

Abstract
Activation of different receptors that act by generating the common second messenger cyclic adenosine monophosphate (cAMP) can elicit distinct functional responses in cardiac myocytes. Selectively sequestering cAMP activity to discrete intracellular microdomains is considered essential for generating receptor-specific responses. The processes that control this aspect of compartmentalized cAMP signaling, however, are not completely clear. Over the years, technological innovations have provided critical breakthroughs in advancing our understanding of the mechanisms underlying cAMP compartmentation. Some of the factors identified include localized production of cAMP by differential distribution of receptors, localized breakdown of this second messenger by targeted distribution of phosphodiesterase enzymes, and limited diffusion of cAMP by protein kinase A (PKA)-dependent buffering or physically restricted barriers. The aim of this review is to provide a discussion of our current knowledge and highlight some of the gaps that still exist in the field of cAMP compartmentation in cardiac myocytes.
Keywords: cAMP compartmentation, cardiac myocytes, beta-adrenergic receptor, prostaglandin receptor, phosphodiesterase, protein kinase A, restricted spaces, buffering
1. INTRODUCTION
A variety of G-protein coupled receptors (GPCRs) act by stimulating cAMP production in virtually every cell in our bodies. This ubiquitous second messenger is involved in regulating a multitude of cellular functions, including metabolism, proliferation, development, and gene transcription. In cardiac myocytes, cAMP plays a particularly important role in regulating excitation-contraction coupling. Yet, despite the fact that multiple GPCRs are able to stimulate cAMP production in cardiac myocytes, not all produce the same functional responses. This can be attributed to the fact that this diffusible second messenger does not move freely throughout the cell.
The β-adrenergic receptor (βAR) is the classic example of a GPCR that elicits responses through the production of cAMP. Following agonist binding, the receptor acts via the stimulatory G protein Gs, which then elicits production of cAMP by adenylyl cyclase (AC). In cardiac myocytes, cAMP then mediates downstream functional responses by activating effector proteins that include protein kinase A (PKA), exchange protein activated by cAMP (Epac), and cyclic nucleotide gated ion channels. This sequence of events is common to a number of different GPCRs, but the resulting functional response can vary. For example, it was recognized early on that βARs as well as E-type prostaglandin receptors (EPRs) act via production of cAMP, but only βAR stimulation led to an increase in cardiac contractility [1–4]. To explain this apparent paradox, it was proposed that cAMP signaling must be compartmentalized, with different receptors producing spatially segregated pools of cAMP in discrete subcellular locations. Initial studies divided these pools into soluble and particulate fractions of cell or tissue homogenates. Consistent with this idea, βARs as well as EPRs led to generation of cAMP in soluble fractions, while only βARs stimulated cAMP production in particulate fractions. But how this related to what happens in the intact cell was unclear.
Differences in the cAMP-dependent responses produced by different subtypes of βARs is another example of compartmentalized cAMP signaling in cardiac myocytes. In ventricular myocytes, β1AR stimulation leads to the PKA-dependent phosphorylation of multiple target proteins, including L-type Ca2+ channels (LTCCs) and phospholamban (PLB). However, β2ARs produce a cAMP-dependent response that is restricted to the regulation of LTCCs [5–7]. This difference can explain how both β1 and β2ARs can produce an increase in contractility (a positive inotropic response). Yet, only β1ARs consistently produce an increase in the rate of relaxation (a positive lusitropic response). The difference in responses correlates with the location of the target proteins being regulated. LTCCs are typically found in the plasma membrane of the transverse (t) tubules, where they are in close proximity to ryanodine receptors (RyRs) found in the junctional sarcoplasmic reticulum (SR) [8] (see figure 1B). This junctional membrane complex forms a restricted space called the dyadic cleft. PLB, on the other hand, is found outside of the dyadic cleft in the non-junctional SR (see figure 1C). Phosphorylation of PLB removes the inhibitory effect it has on the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), which is responsible for pumping Ca2+ back into the SR following contraction. By pumping more Ca2+ back into the SR, this further enhances the positive inotropic effect of β1AR stimulation. It can also explain the ability of β1ARs to produce a positive lusitropic effect.
Figure 1.
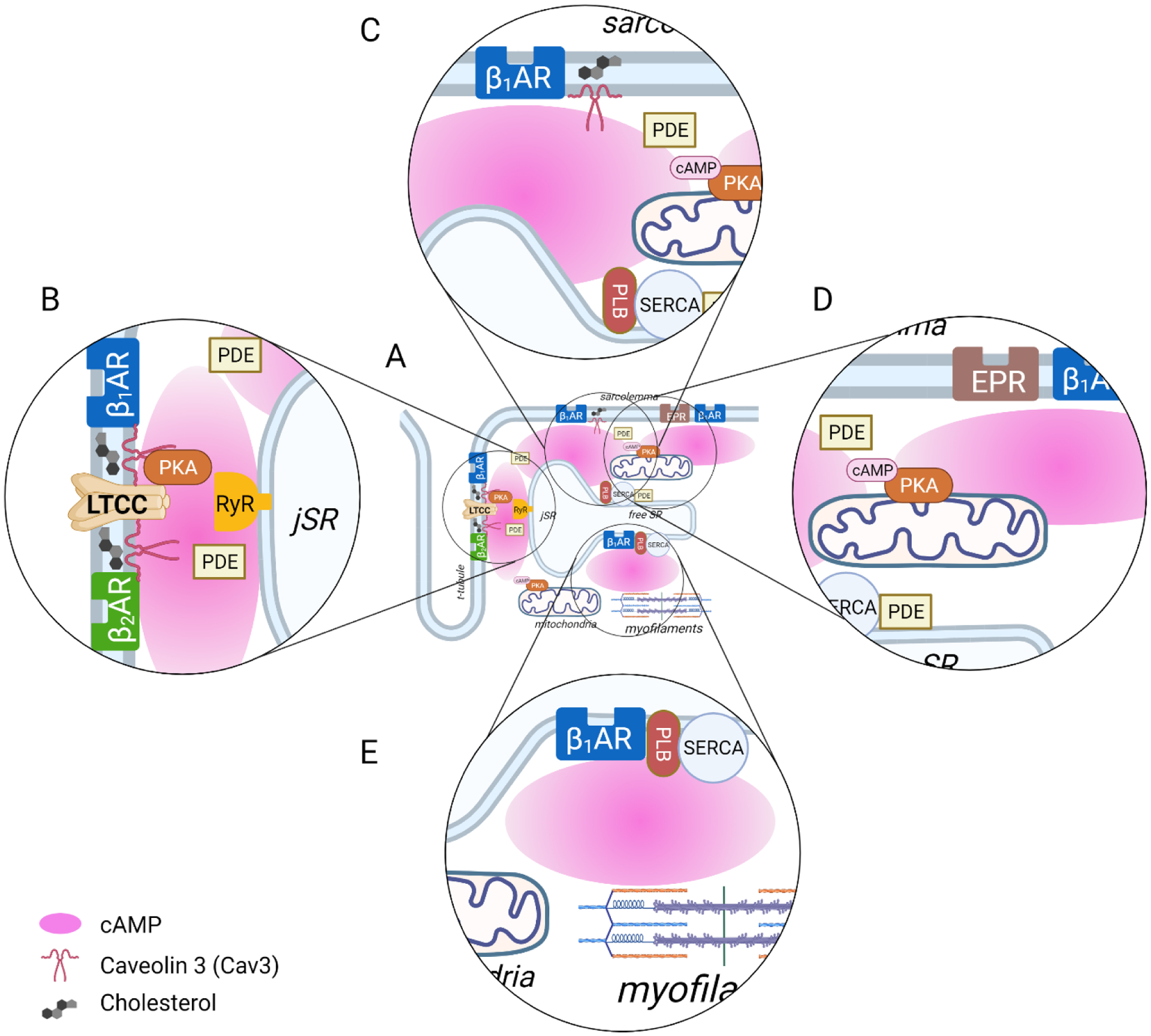
Compartmentalized cAMP signaling by different receptors in adult ventricular myocytes. (A) Distribution of receptors to specific locations, along with targeted PDE activity, buffering of cAMP by protein kinase A (PKA), and physical barriers to diffusion allows for generation of local pools of cAMP. While β1ARs are expressed throughout the cell, β2ARs are confined to the plasma membrane lining the transverse tubules. β2ARs are also found primarily in caveolar lipid raft domains of the plasma membrane; β1ARs can be found in caveolar as well as non-caveolar membrane domains; and EPRs are found primarily in non-caveolar membrane domains. (B) β1ARs and β2ARs form signaling complexes with L-type Ca2+ channels (LTCCs), which are found in dyadic clefts. Both of these receptors produce cAMP that can regulate LTCCs as well as ryanodine receptors (RyRs) within this restricted space. (C) β1ARs are also able to contribute to a distinct pool of cAMP that regulates the activity of the sarco/endoplasmic reticulum ATPase (SERCA) via PKA-dependent phosphorylation of phospholamban (PLB) (D) E type prostaglandin receptors (EPRs) contribute to a pool of cAMP that does not affect PKA regulation of LTCCs or PLB, which are involved in excitation-contraction coupling, although it can produce cardioprotective effects. (E) An intracellular pool of β1ARs found in the SR have also been reported to produce a localized response that leads to phosphorylation of PLB and regulation of SERCA.
A-kinase anchoring proteins (AKAPs) are one important means of ensuring that receptor activation of cAMP production leads to regulation of the correct target protein by PKA, especially in light of evidence that cAMP binding to and activation of PKA does not necessarily result in complete dissociation of catalytic and regulatory subunits of the holoenzyme [9]. Consistent with this idea, disrupting AKAP signaling complexes has been shown in a number of studies to prevent cAMP-dependent regulation of many different PKA-dependent responses [10, 11]. However, AKAPs alone are not sufficient to explain compartmentation of cAMP-dependent signaling. If cAMP can diffuse freely throughout the cell, any receptor capable of stimulating its production would be expected to elicit identical responses due to uniform activation of PKA everywhere, but that is not what happens. In order to produce receptor-specific responses, there must be some way of generating localized pools of cAMP.
For many years, the ability to investigate mechanisms contributing to cAMP compartmentation was limited by the methods available for measuring cAMP activity. This problem was solved with the generation of biosensors that provide a readout of changes in cAMP activity in intact cells. The first such biosensor utilized the principle of fluorescence resonance energy transfer (FRET) [12]. This probe, called FlCRhR, was constructed using the catalytic and regulatory subunits of PKA labeled with donor (fluorescein) and acceptor (rhodamine) fluorophores, respectively. With FRET-based probes, excitation of the donor leads to transfer of energy to the acceptor when the two fluorophores are in close proximity. In this situation, the donor fluorescence is quenched and the acceptor fluorescence is sensitized. When cAMP binds the regulatory subunit, the conformational changes result in a loss of energy transfer that alters the donor-acceptor fluorescence ratio. Thus, these probes can provide a readout of changes in cAMP activity. However, the FlCRhR probe was difficult to synthesize, and it relied on microinjection or dialysis via a patch pipette in order to introduce it into cells [13], limiting its practical utility.
The advent of genetically encoded biosensors made it possible to express probes in a wide range of cell types with relative ease, which opened a whole new era for investigating compartmentalized cAMP signaling. One of the first such probes was actually a modified version of the cyclic nucleotide gated (CNG) ion channel [14]. This probe could be used in cells not expressing endogenous CNG channels, such as adult ventricular myocytes. Changes in cAMP near the plasma membrane could then be detected using the patch clamp technique to measure the membrane current generated when this probe was activated [15–17].
More recently, a number of genetically encoded FRET-based biosensors have been developed using different cAMP binding proteins. These include: PKA-based probes [18, 19], as well as cAMP binding domains from different isoforms of Epac (Epac1-camps and Epac2-camps) [20] and the hyperpolarization activated K+ channel (HCN2-camps) [21]. Some of these, like the type II PKA based biosensor, are inherently targeted to specific subcellular locations through interactions with AKAPs, while others, such as Epac1-camps, Epac2-camps, and HCN-camps, are expressed uniformly throughout the cytoplasm of the cell. However, a number of Epac-based variants have been generated with targeting sequences that enable them to respond to changes in cAMP occurring in specific subcellular microdomains [22]. In addition to FRET-based biosensors that respond directly to cAMP binding, there are also A-kinase activity reporter (AKAR) probes, which respond indirectly to cAMP. They contain a consensus sequence, which when phosphorylated by PKA, undergoes a conformational change affecting FRET donor and acceptor interactions [23].
The present review focuses on how these tools, together with other newly developed approaches, have been used to investigate cAMP compartmentation in cardiac myocytes. Most previous reviews on this subject have focused on the role of localized degradation of cAMP by phosphodiesterases (PDEs). The present review includes an updated discussion of this topic, in addition to an examination of the roles played by localized production of cAMP by different GPCRs, restricted diffusion due to PKA buffering and physically restricted spaces. Finally, we include a brief discussion of how some of these factors are affected in certain disease states.
2. LOCALIZED PRODUCTION BY GPCRs
One mechanism contributing to the generation of receptor-specific pools of cAMP in distinct intracellular microdomains involves segregating receptors into discrete subcellular locations. Some receptors are commonly associated with caveolar signaling complexes in the plasma membrane, others are specifically excluded from these membrane domains, and then there are receptors found in both domains [24–26]. Caveolae are a subset of lipid rafts enriched in cholesterol and sphingolipids and defined by the presence of caveolin, a multifunctional protein, of which there are three isoforms, with caveolin 3 (Cav3) being the predominant subtype present in cardiac myocytes [26].
β1-Adrenergic Receptors
β1ARs represent about 80% of the βARs in cardiac myocytes of most species [27]. These receptors are associated with both caveolar and non-caveolar signaling microdomains (see figure 1B–D) [28–32]. The widespread distribution of β1ARs is associated with their ability to produce a global cAMP response that can be detected throughout the entire cell using FRET-based biosensors [21]. Despite producing a uniform increase in cAMP throughout the cell, there is evidence that β1ARs found in different membrane domains produce compartmentalized responses. For instance, β1ARs associated with caveolar/lipid raft domains appear to modulate myocyte contraction [32]. This conclusion is supported by the observation that disrupting caveolar/lipid raft domains by treatment with cholesterol-depleting agents such as methyl-β-cyclodextrin (MβCD) enhances β1AR-mediated effects. This includes increases in LTCC activity, PLB phosphorylation, intracellular Ca2+ transients, and cell contraction. Consistent with these results, cholesterol depletion was also found to increase the generation of cAMP detected by the type II PKA-based biosensor, which is known to be targeted to the caveolar domains [30, 31]. In contrast, cholesterol depletion did not induce any changes in global cAMP responses detected by the non-targeted Epac2-camps probe. The reason for an increase in cAMP production within caveolar regions following cholesterol depletion is still unclear. However, Cav3 is known to have an inhibitory effect on the proteins that it interacts with [33]. Therefore, it is likely that disruption of caveolae eliminates this inhibitory effect, thereby enhancing AC activity.
Although GPCRs are most often thought to reside in the plasma membrane, recent studies have also shown that intracellular β1ARs also play an important role in producing compartmentalized cAMP responses. This includes local production of cAMP by β1ARs found in the Golgi. Those receptors were found to be responsible for the Epac-dependent activation of phospholipase Cε and hydrolysis of phosphatidylinositol-4-phosphate, which then contributes to the generation of hypertrophic responses [34]. β1ARs found in the SR have also been reported to generate the local production of cAMP that leads to PKA-dependent phosphorylation of PLB and regulation of SERCA-dependent Ca2+ uptake (see figure 1E) [35].
β2-Adrenergic Receptors
β2ARs, which make up most of the remaining βARs found in cardiac myocytes, also act through the production of cAMP. However, as discussed above, the responses they produce are quite different from those elicited by β1ARs. Some investigators attribute these differences to the fact that β2ARs couple not only to Gs, but also to Gi signaling pathways [6, 36, 37]. The other major distinction between the βAR subtypes is in their subcellular distribution. β2ARs are found exclusively in caveolar/lipd raft domains in cardiac myocytes (see figure 1B) [28–30, 38–42]. In addition, the βAR subtypes also vary with respect to their exact placement within a cell.
Adult ventricular myocytes possess a complex intracellular architecture marked by the presence of an extensive t-tubular network that enables the plasma membrane to access the cell interior, facilitating excitation-contraction (EC) coupling. Nikolaev el al. employed scanning ion conductance microscopy to selectively activate β1 and β2ARs at specific cellular locations and measure the resulting response detected by a FRET-based biosensor [43]. With this powerful combination of techniques, the authors demonstrated that β2ARs produced cAMP responses that were confined to t-tubules, while β1ARs produced responses detected across all areas of the plasma membrane, not just the t-tubules. Both β1 and β2ARs have been reported to form signaling complexes with LTCCs [30, 31], which are primarily located in the t-tubules of ventricular myocytes, where they are associated with dyadic clefts [8]. This is consistent with the observation that βAR regulation of LTCC activity is largely limited to t-tubules [44]. It also suggests that βAR production of cAMP associated with t-tubules is most likely occurring within dyadic clefts.
The inability of β2ARs to produce a cAMP response that extends beyond LTCC regulation has been attributed to the fact that these receptors also couple to Gi signaling pathways [6, 7, 36, 45]. Localized responses to β2AR activity may also be regulated by phosphodiesterases (PDEs), enzymes that break down cAMP [46, 47]. Accordingly, a recent study found that activation of β1, but not β2ARs, was able to generate cAMP detected by a FRET-based biosensor targeted to the non-junctional SR, where PLB and SERCA are located. However, β2AR production of cAMP was detected in those distant locations following PDE2 and PDE3 inhibition [48]. PDE4 activity has also been shown to prevent β2AR production of cAMP from reaching RyRs just nanometers away, on the opposite side of the dyadic cleft [49]. The redistribution of PDE4 activity away from this location in cardiac hypertrophy and heart failure results in hyperphosphorylation of RyRs, causing an increase in spontaneous Ca2+ release from the SR and subsequent generation of ventricular arrhythmias.
β3-Adrenergic Receptors
β3ARs are expressed at very low levels in cardiac myocytes of some species, including humans, where they are associated with nitric oxide-dependent production of cGMP and inhibition of contractility [50]. More recently, it has been shown that β3ARs are localized to the t-tubules, where production of cGMP is then able to regulate cAMP levels by stimulating PDE2 activity [51].
Prostaglandin Receptors
Cardiac myocytes express two EPR subtypes, EP2 and EP4, that couple to Gs and stimulate cAMP production [29, 32, 52]. However, the cAMP produced by EPRs has been found to be highly sequestered. Warrier et al. compared cAMP responses detected using a FRET-based biosensors targeted to type II PKA and one expressed globally throughout the cytosol in guinea pig ventricular myocytes [53]. In that study EPR activation produced a cAMP response that could be detected by the globally expressed probe, but not the PKA-targeted probe. They also found that EPR stimulation failed to regulate LTCC activity in these cells, which is consistent with the idea that type II PKA regulates the activity of these channels in cardiac myocytes.
Unlike the results obtained in guinea pig ventricular myocytes, EPR activation was found to elicit cAMP responses detected by the same two probes in rat myocytes, but there were still no changes in LTCC activity [32]. However, disruption of caveolar/lipid raft domains by cholesterol depletion, which enhanced β1AR responses, had no effect on EPR-mediated responses. These results are consistent with the findings that both EP2 and EP4 receptor subtypes are excluded from caveolar membrane fractions [29, 32, 52]. Consistent with this, EPR stimulation was found to produce more pronounced cAMP responses detected by a FRET-based probe targeted specifically to non-raft domains of the plasma membrane [54]. Taken together, these results suggest that even though EPR activation elicits cAMP responses, the targeting of these receptors to non-caveolar membrane domains results in the production of cAMP in subcellular locations that does not regulate excitation or contraction (see figure 1D). Cardiac EPRs do, however, offer protection from ischemia/reperfusion injury in a cAMP-dependent manner [55, 56].
Muscarinic receptors
M2 muscarinic receptors (M2Rs) are also involved in regulation of cAMP production in cardiac myocytes. These receptors regulate AC activity through the inhibitory G protein Gi. However, cAMP responses elicited by M2R exhibit a complex biphasic pattern that involves stimulation as well as inhibition of cAMP production [57]. M2R activation produces a rapid inhibition of cAMP followed by a rebound increase in cAMP upon washout of the agonist [58, 59]. Differences in temporal properties of the stimulatory and inhibitory effects have been suggested to explain this behavior. The inhibitory response turns on and off rapidly. It is also the dominant response observed during receptor activation. However, upon agonist washout, the inhibitory effect turns off rapidly, revealing the stimulatory effect, which turns off more slowly. This type of response may elicit certain types of arrhythmogenic behaviors associated with dynamic changes in parasympathetic tone [60].
An explanation for this type of complex behavior has been attributed to cAMP compartmentation. Cardiac myocytes express different AC subtypes: AC5 and 6 as well as AC4 and 7 [61, 62]. The inhibitory effect can be explained by Gi inhibition of AC5/6 activity, while the stimulatory effect may be due to activation of AC4/7 [57, 59, 63]. While AC5/6 are localized to caveolae, AC4/7 are excluded from those membrane domains [40, 64]. Iancu et al, developed a computational model of compartmentalized cAMP signaling incorporating both M2R and β1AR signaling mechanisms [65, 66]. The modeling results predicted that M2Rs can generate complex cAMP responses by inhibiting AC5/6 in caveolar domains and stimulating AC4/7 in non-caveolar of the plasma membrane in cardiac myocytes.
3. LOCALIZED DEGRADATION BY PHOSPHODIESTERASES
If the production of cAMP is localized, it stands to reason there must be some mechanism to prevent it from freely diffusing throughout the cell in order to preserve targeted functional responses. PDE enzymes, which breakdown cAMP, are believed to play a central role in this process (see figure 1) [67, 68]. At least 4 different PDE families: PDE1, PDE2, PDE3, and PDE4, are found in cardiac myocytes [69]. PDE1 and PDE2 can hydrolyze both cAMP and cGMP, while PDE3 preferentially hydrolyzes cAMP and PDE4 is specific for cAMP. These PDEs also vary in how they are regulated. PDE2 can be allosterically stimulated by cGMP, PDE3 can be competitively inhibited by cGMP, and both PDE3 and PDE4 can be activated by PKA-dependent phosphorylation Selective inhibitors have been used to identify the contribution of the different PDE isozymes to cAMP compartmentation. The exception to this is PDE1, for which the availability of selective inhibitors is limited [70, 71].
PDE2
PDE2 activity is mainly associated with the membrane fractions of cardiac myocytes [72, 73]. Accordingly, the greatest changes in cAMP activity observed following inhibition of PDE2 have been detected by FRET-based biosensors targeted specifically to different regions of the plasma membrane [54]. Regulation of PDE2 by cGMP has also been shown to play an important role in affecting localized cAMP production associated with LTCCs as well as RyRs [74, 75]. In addition, local cAMP signaling involving PDE2 has been shown to regulate mitochondrial function in cardiac myocytes [76]. Furthermore, PDE2-induced cAMP-dependent responses play a significant role in regulating cardiac hypertrophy [77]. Consistent with this hypothesis, PDE2 activity was found to be enhanced in cardiac hypertrophy [78, 79], and the overexpression of PDE2 offered protection against norepinephrine-induced hypertrophy [79]. These studies suggest that the localized control of cAMP microdomains due to PDE2 activity may have therapeutic potential [77, 80]. Additionally, a recent report found evidence suggesting that PDE2 inhibition may enhance cAMP activity associated with PLB/SERCA, which may have a clinical application in augmenting cardiac relaxation in certain types of heart failure [48].
PDE3
Depending on the animal species, PDE3 exhibits a more diverse distribution pattern with activity found in both membrane and soluble fractions [81–83]. Accordingly, inhibition of PDE3 activity led to detection of cAMP responses by FRET-based biosensors that were targeted to sarcolemma as well as bulk cytosol in cardiac myocytes [54]. In neonatal rat ventricular myocytes, PDE3 activity was more prominently confined to cytosolic regions that are occupied by the type I regulatory subunits of PKA [75]. The authors also found that cGMP-mediated inhibition of PDE3 activity modulates localized cAMP responses in these domains.
PDE3 can be divided into two subfamilies, PDE3A and PDE3B. While PDE3A has been shown to be the major subtype found in hearts from most species, both PDE3A and PDE3B are expressed in murine cardiac myocytes [17, 84–86]. In human heart, alternative splicing and post-transcriptional processing generate 3 isoforms of PDE3A [84, 87]. These isoforms exhibit differences in the N-terminus sequence, which has been shown to determine not only the subcellular localization, but also the feedback regulation of these enzymes by PKA or PKB phosphorylation [84, 87].
In mouse heart, knockdown of PI3Kγ was found to increase cAMP levels and enhance contractility [88]. Consistent with these observations, a later study found that PI3Kγ stimulates the activity of PDE3B in a kinase-independent manner [86]. This study found that PI3Kγ acts as an anchoring protein that recruits PDE3B, but not PDE3A, to specific locations in myocytes. In a subsequent study, the authors found that PI3Kγ functions as an AKAP that forms a signaling complex with type II regulatory subunit of PKA and PDE3B [89]. This study demonstrated that localized cAMP levels were regulated by the PKA-dependent activation of PDE3B in cardiac myocytes. However, an ensuing study found that the knockdown of PDE3B did not alter cardiac contractility in the mouse [90]. The authors of that study showed an increase in intracellular Ca2+ transient amplitude as well as SR Ca2 content in ventricular myocytes from PDE3A-deficient mice. The authors’ finding that PDE3A forms a signaling complex with PLB and SERCA2a could explain these results. No change in LTCC currents was apparent in myocytes from PDE3A knockout mice suggesting that the PDE3A may represent the predominant isoform in most species and its activity is preferentially located close to SERCA2a and PLB within non-junctional SR domain.
While PDE3 may selectively regulate cAMP around PLB and SERCA under basal conditions, others have found evidence for PDE3-dependent regulation of LTCC activity in ventricular myocytes following βAR stimulation [91]. Notably, studies that monitored changes in βAR stimulation of cAMP responses within regions associated with the type II subunit of PKA or bulk cytosol found that PDE3 activity may not be as important as other PDE isoforms, at least in adult mouse and neonatal rat myocytes [21, 92].
PDE4
Several studies have shown that inhibition of basal PDE4 activity leads to an increase in cAMP, especially in membrane and type II PKA-associated microdomains [54, 92, 93]. However, inhibition of PDE4 alone has not been found to alter downstream functional effects such as LTCC activity in resting cells [16, 93, 94]. Following βAR stimulation, on the other hand, the role of PDE4 in mediating cAMP dependent responses becomes markedly more prominent, suggesting that PDE4 is involved in regulating receptor mediated cAMP-dependent effects, including LTCC activity [16, 21].
While there are four subfamilies of PDE4, only three: PDE4A, PDE4B, and PDE4D are believed to be expressed in cardiac tissue [94–96]. PDE4D and PDE4B together have been shown to provide about 90% of the total PDE activity in neonatal rat cardiac myocytes [92]. Alternative splicing and the use of different promoters results in an even greater number of isoforms [97–99]. Diversity in the N-terminal domains is responsible for targeted expression of the PDE4 variants to different microdomains [97, 100]. However, due to the high sequence homology of the catalytic site among the different PDE4 isoforms, subtype-selective pharmacological inhibitors are not readily available. To overcome this limitation, transgenic mice have been used to examine the role of specific PDE4 isoforms.
In order to distinguish between the roles of PDE4B and PDE4D in regulating compartmentalized cAMP signaling, Leroy et al. generated PDE4B or PDE4D knockout mice [101]. The authors found that both are part of a signaling complex that includes the LTCC. However, only knockdown of PDE4B, but not PDE4D, increased the Ca2+ current following βAR stimulation. Moreover, PDE4B knockdown also augmented the susceptibility to arrhythmias in mice.
The role of PDE4B in regulating receptor-mediated compartmentalized cAMP responses was further characterized in a study using FRET-based biosensors and biochemical methods [102]. Upon β1AR stimulation, cAMP responses were found to be enhanced specifically in plasma membrane domains in cardiac myocytes obtained from PDE4B-deficient neonatal cardiac myocytes. No change in cAMP responses were seen in the bulk cytosolic compartment. It is likely that the rise in membrane-associated cAMP was localized to the dyadic cleft, since the only proteins resident within this region, LTCCs and RyRs, were phosphorylated. Phosphorylation of TnI and PLB remained unchanged. These effects appear to be specific for β1AR, since the authors found no difference in cAMP responses detected in bulk cytosolic or membrane domains following activation of β2AR or EPR between the wildtype and PDE4B knockdown myocytes.
PDE4D3 has been shown to be a component of a macromolecular signaling complex that includes the RyR in mouse heart [103]. This correlates with the finding that, following βAR stimulation, cAMP levels were significantly higher only in the localized regions associated with the Z lines, but not bulk cytosol, in PDE4D3 knockout mice. Consistent with this finding, RyR2s were found to be hyper-phosphorylated, which in turn reduced their interaction with calstabin2, increasing the leakage of Ca2+ and the frequency of arrhythmias. These findings may provide a mechanistic explanation to the observation that there is a down regulation of PDE4D3 and an increased incidence of arrhythmias and sudden cardiac death in patients with heart failure [103].
PDE4D5 has been shown to be involved in regulating hypertrophic responses induced by chronic βAR stimulation in neonatal mouse cardiac myocytes [104]. In that study, PDE4D5 was reported to directly interact with HSP20, a small heat shock protein known to have cardioprotective properties. This close association afforded the regulation of HSP20 by cAMP/PKA-mediated phosphorylation. Using targeted FRET-based biosensors, pharmacologic inhibition of PDE4 was found to elicit a greater rise in cAMP levels in domains associated with HSP20 as compared to the bulk cytosol.
PDE4D5 also plays a role in regulating β2AR signaling. Following activation, these receptors can undergo phosphorylation by G-protein coupled receptor kinases (GRKs). This results in the binding of β-arrestin and the recruitment of PDE4D5 [46, 47], which can downregulate cAMP activity near the receptor. This is supported by the fact that inhibition of PDE4D5 activity selectively enhances PKA phosphorylation of the receptor, facilitating its ability to couple to Gi inhibition of AC as well as activation of ERK1/2 signaling [105]. Inhibition of GRK2 phosphorylation of the β2AR has also been shown to block PDE4D5 (and PDE4D3) association with the receptor, facilitating its ability to regulate myocyte contraction [106]. This is different from the β1AR, which is associated with PDE4D8 under baseline conditions, but then dissociates following receptor activation [107].
PDE activity plays an essential role in regulating the compartmentalized cAMP signaling in cardiac myocytes. PDEs are often viewed as functional barriers that prevent cAMP from moving between microdomains [4]. However, experimental evidence indicates that cAMP concentrations may be quite high throughout most of the cell, even under baseline conditions [54, 108, 109]. Under these circumstance, PDEs may actually act as “sinks” that deplete cAMP in localized regions [65, 110, 111]. However, PDE activity by itself may not be sufficient to explain cAMP compartmentation [112]. Because the turnover rate for PDE hydrolysis of cAMP is slow, assuming that cAMP moves at a rate equal to free diffusion, the amount of PDE activity found in a typical myocyte is not sufficient to prevent cAMP from moving unimpeded throughout the cell [110]. These results suggest that PDEs can only generate compartments if the rate of cAMP diffusion is sufficiently restricted by some other mechanism(s).
4. LIMITED DIFFUSION
PKA Buffering
The actual diffusion coefficient for cAMP in cardiac myocyte has been estimated using raster image correlation spectroscopy (RICS). RICS is a technique based on the principles of fluorescence correlation spectroscopy, which calculates diffusion coefficients from the fluctuations in fluorescence intensity within small confocal volumes [113]. Using the fluorescently-labeled cAMP derivative, 8-[Pharos-450]-cAMP (φ450-cAMP) [114], it was determined that the diffusion coefficient is approximately 10 μm2/s. This is markedly slower than the rate of free diffusion predicted for a molecule that size, which is 300 μm2/s. The slow rate of diffusion could not be ascribed to an effect of PDE activity, since φ450-cAMP is resistant to hydrolysis [115]. However, addition of the fluorophore did not alter its affinity for PKA binding.
PKA is believed to be the primary effector for the actions of cAMP in cardiac myocytes. This kinase exists a heterotetrameric complex with two catalytic and two regulatory subunits [116]. Binding of two cAMP molecules to each regulatory subunit leads to activation of the catalytic subunits, which then regulate the activity of downstream proteins through phosphorylation. The fact that the regulatory subunits of PKA bind cAMP raises the possibility that they may also function as a buffer for cAMP, slowing its diffusion throughout the cell (see figure 1). In fact, previous computational modeling studies have predicted such a scenario [14, 112].
The idea that buffering by PKA could explain the slow rate of diffusion measured by RICS was supported by the images of cells loaded with φ450-cAMP. It displayed a nonuniform, highly localized fluorescence pattern, suggesting that φ450-cAMP was not diffusing freely throughout the cell, but instead was binding to something. The fluorescence pattern was actually consistent with the pattern for distribution of mitochondria, which occupy ≥30% of the volume of a cardiac myocyte, and φ450-cAMP co-localized with a fluorescent label for mitochondria. Labeled cAMP also co-localized with a fluorescently labeled type II regulatory subunit of PKA, and this pattern of fluorescence was disrupted by treating the cells with Ht31, a peptide that interferes with the interaction between the type II regulatory subunit of PKA and A kinase anchoring proteins (AKAPs). The Ht31 peptide also increased the φ450-cAMP diffusion coefficient measured using RICS. This suggest the rate of cAMP diffusion in cardiac myocytes is slowed due to the buffering effect of binding and unbinding to PKA regulatory subunits anchored to the outer membrane of mitochondria. It has recently been determined that there are at least three different mitochondrial AKAPs involved, D-AKAP1, D-AKAP2, and ACBD3 [117]. Similar diffusion coefficients were also found in morphologically simpler HEK293 cells, suggesting that this behavior is not unique to cardiac myocytes [114]. However, Bock et al. [118] have argued that the slow movement of cAMP in HEK293 cells may not involve PKA buffering.
Consistent with the idea that AKAPs and PKA buffering contribute to cAMP compartmentation, treatment of cardiac myocytes with the Ht31 peptide has also been reported to allow β2AR production of cAMP to propagate throughout the cell [43]. The cAMP buffering capacity of PKA in cardiac myocytes has been estimated at about 1.2 μM [119, 120]. This is similar to estimates of the basal concentration of cAMP present in these cells [108], and would be consistent with previous studies suggesting that most cAMP is bound to PKA under basal conditions [119, 121]. It is also interesting to note that a recent study reported that the ratio of regulatory to catalytic subunits is 6:1 in cardiac myocytes [122]. This suggests the possibility that the excess regulatory subunits may be acting as a buffer to limit the free diffusion of cAMP throughout the cell.
Physically Restricted Spaces
Electron microscopy reveals that intracellular structures, including myofilament proteins, SR, and mitochondria, occupy as much as 90% or more of the volume of an adult ventricular myocyte [123]. This implies that there is very little free space for the movement of solutes such as cAMP. This is particularly true in certain locations such as the dyadic cleft (figure 1). The question then is whether or not these restricted spaces contribute to cAMP compartmentation. A recent in silico study took advantage of cryo-transmission electron microscopy images to generate a realistic 3D framework in order to explore this question as it relates specifically to the dyadic cleft [110]. Besides its critical role in EC coupling, the dyadic cleft is also a locus of cAMP synthesis by βARs. In this model, structures such as t-tubules, the SR, and mitochondria acted as impenetrable barriers to cAMP, limiting its free space for diffusion. When physiologically relevant concentrations of PDE activity were used to create a “barrier” to cAMP movement into and out of the dyadic cleft, the model predicted that cAMP gradients could be generated within this confined space, but only when the diffusion coefficient was slowed due to values previously attributed to the effects of PKA buffering (10 μm2/s).
The complexity of the arrangement of various structures such as mitochondria, t-tubules, myofilaments and the SR, allows formation of other restricted spaces within cardiac myocytes as well. Similarly, it is likely that the same principles that limit the movement of cAMP within the dyadic cleft also apply to other areas of the cell. In a recent study the mobility of cAMP was measured in cardiac myocytes expressing FRET-based cAMP biosensor [124]. Use of a micro-perfusion system allowed the authors to apply a βAR agonist to one-half of the cell, while measuring the time course of changes in cAMP activity in the other half. They fit the results with a diffusion-reaction equation taking into account the predicted rates of cAMP synthesis and degradation. From this, they estimated the diffusion coefficient of cAMP to be about 35 μm2/sec. Consistent with the study by Agarwal et al. [114], this value is much slower than the predicted rate of free diffusion. However, the reason for slower movement was ascribed to the intracellular tortuosity created by the complex internal structure in cardiac myocytes rather than the buffering due to PKA binding. Using fluorescence recovery after photobleaching (FRAP) technique, this study found that the movement of fluorescein-conjugated cAMP, a fluorescent analog of cAMP, was 3–4 times higher in neonatal as compared to adult cardiac myocytes. The authors argued that the reason is due to lower structural tortuosity/complexity because of fewer mitochondria in neonatal cells. Direct experimental evidence in support of this conclusion awaits further research.
5. DISEASE INDUCED CHANGES IN cAMP COMPARTMENTATION
A number of changes in compartmentalized cAMP responses have been associated with disease states such as cardiac hypertrophy and heart failure. This includes factors affecting the localized production of cAMP. Nikolaev et al [43] found that β2AR production of cAMP changed from being specific to t-tubules regions under normal conditions, to producing changes in cAMP that could be detected in all areas of the cell. In addition, changes in AC activity have been associated with certain disease states. Although AC5/6 are the predominant isoforms expressed in the heart, there is also evidence that AC9 plays an important role in local regulation of certain cAMP-dependent responses. For example, genetic deletion of AC9 in mice leads to the development of bradycardia and heart failure [125]. AC9 is also part of a signaling complex with KCNQ1 K+ channels [126, 127], and disruption of this interaction leads to arrhythmogenesis [128].
Hypertrophy and heart failure have also been associated with many changes affecting localized degradation of cAMP by PDEs. Loss of PDE4D activity was shown to cause a localized increase in cAMP activity that leads to hyper-phosphorylation of RyRs, resulting in altered SR Ca2+ release and arrhythmogenesis associated with heart failure [103]. Similarly, the loss of PDE4 activity associated with heart failure resulted in an increase in the local production of cAMP by β2ARs, leading to PKA-dependent phosphorylation of RyRs and an increase in arrhythmogenesis [49]. Regulation of a local pool of cAMP by PDE2 activity has also been implicated in the development of hypertrophy [77, 79]. In addition, changes in PDE2- and PDE3-dependent regulation of local cAMP responses have been reported to contribute to altered βAR responses associated with cardiac hypertrophy [79, 80]. Changes in β3AR production of cGMP have also been reported to affect local regulation of cAMP activity by PDE2 [51].
Heart failure is also associated with changes that might be expected to affect PKA buffering and physically restricted spaces. These include the loss of mitochondrial AKAPs [129], loss of mitochondrial organization [130], and disruption of dyadic clefts [131]. Although one might predict that these changes could affect cAMP compartmentation by altering diffusion, proof awaits further investigation.
6. CONCLUSIONS AND FUTURE DIRECTIONS
This review focuses on four general mechanisms believed to play a role in ensuring the specificity of the receptor-mediated functional responses by compartmentalizing cAMP activity in cardiac myocytes. These include localized production of cAMP by different receptors, localized degradation of cAMP by PDEs, and limited diffusion due to the effects of PKA buffering and physically restricted spaces.. Most previous work has focused on demonstrating the functional significance of either localized production or localized degradation of cAMP. However, our understanding of the role that limited diffusion of cAMP plays in compartmentation and its contribution to functional responses is largely undeveloped. We can directly measure the diffusion of cAMP, so we know that it does not move freely throughout the cell. We also have direct evidence that this is due, at least in part to the effects of PKA buffering. Yet, the details of how this works are not fully understood. For example, why does cAMP appear to preferentially associate with PKA associated with mitochondria? Is it because PKA is concentrated there, or is there something different about the PKA located there? What is the role of type I vs type II PKA in buffering the movement of cAMP? What about the role of excess regulatory subunits? Free subunits bind cAMP with an affinity in the nM range, while the cAMP affinity of the holoenzyme is in the μM range [132]. Could it be that the free regulatory subunits function primarily as a buffer for cAMP? Furthermore, evidence that limited diffusion, due to PKA buffering or physically restricted spaces, contributes to cAMP compartmentation is largely based on theoretical predictions. While computational studies suggest that slow diffusion can contribute to the generation of cAMP gradients in spaces as small as the dyadic cleft (see figure 1), experimental evidence supporting these predictions is lacking. A clearer picture of the role that PKA buffering and/or physically restricted spaces play in affecting functional responses awaits a better understanding of the factors involved.It is also important to recognize that while the majority of studies looking at the mechanisms of cAMP compartmentation in the heart have been conducted using ventricular myocytes, the same basic principles are likely to apply in other cell types, such as atrial myocytes, where there are a number of structural and organizational differences [133]. For example, atrial myocytes are enriched in caveolae, which could provide a basis for segregation of receptors similar to what is seen in ventricular myocytes [134]. However, atrial myocytes have also been reported to exhibit a wide variation in the organization of t-tubules [134]. This could affect the location of cAMP production by different βARs. Atrial preparations have also been reported to vary in the relative expression and distribution of PDE isoforms [135]. This too is likely to influence subcellular cAMP gradients. However, atrial myocytes also exhibit a lower density of mitochondria [136], which together with a less well developed t-tubule system might affect factors limiting cAMP diffusion. It is unclear how all of these differences collectively might be predicted to affect cAMP compartmentation in atrial myocytes. The answer to this question awaits future investigation.
A key to ensuring that the receptor-specific functional effects are elicited is by isolating appropriate downstream signaling partners together into subcellular signaling complexes in proximity to the pools of cAMP produced by that receptor. Technological advances have significantly advanced our understanding of the fundamental mechanisms that control spatiotemporal dynamics of cAMP activity. A major breakthrough was provided by the development of genetically-encoded biosensors that can be expressed in specific subcellular microdomains. Furthermore, computational modeling has provided a means to test the feasibility of various hypotheses and generate new predictions. Advanced imaging techniques are continuously being tested to gain better understanding of the role that the localized cAMP responses play in health and diseases. Novel strategies are likely to continue to evolve from the use of these tools and provide insights directed at developing new therapeutic interventions to combat cardiac disease.
HIGHLIGHTS.
Localized production of cAMP by different GPCRs is often involved.
Localized degradation of cAMP by phosphodiesterases alone may not be sufficient.
Restricted diffusion of cAMP due buffering by protein kinase A is a likely factor.
Diffusion of cAMP limited by physical barriers may also contribute.
Multiple factors affecting cAMP compartmentation are altered by disease.
Acknowledgements
This work was supported by grants R01 HL145778, R01 GM 107094, OT2 OD023848, and the Center of Biomedical Research Excellence P20 GM130459 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hayes JS, Brunton LL, Mayer SE, Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1, J. Biol. Chem 255 (1980) 5113–5119. [PubMed] [Google Scholar]
- [2].Hayes JS, Brunton LL, Brown JH, Reese JB, Mayer SE, Hormonally specific expression of cardiac protein kinase activity, Proc. Natl. Acad. Sci. USA 76 (1979) 1570–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brunton LL, Hayes JS, Mayer SE, Hormonally specific phosphorylation of cardiac troponin I and activation of glycogen phosphorylase, Nature 280 (1979) 78–80. [DOI] [PubMed] [Google Scholar]
- [4].Steinberg SF, Brunton LL, Compartmentation of G protein-coupled signaling pathways in cardiac myocytes, Annu. Rev. Pharmacol. Toxicol 41 (2001) 751–773. [DOI] [PubMed] [Google Scholar]
- [5].Xiao RP, Hohl C, Altschuld R, Jones L, Livingston B, Ziman B, Tantini B, Lakatta EG, Beta 2-adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation, J. Biol. Chem 269 (1994) 19151–19156. [PubMed] [Google Scholar]
- [6].Xiao RP, Ji X, Lakatta EG, Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes, Mol. Pharmacol 47 (1995) 322–329. [PubMed] [Google Scholar]
- [7].Kuschel M, Zhou YY, Spurgeon HA, Bartel S, Karczewski P, Zhang SJ, Krause EG, Lakatta EG, Xiao RP, Beta2-adrenergic cAMP signaling is uncoupled from phosphorylation of cytoplasmic proteins in canine heart, Circulation 99 (1999) 2458–2465. [DOI] [PubMed] [Google Scholar]
- [8].Scriven DR, Dan P, Moore ED, Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes, Biophys. J 79 (2000) 2682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, Scott JD, Local protein kinase A action proceeds through intact holoenzymes, Science 356 (2017) 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Scott JD, Dessauer CW, Tasken K, Creating Order from Chaos: Cellular Regulation by Kinase Anchoring, Annu. Rev. Pharmacol. Toxicol 53 (2013) 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dodge-Kafka KL, Langeberg L, Scott JD, Compartmentation of cyclic nucleotide signaling in the heart: the role of A-kinase anchoring proteins, Circ.Res 98 (2006) 993–1001. [DOI] [PubMed] [Google Scholar]
- [12].Adams SR, Harootunian AT, Buechler YJ, Taylor SS, Tsien RY, Fluorescence ratio imaging of cyclic AMP in single cells, Nature 349 (1991) 694–697. [DOI] [PubMed] [Google Scholar]
- [13].Goaillard JM, Vincent PV, Fischmeister R, Simultaneous measurements of intracellular cAMP and L-type Ca2+ current in single frog ventricular myocytes, J. Physiol. (Lond.) 530 (2001) 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW, Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion, J. Gen. Physiol 116 (2000) 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM, Fischmeister R, Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels, J. Biol. Chem 279 (2004) 52095–52105. [DOI] [PubMed] [Google Scholar]
- [16].Rochais F, bi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G, A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes, Circ. Res 98 (2006) 1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel JL, Lugnier C, Conti M, Fischmeister R, Vandecasteele G, Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals, Circ. Res 105 (2009) 784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zaccolo M, De Giorgi F, Cho CY, Feng L, Knapp T, Negulescu PA, Taylor SS, Tsien RY, Pozzan T, A genetically encoded, fluorescent indicator for cyclic AMP in living cells, Nat. Cell Biol 2 (2000) 25–29. [DOI] [PubMed] [Google Scholar]
- [19].Zaccolo M, Pozzan T, Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes, Science 295 (2002) 1711–1715. [DOI] [PubMed] [Google Scholar]
- [20].Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ, Novel single chain cAMP sensors for receptor-induced signal propagation, J. Biol. Chem 279 (2004) 37215–37218. [DOI] [PubMed] [Google Scholar]
- [21].Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S, Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta-1 adrenergic but locally confined beta-2 adrenergic receptor-mediated signaling, Circ. Res 99 (2006) 1084–1091. [DOI] [PubMed] [Google Scholar]
- [22].Schleicher K, Zaccolo M, Using cAMP Sensors to Study Cardiac Nanodomains, Journal of cardiovascular development and disease 5 (2018) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang J, Ma Y, Taylor SS, Tsien RY, Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering, Proc. Natl. Acad. Sci. USA 98 (2001) 14997–15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brown DA, Lipid rafts, detergent-resistant membranes, and raft targeting signals, Physiology 21 (2006) 430–9. [DOI] [PubMed] [Google Scholar]
- [25].Allen JA, Halverson-Tamboli RA, Rasenick MM, Lipid raft microdomains and neurotransmitter signalling, Nat. Rev. Neurosci 8 (2007) 128–140. [DOI] [PubMed] [Google Scholar]
- [26].Harvey RD, Calaghan SC, Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology, J. Mol. Cell. Cardiol 52 (2012) 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Steinberg SF, The molecular basis for distinct beta-adrenergic receptor subtype actions in cardiomyocytes, Circ. Res 85 (1999) 1101–1111. [DOI] [PubMed] [Google Scholar]
- [28].Rybin VO, Xu X, Lisanti MP, Steinberg SF, Differential targeting of beta -adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway, J. Biol. Chem 275 (2000) 41447–41457. [DOI] [PubMed] [Google Scholar]
- [29].Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA, Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase, J. Biol. Chem 276 (2001) 42063–42069. [DOI] [PubMed] [Google Scholar]
- [30].Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ, Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation, Proc. Natl. Acad. Sci. USA 103 (2006) 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS, Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels, Circ. Res 107 (2010) 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Agarwal SR, Macdougall DA, Tyser R, Pugh SD, Calaghan SC, Harvey RD, Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes, J. Mol. Cell. Cardiol 50 (2011) 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Razani B, Woodman SE, Lisanti MP, Caveolae: from cell biology to animal physiology, Pharmacol. Rev 54 (2002) 431–467. [DOI] [PubMed] [Google Scholar]
- [34].Nash CA, Wei W, Irannejad R, Smrcka AV, Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy, Elife 8 (2019) e48167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Y, Shi Q, Li M, Zhao M, Reddy Gopireddy R, Teoh JP, Xu B, Zhu C, Ireton KE, Srinivasan S, Chen S, Gasser PJ, Bossuyt J, Hell JW, Bers DM, Xiang YK, Intracellular β(1)-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility, Circ. Res 128 (2021) 246–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kuschel M, Zhou YY, Cheng H, Zhang SJ, Chen Y, Lakatta EG, Xiao RP, G(i) protein-mediated functional compartmentalization of cardiac beta(2)-adrenergic signaling, J. Biol. Chem 274 (1999) 22048–22052. [DOI] [PubMed] [Google Scholar]
- [37].Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ, Lakatta EG, Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes, Circ. Res 84 (1999) 43–52. [DOI] [PubMed] [Google Scholar]
- [38].Ostrom RS, Violin JD, Coleman S, Insel PA, Selective enhancement of beta-adrenergic receptor signaling by overexpression of adenylyl cyclase type 6: colocalization of receptor and adenylyl cyclase in caveolae of cardiac myocytes, Mol. Pharmacol 57 (2000) 1075–1079. [PubMed] [Google Scholar]
- [39].Xiang Y, Rybin VO, Steinberg SF, Kobilka B, Caveolar localization dictates physiologic signaling of beta 2-adrenoceptors in neonatal cardiac myocytes, J. Biol. Chem 277 (2002) 34280–34286. [DOI] [PubMed] [Google Scholar]
- [40].Ostrom RS, Insel PA, The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology, Br. J. Pharmacol 143 (2004) 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA, G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes, J. Biol. Chem 280 (2005) 31036–31044. [DOI] [PubMed] [Google Scholar]
- [42].Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA, Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components, J. Biol. Chem 281 (2006) 26391–26399. [DOI] [PubMed] [Google Scholar]
- [43].Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J, Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation, Science 327 (2010) 1653–1657. [DOI] [PubMed] [Google Scholar]
- [44].Orchard C, Brette F, t-Tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes, Cardiovasc. Res 77 (2008) 237–244. [DOI] [PubMed] [Google Scholar]
- [45].Macdougall DA, Agarwal SR, Stopford EA, Chu H, Collins JA, Longster AL, Colyer J, Harvey RD, Calaghan S, Caveolae compartmentalise beta2-adrenoceptor signals by curtailing cAMP production and maintaining phosphatase activity in the sarcoplasmic reticulum of the adult ventricular myocyte, J. Mol. Cell. Cardiol 52 (2012) 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ, Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins, Science 298 (2002) 834–836. [DOI] [PubMed] [Google Scholar]
- [47].Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD, beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi, Proc Natl Acad Sci USA 100 (2003) 940–945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [48].Rudokas MW, Post JP, Sataray-Rodriguez A, Sherpa RT, Moshal KS, Agarwal SR, Harvey RD, Compartmentation of β(2) -adrenoceptor stimulated cAMP responses by phosphodiesterase types 2 and 3 in cardiac ventricular myocytes, Br. J. Pharmacol 178 (2021) 1574–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Berisha F, Götz K, Wegener JW, Brandenburg S, Subramanian H, Molina CE, Rueffer A, Petersen J, Bernhardt A, Girdauskas E, Jungen C, Pape U, Kraft AE, Warnke S, Lindner D, Westermann D, Blankenberg S, Meyer C, Hasenfuß G, Lehnart SE, Nikolaev VO, cAMP Imaging at Ryanodine Receptors Reveals β2-Adrenoceptor Driven Arrhythmias, Circ. Res 129 (2021) 81–94. [DOI] [PubMed] [Google Scholar]
- [50].Gauthier C, Tavernier G, Charpentier F, Langin D, Le MH, Functional beta3-adrenoceptor in the human heart, J. Clin. Invest 98 (1996) 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Schobesberger S, Wright PT, Poulet C, Sanchez Alonso Mardones JL, Mansfield C, Friebe A, Harding SE, Balligand JL, Nikolaev VO, Gorelik J, β(3)-Adrenoceptor redistribution impairs NO/cGMP/PDE2 signalling in failing cardiomyocytes, Elife 9 (2020) e52221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ostrom RS, Bundey RA, Insel PA, Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes, J. Biol. Chem 279 (2004) 19846–19853. [DOI] [PubMed] [Google Scholar]
- [53].Warrier S, Ramamurthy G, Eckert RL, Nikolaev VO, Lohse MJ, Harvey RD, cAMP microdomains and L-type Ca2+ channel regulation in guinea-pig ventricular myocytes, J. Physiol. (Lond.) 580 (2007) 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Agarwal SR, Gratwohl J, Cozad M, Yang PC, Clancy CE, Harvey RD, Compartmentalized cAMP Signaling Associated With Lipid Raft and Non-raft Membrane Domains in Adult Ventricular Myocytes, Front. Pharmacol 9 (2018) 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi T, Narumiya S, Ushikubi F, Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4, Circulation 109 (2004) 2462–2468. [DOI] [PubMed] [Google Scholar]
- [56].Pang L, Cai Y, Tang EH, Irwin MG, Ma H, Xia Z, Prostaglandin E Receptor Subtype 4 Signaling in the Heart: Role in Ischemia/Reperfusion Injury and Cardiac Hypertrophy, J. Diabetes. Res 2016 (2016) 2016:1324347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Harvey RD, Belevych AE, Muscarinic regulation of cardiac ion channels, Br. J. Pharmacol 139 (2003) 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zakharov SI, Harvey RD, Rebound stimulation of the cAMP-regulated Cl- current by acetylcholine in guinea-pig ventricular myocytes, J. Physiol. (Lond.) 499 (1997) 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Belevych AE, Sims C, Harvey RD, ACh-induced rebound stimulation of L-type Ca(2+) current in guinea-pig ventricular myocytes, mediated by Gbetagamma-dependent activation of adenylyl cyclase, J. Physiol. (Lond.) 536 (2001) 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Song Y, Shryock JC, Belardinelli L, Potentiating effect of acetylcholine on stimulation by isoproterenol of L-type Ca2+ current and arrhythmogenic triggered activity in guinea pig ventricular myocytes, J. Cardiovasc. Electrophysiol 9 (1998) 718–726. [DOI] [PubMed] [Google Scholar]
- [61].Ishikawa Y, Homcy CJ, The adenylyl cyclases as integrators of transmembrane signal transduction, Circ. Res 80 (1997) 297–304. [DOI] [PubMed] [Google Scholar]
- [62].Defer N, Best-Belpomme M, Hanoune J, Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase, Am. J. Physiol 279 (2000) F400–F416. [DOI] [PubMed] [Google Scholar]
- [63].Warrier S, Belevych AE, Ruse M, Eckert RL, Zaccolo M, Pozzan T, Harvey RD, Beta-adrenergic and muscarinic receptor induced changes in cAMP activity in adult cardiac myocytes detected using a FRET based biosensor, Am. J. Physiol. Cell Physiol 289 (2005) C455–C461. [DOI] [PubMed] [Google Scholar]
- [64].Willoughby D, Cooper DM, Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains, Physiol. Rev 87 (2007) 965–1010. [DOI] [PubMed] [Google Scholar]
- [65].Iancu RV, Jones SW, Harvey RD, Compartmentation of cAMP signaling in cardiac myocytes: a computational study, Biophys. J 92 (2007) 3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Iancu RV, Ramamurthy G, Harvey RD, Spatial and temporal aspects of cAMP signalling in cardiac myocytes, Clin. Exp. Pharmacol. Physiol 35 (2008) 1343–1348. [DOI] [PubMed] [Google Scholar]
- [67].Conti M, Beavo J, Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling, Annu. Rev. Biochem 76 (2007) 481–511. [DOI] [PubMed] [Google Scholar]
- [68].Francis SH, Blount MA, Corbin JD, Mammalian Cyclic Nucleotide Phosphodiesterases : Molecular Mechanisms and Physiological Functions, Physiol. Rev 91 (2011) 651–690. [DOI] [PubMed] [Google Scholar]
- [69].Osadchii OE, Myocardial phosphodiesterases and regulation of cardiac contractility in health and cardiac disease, Cardiovasc. Drugs Ther 21 (2007) 171–194. [DOI] [PubMed] [Google Scholar]
- [70].Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X, Xu H, Florio V, Rybalkin SD, Beavo JA, Chen YF, Li JD, Blaxall BC, Abe J, Yan C, Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy, Circ. Res 105 (2009) 956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, Radwanski PB, Florio V, Movsesian MA, Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes, J. Biol. Chem 282 (2007) 32749–57. [DOI] [PubMed] [Google Scholar]
- [72].Simmons MA, Hartzell HC, Role of phosphodiesterase in regulation of calcium current in isolated cardiac myocytes, Mol. Pharmacol 33 (1988) 664–671. [PubMed] [Google Scholar]
- [73].Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M, Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway, Circ. Res 98 (2006) 226–234. [DOI] [PubMed] [Google Scholar]
- [74].Mohamed TMA, Oceandy D, Zi M, Prehar S, Alatwi N, Wang Y, Shaheen M.a., Abou-Leisa R, Schelcher C, Hegab Z, Baudoin F, Emerson M, Mamas M, Di Benedetto G, Zaccolo M, Lei M, Cartwright EJ, Neyses L, Plasma membrane calcium pump (PMCA4)/neuronal nitric oxide synthase complex regulates cardiac contractility through modulation of a compartmentalized cyclic nucleotide microdomain, J. Biol. Chem 286 (2011) 41520–41529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G, Zaccolo M, cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes, Circ. Res 108 (2011) 929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liu D, Wang Z, Nicolas V, Lindner M, Mika D, Vandecasteele G, Fischmeister R, Brenner C, PDE2 regulates membrane potential, respiration and permeability transition of rodent subsarcolemmal cardiac mitochondria, Mitochondrion 47 (2019) 64–75. [DOI] [PubMed] [Google Scholar]
- [77].Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I, Zaccolo M, Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2, Circ Res 117 (2015) 707–19. [DOI] [PubMed] [Google Scholar]
- [78].Perera RK, Sprenger JU, Steinbrecher JH, Hubscher D, Lehnart SE, Abesser M, Schuh K, El-Armouche A, Nikolaev VO, Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy, Circ. Res 116 (2015) 1304–11. [DOI] [PubMed] [Google Scholar]
- [79].Mehel H, Emons J, Vettel C, Wittkopper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechene P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R, El-Armouche A, Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes, Journal of the American College of Cardiology 62 (2013) 1596–606. [DOI] [PubMed] [Google Scholar]
- [80].Bastug-Ozel Z, Wright PT, Kraft AE, Pavlovic D, Howie J, Froese A, Fuller W, Gorelik J, Shattock MJ, Nikolaev VO, Heart failure leads to altered beta2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain, Cardiovasc. Res 115 (2019) 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lugnier C, Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents, Pharmacol. Ther 109 (2006) 366–398. [DOI] [PubMed] [Google Scholar]
- [82].Muller B, Stoclet JC, Lugnier C, Cytosolic and membrane-bound cyclic nucleotide phosphodiesterases from guinea pig cardiac ventricles, Eur. J. Pharmacol 225 (1992) 263–272. [DOI] [PubMed] [Google Scholar]
- [83].Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS, Jimmo SL, Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system, Mol. Pharmacol 64 (2003) 533–546. [DOI] [PubMed] [Google Scholar]
- [84].Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC, Movsesian MA, Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes, J. Biol. Chem 277 (2002) 38072–38078. [DOI] [PubMed] [Google Scholar]
- [85].Weishaar RE, Kobylarz-Singer DC, Kaplan HR, Subclasses of cyclic AMP phosphodiesterase in cardiac muscle, J Mol Cell Cardiol 19 (1987) 1025–36. [DOI] [PubMed] [Google Scholar]
- [86].Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin SD, Silengo L, Altruda F, Wetzker R, Wymann MP, Lembo G, Hirsch E, PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects, Cell 118 (2004) 375–387. [DOI] [PubMed] [Google Scholar]
- [87].Hambleton R, Krall J, Tikishvili E, Honeggar M, Ahmad F, Manganiello VC, Movsesian MA, Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to camp hydrolytic activity in subcellular fractions of human myocardium, J. Biol. Chem 280 (2005) 39168–39174. [DOI] [PubMed] [Google Scholar]
- [88].Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM, Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways, Cell 110 (2002) 737–749. [DOI] [PubMed] [Google Scholar]
- [89].Perino A, Ghigo A, Ferrero E, Morello F, Santulli G, Baillie GS, Damilano F, Dunlop AJ, Pawson C, Walser R, Levi R, Altruda F, Silengo L, Langeberg LK, Neubauer G, Heymans S, Lembo G, Wymann MP, Wetzker R, Houslay MD, Iaccarino G, Scott JD, Hirsch E, Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma, Mol. Cell 42 (2011) 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Beca S, Ahmad F, Shen W, Liu J, Makary S, Polidovitch N, Sun J, Hockman S, Chung YW, Movsesian M, Murphy E, Manganiello V, Backx PH, Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart, Circ. Res 112 (2013) 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R, Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes, Br. J. Pharmacol 127 (1999) 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M, Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases, Circ. Res 95 (2004) 67–75. [DOI] [PubMed] [Google Scholar]
- [93].Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G, Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases, Circ. Res 102 (2008) 1091–100. [DOI] [PubMed] [Google Scholar]
- [94].Mika D, Leroy J, Vandecasteele G, Fischmeister R, PDEs create local domains of cAMP signaling, J. Mol. Cell. Cardiol 52 (2012) 323–329. [DOI] [PubMed] [Google Scholar]
- [95].Kostic MM, Erdogan S, Rena G, Borchert G, Hoch B, Bartel S, Scotland G, Huston E, Houslay MD, Krause EG, Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart, J. Mol. Cell. Cardiol 29 (1997) 3135–3146. [DOI] [PubMed] [Google Scholar]
- [96].Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M, Conserved expression and functions of PDE4 in rodent and human heart, Basic research in cardiology 106 (2011) 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C, Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling, J. Biol. Chem 278 (2003) 5493–5496. [DOI] [PubMed] [Google Scholar]
- [98].Houslay MD, Adams DR, PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization, Biochem. J 370 (2003) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bolger GB, Erdogan S, Jones RE, Loughney K, Scotland G, Hoffmann R, Wilkinson I, Farrell C, Houslay MD, Characterization of five different proteins produced by alternatively spliced mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene, Biochem. J 328 (1997) 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Baillie GS, Houslay MD, Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes, Curr. Opin. Cell Biol 17 (2005) 129–134. [DOI] [PubMed] [Google Scholar]
- [101].Leroy J, Richter W, Mika D, Castro LR, Abi-Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Mateo P, Westenbroek R, Catterall WA, Charpentier F, Conti M, Fischmeister R, Vandecasteele G, Phosphodiesterase 4B in the cardiac L-type Ca(2)(+) channel complex regulates Ca(2)(+) current and protects against ventricular arrhythmias in mice, J. Clin. Invest 121 (2011) 2651–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Mika D, Richter W, Westenbroek RE, Catterall WA, Conti M, PDE4B mediates local feedback regulation of beta(1)-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes, J. Cell Sci 127 (2014) 1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR, Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias, Cell 123 (2005) 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Sin YY, Edwards HV, Li X, Day JP, Christian F, Dunlop AJ, Adams DR, Zaccolo M, Houslay MD, Baillie GS, Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the beta-agonist induced hypertrophic response in cardiac myocytes, J. Mol. Cell. Cardiol 50 (2011) 872–83. [DOI] [PubMed] [Google Scholar]
- [105].Daaka Y, Luttrell LM, Lefkowitz RJ, Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A, Nature 390 (1997) 88–91. [DOI] [PubMed] [Google Scholar]
- [106].Salazar NC, Vallejos X, Siryk A, Rengo G, Cannavo A, Liccardo D, De Lucia C, Gao E, Leosco D, Koch WJ, Lymperopoulos A, GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility, Cell. Commun. Signal 11 (2013) 11:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SG, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M, Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4, Embo j 27 (2008) 384–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Iancu RV, Ramamurthy G, Warrier S, Nikolaev VO, Lohse MJ, Jones SW, Harvey RD, Cytoplasmic cAMP concentrations in intact cardiac myocytes, Am. J. Physiol. Cell Physiol 295 (2008) C414–C422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Borner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, Nikolaev VO, FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells, Nat. Protoc 6 (2011) 427–438. [DOI] [PubMed] [Google Scholar]
- [110].Yang PC, Boras BW, Jeng MT, Docken SS, Lewis TJ, McCulloch AD, Harvey RD, Clancy CE, A computational modeling and simulation approach to investigate mechanisms of subcellular cAMP compartmentation, PLoS Comput. Biol 12 (2016) e1005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Houslay MD, Baillie GS, Maurice DH, cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling, Circ. Res 100 (2007) 950–66. [DOI] [PubMed] [Google Scholar]
- [112].Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, McCulloch AD, Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes, Proc. Natl. Acad. Sci. USA 103 (2006) 12923–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Rossow MJ, Sasaki JM, Digman M.a., Gratton E, Raster image correlation spectroscopy in live cells, Nat. Protoc 5 (2010) 1761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Agarwal SR, Clancy CE, Harvey RD, Mechanisms restricting diffusion of intracellular cAMP, Sci. Rep 6 (2016) 6:19577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Moll D, Prinz A, Brendel CM, Berrera M, Guske K, Zaccolo M, Genieser HG, Herberg FW, Biochemical characterization and cellular imaging of a novel, membrane permeable fluorescent cAMP analog, BMC Biochem. 9 (2008) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Taylor SS, Ilouz R, Zhang P, Kornev AP, Assembly of allosteric macromolecular switches: lessons from PKA, Nat. Rev. Mol. Cell Biol 13 (2012) 646–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Sherpa RT, Fiore C, Moshal KS, Wadsworth A, Rudokas MW, Agarwal SR, Harvey RD, Mitochondrial A-kinase anchoring proteins in cardiac ventricular myocytes, Physiol. Rep 9 (2021) e15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Bock A, Annibale P, Konrad C, Hannawacker A, Anton SE, Maiellaro I, Zabel U, Sivaramakrishnan S, Falcke M, Lohse MJ, Optical Mapping of cAMP Signaling at the Nanometer Scale, Cell 182 (2020) 1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Corbin JD, Sugden PH, Lincoln TM, Keely SL, Compartmentalization of adenosine 3’:5’-monophosphate and adenosine 3’:5’-monophosphate-dependent protein kinase in heart tissue, J. Biol. Chem 252 (1977) 3854–3861. [PubMed] [Google Scholar]
- [120].Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD, Modeling beta-adrenergic control of cardiac myocyte contractility in silico, J. Biol. Chem 278 (2003) 47997–48003. [DOI] [PubMed] [Google Scholar]
- [121].Beavo JA, Bechtel PJ, Krebs EG, Activation of protein kinase by physiological concentrations of cyclic AMP, Proc Natl Acad Sci USA 71 (1974) 3580–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Walker-Gray R, Stengel F, Gold MG, Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches, Proc. Natl. Acad. Sci. USA 114 (2017) 10414–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Barth E, Stämmler G, Speiser B, Schaper J, Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man, J. Mol. Cell. Cardiol 24 (1992) 669–681. [DOI] [PubMed] [Google Scholar]
- [124].Richards M, Lomas O, Jalink K, Ford KL, Vaughan-Jones RD, Lefkimmiatis K, Swietach P, Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes, Cardiovasc. Res 110 (2016) 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Li Y, Baldwin TA, Wang Y, Subramaniam J, Carbajal AG, Brand CS, Cunha SR, Dessauer CW, Loss of type 9 adenylyl cyclase triggers reduced phosphorylation of Hsp20 and diastolic dysfunction, Sci. Rep 7 (2017) 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Li Y, Chen L, Kass RS, Dessauer CW, The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart, J. Biol. Chem 287 (2012) 29815–29824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Li Y, Hof T, Baldwin TA, Chen L, Kass RS, Dessauer CW, Regulation of I(Ks) Potassium Current by Isoproterenol in Adult Cardiomyocytes Requires Type 9 Adenylyl Cyclase, Cells 8 (2019) 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS, Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel, Science 295 (2002) 496–499. [DOI] [PubMed] [Google Scholar]
- [129].Marin W, A-kinase anchoring protein 1 (AKAP1) and its role in some cardiovascular diseases, J. Mol. Cell. Cardiol 138 (2020) 99–109. [DOI] [PubMed] [Google Scholar]
- [130].Miragoli M, Sanchez-Alonso JL, Bhargava A, Wright PT, Sikkel M, Schobesberger S, Diakonov I, Novak P, Castaldi A, Cattaneo P, Lyon AR, Lab MJ, Gorelik J, Microtubule-Dependent Mitochondria Alignment Regulates Calcium Release in Response to Nanomechanical Stimulus in Heart Myocytes, Cell Rep. 14 (2016) 140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zhang HB, Li RC, Xu M, Xu SM, Lai YS, Wu HD, Xie XJ, Gao W, Ye H, Zhang YY, Meng X, Wang SQ, Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure, Cardiovasc. Res 98 (2013) 269–276. [DOI] [PubMed] [Google Scholar]
- [132].Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A, Doskeland SO, Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition, J. Biol. Chem 281 (2006) 21500–21511. [DOI] [PubMed] [Google Scholar]
- [133].Bhogal NK, Hasan A, Gorelik J, The Development of Compartmentation of cAMP Signaling in Cardiomyocytes: The Role of T-Tubules and Caveolae Microdomains, J. Cardiovasc. Dev. Dis 5 (2018) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Glukhov AV, Balycheva M, Sanchez-Alonso JL, Ilkan Z, Alvarez-Laviada A, Bhogal N, Diakonov I, Schobesberger S, Sikkel MB, Bhargava A, Faggian G, Punjabi PP, Houser SR, Gorelik J, Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes, Circulation 132 (2015) 2372–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hua R, Adamczyk A, Robbins C, Ray G, Rose RA, Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium, PLoS One 7 (2012) e47652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Seguchi H, Ritter M, Shizukuishi M, Ishida H, Chokoh G, Nakazawa H, Spitzer KW, Barry WH, Propagation of Ca2+ release in cardiac myocytes: role of mitochondria, Cell Calcium 38 (2005) 1–9. [DOI] [PubMed] [Google Scholar]