

Summary
Metabolic cross-feeding frequently underlies mutualistic relationships in natural microbial communities and is often exploited to assemble synthetic microbial consortia. We systematically identified all single-gene knockouts suitable for imposing cross-feeding in Escherichia coli and used this information to assemble syntrophic communities. Most strains benefitting from shared goods were dysfunctional in biosynthesis of amino-acids, nucleotides, and vitamins or mutants in central carbon metabolism. We tested cross-feeding potency in 1,444 strain pairs and mapped the interaction network between all functional groups of mutants. This network revealed that auxotrophs for vitamins are optimal cooperators. Lastly, we monitored how assemblies composed of dozens of auxotrophs change over time and observed they rapidly and repeatedly coalesced to seven strain consortia composed primarily from vitamin auxotrophs. The composition of emerging consortia suggests they were stabilized by multiple cross-feeding interactions. We conclude that vitamins are ideal shared goods since they optimize consortium growth while still imposing member co-dependence.
Graphical Abstract
Introduction
Microorganisms in natural habitats are frequently found in complex communities composed of multiple species. Such communities feature a myriad of interactions that extend beyond just competition and predation and include many social-like interactions such as cooperation, communication, and synchronization(West et al., 2007). In such communities, the fitness of an individual cell depends not only on its own capabilities but also on its interactions with cells from other strains and species. Nutritional cross-feeding between organisms is a key mechanism for maintaining the stability and function in microbial communities in natural environments(Seth and Taga, 2014). These metabolic cooperative interactions rely on the secretion of metabolites that support mutualistic co-dependence through the exchange of shared goods. Experimental and theoretical works suggest that in many cases the release of shared goods in natural communities does not directly benefit the giver but instead facilitates closed cooperative loops between multiple members that assist the entire multi-species community(Freilich et al., 2011; Goldford et al., 2018; Louca et al., 2018; Zengler and Zaramela, 2018).
The observations made in natural microbial communities allowed uncovering the fundamental principles of cross-feeding mutualism and facilitated the assembly of microbial consortia in-vitro(Fredrickson, 2015; Giri et al., 2020). The experiments in-vitro, in turn, allowed to further study and refine the rules of interaction that may take place in natural communities(Campbell et al., 2015; Dolinšek et al., 2016; LaSarre et al., 2020; Pierce et al., 2021; Ponomarova et al., 2017). However, beyond their value as models for natural communities, synthetic consortia have also emerged as valuable chassis for diverse biotechnological applications, as biofuel production, biomedicine, and bioremediation (Hays et al., 2015; Jagmann and Philipp, 2014; Zomorrodi and Segrè, 2015). In many cases, synthetic consortia were assembled from genetically trackable model organisms that were modified to impose co-dependence between the consortia members(Cavaliere et al., 2017) . This obligatory syntrophic interaction ensures a robust consortium that maintains its member composition over time and therefore allows to distribute a complex task in a cooperative manner across multiple consortium members.
E. coli is a valuable model system for studying the design principles underlying cross-feeding in natural communities and is an ideal chassis for engineering synthetic consortia for biotechnological applications. However, to date there was no study that exhaustively identified all E. coli mutants that can benefit from metabolites secreted by neighboring cells. Consequentially, we are also missing information on cross-feeding interactions between such strains and their potential to balance the tradeoff between neighbor dependency and overall fast growth. Such co-optimization is likely key for successful mutualism in both natural communities and robust synthetic consortia that can be utilized for biotechnology. Previous studies on the E. coli model system heavily relied on auxotroph strains that are mutated in a handful of central and essential metabolic pathways, as those for the biosynthesis of different amino-acids (e.g. (Kerner et al., 2012; Lloyd et al., 2019; Mee et al., 2014; Pande et al., 2014; Wintermute and Silver, 2010; Zhang and Reed, 2014; Ziesack et al., 2019)). Systematic characterization of these strains revealed that many of them are suboptimal as members of a synthetic consortia due to slow growth and frequent disappearance in multi-member consortia(Mee et al., 2014; Wintermute and Silver, 2010). Overcoming these limitations, at least partially, can be achieved through additional genetic changes(Ziesack et al., 2019). Interestingly, studies that used biochemical and computational approaches identified additional secreted metabolites, beyond the typically used amino-acids, that can potentially be exploited as shared goods for imposing mutualism(Carneiro et al., 2011; Paczia et al., 2012; Thommes et al., 2019). Taken together, previous works suggest that even in the well-studied E. coli system, current understanding of cross-feeding may be incomplete.
Here we propose that new insights on optimality in cross-feeding interactions can be gained by mapping all possible cross-feeding interactions in E. coli and identifying the top beneficiaries of such interactions. In contrast to studies of cross-feeding that rely on natural isolates and comparative genomics, a grounds-up systematic approach that relies on a single species can clarify which cross-feeding interactions are more beneficial when everything, except the pathways in question, is kept constant. For this study, we establish a generally applicable approach that uses a collection of single-gene knockouts to unbiasedly identify all loss-of-function mutations that can be complemented by cross-feeding. We conducted a genetic screen with a library of 3,680 single-gene deletion strains and identified all strains that benefited from nutrient-poor media that was conditioned by the wild-type lab strain prior to the screen. Beyond the known strict auxotroph strains that arrest without external nutrients, we identified a class of “permissive” auxotroph strains – cells that grow well on nutrient-poor media but further benefit from nutrients in conditioned media. By monitoring the growth of individual strains on conditioned and unconditioned media, we predicted which auxotroph strains will be ideal collaborators, a prediction we later tested across 1,444 strain pairs. We used the results from the pairwise interaction experiments to infer the interaction network between auxotrophy groups. This network revealed that auxotrophs for specific vitamins and co-factors are ideal cooperators and simultaneously support fast growth while still maintaining strain co-dependency. Lastly, we tested how the composition of consortia assembled from dozens of auxotroph strains changes over time. We observed that these consortia rapidly and repeatedly coalesced to lower-complexity communities, typically composed of seven dominant strains. The interaction network we inferred from pairwise interactions supports the hypothesis that the emerging consortia are held together by multiple strong interactions between individual strain pairs.
Results
Sequencing based approach identifies auxotroph strains
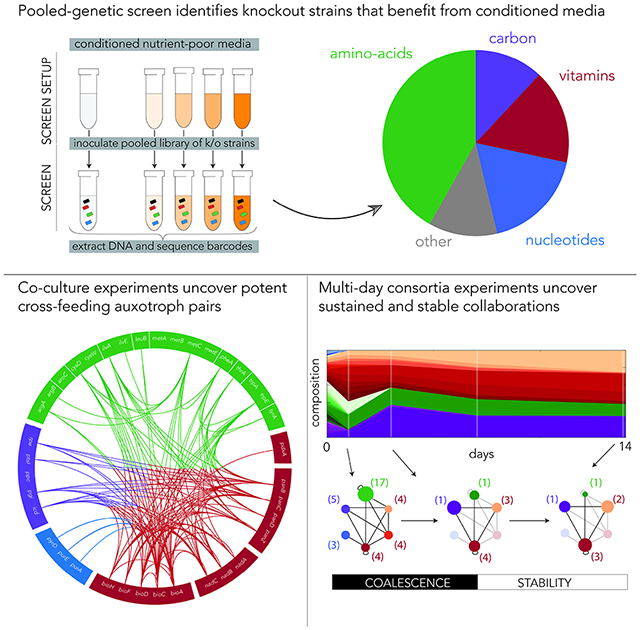
We first aimed to systematically identify all single-gene knockout strains in E. coli that can considerably benefit from metabolites that are secreted by other cells. We used a pooled screen methodology similar to the one we recently used to study drug resistance(Rosener et al., 2020). Towards this aim, we utilized conditioned media, media filtered after growing in it the wild-type strain. We reasoned that the identification of strains that can benefit from by-products that are present in conditioned media will help identifying candidates that can successfully participate in mutualistic cross-feeding interactions since such “ecological beneficiaries” can be paired together to rescued each other. Figure 1A depicts the approach of our screen. The wild-type strain was inoculated into nutrient-poor media at four different concentrations, ranging from 107 to 5*109 cells/mL, to allow secretion of shared goods. After 3.5 hours of growth, we filtered the culture and replenished the carbon, nitrogen, phosphorous and sulfur in the spent media. As a control we used unconditioned nutrient-poor media. Lastly, we inoculated a pooled collection of 3,680 barcoded single-gene knockout strains into the filtered media and grew cultures for 8 hours before extracting genomic DNA. We did not observe considerable differences in growth rates of inoculums, indicating that the majority of strains in the pooled collection grew similarly in all four different conditioned media and the unconditioned media control.
Figure 1. Pooled genetic screen of knockout strains in conditioned media.

(A) Overview of screen approach. We inoculated the wild-type E. coli strain at four different concentrations into nutrient-poor media and grew it for 3.5 hours before filtering. We replenished the carbon and nitrogen sources and then inoculated a pooled collection of 3,680 knockout strains into the filtered media (marked by cells with different colors). We grew the cultures for 7 hours before we extracted genomic DNA and amplified the barcode region. Lastly, we sequenced the amplicon and calculated the frequency of individual barcodes that correspond to individual knockout strains. The orange gradient color marks conditioned media filtered from cultures inoculated at different densities. (B) Inference of conditioned-media impact on individual knockout strains by monitoring changes in barcode frequency. We calculated the frequencies of each knockout strain (barcode) across all screen conditions. We calculated the fold-change in strain (barcode) frequency in conditioned-media (orange tube) relative to the unconditioned-media control (grey tube). We identified strains unaffected by conditioned media (red bars), strains benefiting from conditioned media (green bars), and strains compromised by conditioned media (blue bars). (C) We used the fold-change in knockout strain frequency to measure impact of conditioned media on knockout strains. (D) The genetic sequencing yielded similar barcode coverage across all experiments. The violin plots show the number of times each knockout strain (individual barcode) was detected in each screen experiment. The table above shows the number of unique strains (barcodes) we detected in each screen condition and the total of number barcodes we identified in DNA reads. (E) High correlation between strain frequency in biological duplicates. The scatter plots show the strain (barcode) frequency after normalization to reads-per-million (RPM) in biological duplicates. The Pearson correlation coefficient is shown in the upper left of each scatter plot.
In order to evaluate the fitness of each knockout strain, we amplified the unique nucleotide region that was cloned into the deletion locus during library construction. These 20 nucleotide long barcodes allowed us to identify the frequency of each individual knockout strain by deep sequencing of the DNA amplified from the pooled culture. Figure 1B shows the three categories of barcode frequencies we expected to see in the screen. We expected that the relative frequency of the majority of strains will not change in different screen conditions. This pattern is indicative of knockout strains that were unaffected by extracellular goods (red bars in Figure 1B). We expected to see a monotonic increase in frequency with denser conditioned media only for a minority of strains (green bars in Figure 1B). This pattern is indicative of knockout strains that were rescued by extracellular shared goods. Lastly, we also considered the possibility that strains may experience a decline in fitness in conditioned media, such events will be characterized by monotonic decrease in frequency with denser conditioned media (blue bars in Figure 1B). We expected that decrease in frequency will be rare since we replenished the metabolites that were consumed during conditioned media preparation. Since the key metric indicative of strain rescue is change in its frequency relative to unconditioned media, we normalized each strain’s frequency to its frequency in the unconditioned media control (Figure 1C). The normalization yields a fold-change in strain frequency that marks the strain’s fitness improvement relative to its own fitness in unconditioned media.
Figure 1D shows the distribution of individual barcode reads in each screen experiment. Overall, barcode distributions were extremely similar across all conditions, with roughly 3,500 identified individual barcodes in about 1 million sequencing reads in each condition (table above Figure 1D). We identified more than 10 reads for the vast majority of barcodes (>96%). The median number of reads per individual barcode was 181. Figure 1E shows scatter plots for each biological duplicate after normalizing barcode counts to reads per million. As the figure shows, biological replicates were highly correlated in all conditions (Pearson correlation r > 0.97). Taken together, we observed a similar barcode coverage across all experiments and a very high correlation between biological replicates. These observations are reassuring since they suggest that differences in strain frequency between conditions most likely reflect changes in strain fitness rather than biological variation between replicates or technical noise.
We next aimed to identify all single-gene knockouts that were either positively or negatively affected by conditioned media (second and third cases in Figure 1C) with a statistical approach that can leverage on biological replicates and the sequencing depth of each barcode. Towards this end we used an approach that is similar to the one typically used for detecting differentially expressed genes. However, in our implementation we used barcode counts instead of gene transcript counts. Specifically, we used the DESeq2 tool(Love et al., 2014) that was adjusted for pooled barcode libraries with the DEBRA package(Akimov et al., 2020). Figure 2A shows volcano plots representing fold-change in strain frequency and associated p-values (false-discovery rate adjusted) across all screen conditions (the values also appear in supplementary table 1). As Figure 2A shows, we observed a monotonic increase in the number of differently represented strains with conditioned media filtered from denser cultures (from only 2 differently represented strains in the least dense conditioned media to 69 in the densest conditioned media). This observation is compatible with the expectation that conditioned media filtered from a denser cell population will contain a higher concentration of shared goods and will therefore rescue more knockout strains. We also noted that the number of differently represented strains increased sharply with media density, for example by almost 7-fold between media prepared from 108 cells/mL to media prepared from 109 cells/mL. This considerable increase highlights how impactful the density of secreted metabolites is for cross-feeding rescue. The analysis also revealed that the vast majority of differently represented strains are over-represented (second case in Figure 1C), rather than under-represented (third case in Figure 1C), in conditioned media (67 of 69 hits are over-represented in the densest media). This pattern indicates that conditioned media typically benefits loss-of-function mutants and only very rarely inhibits them.
Figure 2. Knockout strains dysfunctional in biosynthesis of amino-acids, nucleotides, vitamins and co-factors, and central carbon metabolism benefit from condition media.

(A) Analysis of fold-change in strain representation and the statistical significance across the four screen conditions. The number of significantly enriched or depleted strains are shown on the upper corners (the vertical and horizontal black lines mark the fold-change and p-value cutoffs we used). The vast majority of hits were enriched, rather than depleted, in conditioned media. The total number of hits increased with conditioned media filtered from denser cultures. (B) Only a subset of auxotrophs that were enriched on nutrient-rich media also benefited from conditioned media. The Venn diagram shows the overlap between hits found to be enriched on nutrient-rich media (pink) and those found to be enriched on conditioned-media (orange). (C) A comparison of fold-change in knockout strain frequency on nutrient-rich media and on conditioned media. The fold-enrichment of common hits is correlated in the two screens (colors as in B). (D) The conditioned media enriched knockout strains are associated with specific metabolic functions. The colored bars show the fold-enrichment of hit strains in conditioned media filtered from the densest culture. 59 of 67 of the strains can be annotated as dysfunctional in biosynthesis of amino acids (green), nucleotides (blue), vitamins and co-factors (red), and central carbon metabolism (purple). Eight of the enriched strains belong to other biological categories (gray). (E) Analysis of KEGG pathway enrichment across all conditions. The circle plots show the number of genes belonging to the enriched pathway and gray-scale color marks the statistical significance.
We next evaluated whether conditioned-media rescued strains can also be rescued by nutrient-rich media. We reasoned that this comparison could independently validate some of the screen results by showing that the hit strains indeed benefited from extracellular metabolites rather than increased their growth due to bacterial signaling molecules or secreted enzymes (expected to be present in conditioned media but absent from unconditioned nutrient-rich media). Towards this aim we used barcode counts from a previous genetic screen we conducted(Rosener et al., 2020) and calculated the fold-change of strain frequency in nutrient-rich media relative to nutrient-poor media (supplementary Figure 1 and supplementary table 4). Figure 2B shows a Venn diagram of the overlap between the hits of the two screens. We observed that most of the conditioned-media enriched strains were also enriched in nutrient-rich media (61 of 67). The majority of conditioned-media hits that were not hits in nutrient-rich media (5 of 6) were found to be enriched in rich media but were not characterized as hits since they did not pass the fold-enrichment cutoff we selected. However, the comparison also revealed that conditioned-media rescued strains are only a small subset of all hits identified in nutrient-rich media (61 of 503). Figure 2C shows a comparison of strain fold-enrichment in the two screens. The comparison revealed that the enrichment of common hits is highly correlated (orange dots in Figure 2C, Pearson r = 0.79, p = 3e−14) and that a few additional strains might benefit from conditioned media although they were not characterized as hits in the screen (pink dots in the upper-right quadrant). These false-negative strains were not characterized as hits due to the conservative choice we made for the cutoff by p-value. Thus, we conclude that while at least 503 knockout strains can be rescued by extracellular metabolites, only a small subset of them can be rescued by metabolites secreted by other cells. A plausible explanation for this observation is that many of the rescuing metabolites are either not secreted or are secreted and quickly consumed by other strains in the consortia and therefore never accumulate to concentrations required to impact dysfunctional cells.
To investigate the cellular pathways that were rescued by conditioned media we assigned hits into broad functional categories according to the biological annotation in the EcoCyc database(Keseler et al., 2017) (Figure 2D). Most hits (59 of 67) could be assigned to four categories: Carbon metabolism, amino acids biosynthesis, nucleotide biosynthesis, and biosynthesis of co-factors and vitamins. Supplementary table 2 shows the functional affiliation of the hits from all screens. Reassuringly, we frequently identified multiple genes belonging to the same biosynthesis pathway (e.g., the cys genes in Figure 2D). While many of the pathways we identified were previously used to engineer synthetic E. coli consortia(Mee et al., 2014; Wintermute and Silver, 2010), some of them are potentially novel targets for this application. Among them are the pathways for biotin, folate, and Pyridoxal 5'-phosphate biosynthesis (supplementary table 2). We next used the gene set enrichment analysis tool GAGE(Luo et al., 2009) to test for functional enrichment using the Gene Ontology (GO)(Ashburner et al., 2000; Carbon et al., 2018) and Kyoto Encyclopedia of Genes and Genomes (KEGG)(Kanehisa and Goto, 2000) databases. This analysis is complementary to the previous analysis, since it considers the enrichment values from all strains, rather than only the limited set of hit strains obtained by imposing fold-change and p-value cutoffs. Figure 2E shows the functional enrichment by KEGG pathways (enrichment is indicative of a pathway that can be rescued by secreted metabolites). Reassuringly, we observed a high agreement between the functional enrichment by GAGE and our annotation of hits into broad functional categories (Figure 2D). However, the GAGE analysis provided information on a finer resolution. For example, it indicated that all amino-acid synthesis pathways are enriched in conditioned media with the exception of Histidine and Proline. This observation supports the previously raised claim that cross-feeding is nonproductive with auxotrophs for Histidine and Proline(Mee et al., 2014). The full list of GO and KEGG enriched categories is presented in supplementary table 3.
To summarize, our screening approach that relied on improved growth on conditioned media revealed dozens of auxotrophs that benefit from secreted metabolites. The results allowed us to compile, for the first time, an exhaustive catalog of all single-gene knockout E. coli strains that can be potentially utilized for mutualistic cross-feeding interactions (in the nutrient-poor media and the aerobic growth conditions we tested). At this stage of the analysis, we can only determine that these strains can be rescued by the wild-type strain, yet we cannot determine if they can indeed rescue other co-cultured auxotrophs. While many of these mutants were previously utilized for assembling cross-feeding consortia, our unbiased screen identified mutants from additional pathways that are novel targets for this application. Lastly, a comparison to a nutrient-rich media screen suggests that the majority of auxotrophs may not be suited for mutualistic cross-feeding interactions since they are rescued by secreted metabolites.
Strict and permissive auxotrophy
We identified 67 strains that significantly benefited from the densest conditioned media. However, since the screen relies on strain frequency change relative to the unconditioned media control (Figure 2A), it was insufficient for determining how well the strains actually grow. The growth rate measurements are essential for determining which strains balance the tradeoff between neighbor dependency and overall fast growth which is a likely prerequisite for fit and stable cross-feeding interactions. We therefore complemented our screen by monitoring the growth of top hits when they grew as pure strains in conditioned and unconditioned media. We decided to use the area under the growth curve (AUC) as the metric of fitness (see methods). We reasoned that AUC measurement will integrate multiple fitness parameters, such as growth rate, lag period, and stationary loading capacity. Figure 3A shows representative growth curves for the wild-type and the pgi knockout strains on the two media types. As the figure shows, the growth of the pgi knockout strain is considerably improved in conditioned media while the growth of the wild-type strain was largely unaffected.
Figure 3. Absolute fitness measurements suggest that strains dysfunctional for specific pathways will be optimal collaborators for cross-feeding interactions.

(A) Growth curves of the wild-type and pgi knockout strains in conditioned and unconditioned media. The area under the curve (AUC), shown as a shaded area, was used as the fitness metric. The fitness of the pgi knockout strain considerably increased on conditioned media. (B) Fitness measurements of 67 knockout strains in conditioned and unconditioned media revealed that strains occupy diverse positions on the fitness plane. The experiment shows that many of the validated hits (strains above the diagonal line) grow reasonably well even in unconditioned media (permissive auxotrophs). The colors mark the biological function of the gene knockout (as in Fig. 2). Knockout strains that are not observed to benefit from conditioned media are marked in white. The star marker indicates the position of a theoretical ideal collaborator, a strict auxotroph that completely arrests on unconditioned media and grows fast on conditioned media. The Euclidean distance from the ideal collaborator, shown as a red segment, was used to calculate the collaboration potential score for each strain. (C) The collaboration optimality score, calculated by each strain’s distance from the ideal collaborator, shows a bias towards some biological functions. The bar graph marks the collaboration optimality score of each knockout strain. Strains scoring high in their collaboration potential are mostly strains dysfunctional for vitamin and co-factor biosynthesis or mutants for carbon metabolism. The inset shows the inverse relationship between the collaboration score and the fold-enrichment measured in the genetic screen.
Figure 3B shows the fitness measured for all tested strains in the two media types as a scatter plot (the growth curves are presented in supplementary Figure 2). We observed that strains varied considerably in their position on this fitness plane, indicating they are characterized by a wide range of fitness values in the two media types. First, we observed that 80% of the strains (51 of 67) were positioned above the diagonal marking equal fitness in both media types. The position above the diagonal indicates that these strains benefited from conditioned media and are therefore validated are recused independently from the screen. Most of the unvalidated strains (12 of 16) are characterized by very low fitness in both media types. This observation raises the possibility that experimental noise may be a confounding factor for validation with this approach. As expected, we observed that multiple hit strains grow reasonably well even without rescue. This observation highlights that in our experimental setup, rescued strains are not always completely arrested in unconditioned media. This lack of complete arrest can be explained if the missing metabolite is either nonessential or if cells need only very small amounts of the metabolite to grow. In the latter case, trace amounts of the metabolite left intracellularly after washing and diluting the overnight culture (grown in nutrient-rich media) may be sufficient for multiple doublings even without additional supply of the missing nutrient.
The overall goal of our approach was to identify the strains that can participate in robust mutualistic cross-feeding interactions. We assumed that under a potent cross-feeding interaction, collaborators grow vigorously in a co-culture yet are completely co-dependent, thus ensuring the co-culture is highly fit and that a single member will be unable to take over the entire culture. However, our measurements clearly showed that many of the fast-growing strains (potential collaborators) were also “permissive” auxotrophs that grew reasonably well on unconditioned media (Figure 3B). We reasoned that we could predict if individual strains potentially match both requirements for collaboration optimality by factoring in their fitness on both conditioned and unconditioned media: An optimal collaborator strain will be characterized by fast growth on conditioned media and complete growth arrest on unconditioned media (marked as a star in the fitness plane of Figure 3B). We therefore measured the Euclidean distance of each of the tested strains from this ideal collaborator and calculated how the distance compares to the furthest distance we observed in our experiments. Using this method, scores range from 0 (worst collaborator) to 1 (ideal collaborator). An alternative collaboration score we tested relied on the distance of each strain from the diagonal and we found it to be highly correlated with the collaboration optimality score we used (supplementary Figure 3A).
Figure 3C shows the calculated collaboration optimality score for the 51 validated knockouts. As the figure shows, our approach revealed a considerable bias in the score of different biological functions. Low scoring strains were almost exclusively amino-acids and nucleotides auxotrophs and high scoring strains being mostly auxotrophs for vitamins and co-factors. We next tested if our optimality scores were correlated with fold-enrichment in the genetic screen (Figure 3C, inset). We observed that the two metrics are inversely correlated (r = −0.41, p = 0.0027) indicating that highly enriched auxotrophs in our genetic screen are likely to be suboptimal members for cross-feeding interactions (these strains optimize co-dependency at the expense of fast growth). This is an important observation given that many of the amino-acid auxotrophs, that rank low on our optimality score, are frequently used to engineer cross-feeding consortia(Kerner et al., 2012; Mee et al., 2014; Pande et al., 2014; Wintermute and Silver, 2010; Ziesack et al., 2019). To summarize, fitness measurements in pure strains suggest that vitamins and co-factors auxotrophs are ideal candidate collaborators for sustaining robust cross-feeding interactions. In contrast, strains dysfunctional for biosynthesis of amino acids and nucleotides can still be used to successfully impose co-dependence but will be characterized by overall slow growth. Since these observations were based on monitoring the rescue of strains on conditioned media, and not testing mutualistic cross-feeding, we still viewed these auxotrophs as candidate collaborators.
Cross-feeding in strain pairs
The collaboration optimality score predicted that many of the hit strains identified by the screen will be suboptimal for cross-feeding since they grow slowly even if their metabolic needs are met by goods secreted from a dense culture. The effect of slow growth of even one pair member is expected to be far enhanced in a truly collaborating co-culture since cross-feeding pairs are highly co-dependent and are not necessarily present at a high density. We therefore decided to directly test cross-feeding competency in strain pairs by monitoring their co-culture growth on nutrient-poor media. These experiments can also confirm that the growth is due to secreted metabolites and not due to metabolites that may have been released from cells that were lysed during the filtration of conditioned media. For these experiments we used 30 strains that were representative of all hit pathways and added 8 additional knockout strains that were either functionally relevant to our top hits or were previously suggested by others(Mee et al., 2014; Wintermute and Silver, 2010) (see methods). In order to confirm that the knockout strains we chose are annotated correctly for their autotrophy, we validated that their growth can indeed be rescued by the end-product metabolite of the mutated pathway (supplementary Figure 4).
Overall, we monitored 1,444 strain pairs. Figure 4A shows representative growth curves of a potent cross-feeding pair that is composed of knockout strains from different functional groups, rpe (carbon metabolism) and panZ (vitamin biosynthesis) knockout strains. As the figure shows, while each strain grew poorly by itself, their co-culture grew substantially faster. As we did previously, we quantified fitness using the area under the growth curve. The conditions we used for this experiment were slightly different from the ones used for the genetic screen and the hit validation experiments. We therefore confirmed that the growth volume and choice of culturing time did not considerably impact the growth of single-cultured auxotrophs or auxotroph pairs (supplementary figure 5). Figure 4B shows the calculated fitness for all tested pairs. Reassuringly, we observed that duplicate experiments (reciprocal elements in the heatmap) were highly correlated (Pearson correlation r = 0.945, p < 0.0001). As expected, the experiments showed that potency of pair collaboration was highly variable (marked by the wide range of fitness values in the heat map) and that many of the co-cultures were likely benefiting from cross-feeding (off-diagonal interactions are typically brighter than their matching diagonal interactions indicative of single strain growth). Supplementary figure 5B shows the histogram of diagonal and non-diagonal AUC measurements. Nevertheless, since many of the strains grew reasonably well on their own, interpretation of cross-feeding potency solely by these fitness measurements can be misleading.
Figure 4. Fitness measurements in co-cultured strain pairs uncovers the underlying principles for pairwise cross-feeding.

(A) The panels show growth curves of rpe and panZ knockout strains inoculated individually or together as a co-culture. (B) The systematic measurement of fitness in co-cultured pairs reveals multiple instances of likely cross-feeding. The heatmap shows the area under the growth curve measured during 20 hours of growth across all 1,444 co-culture experiments. The thick colored frames mark similar biological function. The thin colored frames marks members belonging to the same biological pathway. (C) A histogram of all growth improvements calculated from pair co-culturing experiments. We used a cutoff of 2 to identify potent cross-feeding interactions that yield vigorous growth. (D) The growth improvement inferred across all co-culture experiments reveal that strains dysfunctional for vitamin and co-factor biosynthesis are potent collaborators (bright columns and rows). The heatmap shows the growth improvement calculated for all co-culture experiments. (E) The number of strong interactions each knockout strain participates in. (F) The network of cross-feeding interactions reveals the rules underlying potent collaborations. The graph shows a compacted version of the full interaction network (supplementary figure 6) after merging strains with a similar biological function into a single network node. The pie chart of each node shows how many strains from the node participate in potent cross-feeding interactions. The edge width marks the number of observed interactions between nodes normalized to total number of possible interactions given the number of strains in each of the two connected nodes.
In order to untangle the cross-feeding competency of pairs from the basal growth of their individual members we decided to use the pair’s growth improvement relative to the growth of the individual strains as our metric for cross-feeding potency (see methods). Since this method is based on measurement of absolute fitness, a high score reflects pairs that were considerably more fit when they were co-cultured as compared to their growth as pure cultures. Figure 4C shows a histogram of fitness improvement for all tested pairs. As the panel shows, we observed a unimodal long tail distribution for this metric. We decided to use a score of 2 as a threshold for determining which cross-feeding interactions were highly potent. Figure 4D shows the fitness improvement by pair identity as a heat map. As the figure shows, growth improvement is highly biased towards specific strains and biological functions. For example, most strains dysfunctional for vitamins and co-factors formed potent collaborations with multiple strains from their own functional group and from other groups (marked by bright rows and columns in the heatmap). This observation is not entirely surprising given the metric we chose. For example, vitamins auxotrophs are characterized by a high basal growth (Figure 3B) and therefore their gain from collaboration is large in absolute fitness terms (added AUC) even if their fold-improvement was modest. Reassuringly, strains from a single pathway, marked by thin frames in the heatmap, tended to collaborate more with strains belonging to other pathways, rather than strains from their own pathway (biotin synthesis is an exception to this trend). We also noticed that strains dysfunctional for the same pathway sometimes differed in their collaboration potential. For example, the trpE knockout strain was a better overall collaborator than the trpA knockout strain.
In order to distil the numerous cross-feeding interactions we detected into a concise format, we used the threshold value we previously chose to separate strong and weak interactions (Figure 4C) in order to transform the interaction matrix into a logical one. This logical matrix can be represented as a network graph with 38 nodes, representing the knockout strains, and 127 edges, representing strong cross-feeding interactions (supplementary figure 6). The node degree in this graph represents the number of strong cross-feeding interactions a strain participates in. Figure 4E shows the number of strong interactions we observed for each strain. Compatible with the interaction matrix, we observed that vitamins and cofactors auxotrophs tended to participate in multiple interactions, while nucleotides and amino acids auxotrophs tended to have fewer interactions. We also observed some notable exceptions to this trend, such as the knockouts strains for metC and trpE (amino-acid biosynthesis) that participated in multiple strong collaborations. Reassuringly, we also observed a positive correlation between the collaboration optimality score, calculated by growing individual auxotrophs on conditioned media, and the number of strong interactions an auxotroph can support (supplementary Figure 3B).
Figure 4F shows a compact representation of the interaction network after grouping strains into functional groups and weighing the number of interactions between groups to denote the edge strength. The network representation highlighted the key principles underlying cross-feeding interactions that can support vigorous growth. First, we observed that knockouts strains from the biotin, nicotinamide adenine dinucleotide (NAD+) and pantothenate biosynthesis pathways can form potent interactions with almost any other functional groups (node degrees 4-5). In contrast, strains dysfunctional for nucleotide biosynthesis can only interact with strains dysfunctional for biotin biosynthesis. Strains dysfunctional for amino-acid biosynthesis and mutants in central carbon metabolism interact mostly with strains dysfunctional for vitamin and co-factors biosynthesis (node degree 3). To further probe the interactions between the different functional groups we decided to measure the relative proportion of representative strains in pair cross-feeding experiments by co-culturing pairs with different antibiotic resistance cassettes and plating the co-cultures on agar plates after 20 hours. We tested a total of 15 pairs and found that pairs can range from extreme proportions such as 0.9:0.1 to a balanced 0.5:0.5 composition (supplementary Figure 7A). In agreement with the predicted optimality of vitamin auxotrophs, we observed that that uneven proportions were biased towards vitamin auxotrophs over other functional groups (supplementary Figure 7B).
Taken together, the observations from pair cross-feeding experiments revealed that multiple strains can indeed collaborate when they are co-inoculated at low density into nutrient-poor media. The results supported the predictions from single strain experiments on conditioned media and showed that strains dysfunctional for biosynthesis of vitamins and co-factors are optimal for imposing cross-feeding interactions with an overall fast growth. Such strains can form cross-feeding interactions with knockout strains from practically any other auxotrophy group.
Synthetic multi-member consortia
Previous studies showed that pairwise cross-feeding interactions can sustain multi-member natural communities and synthetic engineered consortia(Freilich et al., 2011; Goldford et al., 2018; Mee et al., 2014). An open question is therefore whether an assembly of strains that are co-cultured together will coalesce into a co-dependent strain group that is stabilized by cross feeding interactions (termed a syntrophic consortium). Previous work in E. coli demonstrated that an assembly of 14 strict amino-acid auxotrophs converged into a cross-feeding consortia within a few days of growth(Mee et al., 2014). However, it remains unclear if an assembly that is composed of both strict and permissive auxotrophs will similarly converge to a syntrophic consortium or whether a single permissive auxotroph, that can grow reasonably well even without collaboration, will take over the culture. Addressing this question is important in order to know if the permissive auxotrophs we identified can indeed be members of stable communities (in nature or in-vitro). We therefore decided to directly test a multi-strain assembly that was composed of dozens of strict and permissive auxotrophs in a long-term multi-day experiment. Since the members of the initial assembly grew at very different rates even without collaboration, we reasoned that the hallmark of a consortium that is stabilized by potent cross-feeding interactions would be a composition that is stable over time and is similar across independent replicates. We also expected that the stable consortium will be composed of auxotrophs that we identified as optimal collaborators and the co-occurrence of strains we previously identified as potent cross-feeding collaborators (connected strain in the network presented in supplementary Figure 6). In contrast, a complete takeover by a single fast-growing strain or highly different strains composition over replicates would indicate that the assembly failed to coalesce into a stable syntrophic consortium.
We inoculated the 38 top auxotroph strains at equal proportions on nutrient-poor media and serially transferred and diluted this assembly every day for a period of 14 days. We sampled the culture at the end of days 1, 3, 7, and 14 and monitored the strain composition by deep sequencing. We conducted the experiment with six independent biological replicates in order to detect if similar composition dynamics emerged repeatedly. Figure 5A shows how the composition in these assemblies changed during the two weeks of the experiment. Remarkably, we observed almost identical dynamics across all six biological replicates. The qualitative common characteristic, that could be readily detected by eye, were fast convergence to lower-complexity consortia, relative stable strain composition throughout the remaining period, and almost identical member composition post-stabilization (Figure 5A).
Figure 5. Assemblies composed of 38 knockout strains quickly coalesce into lower-complexity consortia with highly consistent member composition that are stable over time.

(A) Changes in strain composition across biological replicates revealed highly similar dynamics. The panels show the inferred member composition of individual assemblies over 14 days of serial transfer. The assemblies initially had equal proportions of all knockout strains. The member composition was measured by deep sequencing samples from days 1, 3, 7, and 14. (B) Strain diversity in serially transferred assemblies rapidly decreased and then stabilized. The graphs show the mean normalized entropy across the six biological replicates, the bars mark the standard deviation. The normalized entropy for individual assemblies is marked by different shapes (typically indistinguishable from the mean). The lower panel shows the member composition overlayed on the interaction network and highlights the two phases taking place over time. The networks show the strain composition (averaged over replicates) at days 0, 3 and 14. Most changes in the assemblies conclude by day 3. This period is characterized by loss of many strains and a shift in the proportion among remaining strains (shown by the node size). The numbers next to the node indicate the number of strains remaining in the node. The assembly composition remains highly conserved from day 3 to day 14. The node area marks the total proportion of all strains belonging to its functional group from all detected strains. (C) The composition of assemblies at day 14 reveals multi-member consortia that are likely stabilized by multiple strong pairwise cross-feeding interactions. The networks show the strain composition (averaged over replicates). The node area marks the total proportion of all strains belonging to its functional group from all detected strains. The identity of dominant strains within each node is shown next to the node, the numbers mark the proportion of the strain from the entire assembly. (D) Strains retained within each biological pathway were likely not a random subset of all auxotrophs from this pathway. The pie charts show the composition of dominant strains in each biological replicate at day 14. We observed a high consistency in the knockouts dominating each dysfunctional pathway. A list of the individual dominant strains and their proportions is presented to the right of each pie chart.
In order to quantify the reduction in strain complexity over time and compare the replicates, we calculated the entropy of consortia composition, or Shannon diversity, as a function of time and normalized it to the maximal entropy possible for a 38-strain consortium. We reasoned that the normalized entropy would be a good metric for diversity since it takes into account the number of strains in the assembly and the skewness in their proportion while remaining oblivious to the biological function of the gene knockouts. The normalized entropy is maximized (=1) if all strains are present at equal proportions (as in the initial inoculum) and is minimized (=0) if the assembly is completely dominated by a single strain. Figure 5B shows the changes in normalized entropy over time after averaging across all biological replicates (normalized entropy dynamics in individual replicates were almost indistinguishable from one another). As the graph shows, we observed that consortium complexity rapidly decreased in the first three days and then stabilized at a normalized entropy of 0.5. This stabilization, that was remarkably similar across all replicates, indicated that all assemblies rapidly converged to consortia that were still relatively complex.
We next investigated if the consortia composition can be explained by known cross-feeding interactions. Since we knew what the strain identity at each of the tested time points was, we were able to impose the member composition on the interaction network we previously inferred from pairs (Figure 4D) and test if the assembly we observed can be explained by multiple known strong pairwise interactions. The lower panel of Figure 5B shows the inferred interaction network at three key time points in this experiment. The initial inoculum, that supports collaborations between all six functional groups, coalesced to a lower complexity assembly within three days. This assembly included strains from four functional groups after extinction of auxotrophs for nucleotides and the co-factor NAD+. In this initial period of coalescence, we also observed a substantial contraction in the number of strains belonging to each functional group. For example, we observed that the amino-acids auxotroph group contracted from 17 strains to a single strain (cysW knockout). The period from day 3 to day 14 is characterized by overall stability in member composition. Although terminal assembly was still composed of members from the same 4 functional groups as the day 3 assembly, we observe a further contraction in the number of strains from each functional group. For example, the pantothenate biosynthesis group contracted from 3 to 2 strains. Notably, even after intra-group contraction, the consortium composition could still be explained by previously characterized strong pairwise interactions.
Figure 5C shows the composition of the consortium in the last day of the experiment (averaged over all replicates). As the figure shows, the 38-strain assembly stabilized on a consortium that is composed of 7 dominant strains (bioC, bioF, bioH, cysW, icd, panD, and panZ knockouts). This terminal assembly spans multiple functional groups – biotin, pantothenate, and amino-acid biosynthesis and carbon metabolism mutants – and is largely dominated by strains that were predicted to be top potential collaborators (70% of cells and 5 of 7 dominant strains were vitamin auxotrophs). It is also important to highlight the consistencies across biological replicates. Figure 5D shows the consortium composition on the last day of the experiment in each of the six replicates. As the figure shows, the strains retained within each biological pathway were likely not a random subset of the auxotrophs from this pathway. For example, in all replicates we observed dominance of the panZ knockout over all pantothenate auxotrophs and dominance of bioH knockout over all biotin auxotrophs. This observation suggests that even within a single pathway, auxotrophs can be prioritized by their cross-feeding competency.
While many of the consortia characteristics matched the predictions made according to strain-pair experiments, it is also important to note that some of the characteristics were unpredictable. For example, only one amino-acid auxotroph was retained in the stable consortia (cysW knockout). While cysW knockout is among the top collaborators from the amino-acid auxotroph group, other amino-acids auxotrophs, as the trpE knockout, were predicted to be even more potent collaborators. A similar observation can be made for the icd knockout from the carbon metabolism group. Different explanations may underly these discrepancies. First, it is possible that the measurements we made over short time scales to estimate the collaboration capacity of strains were limited predictors for technical reasons, either due to trace amounts of metabolites available intracellularly in the first hours of growth or due to slightly different protocols in the different experiments (e.g., different culture volume were used for the two experiments and these can impact multiple parameters of growth such as oxygen availability). It is also possible that discrepancies arose due to biological reasons, including possible higher-order interactions between knockout strains that were not captured in pairwise experiments or that evolutionary adaptation took place during the multi-day community experiment(Lloyd et al., 2019; Zhang and Reed, 2014).
The observation that the emerging consortia was fully supported by pairwise cross-feeding interaction is reassuring, yet it remains unclear whether such a consortium is also likely to emerge by random when multiple strains are co-cultured together. To evaluate the statistical significance of our experimental observations we mathematically calculated what fraction of all possible alternative consortia are fully consistent with the pairwise interaction network. We considered a total ~3*1011 alternative assemblies with 2-38 members (supplementary figure 8A). We sampled 100,000 random assemblies for each assembly size and tested if each one was fully supported by the pairwise cross-feeding network in order to infer an empirical p-value for that assembly size (supplementary figure 8B). Lastly, we integrated across all network sizes and weighted the empirical p-values by the number of alternative networks to calculate the p-value of interest (supplementary figure 8C). Since our simulations revealed that only a very small fraction of randomly selected assemblies (<0.0042) is fully supported by the pairwise cross-feeding network we concluded that the observed community emerging in our experiments is nonrandom and is likely selected by pairwise cross-feeding interactions. Lastly, as another test of our predictions we repeated the community experiment while growing the same set of 38 strains in nutrient-rich media. We reasoned that since in these conditions cross-feeding is nonessential (auxotrophs are rescued by media metabolites) the composition of emerging assemblies will be dictated by other forces. Specifically, given our mathematical results we predicted that the emerging assemblies mostly likely will not be fully supported by the pairwise interaction network. Reassuringly, we observed multiple differences in the dynamics of these assemblies and their end composition when growing on nutrient-poor and nutrient-rich media (supplementary Figure 9). In agreement with our prediction, none of the emerging consortia was fully consistent with pairwise interactions network and the end compositions of emerging consortia were not highly correlated with one-another.
Taken together we conclude that the auxotroph strains we identified as potential cross-feeders can indeed support a sustainable multi-member consortium with a stable composition. While this observation was previously made for strict amino acids auxotrophs(Mee et al., 2014), we can now expand it to include permissive auxotrophs. Since some of these strains are characterized by high growth rate, it is possible they will support a community that can quickly grow even at low confluence (systematic experiments with dozens of communities with different compositions are required to rigorously test this possibility). The stable consortia consisted of auxotrophs from 4 biological pathways and featured co-occurrence of individual strains that were known to strongly interact as pairs. The emerging strain composition of the consortia substantiates our claim that vitamin auxotrophs are ideal collaborators by showing that 5 of 7 dominant members and 70% of all cells, were indeed vitamin auxotrophs.
Discussion
Metabolic cross-feeding frequently underlies mutualistic relationships in natural microbial communities and is often exploited for assembling synthetic consortia. Previous studies of cross-feeding interactions in E. coli knockout strains provided insights into the design principles underlying mutualism. However, most studies that used E. coli relied on existing knowledge of well-characterized auxotroph mutations in a handful of metabolic pathways(Kerner et al., 2012; Lloyd et al., 2019; Mee et al., 2014; Pande et al., 2014; Wintermute and Silver, 2010; Zhang and Reed, 2014; Ziesack et al., 2019). Here, we hypothesized that improvement can be made by developing an unbiased approach for identifying all knockout strains that can participate in cross-feeding interactions. We expected that an unbiased systematic approach will not only assist in identifying new auxotroph strains but will also help prioritizing which auxotrophs are optimal for imposing stable and potent cross-feeding interactions that are characterized by overall fast growth.
We developed a pooled genetic screen approach to identify all single-gene knockouts that benefited from conditioned media. This approach allowed us to circumvent the formidable challenge of testing all possible knockout pairs (~7 million pairs) and to systematically identify the small group of strains that benefit from goods secreted by co-cultured cells. We find that strains that benefit from conditioned media are a small subset of all auxotrophs that can be rescued by extracellular metabolites in rich-nutrient media (Figure 2B). A functional enrichment analysis of the screen results revealed that conditioned media enriches for strains that are dysfunctional for very specific metabolic pathways (many, but not all, of the amino-acids, nucleotides, specific vitamins and co-factors, and central carbon metabolism). While most of these results agree with the previous studies, some hits are potentially new targets for imposing mutualism in E. coli (biotin, folate, and Pyridoxal 5'-phosphate biosynthesis).
Amino-acids and nucleotides auxotrophs are frequently used to successfully impose cross-feeding mutualism in E. coli and other organisms(Kerner et al., 2012; Mee et al., 2014; Pande et al., 2014; Shou et al., 2007; Wintermute and Silver, 2010; Ziesack et al., 2019). While our study does not negate that they can cross-feed, it indicates that pairs composed of these auxotrophs may be characterized by an overall slow growth rate. A very plausible explanation for the slow growth is that amino-acids and nucleotides are cellular building blocks and are therefore required in large amounts for rescue. In contrast, vitamins and co-factors are only needed in trace amounts. Here, we assumed that many applications that rely on cross-feeding interactions will require vigorous growth and therefore designed a scoring metric that considers the resemblance to an “ideal” fast-growing collaborator. Strains that scored high using this metric were typically auxotrophs for vitamins and co-factors and strains with altered carbon metabolism. Later experiments with over a thousand strain pairs further supported this prediction and showed that auxotrophs for vitamins and co-factors tended to participate in a greater number of interactions that yielded fast growth (Figure 4D). The systematic testing of so many strain pairs allowed us to infer the rules of interaction and deduce which functional groups collaborate well (Figure 4F). Taken together, our experiments in individual strains and in cross-feeding pairs provide a catalog of all auxotrophs that can participate in cross-feeding interactions and highlight which strain pairs grow vigorously when they are co-cultured. It will be interesting to test if the interaction rules we deduced for E. coli also apply to other microorganisms. We expect that addressing this question in model microorganisms that are genetically tractable, as the model eukaryote Saccharomyces cerevisiae, will be straightforward using a genetic screen approach(Giaever et al., 2002), similar to the one described here, or using a co-culturing approach recently described for S. cerevisiae and Pseudomonas(N’guyen et al., 2020).
Previous studies suggested that metabolite exchange in natural and synthetic communities can facilitate closed cooperative loops that benefit a multi-member community(Freilich et al., 2011; Goldford et al., 2018; Louca et al., 2018; Zengler and Zaramela, 2018). We therefore decided to test if the auxotrophs we identified as potential collaborators will coalesce into a multi-member consortium with a stable composition during a long-term serial transfer experiment. While a previous study with auxotrophs for amino acids demonstrated emergence of stable multi-strain consortia(Mee et al., 2014), it was unclear if stable consortia will emerge when including permissive auxotrophs. The results from our community experiments revealed that permissive auxotrophs can indeed be used to engineer a stable multi-member consortium. Moreover, the emerging strain composition substantiates our key claim that vitamin auxotrophs are ideal collaborators by showing that most of the stable consortium members were auxotrophs for pantothenate and biotin. The experiments also uncovered a remarkable degree of consistency across biological replicates and highlighted very similar temporal dynamics – rapid convergence into a lower-complexity consortium that remains stable over multiple days. We therefore expect that the 7-strain consortium that emerged from our serial transfer experiment may be useful for biotechnological applications that require division of labor between members of a synthetic multi-strain consortium(Pandhal and Noirel, 2014; Roell et al., 2019; Zhou et al., 2015).
The study of metabolic cross-feeding in model organisms is important for shedding light on mutualism in natural microbial communities. Previous works suggest that cross-feeding in diverse natural communities, such as the gut microbiome(Magnúsdóttir et al., 2015; Soto-Martin et al., 2020), marine ecosystems(Croft et al., 2005; Gómez-Consarnau et al., 2018; Seymour et al., 2017), and production of fermented milk products(Blasche et al., 2021; Ponomarova et al., 2017) can rely on the exchange of amino-acids, vitamins, and carbon sources. However, these conclusions should also be treated with caution for multiple reasons: First, many works in the field rely on computational analysis and comparative genomics to infer that some metabolic pathways are missing. However, results from computational approaches can be highly misleading due to imperfect gene annotation (as elegantly demonstrated here(Price et al., 2018)). Second, in many natural communities it remains unclear if auxotrophs are rescued by metabolites secreted by other community members or by high nutrient availability in a rich extracellular environment, e.g., the host diet supplying nutrients to the gut microbiome(Soto-Martin et al., 2020). Lastly, multiple additional selective pressures, beyond the ability to cross-feed, determine the member composition in natural communities, as demonstrated by the varying taxonomic composition in the gut microbiota across hosts(Louca et al., 2018). The work we present here circumvents these inherent problems for evaluating the selective advantage of cross-feeding, by allowing us to quantify the fitness value of interactions in a well characterized model system where everything, except the pathways in question, is kept constant (media formulation, aeration, temperature, genetic background). Our systematic work reveals that although cross-feeding interactions can indeed rely on the exchange of multiple metabolites, some interactions are more potent than others in sustaining fast cross-feeding pairs. Moreover, our community experiments revealed that auxotrophs relying on these metabolites eventually prevail over other auxotrophs and persist in a multiple strain synthetic community. Thus, our work allows to determine that at least in the environment we tested, well-mixed aerated liquid media, vitamin autotrophy is superior to the exchange of amino-acids, nucleotides, and carbon sources.
An additional important observation of our study is that many of the top collaborating auxotrophs are permissive ones (auxotrophs that can grow reasonably well even in poor media). While it is very plausible that such auxotrophs will not be able to sustain growth indefinitely on nutrient-poor media since they lack essential vitamins and co-factors, they possibly maintain their initial growth due to sufficient intracellular reservoirs and possible scavenging of metabolites from lysed cells and cellular debris. The observation of permissive auxotrophs is reminiscent of a recent study of soil microbiomes that revealed that isolates growing reasonably well on their own on nutrient-poor media can still benefit from cross-feeding interactions(Goldford et al., 2018). These interactions were shown to be strong enough to support a sustainable three-species consortia. Although the identity of the secreted goods is unknown in this system, the observation that the three species can grow as pure cultures on nutrient-poor media but improve when collaborating and our observations of superiority of permissive auxotrophs, together suggest that collaboration that relies on permissive auxotrophy may be selected for in nature. Moreover, since the natural isolates grew on nutrient-poor media that is similar to the one we used for our study, we expect that the genetic approach we used will be instrumental for identifying what are the secreted nutrients that support this multi-species community. In these experiments, conditioned media filtered from each isolate species would be used to grow the pooled E. coli strain collection and analysis of fold-enrichment of knockout strains could be used to infer the identity of the secreted metabolites (and possibly even their extracellular concentration).
Work on cross-feeding in E. coli strains continues to provide fundamental insights into design principles governing complex multi-species microbial systems as well as to allow improved design of biotechnological and biomedical applications. The results presented here provide a systematic catalog of all the knockout strains that can be utilized for cross-feeding applications and offer detailed information on their growth competency as pure strains, as part of a collaborating pair, and as members of a multi-strain consortia. Our work revealed that auxotrophs for biotin and pantothenate are ideal strains for assembling cross-feeding cultures since they support fast growth while maintaining strain co-dependence.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amir Mitchell (amir.mitchell@umassmed.edu).
Materials availability
This study did not generate new unique reagents.
Data and Code availability
The sequencing datasets generated during this study have been deposited at the NCBI Sequence Read Archive (SRA) and are publicly available under the accession numbers: PRJNA727839 (auxotroph screen), PRJNA727852 (community composition in nutrient-poor media), and PRJNA727847 (community composition in nutrient-rich media).
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| E. coli barcoded deletion library | Laboratory of Hirotoda Mori | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| AMPure XP bead | Beckman Coulter | Cat#A63881 |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems | Cat#KK2602 |
| Difco M9 Minimal Media Salts, 5X | BD | Cat#248510 |
| Protein hydrolysate amicase | Sigma Aldrich | Cat#82514 |
| Tryptophan | Sigma Aldrich | Cat#T0254 |
| Uridine | Alfa Aesar | Cat#A15227-06 |
| Adenine sulfate | USBiological | Cat#A0865 |
| Biotin | Sigma Aldrich | Cat#B4639 |
| Niacin | Spectrum | Cat#NI100 |
| D-pantothenic acid | Sigma Aldrich | Cat#P5155 |
| Critical commercial assays | ||
| BioAnalyzer/Agilent High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| Qubit dsDNA high sensitivity assay | Thermo-fisher | Cat#Q32854 |
| Quick DNA Miniprep Plus Kit | Zymo | Cat#D4068 |
| Nextera XT Index Kit | Illumina | Cat#FC-131-1024 |
| MiniSeq High Output Reagent Kit, 75-cycles | Illumina | Cat#FC-420-1001 |
| MiniSeq High Output Reagent Kit, 150-cycles | Illumina | Cat#FC-420-1002 |
| MiSeq Reagent Kit v3, 150-cycles | Illumina | Cat#MS-102-3001 |
| Deposited data | ||
| Auxotroph screen | This study | PRJNA727839 |
| Assembly dynamics in nutrient-poor media | This study | PRJNA727852 |
| Assembly dynamics in nutrient-rich media | This study | PRJNA727847 |
| Pooled library in nutrient rich media | Rosener et al., 2020 | PRJNA645605 |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Oligonucleotides | ||
| Forward primer for barcoded library: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-(4-6xN)-TGTAGGCTGGAGCTGCTTCG 3' | Rosener et al., 2020 | N/A |
| Reverse primer for barcoded library: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-GCAAATATTATACGCAAGGCGACAAG 3’ | Rosener et al., 2020 | N/A |
| Recombinant DNA | ||
| Software and algorithms | ||
| Cystoscape | Shannon et al., 2003 | https://cytoscape.org |
| Matlab(R2020a) | Mathworks | https://www.mathworks.com/downloads/ |
| Circa | OMGenomics | https://omgenomics.com/circa/ |
| DEBRA, R package | Akimov et al., 2020 | https://github.com/YevhenAkimov/DEBRA |
| GAGE, R package | Luo et al., 2009 | https://bioconductor.org/packages/release/bioc/html/gage.html |
| DESeq2, R package | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Code to analyze data and generate plot | This study | DOI: 10.5281/zenodo.5106763 |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Barcoded strain library
The E. coli barcoded deletion library was developed and provided by Dr. Hirotoda Mori from the Nara Institute of Science and Technology in Japan. More details on this resource appear here(Rosener et al., 2020). The parent strain of this library is BW38028 with the genotype Δ(araD-araB)567 lacZp-4105(UV5)-lacYλ− hsdR514, rph+(Conway et al., 2014). The strain collection has 3,680 individual knockout strains. In each strain, the open reading frame of a single gene was replaced in-frame with a fragment containing turbo GFP, a chloramphenicol resistance cassette and a unique 20bp sequence that serves as an identification barcode. Since the barcode is the only variable region across strains, it can be amplified from a mixed culture of strains with a single pair of primers.
METHOD DETAILS
Media and growth conditions.
All experiments were performed in either nutrient-rich media (Luria Broth, LB), nutrient-poor media (M9 salts supplemented with 2mM MgSO4, 0.1 mM CaCl2 and 0.5% glucose), or nutrient-poor conditioned media. Conditioned media was prepared by growing the barcoded ybcN strain overnight in nutrient-poor media supplemented with 20 μg/mL chloramphenicol. When the overnight culture reached an OD600 (optical density at 600 nm) of 1.2, cells were washed twice in water containing M9 salts and diluted to multiple inoculating densities. Conditioned media was obtained by inoculating the following cell densities: 5*109, 109, 108, and 107 cells/mL, and growing them in nutrient-poor media for 3.5 hours. Cultures were then filtered with a 0.22 micron filter. We replenished the spent media by adding nitrogen, phosphorous and carbon sources by adding x5 concentrated stocks of the M9 salts and glucose. All experiments were performed at 37 °C, and overnight cultures were shaken in 14 mL culture tubes (orbital) at 200 rpm. Experiments in multi-well plates were conducted with an automated plate reader (Tecan Spark/BioTek Eon MicroPlate) at 37 °C with double orbital shaking at 180 rpm.
Pooled Genetic screen – nutrient-poor vs conditioned nutrient-poor media
For the screen, 50 μL of a frozen glycerol stock of the pooled E. coli barcoded strain library was inoculated into 7 mL of nutrient-rich media (LB) supplemented with 20 μg/mL chloramphenicol and grown over night. This high inoculum was used to maintain a large population size and minimize the sampling bottleneck in the thawing stage. Cells were washed twice in water containing M9 salts and inoculated at a density of 5*106 cells/mL in 7 mL of nutrient-poor media or conditioned media. The screen was performed at 37 °C in biological triplicates (nutrient-poor media) or duplicates (conditioned media). Growth was halted by flash freezing the cultures when they reached the OD600 range of 0.2-0.3 (typically after about 8 hours of growth). Samples were stored at −80 °C for later DNA extraction (Zymo Quick DNA Miniprep Plus Kit, Cat#D4068).
Pooled Genetic screen – nutrient-poor vs nutrient-rich media
Full details on the genetic screen conducted in nutrient-rich media can be found in our previous study(Rosener et al., 2020). For that nutrient-rich media screen, 15 μL of the frozen glycerol stock of the pooled strain library was inoculated into 25 ml of the two media types supplemented with 20 μg/ml chloramphenicol for overnight growth. The culture was then diluted to OD600 of 1, and again diluted 1:200 into 7ml of nutrient-rich media (LB). The screen was performed at 37 °C in biological duplicates. The optical density of cultures was monitored throughout the screen. When the cultures reached OD600 of 0.6, the experiment was stopped and DNA was immediately extracted (Zymo Quick DNA Miniprep Plus Kit, Cat#D4068).
Limitations of pooled genetic screening
The pooled genetic approach we used for this study is relatively easy to implement and is powerful for the identification of auxotrophs that can be rescued by secreted metabolites. However, it is also important to note that pooled screening has an inherent limitation in this context. A pooled approach its inadequate for detecting unique interactions that only transpire between very specific strain pairs. For example, it is plausible that some knockout strains may be characterized by altered metabolite secretion profiles (e.g., they secrete metabolites that are not typically secreted by most strains in our library) and therefore possibly rescue auxotrophs that are not typically rescued by media that was conditioned by the wild-type strain. Such pair specific interactions will not be detected by pooled screening. However, while an ordered genetic screen that systematically tests the interactions of all individual strain pairs can likely uncover such interactions, the experimentation scale it requires makes it highly impractical since it requires monitoring the growth of over 7 million interacting pairs (3,6802/2 different pairs).
Barcode Sequencing and Analysis
The genomic DNA (gDNA) concentration of each screen sample was measured with Qubit dsDNA high sensitivity assay kit (Thermo-fisher, Cat#Q32854) and 6.25 ng of gDNA was used to prepare DNA libraries. A region of ~350 bp around the barcode locus was amplified with custom forward and reverse primers using 2x KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Cat#KK2602). The primers sequences were: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-(4-6xN)-TGTAGGCTGGAGCTGCTTCG 3' 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-GCAAATATTATACGCAAGGCGACAAG 3’ The Following thermocycler protocol was used: initial denaturation at 95° C for 3 minutes, 23 cycles of 95 °C for 30 seconds, 55 °C for 30 seconds, 72 °C for 30 seconds, followed by a final extension at 72 °C for 5 minutes. PCR products were purified using standard AMPure XP bead protocol (Beckman Coulter, Cat#A63881) with beads added at 0.9x volume. The standard Nextera XT Index Kit protocol (Illumina, Cat#FC-131-1024) was used to add indices and Illumina sequencing adapters to each PCR sample followed by the same AMPure XP bead purification protocol. The libraries were then run on a 2.5% Agarose gel with the product extracted using NEB Monarch DNA Gel Extraction Kit standard protocol (NEB, Cat# T1020L). Quality control of libraries was performed using BioAnalyzer/Agilent High Sensitivity DNA Kit (Agilent Technologies, Cat# 5067-4626). Library concentrations were assessed by Qubit dsDNA high sensitivity assay. Libraries were normalized to the same concentration, denatured, and diluted according to Illumina MiniSeq System Denature and Dilute Libraries Guide. Sequencing was performed using MiniSeq High Output Reagent Kit, 75-cycles (Illumina, Cat# FC-420-1001) on Illumina MiniSeq device.
Growth of auxotroph strains
We individually grew the 67 rescued hits form the 5*109 cells/mL conditioned media overnight in nutrient-rich media (LB) supplemented with 20 μg/mL chloramphenicol. We washed them twice in water containing M9 salts and seeded them at a density of around 5*106 cells/mL in either nutrient-poor media or conditioned media filtered from a 109 cells/mL culture. We grew them in 75 μL in 384-well plates and monitored by measuring absorbance at 600 nm (A600) every half hour with an automated plate reader (BioTek Eon MicroPlate). Plates were shaken and maintained at 37 °C between the reads.
Cross-feeding competence in strain pairs
We tested cross-feeding competence for a total of 38 strains. 27 of these strains were in the top 32 collaborators according to our collaboration score (five cys strains were omitted to avoid saturation of strains from the same pathway). We also included four additional strains belonging to the top 42 of collaborators due to their high log2(fold-change) in the genetic screen. Additionally, we included seven strains despite not being characterized as hits on our screen in conditioned media from 5*109 cells/mL condition (six of these strains were found to be enriched in our screen but did not qualify as hits according to our enrichment cutoff). The additional strains we included according to this reasoning: bioD and bioF were included to complement the auxotrophs belonging to the biotin biosynthesis pathway and because they were significantly enriched in conditioned media from 109 cells/mL, purA, ilvE, and pheA, were included because they were shown to participate in successful cross-feeding pairs in previous works(Mee et al., 2014; Wintermute and Silver, 2010). We chose the other three knockout strains nadA, metE and rpe to complement hit strains from the same biological pathways of strains with high collaboration scores. All the added strains were still found to be highly enriched in conditioned media but were not characterized as hits in our screen due to our conservative choice for cutoff values for the fold-enrichment or adjusted p-value (supplementary table 1). We picked these 38 strains from a glycerol stock stored at −80 °C and grew them overnight in nutrient-rich media (LB) supplemented with 20 μg/mL chloramphenicol. After washing twice in water containing M9 salts, we diluted the individual cultures to a density of 3x108 cells/mL. We combined equal volumes and inoculated them into 75 μL of nutrient-poor media at a final density of 4x107 cells/mL. We grew them in 384-well plates and monitored growth by measuring A600 every half hour with an automated plate reader (Tecan Spark).
Growth of auxotroph strains in supplemented nutrient-poor media
We supplemented media with amino acids, purines, pyrimidines, biotin, pantothenic acid, or niacin in order to test for auxotroph rescue. For the amino acid auxotrophs, the nutrient-poor media was supplemented with protein hydrolysate amicase (Sigma Aldrich, Cat#82514) 0.2% (weight/volume). For rescue of tryptophan auxotrophy we supplemented the media with tryptophan (Sigma Aldrich, Cat#T0254) 0.2% (weight/volume), we did this because tryptophan concentration in the amicase is too low (<0.5 mg/g) to rescue tryptophan auxotrophs. For the purine auxotrophs, we supplemented the nutrient-poor media with adenine sulfate (USBiological, Cat#A0865) 200 μM. For the pyrimidine auxotrophs, we supplemented the nutrient-poor media with uridine (Alfa Aesar, Cat#A15227-06) 200 uM. For the nicotinamide auxotrophs, we supplemented the nutrient-poor media with niacin (Spectrum, Cat#NI100) 0.001 g/L. For the biotin auxotrophs, we supplemented the nutrient-poor media with biotin (Sigma Aldrich, Cat#B4639) 0.001 g/L. For the pantothenic acid auxotrophs, we supplemented the nutrient-poor media with pantothenic acid (Sigma Aldrich, Cat#P5155) 0.001 g/L. We tested the rescue of 31 of the 38 strains used to test cross-feeding (we did not test rescue of mutants in carbon metabolism since it was not clear which exact carbon source can be used for rescue). We grew the 31 strains overnight in nutrient-rich media supplemented with 20 μg/mL chloramphenicol. We washed them twice in water containing M9 salts and seeded them at a density of around 5*106 cells/mL in nutrient-poor media or nutrient-poor media supplemented with the rescuing metabolite (the end-product of the mutated pathway). We grew them in 5 replicates in a volume of 75 μL in 384-well plates and monitored them by measuring A600 every hour for 24 hours with an automated plate reader (Tecan Spark). Plates were shaken and maintained in 37 °C between the reads.
Growth of auxotroph pairs in different culture conditions
We selected 24 auxotrophs that form 16 pairs ranging from poor to successful cross-feeding interactions to test if growth conditions (culture volume and culture container) considerably impact the ability of pairs to cross-feed. We inoculated the 24 strains from a glycerol stock stored at −80 °C and grew them overnight in nutrient-rich media supplemented with 20 μg/mL chloramphenicol. After washing twice in water containing M9 salts, we diluted the individual cultures to OD600 of 1. We combined equal volumes and inoculated them into 30 mL of nutrient-poor media to reach a final density of 4*107 cells/mL. We split each co-culture into culture tubes (7 mL), 96 deep well plates (1 mL), and 384 well plates in triplicates. We grew 384-well plates and monitored their growth by measuring A600 every hour with an automated plate reader (Tecan Spark). Plates were shaken double orbital at 180 rpm and maintained in 37 °C between the reads. We grew the 96 deep-well plate and culture tubes at 37 °C and with orbital shaking at 200 rpm. We took 200 μL aliquots from the 7 mL cultures every 3 hours and measured A600 in an automated plate reader (Tecan Spark). We took 50 μL aliquots from the 1 mL cultures every 3 hours, diluted into 150 μL of PBS, and measured A600 in an automated plate reader (Tecan Spark).
Proportion of auxotrophs in representative cross-feeding pairs
For each functional group, we selected an auxotroph with a high number of interactions, nadC, panC, bioC, purE, icd and metC. With those 6 auxotrophs, a total of 15 different pairs can be formed. To be able to grow auxotroph pairs and then calculate the relative proportion of each strain from the pair, we decided to take one auxotroph from the barcoded library, which is resistant to chloramphenicol, and one auxotroph from the Keio knockout strain collection(Baba et al., 2006), which is resistant to kanamycin. The pairs used are shown in the following table:
| Kanamycin | Chloramphenicol |
|---|---|
| nadC | panC |
| nadC | bioC |
| nadC | purE |
| nadC | icd |
| nadC | metC |
| panC | bioC |
| panC | purE |
| panC | icd |
| panC | metC |
| bioC | purE |
| bioC | icd |
| bioC | metC |
| purE | icd |
| purE | metC |
| icd | metC |
We picked the strains from a glycerol stock stored at −80 °C and grew them overnight in nutrient-rich media supplemented with either 20 μg/mL chloramphenicol or 50 μg/mL kanamycin. After washing twice in water containing M9 salts, we diluted the individual cultures to OD600 of 1. We combined equal volumes and diluted 1:100 into 1 mL of nutrient-poor media at a final density of 107 cells/mL. We grew every co-culture in triplicates, at 37 °C and with orbital shaking at 200 rpm. After 20 hours, we plated 1/103 dilutions on selective LB agar plates (either 20 μg/mL chloramphenicol or 50 μg/mL kanamycin). After 24 hours, we imaged the plates with an EOS Rebel T3i camera, and we counted colonies were using an automated MATLAB script. We used the CFUs to estimate the proportion of each auxotroph in the pair.
Temporal dynamics of synthetic consortia
We individually picked the same 38 strains used to test cross-feeding from a glycerol stock stored at −80 °C and grew them overnight in nutrient-rich media (LB) supplemented with 20 μg/mL chloramphenicol. After washing twice in water supplemented with M9 salts, we diluted the individual cultures to a density of 109 cells/mL. We combined equal volumes and diluted 1:100 into 1 mL of nutrient-poor media at a final density of 107 cells/mL. We grew the assemblies in a 96 deep-well plate containing 1 mL of nutrient-poor media per well at 37 °C with orbital shaking at 200 rpm. After 24 hours, we thoroughly mixed the assemblies and transferred 5 μL to 995 μL of fresh nutrient-poor media. We stored the remaining culture at −80 °C for later DNA extraction. We repeated this 24-hour cycle for 14 days to track changes in the assembly composition over time.
As a control, we repeated the experiment while growing the assemblies in nutrient-rich media. We picked the same 38 strains from a glycerol stock stored at −80°C and grew them overnight in nutrient-rich media (LB) supplemented with 20 μg/mL chloramphenicol. We diluted the individual cultures to a density of 109 cells/mL. We combined equal volumes and diluted 1:100 into 1 mL of nutrient-poor media at a final density of 107 cells/mL. We grew the assemblies in a 96 deep-well plate containing 1 mL of nutrient-poor media per well at 37 °C with orbital shaking at 200 rpm. After 12 hours, we thoroughly mixed the assemblies and transferred 5 μL to 995 μL of fresh nutrient-rich media (LB). We stored the remaining culture at −80 °C for later DNA extraction. We repeated this 12-hour cycle 14 times to track changes in the assembly composition over time.
We extracted DNA from time points 1, 3, 7 and 14 and prepared it for sequencing as detailed above for the genetic screen. For the nutrient-poor media experiment, sequencing was performed using MiSeq Reagent Kit v3, 150-cycles (Illumina, Cat# MS-102-3001) on an Illumina MiSeq device. For the nutrient-rich media experiment, sequencing was performed using MiniSeq High Output Reagent Kit, 150-cycles (Illumina, Cat#FC-420-1002) on an Illumina MiniSeq device.
Random simulation of synthetic consortia
Using a custom Matlab (MathWorks) script, we developed a mathematical simulation that allowed us to estimate the likelihood of obtaining a strain assembly that is fully supported by the network of pairwise cross-feeding interactions that we inferred experimentally. We reasoned that an assembly compatible with our pairwise interaction network would be one were all the member strains (nodes) can be rescued (edges) by at least one other member in the assembly. Mathematically, a network that is fully supported by pairwise interactions is defined as a getting a connected graph when selecting the subset of nodes from the full pairwise interaction graph. In our simulation, for each assembly with a given size (2 to 38 strains), we sampled without replacement 100,000 random assemblies. We then counted how many times, out of 100,000 generated assemblies, resulted in a connected graph. This fraction represents an empirical p-value for the likelihood of obtaining a strain assembly that is fully explained by the pairwise interaction network by random chance for a given assembly size. We also calculated separately the total number of unique strain assemblies that can be formed from 38 auxotrophs. Finally, we weighed empirical p-values (calculated by our simulation) by the number of alternative networks for each assembly size. This p-value represents the likelihood of obtaining an assembly with 2-38 strains that is that is fully explained by the pairwise interaction network by random chance. The weight of each network size is given by the following equation:
where ni marks the number of unique alternative subsets with i strains that are chosen out of 38 strains without replacement. The empirical p-value can be calculated by summing the weighted empirical p-value across all possible network sizes (2-38) with the following equation:
where, pi is the empirical p-value of getting an assembly of size i that is supported by the pairwise interaction network and wi is the number of unique alternative subsets with i strains that can be chosen without replacement out of 38 strains.
Extracellular metabolites due to cell lysis of debris
Multiple experiments throughout this study relied on conditioned media that was filtered and may have likely also contained cellular debris and metabolites that originated from cell lysis during sample handling. To minimize cell lysis during conditioned media preparation we used a filtering method that relied on a weak vacuum pressure (250 mL vacuum-based filter flasks). We avoided filtering media by more aggressive methods (e.g., by applying pressure on syringe filters). Moreover, the conditioned media experiments were used only for the initial steps of this work that focused on the identification of candidate collaborators. All candidate collaborators we used for later experiments were tested by monitoring their growth in co-culture experiments (as pairs or in a community). Such co-culture experiments did not include any filtering step. We therefore reasoned that auxotroph growth in these experiments reflects predominantly rescue due to metabolites secreted by live cells and not due to cell lysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Barcode Sequencing and Analysis
We converted the raw reads to barcode counts using a custom MatLab (MathWorks) script that searched for exact match of barcodes with 15-25bp in each individual read. We masked any nucleotide with a quality score of 10 or less. We identified knockouts that influenced bacterial growth in conditioned media by comparing the relative frequency of individual barcodes when the pooled library grew in conditioned media and in unconditioned media. We identified knockouts that influenced bacterial growth in nutrient-rich media by comparing the relative frequency of individual barcodes when the pooled library grew in nutrient-rich media (PRJNA645605) and in unconditioned media (this study). For both analyses, we used the barcode counts to identify strains with significant changes in their relative frequency with DEBRA(Akimov et al., 2020). DEBRA is an R package that allows the run of DESeq2(Love et al., 2014) analysis on barcode libraries. For both analyses we chose the Wald statistical test and cutoffs of 3 log2-fold for enrichment and false-discovery-rate adjusted p-value of 0.05. We discarded barcodes with less than 10 counts in a single screen condition. We performed gene-set enrichment analysis with GAGE(Luo et al., 2009) with a false-discovery-rate adjusted p-value of 0.1.
Growth of auxotroph strains
To assess absolute fitness in each condition, we used the area under the growth curve (AUC) after subtracting the absorbance of the first time point. We used fitness measurements from both conditioned and unconditioned media to predict which strains are likely to be potent collaborators that can support fast growth. For the purpose of this study, we defined an ideal collaborator as an auxotroph that will be unable to grow on unconditioned media and will grow rapidly on conditioned media. We assigned the minimal nutrient-poor AUC value measured in nutrient-poor media and the maximum AUC value measured in conditioned media to this ideal collaborator. Then, we measured the Euclidean distance of each knockout strain from this ideal collaborator. To define a collaboration optimality score, we subtracted the Euclidean distance value of each strain from the maximum distance value of all the validated strains, then we normalized the result to that same maximum distance. This gives a collaboration optimality score that ranges between 0 and 1, where one would be the score of an ideal collaborator and 0 is the score of the worse collaborator.
Cross-feeding competence in strain pairs
To assess the interaction competency between strains, we calculated the difference between the AUC of the pair and the max AUC of its individual members. We chose the minimum AUC value of all the pairs containing each strain as the representative AUC of that individual strain, based on the assumption that this growth curve represents growth when there is no interaction. We chose that measurement instead of the AUC of the culture of the individual strains because, given the experimental setting, the single strain inoculum initially has twice the number of cells of a single strain than the number of cells that same strain starts with in a pair. The cross-feeding potency can be described by the following equation:
where i and j represent unique strains and n is the total number of strains. This value was used to build a heatmap of cross-feeding potency. We set the diagonal, corresponding to cultures of single strains, to zero. We ignored the diagonal since it marks experiments that started from an inoculum that had twice the number of cells from a single strain (relative to all co-culturing experiments that had two strains). We considered nteractions with a potency value greater than 2 positive and we used them to build an interaction network using the Circa software. To build a compact version of the same network, we pooled together all the strains that belong to the same functional group into a single node and plotted the graph with Cytoscape(Shannon et al., 2003). We then counted how many interactions exist between the nodes (functional groups) to calculate the interaction strength. We weighted each edge according to the following equation:
where a and b represent different functional groups, Inta,b represents the number of positive interactions between members of functional groups a and b, Sa represents the number of deletion strains belonging to functional group a, and Sb represents the number of deletion strains belonging to functional group b. In this way, every edge is weighted by the total number of possible interactions between the deletion strains of each group.
Temporal dynamics of synthetic consortia
As previously described for the genetic screen, we concerted raw reads to barcode counts using a custom MatLab (Mathworks) script. After that, we calculated the frequency of each strain in the community. We used the Shannon entropy (H) to quantify the diversity of each community. Shannon entropy is defined as:
where p(xi) is the proportion of strain i in the sample. We calculated the normalized entropy by dividing the entropy of each day by the entropy of the initial strain assembly (where all the strains were equally represented). We defined a proportion cutoff of 0.02 to estimate the number of dominant strains in a consortium at a given point in time. Strains appearing at lower proportions were still used for the normalized entropy calculation, but we not considered as dominant strains.
Supplementary Material
Supplementary table 1 – Screen results in conditioned media (fold-change and p-value)
Supplementary table 2 – List of hits from all screen conditions and their pathway affiliation
Supplementary table 3 – KEGG and GO Pathway enrichment in conditioned media
supplementary table 4 – Screen results in nutrient-rich media (fold-change and p-value)
Supplementary table 5 – KEGG and GO Pathway enrichment in nutrient-rich media
Genetic screens uncovered all E. coli knockouts benefiting from conditioned media.
Co-culture experiments identified potent cross-feeding auxotroph strain pairs.
Multi-day consortia experiments revealed persistent mutualistic collaborations.
Vitamins emerged as ideal shared goods that co-optimize growth and co-dependence.
Acknowledgements
We thank Dr. Hirotoda Mori from Nara Institute of Science and Technology in Japan for providing us the barcoded strain collection. We thank Hyun Youk for commenting on our manuscript. This work was supported by NIGMS (AM NIGMS GM133775).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Akimov Y, Bulanova D, Timonen S, Wennerberg K, Aittokallio T, 2020. Improved detection of differentially represented DNA barcodes for high-throughput clonal phenomics. Mol Syst Biol 16, e9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H, 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasche S, Kim Y, Mars RAT, Machado D, Maansson M, Kafkia E, Milanese A, Zeller G, Teusink B, Nielsen J, Benes V, Neves R, Sauer U, Patil KR, 2021. Metabolic cooperation and spatiotemporal niche partitioning in a kefir microbial community. Nat Microbiol 6, 196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K, Vowinckel J, Mülleder M, Malmsheimer S, Lawrence N, Calvani E, Miller-Fleming L, Alam MT, Christen S, Keller MA, Raiser M, 2015. Self-establishing communities enable cooperative metabolite exchange in a eukaryote. Elife 4, e09943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Douglass E, Dunn N, Good B, Harris NL, Lewis SE, Mungall CJ, Basu S, Chisholm RL, Dodson RJ, Hartline E, Fey P, Thomas PD, Albou LP, Ebert D, Kesling MJ, Mi H, Muruganujan A, Huang X, Poudel S, Mushayahama T, Hu JC, LaBonte SA, Siegele DA, Antonazzo G, Attrill H, Brown NH, Fexova S, Garapati P, Jones TEM, Marygold SJ, Millburn GH, Rey AJ, Trovisco V, Santos G. dos, Emmert DB, Falls K, Zhou P, Goodman JL, Strelets VB, Thurmond J, Courtot M, Osumi-Sutherland D, Parkinson H, Roncaglia P, Acencio ML, Kuiper M, Lægreid A, Logie C, Lovering RC, Huntley RP, Denny P, Campbell NH, Kramarz B, Acquaah V, Ahmad SH, Chen H, Rawson JH, Chibucos MC, Giglio M, Nadendla S, Tauber R, Duesbury MJ, Del-Toro N, Meldal BHM, Perfetto L, Porras P, Orchard S, Shrivastava A, Xie Z, Chang HY, Finn RD, Mitchell AL, Rawlings ND, Richardson L, Sangrador-Vegas A, Blake JA, Christie KR, Dolan ME, Drabkin HJ, Hill DP, Ni L, Sitnikov D, Harris MA, Oliver SG, Rutherford K, Wood V, Bahler J, Lock A, Bolton ER, Pons JD, Dwinell M, Hayman GT, Laulederkind SJF, Shimoyama M, Tutaj M, Wang S-J, D’Eustachio P, Matthews L, Balhoff JP, Aleksander SA, Binkley G, Dunn BL, Cherry JM, Engel SR, Gondwe F, Karra K, MacPherson KA, Miyasato SR, Nash RS, Ng PC, Sheppard TK, VP AS, Simison M, Skrzypek MS, Weng S, Wong ED, Feuermann M, Gaudet P, Bakker E, Berardini TZ, Reiser L, Subramaniam S, Huala E, Arighi C, Auchincloss A, Axelsen K, Argoud-Puy G, Bateman A, Bely B, Blatter M-C, Boutet E, Breuza L, Bridge A, Britto R, Bye-A-Jee H, Casals-Casas C, Coudert E, Estreicher A, Famiglietti L, Garmiri P, Georghiou G, Gos A, Gruaz-Gumowski N, Hatton-Ellis E, Hinz U, Hulo C, Ignatchenko A, Jungo F, Keller G, Laiho K, Lemercier P, Lieberherr D, Lussi Y, MacDougall A, Magrane M, Martin MJ, Masson P, Natale DA, Hyka-Nouspikel N, Pedruzzi I, Pichler K, Poux S, Rivoire C, Rodríguez-López M, Sawford T, Speretta E, Shypitsyna A, Stutz A, Sundaram S, Tognolli M, Tyagi N, Warner K, Zaru R, Wu C, Chan J, Cho J, Gao S, Grove C, Harrison MC, Howe K, Lee R, Mendel J, Muller H-M, Raciti D, Auken KV, Berriman M, Stein L, Sternberg PW, Howe D, Toro S, Westerfield M, 2018. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro S, Villas-Bôas SG, Ferreira EC, Rocha I, 2011. Metabolic footprint analysis of recombinant Escherichia coli strains during fed-batch fermentations. Mol Biosyst 7, 899–910. [DOI] [PubMed] [Google Scholar]
- Cavaliere M, Feng S, Soyer O, Jimenez JI, 2017. Cooperation in microbial communities and their biotechnological applications. Environ Microbiol 19, 2949–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway T, Creecy JP, Maddox SM, Grissom JE, Conkle TL, Shadid TM, Teramoto J, Miguel PS, Shimada T, Ishihama A, Mori H, Wanner BL, 2014. Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing. Mbio 5, e01442–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG, 2005. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. [DOI] [PubMed] [Google Scholar]
- Dolinšek J, Goldschmidt F, Johnson DR, 2016. Synthetic microbial ecology and the dynamic interplay between microbial genotypes. Fems Microbiol Rev 40, 961–979. [DOI] [PubMed] [Google Scholar]
- Fredrickson JK, 2015. Ecological communities by design. Science 348, 1425–1427. [DOI] [PubMed] [Google Scholar]
- Freilich S, Zarecki R, Eilam O, Segal ES, Henry CS, Kupiec M, Gophna U, Sharan R, Ruppin E, 2011. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun 2, 589. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, Bakkoury ME, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian K-D, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang C, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M, 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. [DOI] [PubMed] [Google Scholar]
- Giri S, Shitut S, Kost C, 2020. Harnessing ecological and evolutionary principles to guide the design of microbial production consortia. Curr Opin Biotech 62, 228–238. [DOI] [PubMed] [Google Scholar]
- Goldford JE, Lu N, Bajić D, Estrela S, Tikhonov M, Sanchez-Gorostiaga A, Segrè D, Mehta P, Sanchez A, 2018. Emergent simplicity in microbial community assembly. Sci New York N Y 361, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Consarnau L, Sachdeva R, Gifford SM, Cutter LS, Fuhrman JA, Sañudo-Wilhelmy SA, Moran MA, 2018. Mosaic patterns of B-vitamin synthesis and utilization in a natural marine microbial community. Environ Microbiol 20, 2809–2823. [DOI] [PubMed] [Google Scholar]
- Hays SG, Patrick WG, Ziesack M, Oxman N, Silver PA, 2015. Better together: engineering and application of microbial symbioses. Curr Opin Biotech 36, 40–49. [DOI] [PubMed] [Google Scholar]
- Jagmann N, Philipp B, 2014. Design of synthetic microbial communities for biotechnological production processes. J Biotechnol 184, 209–218. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28, 27 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner A, Park J, Williams A, Lin XN, 2012. A Programmable Escherichia coli Consortium via Tunable Symbiosis. Plos One 7, e34032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martínez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Peralta-Gil M, Subhraveti P, Velázquez-Ramírez DA, Weaver D, Collado-Vides J, Paulsen I, Karp PD, 2017. The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res 45, D543 D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSarre B, Deutschbauer AM, Love CE, McKinlay JB, 2020. Covert Cross-Feeding Revealed by Genome-Wide Analysis of Fitness Determinants in a Synthetic Bacterial Mutualism. Appl Environ Microb 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd CJ, King ZA, Sandberg TE, Hefner Y, Olson CA, Phaneuf PV, O’Brien EJ, Sanders JG, Salido RA, Sanders K, Brennan C, Humphrey G, Knight R, Feist AM, 2019. The genetic basis for adaptation of model-designed syntrophic co-cultures. Plos Comput Biol 15, e1006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louca S, Polz MF, Mazel F, Albright MBN, Huber JA, O’Connor MI, Ackermann M, Hahn AS, Srivastava DS, Crowe SA, Doebeli M, Parfrey LW, 2018. Function and functional redundancy in microbial systems. Nat Ecol Evol 2, 936–943. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ, 2009. GAGE: generally applicable gene set enrichment for pathway analysis. Bmc Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir S, Ravcheev D, Crécy-Lagard V. de, Thiele I, 2015. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Frontiers Genetics 6, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee MT, Collins JJ, Church GM, Wang HH, 2014. Syntrophic exchange in synthetic microbial communities. P Natl Acad Sci Usa 111, E2149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’guyen GQ, Jain M, Landry CR, Filteau M, 2020. Mapping Gene-Microbe Interactions: Insights from Functional Genomics Co-culture Experiments between Saccharomyces cerevisiae and Pseudomonas spp. Biorxiv 2020.06.01.127472 [Google Scholar]
- Paczia N, Nilgen A, Lehmann T, Gätgens J, Wiechert W, Noack S, 2012. Extensive exometabolome analysis reveals extended overflow metabolism in various microorganisms. Microb Cell Fact 11, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande S, Merker H, Bohl K, Reichelt M, Schuster S, Figueiredo L.F. de, Kaleta C, Kost C, 2014. Fitness and stability of obligate cross-feeding interactions that emerge upon gene loss in bacteria. Isme J 8, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandhal J, Noirel J, 2014. Synthetic microbial ecosystems for biotechnology. Biotechnol Lett 36, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Pierce EC, Morin M, Little JC, Liu RB, Tannous J, Keller NP, Pogliano K, Wolfe BE, Sanchez LM, Dutton RJ, 2021. Bacterial–fungal interactions revealed by genome-wide analysis of bacterial mutant fitness. Nat Microbiol 6, 87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarova O, Gabrielli N, Sévin DC, Mülleder M, Zirngibl K, Bulyha K, Andrejev S, Kafkia E, Typas A, Sauer U, Raiser M, Patil KR, 2017. Yeast Creates a Niche for Symbiotic Lactic Acid Bacteria through Nitrogen Overflow. Cell Syst 5, 345–357.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Zane GM, Kuehl JV, Melnyk RA, Wall JD, Deutschbauer AM, Arkin AP, 2018. Filling gaps in bacterial amino acid biosynthesis pathways with high-throughput genetics. Plos Genet 14, e1007147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roell GW, Zha J, Carr RR, Koffas MA, Fong SS, Tang YJ, 2019. Engineering microbial consortia by division of labor. Microb Cell Fact 18, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosener B, Sayin S, Oluoch PO, González APG, Mori H, Walhout AJ, Mitchell A, 2020. Evolved bacterial resistance against fluoropyrimidines can lower chemotherapy impact in the Caenorhabditis elegans host. Elife 9, e59831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth EC, Taga ME, 2014. Nutrient cross-feeding in the microbial world. Front Microbiol 5, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour JR, Amin SA, Raina J-B, Stocker R, 2017. Zooming in on the phycosphere: the ecological interface for phytoplankton–bacteria relationships. Nat Microbiol 2, 17065. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, 2003. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou W, Ram S, Vilar JMG, 2007. Synthetic cooperation in engineered yeast populations. Proc National Acad Sci 104, 1877 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Martin EC, Warnke I, Farquharson FM, Christodoulou M, Horgan G, Derrien M, Faurie J-M, Flint HJ, Duncan SH, Louis P, 2020. Vitamin Biosynthesis by Human Gut Butyrate-Producing Bacteria and Cross-Feeding in Synthetic Microbial Communities. Mbio 11, e00886–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommes M, Wang T, Zhao Q, Paschalidis IC, Segrè D, 2019. Designing Metabolic Division of Labor in Microbial Communities. Msystems 4, e00263–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SA, Diggle SP, Buckling A, Gardner A, Griffin AS, 2007. The Social Lives of Microbes. Annu Rev Ecol Evol Syst 38, 53–77. [Google Scholar]
- Wintermute EH, Silver PA, 2010. Emergent cooperation in microbial metabolism. Mol Syst Biol 6, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengler K, Zaramela LS, 2018. The social network of microorganisms — how auxotrophies shape complex communities. Nat Rev Microbiol 16, 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Reed JL, 2014. Adaptive Evolution of Synthetic Cooperating Communities Improves Growth Performance. Plos One 9, e108297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Qiao K, Edgar S, Stephanopoulos G, 2015. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat Biotechnol 33, 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziesack M, Gibson T, Oliver JKW, Shumaker AM, Hsu BB, Riglar DT, Giessen TW, DiBenedetto NV, Bry L, Way JC, Silver PA, Gerber GK, 2019. Engineered Interspecies Amino Acid Cross-Feeding Increases Population Evenness in a Synthetic Bacterial Consortium. Msystems 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomorrodi AR, Segrè D, 2015. Synthetic Ecology of Microbes: Mathematical Models and Applications. J Mol Biol 428, 837–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1 – Screen results in conditioned media (fold-change and p-value)
Supplementary table 2 – List of hits from all screen conditions and their pathway affiliation
Supplementary table 3 – KEGG and GO Pathway enrichment in conditioned media
supplementary table 4 – Screen results in nutrient-rich media (fold-change and p-value)
Supplementary table 5 – KEGG and GO Pathway enrichment in nutrient-rich media
Data Availability Statement
The sequencing datasets generated during this study have been deposited at the NCBI Sequence Read Archive (SRA) and are publicly available under the accession numbers: PRJNA727839 (auxotroph screen), PRJNA727852 (community composition in nutrient-poor media), and PRJNA727847 (community composition in nutrient-rich media).
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| E. coli barcoded deletion library | Laboratory of Hirotoda Mori | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| AMPure XP bead | Beckman Coulter | Cat#A63881 |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems | Cat#KK2602 |
| Difco M9 Minimal Media Salts, 5X | BD | Cat#248510 |
| Protein hydrolysate amicase | Sigma Aldrich | Cat#82514 |
| Tryptophan | Sigma Aldrich | Cat#T0254 |
| Uridine | Alfa Aesar | Cat#A15227-06 |
| Adenine sulfate | USBiological | Cat#A0865 |
| Biotin | Sigma Aldrich | Cat#B4639 |
| Niacin | Spectrum | Cat#NI100 |
| D-pantothenic acid | Sigma Aldrich | Cat#P5155 |
| Critical commercial assays | ||
| BioAnalyzer/Agilent High Sensitivity DNA Kit | Agilent | Cat# 5067-4626 |
| Qubit dsDNA high sensitivity assay | Thermo-fisher | Cat#Q32854 |
| Quick DNA Miniprep Plus Kit | Zymo | Cat#D4068 |
| Nextera XT Index Kit | Illumina | Cat#FC-131-1024 |
| MiniSeq High Output Reagent Kit, 75-cycles | Illumina | Cat#FC-420-1001 |
| MiniSeq High Output Reagent Kit, 150-cycles | Illumina | Cat#FC-420-1002 |
| MiSeq Reagent Kit v3, 150-cycles | Illumina | Cat#MS-102-3001 |
| Deposited data | ||
| Auxotroph screen | This study | PRJNA727839 |
| Assembly dynamics in nutrient-poor media | This study | PRJNA727852 |
| Assembly dynamics in nutrient-rich media | This study | PRJNA727847 |
| Pooled library in nutrient rich media | Rosener et al., 2020 | PRJNA645605 |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Oligonucleotides | ||
| Forward primer for barcoded library: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-(4-6xN)-TGTAGGCTGGAGCTGCTTCG 3' | Rosener et al., 2020 | N/A |
| Reverse primer for barcoded library: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-GCAAATATTATACGCAAGGCGACAAG 3’ | Rosener et al., 2020 | N/A |
| Recombinant DNA | ||
| Software and algorithms | ||
| Cystoscape | Shannon et al., 2003 | https://cytoscape.org |
| Matlab(R2020a) | Mathworks | https://www.mathworks.com/downloads/ |
| Circa | OMGenomics | https://omgenomics.com/circa/ |
| DEBRA, R package | Akimov et al., 2020 | https://github.com/YevhenAkimov/DEBRA |
| GAGE, R package | Luo et al., 2009 | https://bioconductor.org/packages/release/bioc/html/gage.html |
| DESeq2, R package | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Code to analyze data and generate plot | This study | DOI: 10.5281/zenodo.5106763 |
| Other | ||