0.42 | -0.51 | -0.47 |
| 15a | -0.27 | -0.31 | -0.32 |
| 15c | -0.30 | -0.34 | -0.32 |
| 15g | -0.26 | -0.32 | -0.26 |
| 15j | -0.32 | -0.36 | -0.34 |
| 15k | -0.28 | -0.33 | -0.33 |
*Kcal/mol
Fig. 5.

The binding modes for SAHA and compounds 15a, 15c, 15g, 15j, and 15k at the active site of HDAC1, according to molecular docking calculations. The stacking interactions are shown as a pink line, hydrogen bonds are displayed as green dotted lines, π-alkyl interaction is displayed as middle pink, Van der Waals attraction is placed as green circles, interaction π-σ is displayed as purple lines, the coordination bond with zinc is shown as a purple circle. The observed distances of interaction between zinc and the closest oxygen of hydroxamic acid moiety of SAHA, 15a, 15c, 15g, 15j, and 15k are 3.9, 12.7, 5.8, 7.2, 5.0, and 14.4 Å, respectively
Fig. 6.

Interactions between compounds and the residue in the active site of HDAC6. The stacking interactions are shown as a pink line, hydrogen bonds are displayed as green dotted lines, Van der Waals attraction is placed as green circles, interaction π-σ is displayed as purple lines, π-sulfur interaction is shown as yellow lines, the coordination bond with zinc are shown as purple lines. The observed distances of interaction between zinc and the closest oxygen of hydroxamic acid moiety of SAHA, 15a, 15c, 15g, 15j, and 15k are 2.0, 2.4, 2.1, 2.8, 2.3, and 2.3 Å, respectively
Fig. 7.

Interactions between compounds and the residue in the active site of HDAC8. The stacking interactions are shown as a pink line, π-alkyl interactions are displayed as middle pink, interaction π-σ are displayed as purple lines, Van der Waals attraction is placed as green circles, π-anion interaction is orange, hydrogen bond is displayed as blue dotted lines and zinc is gray color. The observed distance between the zinc cofactor and the closest oxygen of hydroxamic moiety of SAHA, 15a, 15c, 15 g, 15j, and 15k is 2.3, 3.0, 3.7, 3.2, 3.2, and 3.2 Å, respectively
The results of binding energies indicate a favorable interaction between ligands and HDAC isoforms. The docking between active site HDAC1 and SAHA showed the hydroxamic group inside the pocket is coordinated with the zinc atom in a mono-dentate fashion. However, the interaction at the active site of HDAC1 and the five ligands (15a, 15c, 15 g, 15j, and 15k) does not display a coordination interaction with the zinc atom at all. The compounds 15a, 15 g, and 15k remain on the surface and do not penetrate through the HDAC1 active site pocket, while 15c and 15j came closer to the Zn II cofactor than the rest of the ligands. The ligands 15c, 15j, and 15k display larger interaction energy and ligand efficiency with HDAC1. Such a fact correlates with the finding that these molecules displayed the highest percentage of growth inhibition for PC3 and BT-747 culture cells, which overexpress HDAC1.
Considering the docking of HDAC6, we observe that compounds 15a, 15c, 15 g, and 15k display a binding mode similar to the displayed by SAHA, a bidentate chelation geometry is observed. Nevertheless, the hydroxamic group of the ligand 15j displays a mono-dentate geometry with the zinc atom inside the enzyme pocket. The docking calculations show that the capping group of five analogs is located in a zone farther to the HDAC6 active site pocket entrance than the SAHA capping group. Besides, it is observed a binding interaction between 15j with histidine-500 HDAC6 residue through an acceptor hydrogen bond. This interaction could be responsible for the fact that the ligand 15j was not sufficiently introduced to form bidentate chelation with the Zn cofactor. In general, we obtained the largest interaction energy with HDAC6, and this is consistent with the antiproliferative effect in the cell line MDA-MB-231 that overexpresses this isoform.
Regarding the docking calculations in the active site of HDAC8, the hydroxamic group of compounds 15a, 15g, and 15k displays bi-dentate chelation with Zn II, unlike 15c and 15j, displaying a mono-dentate interaction. The ligand 15k displays a binding mode pretty similar to the one displayed by SAHA, in which a donor hydrogen-bond interaction is observed with histidine 142 residues. Besides, three π-staking interactions are displayed, those with phenylalanine 152, histidine 108, and phenylalanine 208 residues. Moreover, 15c and 15j show binding interactions with two residues located in the upper part of the HDAC8 active pocket, such resides are lysine 33 and aspartate 101.
It is observed that the differences in the heteroatom of 15j and 15k affect the binding interactions with the enzyme pocket. Also, the properties of the oxygen belonging to the furan of the 15 k display a more effective interaction mode than 15j with the active site cavity of HDAC6 and HDAC8. The fundamental difference between the heteroatom of 15j and 15k is the size and electronegativity; the oxygen displayed by 15k is more electronegative and has a smaller atomic radius than the sulfur of 15j. Consequently, 15 k heteroatom holds the right size and electronegativity to penetrate the active site of HDAC6 and HDAC8 to form more stable interactions with amino acid residues. In recent studies has been suggested that HDAC6 inhibition may block breast cancer tumor metastasis [57]. Experimental and computational results in this study suggest that compounds 15a, 15c, 15 g, 15j, and 15k showed selectivity for the HDAC isoforms, overexpressed in highly metastatic breast cancer (MDA-MB-231). Finally, the general behavior displayed by all the tested molecules, 15a-k, is the improvement of the strength of the interactions of the capping moiety compared with SAHA, pyridine derivatives used in this study hold functional groups with the capability to perform stacking, hydrogen bond, and Van der Waals interactions in an efficient manner to block the active site of HDACs. However, the possible enzymatic inhibition of HDACs by 15a-k deserves future experimental confirmation.
Pharmacokinetic of 15a-k compounds
The determination of the pharmacokinetic and physicochemical properties of SAHA, 15a, 15c, 15g, 15j, and 15k, were calculated through the free server for pharmacokinetic evaluations SwissADME [58]. The calculated pharmacokinetic properties are displayed in Table 5, and the obtained physicochemical properties are shown in Table 6.
Table 5.
Pharmacokinetic parameter of compounds 15a-k
| Structure | HIA | BBB | P-gp substrate |
Bioavailability Score |
|---|---|---|---|---|
| SAHA | High | No | Yes | 0.55 |
| 15a | High | No | No | 0.55 |
| 15c | Low | No | Yes | 0.55 |
| 15g | Low | No | No | 0.55 |
| 15j | Low | No | Yes | 0.55 |
| 15k | Low | No | Yes | 0.55 |
Table 6.
Physicochemical parameter values of compounds 15a-k
| Structure | PSA (Å2) | Log P | Solubility (mg/ml) | Solubility Class |
|---|---|---|---|---|
| SAHA | 108.64 | 1.92 | 0.509 | Soluble |
| 15a | 124.34 | 3.52 | 0.0000783 | Moderately soluble |
| 15c | 170.16 | 3.34 | 0.0000662 | Moderately soluble |
| 15g | 133.57 | 3.49 | 0.0000647 | Moderately soluble |
| 15j | 152.58 | 3.23 | 0.000113 | Soluble |
| 15k | 137.48 | 2.62 | 0.000345 | Soluble |
It is observed that SAHA has a large polar surface area (PSA) and good solubility in water. In respect of the partition coefficient n-octanol/water (log Po/w), the more positive Log Po / w, the more lipophilic properties are displayed. Then, SAHA and 15a-k are within the optimal range (− 0.7, + 5.0); therefore, the compounds 15a, 15c, 15 g, 15j, and 15k are more lipophilic than SAHA. Also, the prediction for passive human gastrointestinal absorption (HIA) was high for SAHA and 15a but low for the rest of the compounds. In addition, it was found that the permeability of the blood–brain barrier (BBB) is negative for SAHA and 15a-k. Besides, it is observed that SAHA and 15a-k do not have permeability at the BBB.
It was found that SAHA, 15c, 15j y 15k, efficient interacts with glycoprotein P (P-gp), while 15a and 15g do not exhibit such behavior. To further clarify the binding to the glycoprotein, we calculate a molecular coupling between analogs and P-gp (code PDB 6QEX) [59]; results are displayed in Table 7 (Figure S2 is placed in the SI). The docking results show that compounds 15g and 15k display the lowest ligand efficiency in the coupling with P-gp. Even though it has a union with the cellular efflux pump, 15g and 15k are the compounds that best inhibited the proliferation of cancer cells, of which, the one with the highest growth inhibition percentage was 15k.
Table 7.
Interaction energies (IE) and ligand efficiency (LE) of molecules docked into the active site of P-gp
| P-gp | ||
|---|---|---|
| Ligand | IE* | LE* |
| SAHA | -9.19 | -0.48 |
| 15a | -10.74 | -0.36 |
| 15c | -11.72 | -0.36 |
| 15g | -11.31 | -0.35 |
| 15j | -10.92 | -0.38 |
| 15k | -10.20 | -0.35 |
*Kcal/mol
Therefore, we can infer that the compounds 15a-k exhibit good absorption, favorable permeability to the cell´s interior.
Conclusions
The synthesis of 15a-k was performed with overall yields from 40 to 71%, the compounds were confirmed by spectroscopic, NMR, and crystallographic experiments. About the biological activity, 15k displays the highest antiproliferative activity for the BT-474 and MDA-MB-231 breast cancer cultures and the PC3 prostate cancer cell line at a concentration of 10 µM. Besides, 15j inhibited cell proliferation in a similar fashion to SAHA in both breast cancer cell lines. Also, 15j and 15k increased p21 mRNA levels and decreased cyclin D1 and p53 gene expression, a similar behavior is displayed by SAHA. Such behavior can be rationalized in the following way: inhibition of cell proliferation induced by 15j and 15k is performed through a cooperative effect, the induction of p21, and downregulation of cyclin D1 and p53 gene expressions. Besides, IC50 values of 15j and 15k are located in the µM scale, which is in the direction to discover candidates for antiproliferative molecules.
Regarding the investigation of the binding properties behind such biological activity, the molecular coupling analysis performed showed an increment in the number of interactions performed by the capping moiety, and consequently in the strength of the capping group interaction, of the tested molecules compared with SAHA. The interactions responsible for improving the caping group interactions are Van der Waals, stacking, and hydrogen bond. Finally, 15j and 15k display the larger bonding interactions with the biological targets, in agreement with the observed performance of the biological evaluation. With reference to the pharmacokinetic properties, it was observed that 15a-k exhibit good absorption and favorable permeability to the cell´s interior. Besides, compounds 15g and 15k display the lowest binding properties with glycoprotein P.
The general conclusion is that the two synthesized pyridine derivatives with the best performance in this study, 2-thiophene (15j) and 2-furan (15k), may serve for the research of new HDAC inhibitors. Such asseveration is confirmed by the displayed inhibition of cell growth, regulation of genes related to cell cycle progression HDACs in highly metastatic cancer cells and the binding properties with HDACs.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors appreciate of the Guanajuato National Laboratory (UG-UAA-CONACyT (#123732) for their generous allocation of analytical and computing resources. We are also thankful the Unit for Industry and Research Support (USAII) at the School of Chemistry at UNAM and Dr. Marcos Flores-Álamo for X-ray crystallography support. Thanks to “Fundación para la Salud y la Educación Dr. Salvador Zubirán” and “FOINS-INCMNSZ” by postdoctoral fellowship to N.S-M (A-307-7). The authors would like to thank the Programa de Investigación en Cáncer de Mama, Universidad Nacional Autónoma de México and Biol. Salvador Ramirez Jiménez, who is responsible of the repository of breast cancer cell lines.
Funding
This research was funded by Consejo Nacional de Ciencia y Tecnología (CONACYT) (Grant A1-S-27694) and DAIP-UG (Grants 034/2021, 131/2021) and by Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT, Grant IN208520), Dirección General de Asuntos del Personal Académico (DGAPA), Universidad Nacional Autónoma de México (UNAM) to R.G-B. F.H.B. and I.M.S acknowledge CONACYT for a graduate scholarships (#482137 and #673473, respectively).
Declarations
Conflicts of Interest
The authors confirm that the content of this article has no conflict of interest.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 2.Wang A, Qu L, Wang L. At the crossroads of cancer stem cells and targeted therapy resistance. Cancer Lett. 2017;385:87–96. doi: 10.1016/j.canlet.2016.10.039. [DOI] [PubMed] [Google Scholar]
- 3.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: Overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 4.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 5.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 7.Raynal NJ-M, Da Costa EM, Lee JT, Gharibyan V, Ahmed S, Zhang H, et al. Repositioning FDA-approved drugs in combination with epigenetic drugs to reprogram colon cancer epigenome. Mol Cancer Ther. 2017;16:397–407. doi: 10.1158/1535-7163.MCT-16-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taby R, Issa J-PJ. Cancer Epigenetics. C A Cancer J Clin. 2010;60:376–92. doi: 10.3322/caac.20085. [DOI] [PubMed] [Google Scholar]
- 9.Chang DK. Cancer epigenetics. Korean J Gastroenterol. 2004;44:1–12. doi: 10.1053/j.gastro.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 10.Mottamal M, Zheng S, Huang TL, Wang G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plumb J, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2003;2:721–728. [PubMed] [Google Scholar]
- 12.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009;280:233–241. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Cao J, Lv W, Wang L, Xu J, Yuan P, Huang S, He Z, Hu J. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 2018;9:817. doi: 10.1038/s41419-018-0788-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget M, Kalita A, Liu J, Lu A–H, Zhou NZ, Robert M, Gillespie J, Wang JJ, Ste-Croix H, Rahill J, Lefebvre S, Moradei O, Delorme D, MacLeod A, Besterman J, Li Z. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–68. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 15.Gediya LK, Belosay A, Khandelwal A, Purushottamachar P, Njar VCO. Improved synthesis of histone deacetylase inhibitors (HDIs) (MS-275 and CI-994) and inhibitory effects of HDIs alone or in combination with RAMBAs or retinoids on growth of human LNCaP prostate cancer cells and tumor xenografts. Bioorg Med Chem. 2008;16:3352–3360. doi: 10.1016/j.bmc.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443:735–746. doi: 10.1042/BJ20111685. [DOI] [PubMed] [Google Scholar]
- 17.Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X, Lu X. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69:901–909. doi: 10.1007/s00280-011-1766-x. [DOI] [PubMed] [Google Scholar]
- 18.Sundarapandian T, Shalini J, Sugunadevi S, Woo LK. Docking-enabled pharmacophore model for histone deacetylase 8 inhibitors and its application in anti-cancer drug discovery. J Mol Graph Model. 2010;29:382–395. doi: 10.1016/j.jmgm.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 20.Huang WJ, Chen CC, Chao SW, Lee SS, Hsu FL, Lu YL, Hung MF, Chang C. Synthesis of N-Hydroxycinnamides Capped with a Naturally Occurring Moiety as Inhibitors of Histone Deacetylase. ChemMedChem. 2010;5:598–607. doi: 10.1002/cmdc.200900494. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Yu N, Song H, Chen D, Zou Y, Deng W, Lye PL, Chang J, Ng M, Blanchard S, Sun ET, Sangthongpitag K, Wang X, Goh KC, Wu X, Khng HH, Lijuan M. N-Hydroxy-1,2-disubstituted-1H-benzimidazol-5-yl acrylamides as novel histone deacetylase inhibitors: Design, synthesis, SAR studies, and in vivo antitumor activity. Bioorg Med Chem Lett. 2009;19:1403–1408. doi: 10.1016/j.bmcl.2009.01.041. [DOI] [PubMed] [Google Scholar]
- 22.Pirali T, Pagliai F, Mercurio C, Boggio R, Canonico PL, Sorba G, Tron GC, Genazzani A. Triazole-Modified Histone Deacetylase Inhibitors As a Rapid Route to Drug Discovery. J Comb Chem. 2008;10:624–627. doi: 10.1021/cc800061c. [DOI] [PubMed] [Google Scholar]
- 23.Salmi-Smail C, Fabre A, Dequiedt F, Restouin A, Castellano R, Garbit S, Roche P, Morelli X, Brunel JM, Collette Y. Modified Cap Group Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitor Derivatives Reveal Improved Selective Antileukemic Activity. J Med Chem. 2010;53:3038–3047. doi: 10.1021/jm901358y. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh B, Zhao W-N, Reis SA, Patnaik D, Fass DM, Tsai L-H, et al. Dissecting structure–activity-relationships of crebinostat: Brain penetrant HDAC inhibitors for neuroepigenetic regulation. Bioorg Med Chem Lett. 2016;26:1265–1271. doi: 10.1016/j.bmcl.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walton JW, Cross JM, Riedel T, Dyson P. Perfluorinated HDAC inhibitors as selective anticancer agents. Org Biomol Chem. 2017;15:9186–9190. doi: 10.1039/c7ob02339a. [DOI] [PubMed] [Google Scholar]
- 26.Hesham HM, Lasheen DS, Abouzid KAM. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med Res Rev. 2018;38:2058–2109. doi: 10.1002/med.21505. [DOI] [PubMed] [Google Scholar]
- 27.García S, Mercado-Sánchez I, Bahena L, Alcaraz Y, García-Revilla MA, Robles J, Santos-Martínez N, Ordaz-Rosado D, García-Becerra R, Vazquez M. Design of Fluorescent Coumarin-Hydroxamic Acid Derivatives as Inhibitors of HDACs: Synthesis, Anti-proliferative Evaluation and Docking Studies. Molecules. 2020;25:5134. doi: 10.3390/molecules25215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bahena L, Cervantes C, Soto-Arredondo KJ, Martínez-Alfaro M, Zarco N, García-Revilla MA, Alcaraz-Contreras, Palma Tirado L, Vázquez MARJ. In silico, synthesis and biological investigations of Pyrrolo-[3,4-c]pyrrole hydroxamic acid derivatives as potential anticancer agents. J Mex Chem Soc. 2017;61:297–308. [Google Scholar]
- 29.Hernández F, Sánchez A, Rendón-Vallejo P, Millán-Pacheco C, Alcaraz Y, Delgado F, Vázquez MA, Estrada-Soto S. Synthesis, ex vivo and in silico studies of 3-cyano-2-pyridone derivatives with vasorelaxant activity. Eur J Med Chem. 2013;70:669–76. doi: 10.1016/j.ejmech.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Hernández F, De la Cruz F, López J, Peña E, Delgado F, Alcaraz Y, Robles J, Martínez-Alfaro M, Vázquez M. A Green Approach to the Production of 2-pyridone Derivatives Promoted by Infrared Irradiation. J Mex Chem Soc. 2014;58:152–158. [Google Scholar]
- 31.Keepers YP, Pizao PE, Peters GJ, van Ark-Otte J, Winograd B, Pinedo HM. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer Clin Oncol. 1991;27:897–900. doi: 10.1016/0277-5379(91)90142-z. [DOI] [PubMed] [Google Scholar]
- 32.Frisch, M J, Trucks, G W, Schlegel, H B, Scuseria, G E, Robb, M A, Cheeseman, J R, Scalmani, G, Barone, V, Mennucci, B, Petersson, G A, Nakatsuji, H, Caricato, M, Li, X, Hratchian, H P, Izmaylov, A F, Bloino, J, Zheng, G, Sonnenberg, J L, Hada, M, Ehara, DJ. Gaussian 09, Revision B.01. Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford CT. 2009.
- 33.Turnbull RE, Fairall L, Saleh A, Kelsall E, Morris KL, Ragan TJ, Savva CG, Chandru A, Millard CJ, Makarova OV, Smith CJ, Roseman AM, Fry AM, Cowley SM, Schwabe J. The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nat Commun. 2020;11:3252. doi: 10.1038/s41467-020-17078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hai Y, Christianson D. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari L. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Varano F, Catarzi D, Squarcialupi L, Betti M, Vincenzi F, Ravani A, et al. Exploring the 7-oxo-thiazolo[5,4-d]pyrimidine core for the design of new human adenosine A3 receptor antagonists. Synthesis, molecular modeling studies and pharmacological evaluation. Eur J Med Chem. 2015;96:105–21. doi: 10.1016/j.ejmech.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Wagaw S, Buchwald SL. The Synthesis of Aminopyridines: A Method Employing Palladium-Catalyzed Carbon−Nitrogen Bond Formation. J Org Chem. 1996;61:7240–7241. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]
- 38.Tan Q, Zhang Z, Hui J, Zhao Y, Zhu L. Synthesis and anticancer activities of thieno[3,2-d]pyrimidines as novel HDAC inhibitors. Bioorg Med Chem. 2014;22:358–365. doi: 10.1016/j.bmc.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 39.Usachova N, Leitis G, Jirgensons A, Kalvinsh I. Synthesis of Hydroxamic Acids by Activation of Carboxylic Acids with N, N′-Carbonyldiimidazole: Exploring the Efficiency of the Method. Synth Commun. 2010;40:927–935. [Google Scholar]
- 40.Deposition Numbers (CCDC): 1569641(11i), 2055316 (14f). www.ccdc.cam.ac.uk/structures.
- 41.Lakshmaiah K, Jacob L, Aparna S, Lokanatha D, Saldanha S. Epigenetic therapy of cancer with histone deacetylase inhibitors. J Cancer Res Ther. 2014;10:469–478. doi: 10.4103/0973-1482.137937. [DOI] [PubMed] [Google Scholar]
- 42.Peng X, Sun Z, Kuang P, Chen J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur J Med Chem. 2020;208:112831. doi: 10.1016/j.ejmech.2020.112831. [DOI] [PubMed] [Google Scholar]
- 43.Kim NH, Kim SN, Kim Y. Involvement of HDAC1 in E-cadherin expression in prostate cancer cells; its implication for cell motility and invasion. Biochem Biophys Res Commun. 2011;404:915–921. doi: 10.1016/j.bbrc.2010.12.081. [DOI] [PubMed] [Google Scholar]
- 44.Huang L, Pardee AB. Suberoylanilide hydroxamic acid as a potential therapeutic agent for human breast cancer treatment. Mol Med. 2000;6:849–866. [PMC free article] [PubMed] [Google Scholar]
- 45.Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, et al. Suberoylanilide Hydroxamic Acid, an Inhibitor of Histone Deacetylase, Suppresses the Growth of Prostate Cancer Cells in Vitro and in Vivo. Cancer Res. 2000;60:5165–5170. [PubMed] [Google Scholar]
- 46.Dai X, Cheng H, Bai Z, Li J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J Cancer. 2017;8:3131–3141. doi: 10.7150/jca.18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Invest Urol. 1979;17:16–23. [PubMed] [Google Scholar]
- 48.Hontecillas-Prieto L, Flores-Campos R, Silver A, de Álava E, Hajji N, García-Domínguez D. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front Genet. 2020;11:1113. doi: 10.3389/fgene.2020.578011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Botrugno OA, Santoro F, Minucci S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 2009;280:134–144. doi: 10.1016/j.canlet.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 50.Natarajan U, Venkatesan T, Radhakrishnan V, Samuel S, Rasappan P, Rathinavelu A. Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated through p21(WAF1/CIP1) and p27(KIP1) in Cancer Cells. Medicina (Kaunas) 2019;55:30. doi: 10.3390/medicina55020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patra N, De U, Kim TH, Lee Y, Ju A, Mee Young Kim ND, Yoon JH, Choi WS, Moon HR, Lee BM, Kim H. A novel histone deacetylase (HDAC) inhibitor MHY219 induces apoptosis via up-regulation of androgen receptor expression in human prostate cancer cells. Biomed Pharmacother. 2013;67:407–15. doi: 10.1016/j.biopha.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Knutson Kekapa’a A, Welsh J, Taylor T, Roy S, Wang Winnie WL, Tenniswood M. Comparative effects of histone deacetylase inhibitors on p53 target gene expression, cell cycle and apoptosis in MCF-7 breast cancer cells. Oncol Rep. 2012;27:849–53. doi: 10.3892/or.2011.1590. [DOI] [PubMed] [Google Scholar]
- 53.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, Chiu J, Guo H, Satagopan J, Wilton A, Gerald W, Basch R, Wang Z, Osman I, Lee P. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J Transl Res. 2009;1:62–71. [PMC free article] [PubMed] [Google Scholar]
- 55.Greer CB, Tanaka Y, Kim, Yoon J, Xie P, Zhang MQ, Park IH, Kim T. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015;13:1444–55. doi: 10.1016/j.celrep.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mravljak J, Ojsteršek T, Pajk S, Sollner DM. Coumarin-based dual fluorescent spin-probes. Tetrahedron Lett. 2013;54:5236–5238. [Google Scholar]
- 57.Ediriweera MK, Tennekoon KH, Samarakoon SR. Emerging role of histone deacetylase inhibitors as anti-breast-cancer agents. Drug Discov Today. 2019;24:685–702. doi: 10.1016/j.drudis.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 58.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alam A, Kowal J, Broude E, Roninson I, Locher K. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science. 2019;363:753–756. doi: 10.1126/science.aav7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.