

Abstract
Purpose
The enzyme Cyclooxygenases (COX-1 and COX-2) catalyze the formation of prostaglandin, a mediator of the inflammatory pathway. Inflammation related pathological conditions may be alleviated by targeting the Cox enzymes.COX-2 inhibitors that are currently available in the market causes undesirable side effects. Our present study focuses on the in-silico inhibition of COX -2 enzyme by the phytocompounds from Albizia amara and Phyla nodiflora.
Methods
The phytochemicals present in Albizia amara and Phyla nodiflora were analyzed for their COX-2 inhibition potential. Eight compounds from Albizia amara and eleven compounds from Phyla nodiflora obtained from GC–MS analysis was used for the current study. Molecular docking was performed using AutoDock vina. The crystal structure of COX-2 (PDB ID: 5IKR) was obtained from Protein data bank. PyMol was used to remove any solvent, organic and inorganic molecules. Energy minimization of the protein was carried out using SPDBV software. Geometrical optimizations of the ligands were performed using Avogadro software. Celecoxib was used as the positive control. ADMET properties of the compounds were analyzed using SwissADME and ProtoxII online servers. Molecular mechanics/generalized born surface area (MM/GBSA) calculations were performed to evaluate the binding efficiency. Molecular dynamics of the protein and protein–ligand complex was studied for about 100 ns using Desmond package of Schrodinger suite.
Results
Among the eighteen compounds, Squalene present in both the plants showed a better binding energy of -7.7 kcal/mol, when compare to other phytocompounds present in the extract. The control celecoxib showed a binding energy of about – 9.4 kcal/mol. The toxicity and ADMET properties of squalene indicated that it is non-toxic and followed Lipinski’s rule. Molecular Dynamics (MD) analysis showed that the binding of squalene to the enzyme was stable.
Conclusion
Squalene could potentially inhibit COX2 and o wing to its properties, squalene can be formulated in gels/creams and could be possibly used for external edema and inflammation
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1007/s40199-021-00408-6.
Keywords: COX-2, Albizia amara, Phyla nodiflora, Anti-inflammatory, Molecular docking, Molecular dynamics
Introduction
Inflammation is the defense mechanism of our body. Inflammation is a process by which the body reacts to infections, auto immune diseases or trauma of the body tissues. Inflammation is a necessary process for the body to recover from infections and the damages caused in the tissues [1]. Inflammation involves a complex array of enzyme activation, mediator release, and extravasation of fluid, cell migration, tissue breakdown, repair responsible for injury in living tissue [2]. The clinical manifestations of inflammation includes swelling, heat, redness and pain [3].Inflammation is caused by the over production of prostaglandins mediated by the action of cyclooxygenase enzymes [4].
Cyclooxygenase enzyme exists as two isoforms i.e. Cyclooxygenase-1 (COX-1) and Cyclooxygenase -2 (COX-2) [5]. During homeostasis, COX-1 is constitutively produced maintaining the basal production of required prostaglandins. At normal conditions, COX-2 is produced at very low levels. But during pathogenic stimuli, prognosis of cancer and inflammatory response, COX-2 produces very high levels of prostaglandins, [6].COX-2 is an inducible enzyme which is triggered by mediators such as growth factors, cytokines, bacterial lipopolysaccharide, phorbol esters and mitogens during high stress circumstances. i.e. inflammation[2, 7]. Both COX-1 and COX-2 have a very high structural and functional similarity. Cox enzymes are heme containing homodimeric proteins [4]. The active site of COX-2 is slightly larger than that of COX-1 enzmye and hydrophobic in nature [2, 8].This is due to the change in the aminoacid residue at position 523. In COX-1, it is Isoleucine whereas in COX-2 it is occupied by valine. This change is generally exploited for the design of COX-2 specific inhibitors [8]. Both the COX proteins carry out the same two separate catalytic functions—oxidation of arachidonate to Prostaglandin G2 (PGG2) and reduction of peroxide [9].
COX-2 pathway is generally inhibited by selective COX-2 inhibitors, antagonists, antibodies to mitogens, cytokines and glucocorticoids. As COX-1 and COX-2 are pharmacologically different, development of selective inhibitors of COX-2 and non-selective inhibitors of COX-1 was essential [2]. Non—steroidal anti-inflammatory drugs (NSAIDs) are drugs that are often employed in the pain management and inflammation. NSAIDs work by inhibitinh the COX enzymes. NSAIDs are extremely effective in managing the pain but causes undesirable side effects [3].This has kindled the need for new anti-inflammatory drugs and compounds from natural sources and as a result phytochemicals were explored [10]. NSAIDs such as ibuprofen, indomethacin and aspirin inhibit both the COX enzymes [11]. Significant analgesic and anti-inflammatory effects of the NSAIDs were managed by the selective inhibition of COX-2. This is due to the fact that prostaglandins are the products of enzymatic catalysis of arachidonic acid by COX-2 enzyme. Therefore, selective inhibition of COX-2 may represent a significant therapeutic advance in the management of pain and inflammation [11].The quest in need for selective COX-2 inhibitors has risen over the past, as non-selective inhibition of COX-1 leads to complications such as gastritis, ulceration, gastrointestinal upset, hemorrhage and sometimes even death [12].
Albizia amara or oil cake tree is locally known as Arappu. It usually grows in the dry areas of Tamilnadu, Andhra and Karnataka in India [13]. The barks are used for mouth inflammation and jaundice. The flowers and leaves are used for swellings, boils, eruptions and used to treat dandruff [14, 15]. Albizia amara has been used as an important folk medicine for the treatment of several diseases like diarrhea, gonorrhea, skin diseases, poisonous bites and leprosy. It contains a wide variety of bioactive compounds such as macrocyclic spermine alkaloids, triterpene saponins, phenols, flavonyl glycosides, tannins and sterols. Additionally, the plant possesses some pharmacological properties like anticancer, anti-hyperlipidimic, anti-inflammatory, anti-microbial, analgesic and anti-oxidant activities [16].
Phyla nodiflora is the fast growing perennial plants located in India, Ceylon, Central America, Srilanka and Tropical Africa [17]. These leaves are used to treat dandruff and the plant is known to possess antifungal, anti-inflammatory, antioxidant and anticancer activities [18]. The aerial parts of the plant are used as anodyne, parasiticide, antibacterial, emmenogogue, and febrifuge. It is also used in the treatment of wounds, piles, asthma, thirst and loss of consciousness. Poultice made from the plant are used as maturant for boils. The infusion of leaves and tender stalks are given to children for indigestion and to women postpartum. In Unani and Ayurveda, the plant is used as aphrodisiac, diuretic, and for the treatment of heart diseases, ulcers, bronchitis, fevers, cold, knee joint pain and in lithiasis [19].
The current study focuses on the in-silico docking and dynamics of the phytocompounds present in the ethanolic extracts of the leaves of Albizia amaraand Phyla nodiflora against COX-2 enzyme.
Materials and methods
GC–MS analysis of the extract
The GC–MS analysis and the phytocompounds present in the ethanolic extract of the leaves of Albizia amara and Phyla nodiflora were previously reported [20]. Briefly, the leaves of the plant were collected and authenticated (Authentication number: BSI/SRC/5/23/2018/Tech/3453). The leaves were then washed, dried and were pulverized to a fine powder. The phytocompunds were extracted from the powder using ethanol. The ethanol extract was then subjected to GC–MS Analysis. GC–MS analysis was performed using Perkin–Elmer GC Clarus 500 system equipped with Elite-5MS fused silica column (5% biphenyl 95% dimethylpolysiloxane, 30 m × 0.25 mm ID × 250 μm df). Helium was used as the carrier gas with a constant flow rate of 1 mL/min. 1μL of sample was injected into the system with an injection temperature of 260 °C. Initially, the oven temperature was kept at 60 °C for 2 min and increased to 300 °C at the rate of 10 °C/min. It was held at the temperature of about 300 °C for 6 min. For mass detector, the transfer line temperature and ion source temperature were 240 °C and 240 °C respectively. The ionization mode electron impact was set as 70 eV with 0.2 s of scan time and 0.1 s of scan interval and from 40 to 600 Da of fragments. The spectrums of the unknown components from the samples were compared with the spectrum database of known components stored in the GC–MS NIST (2008) library. These phytocompounds were used as ligands against cox-2 enzyme (Supplementary table 1).
Ligand preparation
Pubchem database (https://pubchem.ncbi.nlm.nih.gov/) was used to retrieve the structures of the compounds. Avogadro, a free cross-platform molecular editor was used for geometrical optimization of the compounds. Geometrical optimization was carried out using GAFF force field employing steepest descent algorithm with 4 steps per update. Energy optimized phytochemicals were used as ligands for docking [21].
Target protein and preparation
The Structure of Mefenamic Acid Bound to Human Cyclooxygenase-2 (PDB ID: 5IKR) was downloaded from RSCB PDB (https://www.rcsb.org/). The protein structure was solved using X-ray diffraction method and had a resolution of 2.34 Å. The solvent and other bound organic and inorganic moieties were removed using PyMol before docking. Energy minimization of the protein was carried out using SPDBV 4.1. Energy minimization computations were done in-vacuo employing GROMOSS96 43B1 parameter set [22].
Docking analysis
AutoDock Vina was used to perform docking of the phytocompounds against COX-2 enzyme [23]. Computed Atlas of Surface Topography of proteins (CASTp) (http://sts.bioe.uic.edu/castp/calculation.html) was used to predict the probable active site of the protein [24].A grid was constructed with the spacing of 0.375 Å. The grid size was set at 70 × 70 × 70 (x, y, and z) points, and the grid centre was designated at x, y, and z dimensions of 37.532, 3.836, and 56.489, respectively. Estimated free energy of binding was used to evaluate the docking of compounds against the enzyme.
Post-dock analysis were visualized using Protein–Ligand interaction profiler (https://projects.biotec.tu-dresden.de/plip-web/plip) and LigPlot plus which showed the location of binding sites, hydrogen-bond and hydrophobic interactions from an interaction radii of < 5 Å from the position of the docked drug [25, 26].
MM/GBSA calculations
To estimate relative binding affinities of protein ligand complexes, Molecular mechanics with generalized born surface area (MM/GBSA) is employed. Prime MM/GMSA modules were used for calculating the relative binding free energy for each molecule. The protein ligand complexes obtained from AutoDock Vina docking were subjected to MM/GBSA calculations [27, 28].
ADMET analysis
Molecular descriptors and drug-likeliness properties i.e. ADME (Absorption, Distribution, Metabolism and Excretion) properties of the ligands were analyzed using SwissADME online tool (http://www.swissadme.ch/). Toxicities of the compounds were predicted using Protox II online server [29]. Various ADMET associated properties like blood–brain barrier penetration, gastrointestinal absorption, phosphoglycoprotein substrate, Logkp, LogPo/w, LogS, cytochrome 450 substrate or inhibitor, carcinogenicity, cytotoxicity, hepatotoxicity, immunotoxicity and mutagenicity were estimated. ADMET testing helps to characterize promising pharmacological compounds in order to identify both the compounds with potential and those with major disqualifying drawbacks.
Molecular dynamics
MD simulations were carried out using the Desmond software by Schrödinger [30]. The optimized potentials for the liquid simulations (OPLS)-2005 force field were used in this system to determine the protein interactions with ligand molecules, which was solvated with the simple point charged (TIP4P) water model [31]. The orthorhombic water box was used to create a 10 Å buffer region between the protein atoms and box sides. Overlapping water molecules were deleted and the systems were neutralized with Na + and Cl− ions. The OPLS-2005 force field was used for energy calculation. The NPT ensemble was used and the temperature was maintained constant at 310 K, and a 2.0 fs value was obtained in the integration step. MD simulations for the complex structure of the protein as well as the target with position restraints were executed for 100 ns to allow the water molecules to remain in the system. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were computed to monitor the stability [32].
Results and discussion
Molecular docking & MM/GBSA calculations
The ethanolic extracts of the leaves of Albizia amara and Phyla nodiflora were analysed using GC–MS. Gas Chromatography was run for approximately 32 min. The fractions separated from the GC were analyzed in mass spectrometer. The compounds having a molecular weight between 40 and 600 Da were screened and is tabulated in Table 1. These compounds were used as ligands for the docking analysis. A total of eight compounds were identified in the extracts of Albizia amara and eleven compounds were identified in the extracts of Phyla nodiflora. Squalene was present in both the extracts. Celecoxib, a standard NSAID was used as the control (Table 1).
Table 1.
The table represents the binding energies in kcal/mol of phytocompounds from Albiziaamara and Phyla nodifloraupon docking with COX-2 enzyme using Autodockvina
| Source | Compound | Binding Energy (kcal/mol) |
|---|---|---|
| Albizia amara | Methyl-2-O- Methyl.Beta. L- Arabinopyranoside | -5.2 |
| Phytol | -6 | |
| 1-Octen-3-Yl-N- Propionate | -5.1 | |
| Propane-1,1-Diol Diacetate | -5.5 | |
| Methyl 2- Hydroxy- Octadeca-9,12,15- Trienoate | -6 | |
| Methyl 8,11,14- Heptadecatrienoate | -5.5 | |
| Squalene | -7.7 | |
| (3. Alpha.)- 12- Oleanen-3-Yl Acetate | -7.2 | |
| Phyla nodiflora | Trans,Trans-Muconic Acid | -5.3 |
| Cyclohexanone, 2-(1-Methyl-2-Oxopropyl)- | -5.2 | |
| 1H-Imidazole, 2-(Diethoxymethyl)- | -4.7 | |
| Trans,Cis-1,7-Dimethylspiro[4.5]Decane | -6.1 | |
| 1,6-Anhydro-.Alpha.-D-Galactofuranose | -5.6 | |
| Emylcamate | -4.6 | |
| Benzene, 1,1'-[1,4-Butanediylbis(Oxymethylene)]Bis- | -6.8 | |
| Oxalic Acid, CyclobutylPentadecyl Ester | -5.8 | |
| Ethyl 9,12,15-Octadecatrienoate | -6.5 | |
| 1,2-Dioxin-3-Acetic Acid, 6-(8,10-Dodecadienyl)-3,6-Dihydro-6-Methoxy-, Methyl | -5.9 | |
| Standard | Celecoxib—COX2 | -9.4 |
Docking studies were performed to find the ligand efficiency (binding energy) of the phytocompounds towards the COX2 protein. Molecular dynamics were performed to identify the interactions and dynamics of the protein–ligand complex. Docking studies of the eighteen compounds were carried out using AutoDock vina. The active site of the COX-2 enzyme was predicted using Castp and further confirmation was made by reviewing existing literature. As per literature, the COX-2 active site extends from the membrane binding domain to the catalytic domain core. The upper half of this site extending from Arg120 till Tyr385 is where the binding site of arachidonate is located. This is then followed by middle of the channel where Ser530 is located. The active site of COX-2 is comparatively larger than that of COX-1. This is because of the presence of Valine instead of isoleucine at the 523rd position, thereby creating a larger and more accessible channel. This channel is important for COX-2 drug selectivity. Another major difference is the presence of Arginine in COX-2 instead of histidine in COX-1 at the 513th position. Even though it doesn’t impact drug binding site, a change in the chemical environment is seen as Arg513 in COX-2 can interact with polar molecules which also determine the drug selectivity [33]. It can be seen from the docking studies that squalene present in both the leaf extracts gave the least binding energy when compared to the other phytocompounds present. The binding energies of the phytocompounds docked with COX2 are tabulated in Table 1.
Post dock analysis of the protein–ligand complex was performed using Ligplot plus. Post dock analysis was performed to understand the various non-covalent interactions like hydrogen bond, hydrophobic, and charge interactions between the ligand and the protein. Hydrogen bonds are an important factor that influences protein stability in docking complexes.
Celecoxib was bound with the COX-2 enzyme with a binding energy of -9.4 kcal/mol. Celecoxib interacted with Asp125, Gln372, Lys532 via hydrogen bonds whose bond lengths were 3.08 Å, 2.79 Å and 2.88 Å, respectively. Hydrogen bond network plays an integral role in strengthening the interaction between ligand and the protein. This may be due to the bond length distance between amino acid residue [34, 35]. Hydrophobic interaction were observed between the aminoacid residues and the ligand at Ser121, Ser126, Gln370 and Phe371 (Fig. 1).
Fig. 1.

(a) & (b) Interaction of celecoxib with the amino acid residues present in COX-2 enzyme
Squalene was bound with the COX-2 enzyme with a binding energy of -7.7 kcal/mol. It interacted with 13 aminoacid residues via hydrophobic interactions. Squalene showed interactions with the following aminoacid residues Trp139, Phe142, Ser143, Leu145, Leu 224, Gly225,Gln227, Gln374, Asn375, Arg376, Gly533, Gly536 and Asn537 (Fig. 2). The hydrophobic effect also plays a dominant role in protein–ligand binding. It has been understtod that the pockets with more hydrophobic substituents led to an increase in the binding energy. The number of hydrophobic atoms in the active core of the drug-target interface increases the binding affinity of the target-receptor and biological activity of the lead compound [34, 35]. Squalene is an isoprenoid compound that possesses antioxidant, antibacterial, antitumor,pesticide, immuno stimulant, diuretic, anti-cancer and Lipoxygenase inhibitor activities [20].
Fig. 2.

(a) & (b) Interaction of squalene with the amino acid residues present in COX-2 enzyme
Inorder to understand the binding strength, the non-covalent interaction residues and their interaction fraction (IF) during MD simulations were also studied (Supplementary Fig. 1 and 2). Molecular mechanics/generalized born surface area (MM/GBSA) calculations were performed for best ranking molecules. Estimation of relative binding affinity of ligands to the receptor is performed using MM/GBSA calculations. In principle, MM/GBSAis used for free energy based ranking of ligands belonging to a congeneric series. MM/GBSA and docking protocols employed in the present work for assessing ligand affinities to COX2 allow protein flexibility and therefore, give more reliable results [27, 28]. Results of MM/GBSA are tabulated in Table 2. Based on the calculations, ΔG bind of squalene was lower than that of celecoxib.The predominant forces that were observed for squalene binding with COX2 were covalent, Generalized Born electrostatic solvation energy and vanderwaal’s forces.Incase of celecoxib, the predominant forces were coulombic forces and hydrogen bonds.
Table 2.
The table represents the binding energies in kcal/mol calculated based on MM/GBSA studies
| Compounds | ΔG Bind | ΔG Bind Coulomb | ΔG Bind Covalent | ΔG Bind Hbond | ΔG Bind Solv GB | ΔG Bind vdW |
|---|---|---|---|---|---|---|
| Celecoxib | -39.99 | -12.82 | 1.77 | -1.91 | 33.84 | -33.25 |
| Squalene | -45.06 | -1.18 | 14.79 | 0 | 34.2 | -50.26 |
Coulomb represents Coulomb energy; Covalent represents Covalent binding energy; Hbond represents Hydrogen bond; Solv GB represents Generalized Born electrostatic solvation energy; vdW represents Van der Waals energy
ADMET properties
ADME properties of squalene were analyzed using SwissADME server. Squalene has a molecular weight of about 410.72 g/mol. It has about 15 rotatable bonds and does not have any hydrogen bond donor or acceptor groups. The molar refractivity is about 143.48 and the topological polar surface area (TPSA) is around 0Å2. The Log Po/w (MLOGP) is about 7.93 indicating that the molecule is highly lipophilic and it is insoluble in water. On observing the pharmacokinetic properties of squalene, it is observed that the absorption of the molecule in the gastrointestinal tract is low. Squalene does not permeate the blood brain barrier. It is not a substrate for P-glycoprotein and does not show any inhibition towards cytochrome P450 (1A2, 2C19, 2C9, 2D6, 3A4) enzymes. The skin permeation Log Kp value is about -0.58 cm/s indicating it is a very good skin permeant. Bioavailability score for the molecule is 0.55 and it follows Lipinski’s rule.
Upon predicting the toxicity of the compound, it was observed that squalene was not hepatotoxic, Carcinogenic, Immunotoxic, Cytotoxic and Mutagenic indicating that it is safe for human consumption. The LD50 of the compound was predicted to be 5000 mg/Kg. Previous literature on animal studies indicates that squalene is absorbed through the skin and is poorly absorbed from the gastrointestinal tract. The acute animal toxicity of squalene by all routes was low which corroborates with the predicted results. Even at 100% concentration, squalene was non-irritant to rabbit’s skin and eyes indicating squalene is not a significant human skin irritant or sensitizer. Also, Squalene was observed to be non-allergen [36, 37].
Molecular dynamics simulations
MD simulations were conducted to understand the conformational behavior, structural specificities, and ligand–target complexes stability [38, 39]. Molecular docking simulations empowered us to uncover the atomic characteristics of the bio molecular processes along with the stability analysis of protein–ligands complexes [40]. To analyze and understand the molecular interactions at atomic level of the COX-2domain complexed, we performed the molecular docking simulation studies with the compound which shown best binding efficacy (squalene), among the 18 compounds along with celecoxib for 100 ns.
Root mean square deviation (RMSD), Root mean square fluctuations (RMSF), intra molecular hydrogen bonds and the time dependent total energy function of molecular dynamic simulations at each frame of the molecular dynamic trajectories were analyzed. This was performed to understand the stability of the complex and the changes that occur in conformations of the COX-2 domain complexed with the compounds, squalene and celecoxib. RMSD is used to measure the variation that takes place in the backbone of the protein from its initial structural conformation. The protein’s stability relative to its conformation can be calculated by the deviations generated during its simulation. Smaller variations suggest more stable structure of the protein. In order to evaluate the stability of all the systems, RMSD value for the protein backbone was calculated for 100 ns simulations. The average RMSD of the protein (COX-2) for 100 ns simulation was 2.25 Å and fluctuated between 1.17 Å and 2.86 Å. RMSD values for the squalene – COX-2 complex averaged at 2.56 Å and the values ranged between 1.42 Å and 3.386 Å. For the celecoxib-COX-2 complex, two noticeable changes in the RMSD were observed at 39.8 ns and 58.6 ns. RMSD (average) for Celecoxib-COX-2 complex was 2.28 Å and fluctuated between 1.22 Å and 3.016 Å (Fig. 3).
Fig. 3.
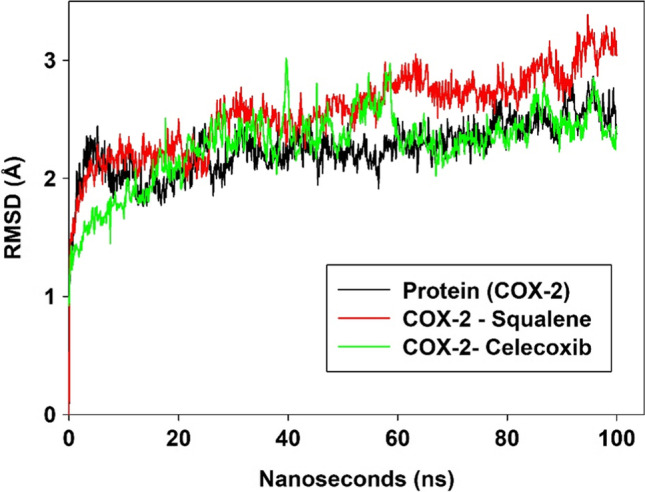
RMSD plot of COX-2 enzyme, COX-2-Squalene complex and COX-2-Celecoxib complex for 100 ns
RMSF plots analysis for the protein–ligand complexes provides details about the versatile regions of the complexes. In proteins, the rings, twists and coils display higher RMS fluctuations as compared to helical and sheet structures. To identify the higher flexible region of the protein, we calculated the fluctuations of each residue and noted its average for entire100 ns simulation time period. In the RMSF graph of the COX-2 –squalene complex, we found that there were high peaks of fluctuations in the range of 150 to 200 positioned residues. Apart from that, residues at 340 and 360 positions showed high peaks of fluctuations. This indicates that the above residues maybe probably involved in the conformational changes in the protein when bound with squalene. For celecoxib, peak fluctuations were found between 45 and 65 residues and minor fluctuations were observed throughout the protein structure (Fig. 4a, b).
Fig. 4.

(a) RMSF plot of COX-2-Squalene complex. (b)RMSF plot of COX-2-Celecoxib complex
Protein secondary structure elements (SSE) like alpha-helices and beta-strands were monitored throughout the simulation. From this study, we found that the COX-2 protein complexed with squalene and celecoxib had maintained 40% of SSE composition of α-helices and β-strands throughout the simulation time period. Also, helices played an important role in maintaining the stability and followed by strands and loops.
Conclusion
Compared to synthetic products, natural products of herbal origin are well-known to exhibit diverse biological activities. Here, in this study 18 phytocompounds from Albizia amara and Phyla nodiflora were tested against COX-2 in order to find potent anti-inflammatory drug molecule. The activities of the phytocompounds were compared with the standard drug celecoxib. Squalene showed potent inhibitory activity against COX-2 protein receptor. The preliminary computational studies like docking, ADMET and molecular simulation studies proved that, squalene has a higher binding affinity towards the targeted COX-2 protein receptor. Further molecular dynamic simulations for 100 ns revealed the protein and ligand stability. MD simulations gave the scope to understand the molecular interactions in atomic level. Overall, the present study suggests that squalene present in both the plants has the capacity to inhibit the COX-2 protein. Being lipophilic, the compound can be formulated in gels/creams and could be possible used for external edema and inflammation. Further in-vitro and in-vivo studies have to be performed in order to further validate the anti-inflammatory and COX-2 inhibition potential of the plant extracts.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Bannari Amman Institute of Technology, Sathyamangalam and Post Graduate Institute of Medical Education and Research (PGIMER), Chandigarh for providing the necessary facilities to carry out the work.
Authors’ contributions
Yukeswaran L, Subhashini T, Shreeranjana S, Suresh Kumar M performed the Protein analysis, active site prediction, docking experiments and ADMET predictions. Dr. Manav Jain performed the molecular dynamic simulations. Moni Philip Jacob designed the workplan and wrote the manuscript. Dr. Tamilselvi S designed the workplan and guided the entire work.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Code availability
1. AutoDock Vina (RRID:SCR_011958)
2. Avogadro (RRID:SCR_015983)
3. PyMol (RRID:SCR_000305)
4. Desmond (RRID:SCR_014575)
5. UCSF Chimera (RRID:SCR_004097)
Declarations
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Joshi T, Sharma P, Joshi T, Chandra S. In silico screening of anti-inflammatory compounds from Lichen by targeting cyclooxygenase-2. J Biomol Struct Dyn. 2020;38(12):3544–3562. doi: 10.1080/07391102.2019.1664328. [DOI] [PubMed] [Google Scholar]
- 2.Vane JR, Botting RM. New insights into the mode of action of anti-inflammatory drugs. Inflamm Res. 1995;44:1–10. doi: 10.1007/BF01630479. [DOI] [PubMed] [Google Scholar]
- 3.Bost J, Maroon A, Maroon J. Natural anti-inflammatory agents for pain relief. Surg Neurol Int. 2010;2010(1):80. doi: 10.4103/2152-7806.73804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajakrishnan V, Manoj VR, Rao GS. Computer-aided, rational design of a potent and selective small peptide inhibitor of cyclooxygenase 2 (cox2) J Biomol Struct Dyn. 2008;25:535–542. doi: 10.1080/07391102.2008.10507200. [DOI] [PubMed] [Google Scholar]
- 5.Hla T, Neilson K. Human cyclooxygenase-2 cDNA. ProcNatl Acad Sci USA. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.das Chagas Pereira de Andrade F, Mendes AN. Computational analysis of eugenol inhibitory activity in lipoxygenase and cyclooxygenase pathways. Sci Rep. 2020;10:1–14. doi: 10.1038/s41598-020-73203-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehman Q, Sack KE. When to try COX-2-specific inhibitors: Safer than standard NSAIDs in some situations. Postgrad Med. 1999;106:95–106. doi: 10.3810/pgm.1999.10.1.704. [DOI] [PubMed] [Google Scholar]
- 8.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H 2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 9.O’Banion MK, Sadowski HB, Winn V, Young DA. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J Biol Chem. 1991;266:23261–23267. doi: 10.1016/s0021-9258(18)54491-4. [DOI] [PubMed] [Google Scholar]
- 10.Gunathilake KDPP, Ranaweera KKDS, Rupasinghe HPV. In vitro anti-inflammatory properties of selected green leafy vegetables. Biomedicines. 2018;6:1–10. doi: 10.3390/biomedicines6040107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehrich EW, Dallob A, De Lepeleire I, Van Hecken A, Riendeau D, Yuan W, Porras A, Wittreich J, Seibold JR, De Schepper P, Mehlisch DR, Gertz BJ. Characterization of rofecoxib as a cyclooxygenase-2 isoform inhibitor and demonstration of analgesia in the dental pain model. Clin Pharmacol Ther. 1999;65:336–347. doi: 10.1016/S0009-9236(99)70113-X. [DOI] [PubMed] [Google Scholar]
- 12.Ballinger A, Smith G. COX-2 inhibitors vs. NSAIDs in gastrointestinal damage and prevention. Expert Opin Pharmacother. 2001;2:31–40. doi: 10.1517/14656566.2.1.31. [DOI] [PubMed] [Google Scholar]
- 13.Rajkumar T, Kumar ES, Sinha BN. Evaluation ofantioxidant properties of Albizia amara leaves. Int J Adv PharmBiolSci. 2010;2:99–106. [Google Scholar]
- 14.Kokila K, Priyadharshini SD, Sujatha V. Phytopharmacological properties of Albizia species: A review. Int J Pharm Pharm Sci. 2013;5:70–73. [Google Scholar]
- 15.Nivetha S, Padmini S, Tamilselvi S. A Review on Phytopharmacological Properties of Arappu. Int Res J Pharm. 2017;8:28–32. doi: 10.7897/2230-8407.0811213. [DOI] [Google Scholar]
- 16.Indravathi G, Reddy RS, Babu PS. Albizia amara - A Potential Medicinal Plant: A Review. Int J Sci Res. 2016;5:621–627. [Google Scholar]
- 17.Jabeen M, Jillani U, Chaudhary BA, Uzair M. Phytochemical and pharmacological studies of Phyla Nodiflora(Verbenaceae): A review. Pak J Pharm Sci. 2012;2:48–54. [Google Scholar]
- 18.Sharma RA, Singh R. A Review on Phyla nodifloraLinn.: A wild wetland medicinal herb. Int J Pharm Sci Rev Res. 2013;20:57–63. [Google Scholar]
- 19.Al-Snafi AE. Pharmacological and therapeutic effects of Lippia nodiflora (Phyla nodiflora) IOSR J Pharm. 2019;9(8):15–25. [Google Scholar]
- 20.Tamilselvi S, Dharani T, Padmini S, Nivetha S, Sangeetha M, Das A, Balakrishnaraja R. GC-MS analysis of Albizia amara and Phyla nodiflora ethanolic leaf extracts. Int J Recent Technol Eng. 2019;7:466–473. [Google Scholar]
- 21.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemicaleditor, visualization, and analysis platform. J Cheminformatics. 2012;4:1–17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Zürich: Vdf Hochschulverlag AG an der ETH; 1996. pp. 1–1042. [Google Scholar]
- 23.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363–W367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 27.Bathini R, Sivan SK, Fatima S, Manga V. Molecular docking, MM/GBSA and 3D-QSAR studies on EGFR inhibitors. J Chem Sci. 2016;128(7):1163–1173. doi: 10.1007/s12039-016-1103-3. [DOI] [Google Scholar]
- 28.Zhang X, Perez-Sanchez H, Lightstone F. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr Top Med Chem. 2017;17(14):1631–1639. doi: 10.2174/1568026616666161117112604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrödinger Release. Desmond molecular dynamics system. Maestro-Desmond interoperability tools. New York: D. E. Shaw Research; 2019.
- 31.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 32.Vijayakumar S, Manogar P, Prabhu S, Singh RAS. Novel ligand-based docking; molecular dynamic Simulations; and absorption, distribution, metabolism, and excretion approach to analyzing potential acetylcholinesterase inhibitors for Alzheimer’s disease. J Pharm Anal. 2018;8:413–420. doi: 10.1016/j.jpha.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zarghi A, Arfaei S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran J Pharm Res. 2011;10:655–683. [PMC free article] [PubMed] [Google Scholar]
- 34.Prabhavathi H, Dasegowda KR, Renukananda KH, Lingaraju K, Naika HR. Exploration and evaluation of bioactive phytocompounds against BRCA proteins by in silico approach. J Biomol Struct Dyn. 2020; 1–15. 10.1080/07391102.2020.1790424 [DOI] [PubMed]
- 35.Daze K, Hof F. Molecular interaction and recognition. In Encyclopedia of physical organic chemistry. Vol 5. Wiley; 2016. 10.1002/9781118468586.epoc3001
- 36.Panel, CIR Expert, Cosmetic Ingredient Review Expert Panel Final report on the safety assessment of squalane and squalene. J Am Coll Toxicol. 1982;1(1982):37–56. [Google Scholar]
- 37.Huang ZR, Lin YK, Fang JY. Biological and pharmacological activities of squalene and related compounds: potential uses in cosmetic dermatology. Molecules. 2009;14(1):540–554. doi: 10.3390/molecules14010540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey T, Shukla R, Shukla H, Sonkar A, Tripathi T, Singh AK. A combined biochemical and computational studies of the rho-class glutathione s-transferase sll1545 of Synechocystis PCC 6803. Int J Biol Macromol. 2017;94:378–385. doi: 10.1016/j.ijbiomac.2016.10.040. [DOI] [PubMed] [Google Scholar]
- 39.Shukla R, Shukla H, Sonkar A, Pandey T, Tripathi T. Structure-based screening and molecular dynamics simulations offer novel natural compounds as potential inhibitors of Mycobacterium tuberculosis isocitrate lyase. J Biomol Struct Dyn. 2018;36:2045–2057. doi: 10.1080/07391102.2017.1341337. [DOI] [PubMed] [Google Scholar]
- 40.ul Hassan SS, Zhang WD, Jin H, Basha SH, Priya SVSS. In-silico anti-inflammatory potential of guaiane dimers from Xylopia vielana targeting COX-2. J Biomol Struct Dyn. 2020; 1–15. 10.1080/07391102.2020.1815579 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
1. AutoDock Vina (RRID:SCR_011958)
2. Avogadro (RRID:SCR_015983)
3. PyMol (RRID:SCR_000305)
4. Desmond (RRID:SCR_014575)
5. UCSF Chimera (RRID:SCR_004097)