

Abstract
Background
Ovarian cancer tends to be chemosensitive and confine itself to the surface of the peritoneal cavity for much of its natural history. These features have made it an obvious target for intraperitoneal (IP) chemotherapy. Chemotherapy for ovarian cancer is usually given as an intravenous (IV) infusion repeatedly over five to eight cycles. Intraperitoneal chemotherapy is given by infusion of the chemotherapeutic agent directly into the peritoneal cavity. There are biological reasons why this might increase the anticancer effect and reduce some systemic adverse effects in comparison to IV therapy.
Objectives
To determine if adding a component of the chemotherapy regime into the peritoneal cavity affects overall survival, progression‐free survival, quality of life (QOL) and toxicity in the primary treatment of epithelial ovarian cancer.
Search methods
We searched the Gynaecological Cancer Review Group's Specialised Register, the Cochrane Central Register of Controlled Trials (CENTRAL) Issue 2, 2011, MEDLINE (1951 to May 2011) and EMBASE (1974 to May 2011). We updated these searches in February 2007, August 2010, May 2011 and September 2015. In addition, we handsearched and cascade searched the major gynaecological oncology journals up to May 2011.
Selection criteria
The analysis was restricted to randomised controlled trials (RCTs) assessing women with a new diagnosis of primary epithelial ovarian cancer, of any FIGO stage, following primary cytoreductive surgery. Standard IV chemotherapy was compared with chemotherapy that included a component of IP administration.
Data collection and analysis
We extracted data on overall survival, disease‐free survival, adverse events and QOL and performed meta‐analyses of hazard ratios (HR) for time‐to‐event variables and relative risks (RR) for dichotomous outcomes using RevMan software.
Main results
Nine randomised trials studied 2119 women receiving primary treatment for ovarian cancer. We considered six trials to be of high quality. Women were less likely to die if they received an IP component to chemotherapy (eight studies, 2026 women; HR = 0.81; 95% confidence interval (CI): 0.72 to 0.90). Intraperitoneal component chemotherapy prolonged the disease‐free interval (five studies, 1311 women; HR = 0.78; 95% CI: 0.70 to 0.86). There was greater serious toxicity with regard to gastrointestinal effects, pain, fever and infection but less ototoxicity with the IP than the IV route.
Authors' conclusions
Intraperitoneal chemotherapy increases overall survival and progression‐free survival from advanced ovarian cancer. The results of this meta‐analysis provide the most reliable estimates of the relative survival benefits of IP over IV therapy and should be used as part of the decision making process. However, the potential for catheter related complications and toxicity needs to be considered when deciding on the most appropriate treatment for each individual woman. The optimal dose, timing and mechanism of administration cannot be addressed from this meta‐analysis. This needs to be addressed in the next phase of clinical trials.
Plain language summary
Intraperitoneal chemotherapy (administered into the peritoneal cavity) for advanced ovarian cancer improves both overall and disease‐free survival
Ovarian cancer commonly spreads through the peritoneal cavity and usually responds to intravenous (IV) chemotherapy. This review compared the effectiveness of IV chemotherapy to chemotherapy administered directly into the peritoneal cavity (intraperitoneal, or IP). The evidence suggests an improvement in survival if some of the chemotherapy is administered via the intraperitoneal route. The disadvantage is an increase in adverse effects principally relating to the presence of a peritoneal catheter, including pain, catheter blockage, gastrointestinal effects and infection.
Summary of findings
Background
Description of the condition
In the United States, epithelial ovarian cancer is the seventh most common cancer among women and ranks fourth in female cancer deaths (ACS 2005). The main treatment for advanced disease includes a combination of surgical removal of all resectable disease, if possible, followed by intravenous chemotherapy, including a platinum‐based drug with or without a taxane. Based on a meta‐analysis of 6885 women (including 81 cohorts of women with stage III and IV ovarian cancer), Bristow and colleagues demonstrated that women with ≤ 25% maximal cytoreduction had a mean weighted median survival of 22.7 months; those with > 75% maximal cytoreduction had a mean weighted median survival of 33.9 months ‐ an increase of 50%. In addition, they showed that each 10% increase in maximal cytoreduction was associated with a 5.5% increase in median survival time (Bristow 2002). This has prompted large trials to evaluate the role of more aggressive surgery and alternative chemotherapy regimes. Further improvements are being sought to specifically target ovarian cancer cells within the peritoneal cavity. These include intraperitoneal antibodies, immunotherapy, radiotherapy and the administration of chemotherapeutic agents directly into the peritoneal cavity before, during or after surgery.
Description of the intervention
Intraperitoneal (IP) chemotherapy for intra‐abdominal cancer has its origins in the 1950s when nitrogen mustard was used intraperitoneally for malignant ascites (Weisberger 1955). This concept was developed in the 1970s when significantly greater concentrations of certain chemotherapeutic drugs were demonstrated in the peritoneal cavity than in the blood (Jones 1978).
Ovarian cancer remains confined to the peritoneal cavity for much of its natural history, spreading by local extension and then by direct spread from the surface of the ovary through the peritoneal cavity. This IP spread is the most common and recognized characteristic of ovarian cancer and makes the disease an ideal candidate for such a drug delivery strategy. The rationale for IP chemotherapy is to eradicate residual disease by concentrating the cytotoxic effect, generating high drug concentrations directly into the peritoneal cavity, and reducing the systemic toxic effects associated with the standard method of intravenous administration.
Intraperitoneal chemotherapy has several foreseeable limitations:
Los 1990 injected radiolabelled cisplatin into the peritoneal cavity of rats; IP concentrations of cisplatin were 10 to 20 times higher than serum levels, but IP tumour penetration depth was limited to 1 to 2 mm. This suggests that the benefit of IP chemotherapy may be limited to women with microscopic or very small volume residual disease after debulking surgery.
Ovarian cancer spreads not only by the transperitoneal route, but also via the lymphatic and venous system and by direct invasion through the diaphragm: these sites may not be targeted as well using IP as IV chemotherapy. In Burghardt 1991, the incidence of positive nodes at primary surgery was reported to be as high as 24% in women with stage I disease, 50% in women with stage II disease, 74% in women with stage III disease, and 73% in women with stage IV disease. Therefore, concern arises as to the appropriateness of targeting the peritoneal cavity.
The peritoneal cavity in women with ovarian cancer is often distorted by adhesions and sanctuary sites may develop, limiting the access of cytotoxic drugs to tumour surfaces in these areas.
Complications of IP drug administration are well recognised and deterioration in QOL scores may be more significant with IP than with standard IV therapy.
Concentrations of IP platinum compounds are high after IV administration and IP instillation may not add therapeutic benefit.
IP chemotherapy administration requires more clinical space, more time, and a team of medical oncologists and nurses confident and experienced at both the technical aspects and management of complications.
Gynaecological oncologists need to be experienced and competent at maximal debulking surgery and insertion and removal of intraperitoneal catheters or intermittent direct transperitoneal infusions.
Why it is important to do this review
Since the 1970s there have been many phase I and phase II clinical trials evaluating the effects of IP chemotherapy, and several have been tested at phase III. In summary these phase I and II trials established the feasibility and safety of this approach (Howell 1982; Lopez 1985; Markman 1999) but did not establish the efficacy of the treatment. By taking a systematic approach to reviewing all relevant phase III studies, this review of available literature focuses on survival, adverse events and QOL comparing IP with IV chemotherapy. This is an updated version of our original review (Jaaback 2007).
Objectives
Primary objective To determine the efficacy of active chemotherapy treatments administered into the peritoneal cavity in the initial management of primary epithelial ovarian cancer. Specifically, we addressed the following questions:
Is administering some of the chemotherapy IP more effective than a pure IV regime in improving survival from ovarian cancer?
Is administering some of the chemotherapy IP more effective than a pure IV regime in delaying recurrence of ovarian cancer?
Secondary objectives
To determine if there are some women with epithelial ovarian cancer who are more or less likely to benefit from this treatment.
To determine if there is any difference in QOL for women treated with chemotherapy involving IP administration versus IV chemotherapy.
Is administering some of the chemotherapy IP safer than an IV regime?
Methods
Criteria for considering studies for this review
Types of studies
Only RCTs regardless of other co‐morbidity were considered for this review. Trials where the method of randomisation was not specified in detail were included but were considered of low quality (see Subgroup analysis and investigation of heterogeneity).
Types of participants
Women with a new diagnosis of primary epithelial ovarian cancer, of any FIGO stage, following primary cytoreductive surgery. No age limit was applied.
Types of interventions
Standard IV chemotherapy was compared with chemotherapy that included a component of IP administration. We accepted trials comparing regimes which included IP treatment with similar regimes that excluded IP treatment. Where interventions differed significantly between studies this was clearly stated and the implications were discussed.
The review excluded the following:
radio colloids;
gene therapy;
biologic therapy;
radio‐isotopes;
vascular growth factors;
immunomodulating drugs;
matrix metalloproteinase inhibitors;
radiolabelled monoclonal antibodies.
Types of outcome measures
Primary outcomes
Progression‐free survival (time to recurrence).
Overall survival (time to death).
The definitions used in the trials to describe what constitutes recurrence are noted in Characteristics of included studies where possible.
Secondary outcomes
Adverse effects as measured by any recognised and validated scoring system. It is acknowledged that the frequency of important long‐term adverse effects may not be adequately captured by information in (small) RCTs.
QOL as measured by any scale recognised and validated for cancer care.
Search methods for identification of studies
Electronic searches
See: Cochrane Gynaecological Cancer Collaborative Review Group search strategy.
The original search was conducted to March 2005 with updated searches conducted on the following databases in February 2007, August 2010, May 2011 and September 2015. We searched the following databases:
Gynaecological Cancer Review Group's Specialised Register
Cochrane Central Register of Controlled Trials (CENTRAL) on The Cochrane Library, Issue 8, 2015
MEDLINE (1951 to week 4, Aug 2015)
EMBASE (1974 to week 36, 2015)
The search strategies for MEDLINE, EMBASE and CENTRAL are listed in Appendix 1, Appendix 2 and Appendix 3 respectively.
Searching other resources
Handsearching
The following journals were handsearched:
Gynecologic Oncology from 1998 to May 2011;
International Journal of Gynecological Cancer from 1998 to May 2011.
We also contacted authors who were active in the field. The "first generation" reference lists of all eligible trials were searched for additional studies. We searched grey literature, in particular conference proceedings and abstracts through ZETOC and Dissertation Abstracts.
We searched for all relevant studies irrespective of language. A French trial (Zylberberg 1986) was translated, with the assistance of a native speaker, and was included in the review.
Data collection and analysis
Selection of studies
For the updated review, Kenneth Jaaback (KJ) and Tess Lawrie (TL) scanned the searches and selected potentially relevant studies. All three authors obtained the full text of potentially relevant studies for independent assessment of eligibility. For the original review, KJ selected potentially relevant studies from the searches and KJ and Nick Johnson (NJ) assessed the full text papers. NJ looked at a random sample of search results to check that nothing relevant had been discarded.
Data extraction and management
For the original review, two review authors independently extracted the data using a specifically designed form (KJ and NJ). For the 2011 updated review, only one new trial was eligible (Yen 2009). As the relevant data from this trial were unpublished, we sent the data extraction form to the trial authors (Dr CM Juang, see Acknowledgements) who completed the form. Additional reports of the GOG 172 trial rendered data on QOL and adverse effects and these were extracted independently by two review authors (KJ, TL). No new studies were identified for inclusion 2015. We resolved any disagreement by discussion. We entered data into Review Manager software (RevMan 2008) and checked for accuracy. When information regarding results, outcomes or methods was unclear, or where data were missing, we attempted to contact authors of the original reports to provide further details. Data extracted included:
participant characteristics (age, stage and postoperative residuum of malignancy);
numbers of participants in each arm of the trial;
length of follow‐up;
withdrawal from treatment protocol;
number of participants who received all, part or none of the planned treatment, or who experienced delays during treatment will be recorded for each treatment arm;
type of intervention and control (chemotherapy agents administered in each arm of the trial and timing of administration in relation to surgery);
data relating to methodological quality ‐ see above;
log(HR) and its variance summarising (i) risk of recurrence of ovarian cancer and (ii) survival;
adverse effects where toxicity was classified as Grade 3 or 4;
QOL data from reliable and validated tools ‐ changes (either improvement or worsening) in symptoms were summarised;
catheter‐related complications.
Assessment of risk of bias in included studies
NJ and KJ independently assessed the risk of bias for each eligible study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2009). We resolved any disagreement by discussion.
(1) Random sequence generation (checking for possible selection bias)
We described for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups. We assessed the method as:
low risk of bias (any truly random process, e.g. random number table;computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth;hospital or clinic record number) or;
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We described for each included study the method used to conceal allocation to interventions prior to assignment and assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment. We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
(3) Blinding of outcome assessment (checking for possible detection bias)
We described for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received. We assessed methods used to blind outcome assessment as:
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We described for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion, where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we re‐included missing data in the analyses we undertook. We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
We described for each included study how we investigated the possibility of selective outcome reporting bias and what we found. We assessed the methods as:
low risk of bias (where it is clear that all of the study’s pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the study’s pre‐specified outcomes have been reported; one or more reported primary outcomes were not prespecified; outcomes of interest were reported incompletely and so cannot be used; study fails to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by 1 to 5 above)
We described for each included study any important concerns we had about other possible sources of bias. We assessed whether each study was free of other problems that could put it at risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there is risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2009). With reference to (1) to (6) above, we assessed the likely magnitude and direction of the bias and whether we considered it likely to impact on the findings. We explored the impact of the level of bias through undertaking sensitivity analyses.
Measures of treatment effect
Dichotomous data
The following outcomes were presented as dichotomous data:
adverse effects grade 3 to 4 including leukopenia, thrombocytopenia, anaemia, gastrointestinal effects, neurologic, renal, pulmonary, cardiovascular, fever, fatigue, infection, metabolic, pain and hearing loss.
For this type of data, we presented results as a summary risk ratio with 95% confidence intervals.
Time‐to‐event data
For time‐to‐event outcomes, we combined hazard ratios (HR) and their associated variances using the generic inverse‐variance method (RevMan 2008). We used this method for:
time to death;
time to recurrence.
Dealing with missing data
For included studies, we noted levels of attrition. We explored the impact of including studies with high levels of missing data in the overall assessment of treatment effect by using sensitivity analysis. For all outcomes, we carried out analyses, as far as possible, on an intention‐to‐treat basis, i.e. we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis using the T², I² and Chi² statistics. We regarded heterogeneity as substantial if I² was greater than 30% and either T² was greater than zero, or there was a low P value (< 0.10) in the Chi² test for heterogeneity.
Assessment of reporting biases
If there were 10 or more studies in the meta‐analysis we planned to investigate reporting biases (such as publication bias) using funnel plots. We planned to assess funnel plot asymmetry visually, and if asymmetry was suggested by a visual assessment, we would perform exploratory analyses to investigate it. However, the largest meta‐analysis in this review consisted of eight studies.
Data synthesis
We carried out statistical analyses using the Review Manager software (RevMan 2008). We used fixed‐effect meta‐analysis for combining data where it was reasonable to assume that studies were estimating the same underlying treatment effect, i.e. where trials were examining similar populations and used similar methods. If survival methods were used but HRs not reported, we estimated HRs using Parmar's methods (Parmar 1998).
Where necessary, we combined different categories of toxicity within trials in order to facilitate pooling of data from different trials. If there was clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if substantial statistical heterogeneity was detected, we used random‐effects meta‐analysis to produce an overall summary if an average treatment effect across trials was considered clinically meaningful. The random‐effects summary was treated as the average range of possible treatment effects and we discuss the clinical implications of treatment effects differing between trials. Where we used random‐effects analyses, the results are presented as the average treatment effect with 95% confidence intervals, and the estimates of T² and I².
Subgroup analysis and investigation of heterogeneity
Where we identified substantial heterogeneity, we investigated it using subgroup analyses and sensitivity analyses. For exploratory purposes, we subgrouped trials according to quality for all outcomes. We assessed the difference between subgroups by interaction tests.
Sensitivity analysis
Sensitivity analyses were performed where there was a risk of bias associated with the quality of some of the included trials.
Results
Description of studies
Results of the search
See Characteristics of included studies and Characteristics of excluded studies.
Ten potentially relevant studies were identified from the search strategy described above. The full text for all of these was obtained. These were then assessed for inclusion/exclusion by two reviewers (KJ, NJ) using the stated criteria.
Included studies
Nine trials consisting of 2119 women, representing 100% of eligible women from known randomised trials, contributed data. Patient accrual varied from 20 to 546.
Three United States trials were inter‐group studies with more than 40 participating institutions in each (Alberts 1996; GOG 172; Markman 2001). The Gadducci 2000 study involved the Gruppo Oncologico Nord‐Ovest in Italy and it was unclear how many centres participated. The Zylberberg 1986 study involved three participating centres. In the Kirmani 1994 trial an unclear number of community hospitals participated in the San Diego area of California. In the remaining three trials one centre participated (Polyzos 1999; Yen 2001; Yen 2009).
Zylberberg 1986 is the earliest RCT on IP chemotherapy for the initial management of epithelial ovarian cancer. They randomised 20 women with stage III disease to receive either IV chemotherapy including doxorubicin, fluorouracil, bleomycin, cisplatin, vinorelbine and ifosfamide, or a combination of IV doxorubicin, fluorouracil, bleomycin, cisplatin, vinorelbine, ifosfamide and IP bleomycin, cisplatin, fluorouracil and doxorubicin. Both groups received maintenance intramuscular chemotherapy with 12 monthly courses of ifosfamide, fluorouracil, and methotrexate. Ten women were randomised to each arm and the outcome of each patient was reported. They described a statistically significant increase in the number of women alive and free of disease in the IP group (P < 0.05) but no further statistics were provided. In the IV group, of the 10 randomised, there were five deaths after 11, 18, 24, 38 and 62 months and five free of relapse after 31, 40, 42, 64 and 72 months. In the IP group there was one death after 29 months, two with residual disease resected at second look laparotomy, free of disease after 23 and 54 months, and the other seven were free of disease after 23, 23, 27, 28, 34, 58 and 59 months. More mature data are unfortunately not available and our attempts to contact the lead author were unsuccessful. We were able to construct a Kaplan‐Meier curve for time to death and these data have been included in the survival meta‐analysis.
Kirmani 1994 randomised 87 women with stage IIC‐IV disease over four years to receive IV cisplatin 100 mg/m² and cyclophosphamide 600 mg/m² or IP cisplatin 200 mg/m² and etoposide 350 mg/m². Of these, 68 were evaluable for toxicity, 62 for survival (33 IV and 29 IP), and 46 for response. Ten were excluded from participation after review of the pathology. Seven women refused their assigned treatment arm (4 IP and 3 IV). For various reasons another eight women were not evaluable for response. The two groups were well balanced with respect to age, stage and residual disease, but more in the IV group had an ECOG performance status of 0 (14 versus 5) and fewer had a status of 2 (3 versus 6). This was the only trial comparing direct IV to IP (without additional IV) chemotherapy. Kaplan‐Meier curves show non‐significantly better overall survival in the IV arm. Disease‐free survival included only those with no clinical evidence of disease at the end of treatment, and also showed no significant difference between the two groups. However, only 25 IV and 21 IP women were evaluable for this assessment. There was also no detectable difference in either haematological or non‐haematological toxicity between the high‐dose IP regime and the standard‐dose IV regime.
The North West Oncology Group trial (Gadducci 2000) was run over 7½ years, and randomised 113 women with 100 evaluable (54 IV and 46 IP) participants. They considered stage II‐IV disease with < 2 cm residual disease after debulking surgery. The chemotherapy arms included IV or IP cisplatin 50 mg/m². Both groups also received IV epidoxorubicin 60 mg/m² + IV cyclophosphamide 600 mg/m². Twenty‐two women did not complete the assigned treatment (2 IV and 20 IP). Significant reasons for treatment change in the IP arm included patient refusal (6), bowel perforation (3) and abdominal pain (2). Patient characteristics were well balanced in the two groups except histotype. Seven women with clear cell histology were randomly allocated to the IV arm. Median disease free survival was 25 and 42 months for the IV and IP arms (P = 0.13) and median overall survival was 51 and 67 months (P = 0.14). Similar survival curves were obtained after exclusion of the women with clear cell histology. The only significant toxicity difference was more grade 3‐4 myelotoxicity in the IP group (52% versus 34%).
Polyzos 1999 randomised 90 women (46 IV and 44 IP) with stage III disease randomised to receive either IV or IP carboplatin 350 mg/m² all of whom were eligible for analysis. Both groups also received IV cyclophosphamide 600 mg/m². There were no statistically significant differences in the distribution of characteristics of the women between the two chemotherapy groups. There was no maximum tumour diameter permitted for entry into this trial. A residual tumour volume ≥ 2 cm was found in 43% of women. There were no significant differences in time to progression or overall survival in the two groups, even after adjusting for residual tumour volume but we were unable to obtain the survival curves to include data in the meta‐analysis. Significantly more women in the IV group had grade 3 or 4 leukopenia (P < 0.01) and non‐significantly more grade 3 or 4 thrombocytopenia (P < 0.09). However, 10% of women receiving IP chemotherapy were reported to have major catheter‐related morbidity, including three cases of chemotherapy infused directly into the large bowel with ensuing massive diarrhoea, and two cases where fluid was infused between layers of the abdominal wall. Weaknesses in this study include the poor statistical power with the low number randomised. Fewer women had tumour volumes < 2 cm (25 IV and 26 IP). The randomisation technique and allocation concealment was unclear, and analysis by intention‐to‐treat was not described.
Yen 2001 randomised 132 women with stage III disease to receive either IV cisplatin 50 mg/m² or IP cisplatin 100 mg/m² and of these 118 were eligible (55 IV and 63 IP). Both groups also received IV adriamycin or epirubicin 50 mg/m² and IV cyclophosphamide 500 mg/m². There were no significant differences between the two study groups with regard to important prognostic factors. All women initially had optimal debulking surgery with residual disease of ≤ 1 cm. The median survival rates were 43 months for the IP group and 48 months for the IV group (P = 0.469). Significantly more women in the IV group had grade 3 or 4 leukopenia but there was no significant increase in thrombocytopenia or anaemia. Catheter‐related complications in the IP group included pain (41.8%) and obstruction (25.5%).
Alberts 1996 coordinated the first large study incorporating investigators from the SWOG and GOG, randomising 654 women with residual disease of ≤ 2 cm to either IP or IV cisplatin (100 mg/m²); 546 women were finally eligible. Both groups also received IV cyclophosphamide 600 mg/m². The trial investigators included a subgroup analysis in January 1991, four and a half years after the study commenced accrual, after consensus emerged that women with a residual tumour size of ≤ 0.5 cm would be most likely to benefit from IP chemotherapy. The study was then extended for an additional year to achieve an adequate (powered) sample size for analysis of this subgroup. There were no significant differences between the two study groups with respect to important prognostic factors. The IP arm had significantly less myelotoxicity and tinnitus but there were no major differences between the two arms in terms of severe toxicity, treatment‐related deaths and removal of women from the study due to adverse events.
Markman 2001 randomised 532 women with 462 evaluable (227 IV and 235 IP) and compared an IV paclitaxel plus cisplatin regime with an experimental combination of two courses of high dose IV carboplatin followed by six courses of IV paclitaxel plus IP cisplatin. The maximum tumour diameter permitted for entry into this trial was 1 cm. Women in the IP arm had a median disease‐free survival of 28 months and a median overall survival of 63 months, both of which were superior to the median 22‐month progression‐free survival and median 52‐month overall survival associated with IV‐administered drugs (P = 0.02 and P = 0.05, respectively). Neutropenia, thrombocytopenia, and gastrointestinal and metabolic toxicities were significantly greater in the IP group such that 18% of these women actually received ≤ 2 cycles of intraperitoneal cisplatin. This was thought to be a consequence of the two initial high dose IV carboplatin treatments (AUC 9), designed to minimize the residual volume of disease prior to IP treatment.
GOG 172 was a similar trial for women with stage III, optimally de‐bulked ovarian cancer. In this trial 429 women were randomised and 14 were ineligible. This left 210 in the IV arm to receive paclitaxel 135 mg/m² plus cisplatin 75 mg/m² and 205 in the IP arm to receive IV paclitaxel 135 mg/m² plus IP cisplatin 100 mg/m² plus IP paclitaxel 60 mg/m² on day 8, with each regime delivered every 21 days for 6 cycles. The principal endpoint was progression‐free interval, with secondary endpoints being overall survival and toxicity and quality of life scores. Data provided by the study chair from the GOG meeting in January 2005 revealed the following: 429 women had entered, well balanced for prognostic factors and completion of treatment in each arm. There were significantly more grade 3 or 4 toxicities in the IP arm, including leukopenia, thrombocytopenia, gastrointestinal, renal, neurologic, fatigue, infection, metabolic and pain scores. This may be expected due to the use of a higher IV cisplatin dose (100 mg/m² v 75 mg/m²) and the addition of IP paclitaxel on day eight. Nevertheless, the median progression‐free interval and time to death for the IP group was prolonged, with the RR of recurrence 0.79 and a RR of death 0.75, suggesting a therapeutic advantage for IP therapy. These data have since been published (Armstrong 2006).
Yen 2009 randomised Stage III ovarian cancer patients to IP versus IV cisplatin or carboplatin after giving all IV paclitaxel. The objective of the trial and published manuscript was to construct a prognostic nomogram addressing the following risk factors: age, CA‐125, IP/IV delivery, stage, histology, and upper abdominal metastases. Out of 367 women recruited, 298 were analysed (152 in IV group and 146 in IP group). Chemotherapy consisted of a three hour infusion of paclitaxel (175mg/m²) on day one to all women. On day two, platinum (either 100 mg/m² cisplatin or 300 mg/m² carboplatin) was administered by either the IP or IV route. This was repeated every three weeks for six cycles provided serum creatinine was less than or equal to 2 mg/dl, WCC > 3000/mm3, and platelet count was > 80,000/mm3. IP therapy was discontinued and shifted to IV chemotherapy if catheter problems occurred. Published data consisted of a nomogram to predict survival, including an odds ratio (OR) for IV versus IP of 2.14 (95% CI 1.93 to 2.41) in favour of IP therapy. The investigators also showed that they could identify the subset of women who are least likely to benefit from IP chemotherapy, i.e. age > 80 years or with baseline CA 125 > 3000 U/ml or Stage IIIc disease or clear cell carcinoma or upper abdominal tumour metastases or colon resection, and may, therefore, be spared the potential complications. We obtained additional methodological information and unpublished data, including survival data and adverse effects (Grade 3/4), from the trial investigators.
Overall survival data were extracted from the reports and unpublished data of all but one of the trials (Polyzos 1999). Five trials provided HRs and covariates in Cox regression analysis (Alberts 1996; GOG 172; Markman 2001; Yen 2001; Yen 2009[unpublished data]). Most included age, tumour type and grade, performance status and residual disease as covariates prior to commencing chemotherapy. Six trials displayed Kaplan‐Meier survival curves (Alberts 1996; Gadducci 2000; GOG 172; Kirmani 1994; Markman 2001; Yen 2001) and we were able to construct a survival curve for the Zylberberg 1986 trial from individual patient data presented. One of these presented sufficient data to allow calculation of the HR using Parmar's methods (Gadducci 2000). The HR of death for Kirmani 1994 and Zylberberg 1986 was estimated from the Kaplan‐Meier survival curves. Only one of the trials (Alberts 1996) included subgroup analysis data, and so meta‐analysis on subgroup data could not be performed.
Disease‐free survival data and Kaplan‐Meier disease‐free survival curves were available from five trials (including unpublished data from Yen 2009). Two presented the calculated HRs (GOG 172; Markman 2001), two trials presented sufficient data to allow calculation of the HR using Parmar's methods (Gadducci 2000; Yen 2009) and the HR of recurrence for Kirmani 1994 was estimated from the Kaplan‐Meier diease‐free survival curves. The definitions of recurrence varied amongst these trials. For example, Kirmani 1994 used the Eastern Cooperative Oncology Group definition, Markman 2001 used date of entry to date of appearance of disease (clinical or radiological) or equated it to survival, Gadducci 2000 used date of entry to date of first progression and GOG 172 used date of randomisation to progression, death, or the date of last contact, whichever came first.
Eight of the nine included trials reported adverse effects with the exception of Zylberberg 1986. We obtained unpublished data from Yen 2009. Toxicity criteria included SWOG, Common toxicity criteria and WHO, all of which were comparable for the toxicities described. Gadducci 2000 used the WHO criteria but included 4 grades of alopecia and these data, therefore, could not be assimilated because the WHO criteria divide alopecia into only 3 grades.
QOL scores were reported in the GOG 172 trial using the Functional Assessment of Cancer Therapy scale with General, Neurotoxicity, Pain and Ovarian cancer sub‐scales (Wenzel 2004; Wenzel 2007).
Only GOG 172 assessed the effect of chemotherapy on QOL issues. Women receiving the higher dose IP therapy experienced more QOL disruption, pain and neurotoxicity when compared with those who received more conventional IV therapy. No other studies addressed this issue and, therefore, no meta‐analysis could be performed.
Median length of follow‐up was reported in eight of the nine trials: Alberts 1996 > 60 months; Gadducci 2000 60 months; GOG 172 > 60 months; Kirmani 1994 46 months; Markman 2001 > 60 months; Yen 2001 74 months; Yen 2009 62 months; and Zylberberg 1986 50 months. Polyzos 1999 did not provide data on length of follow‐up.
Excluded studies
Piccart 2003 compared four doses of maintenance IP chemotherapy compared to observation in 153 women who had had a complete response to conventional platinum‐based intravenous chemotherapy. After a median follow‐up of 8 years, 80 women (52%) had progressed with no difference in the pattern of relapse between the two groups. The respective hazard ratios for PFS and OS with 95% CI were 0.89 (0.59 to 1.33) and 0.82 (0.52 to 1.29). The authors concluded that this was suggestive of a treatment benefit. However, as maintenance therapy did not fit this review's eligibility criteria we excluded this trial.
Risk of bias in included studies
See Risk of Bias Tables in Characteristics of included studies and Figure 1.
1.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
We made attempts to contact lead investigators of trials (Gadducci 2000; Kirmani 1994; Polyzos 1999; Yen 2001; Yen 2009; Zylberberg 1986) where necessary to obtain missing details relating to methodological quality.
Allocation
Three United States multicentre trials (Alberts 1996; GOG 172; Markman 2001) followed guidelines for the conduct of inter‐group trials with adequate randomisation and allocation concealment. Randomisation and allocation concealment was also adequate for the Gadducci 2000 study. Yen 2001 described the use of random number tables but did not confirm a method of allocation concealment. Yen 2009 randomised women using a computer generated table but did not describe allocation concealment. Both randomisation and concealment of allocation were unclear in Kirmani 1994; Polyzos 1999 and Zylberberg 1986.
Blinding
Personnel and patient blinding after allocation was not possible because of the nature of the trials. Therefore, both recipients of care and care providers were aware of their assigned intervention. Assessor blinding was performed in Alberts 1996; GOG 172 and Markman 2001.
Selective reporting
Six studies reported intention‐to‐treat analyses (Alberts 1996; Gadducci 2000; GOG 172; Markman 2001; Yen 2001; Yen 2009). Most trials reported expected outcomes except for Yen 2009 where the stated objective was to construct a nomogram for survival, but additional data was provided by the investigators for the purposes of this review. Zylberberg 1986 did not report adverse effects.
Other potential sources of bias
We sub‐grouped the Alberts 1996; Gadducci 2000; GOG 172; Markman 2001; Yen 2001 and Yen 2009 trials as high quality, and the Kirmani 1994; Polyzos 1999 and Zylberberg 1986 trials as low quality and data from all are included in the meta‐analysis. It was not possible to comprehensively document the numbers of, or reasons for, withdrawal from treatment protocol. The number who received all, part or none of the planned treatment is recorded for each treatment arm in Table 3.
1. Courses of chemotherapy administered.
| Courses of chemotherapy administered | ||||||
| No. (%) women received all assigned chemotherapy |
No. (%) women received part of assigned chemotherapy |
No. (%) women received no assigned chemotherapy |
||||
| Trial | IP | IV | IP | IV | IP | IV |
| Alberts 1996 | 155/267 (58) | 162/279 (58) | 93/267 (35) | 114/279 (41) | 19/267 (7) | 3/279 (1) |
| GOG 172 | 86/205 (42) | 174/210 (83) | 103/205 (50) | 34/210 (16) | 16/205 (8) | 2/210 (1) |
| Gadducci 2000 | 26/46 (57) | 52/54 (96) | 13/46 (28) | 2/54 (4) | 7/46 (15) | 0 (0) |
| Kirmani 1994 | 25/33 (76) | 21/35 (60) | 8/33 (24) | 14/35 (40) | 0 (0) | 0 (0) |
| Markman 2001 | 52/235 (22) | 27/227 (13) | 16/235 (7) | 4/227 (2) | ||
| Yen 2001 | 41/55 (75) | 43/63 (68) | 0 (0) | 0 (0) | ||
| Yen 2009 | 72/146 (49) | 74/146 (51) | 0 (0) | |||
| Polyzos 1999 | No data | |||||
| Zylberberg 1986 | No data | |||||
In Yen 2009, 95/146 women (65%) received five or more cycles. No data were provided for those assigned to IV chemotherapy in this trial.
Effects of interventions
Summary of findings for the main comparison. Summary of survival outcomes.
| IP component therapy compared with IV therapy for initial management of primary epithelial ovarian cancer | ||||
|
Patient or population: Women with newly diagnosed epithelial ovarian cancer Settings: Hospital setting Intervention: Intraperitoneal component therapy Comparison: Intravenous therapy | ||||
| Outcomes | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments |
| Overall survival | HR 0.81 (0.72 to 0.90) |
2026 women (8 trials) |
⊕⊕⊕⊕ high | Homogeneous data. When only the six high quality trials were considered HR = 0.80 (0.72 to 0.90) . |
| Progression‐free survival | HR 0.78 (0.70 to 0.86) | 1311 women (5 trials) | ⊕⊕⊕⊕ high | Homogeneous data. |
| CI: Confidence interval; HR: Hazard Ratio | ||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||
Summary of findings 2. Summary of severe adverse effects.
| IP component therapy compared with IV therapy for initial management of primary epithelial ovarian cancer | ||||||
|
Patient or population: Women with newly diagnosed epithelial ovarian cancer Settings: Hospital setting Intervention: Intraperitoneal component therapy Comparison: Intravenous therapy | ||||||
| Severe adverse effects (G3/4) outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| IV | IP | |||||
| fever | 46 per 1000 | 75 per 1000 (52 to 109) | RR 1.64 (1.13 to 2.38) |
1797 women (5 trials) |
⊕⊕⊕⊕ high | High quality trials only. |
| fatigue | 75 per 1000 | 174 per 1000 (80 to 380) | RR 2.32 (1.06 to 5.07) |
1171 women (3 trials) |
⊕⊕⊕⊕ high | High quality trials only. |
| gastrointestinal AEs | 193 per 1000 | 330 per 1000 (247 to 436) | RR 1.71 (1.28 to 2.26) | 1339 women (5 trials) | ⊕⊕⊕⊕ high | I² = 48%. When only the four high quality trials were considered, data were homogeneous and RR = 1.90, 95% CI 1.57 to 2.30. Corresponding risk = 355 per 1000 (294 to 430) |
| infection | 34 per 1000 | 114 per 1000 (70 to 185) | RR 3.34 (2.06 to 5.43) | 1171 women (3 trials) | ⊕⊕⊕⊕ high | High quality trials only. |
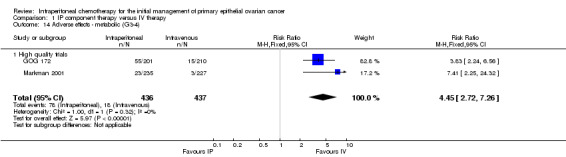
| metabolic AEs | 21 per 1000 | 93 per 1000 (57 to 152) | RR 4.45 (2.72 to 7.26) | 873 women (2 trials) | ⊕⊕⊕⊕ high | High quality trials only. |
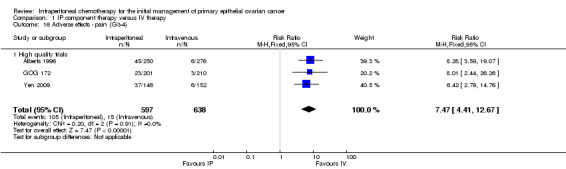
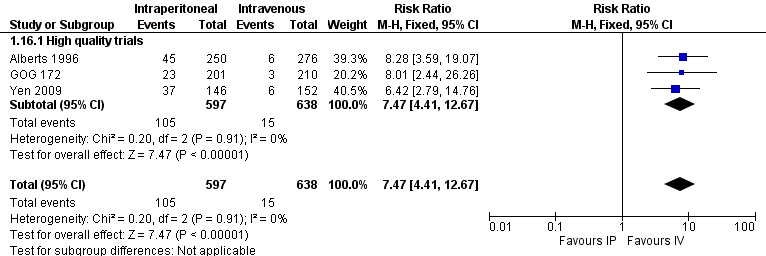
| pain | 24 per 1000 | 179 per 1000 (106 to 304) | RR 7.47 (4.41 to 12.67) | 1235 women (3 trials) | ⊕⊕⊕⊕ high | High quality trials only. |
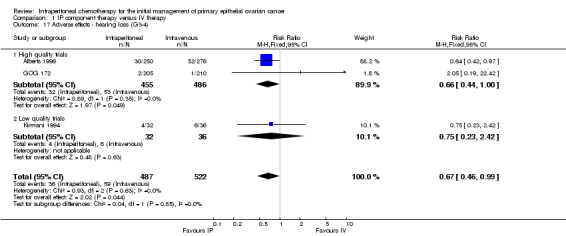
| hearing loss | 113 per 1000 | 76 per 1000 (52 to 112) | RR 0.67 (0.46 to 0.99) | 1009 women (3 trials) | ⊕⊕⊕⊕ high | When only the two high quality trials were considered, the RR (CI) was similar. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; AEs: Adverse effects | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
The assumed risk is the average control group (IV) risk across included studies.
Overall survival (time to death)
Eight trials (2026 women) were included in this meta‐analysis. There was a significant improvement in overall survival in the IP intervention group (HR 0.81, 95% CI 0.72 to 0.90; Analysis 1.1; Figure 2). This remained when only high quality trials were considered (six trials; HR 0.80, 95% CI 0.72 to 0.90). Data were homogeneous and there was no significant difference between subgroups. Some experts have argued that the excluded trial (Piccart 2003) should be included on the basis that the addition of IP chemotherapy after completion of initial surgery and IV chemotherapy may still be described as part of primary treatment for ovarian cancer. If these data are included, the HR for time to death is similar. Furthermore, if the analysis is restricted to trials that used the same chemotherapy regimes in each arm, the HR remains similar (three trials; 0.79, 95%CI 0.67 to 0.92; Analysis 1.2)
1.1. Analysis.
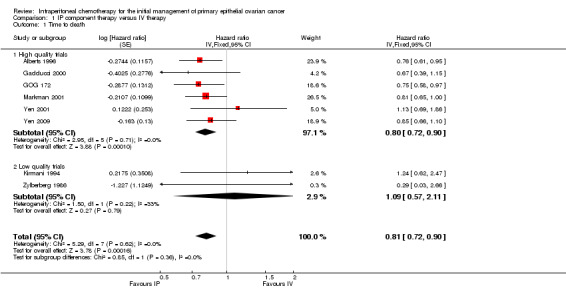
Comparison 1 IP component therapy versus IV therapy, Outcome 1 Time to death.
2.
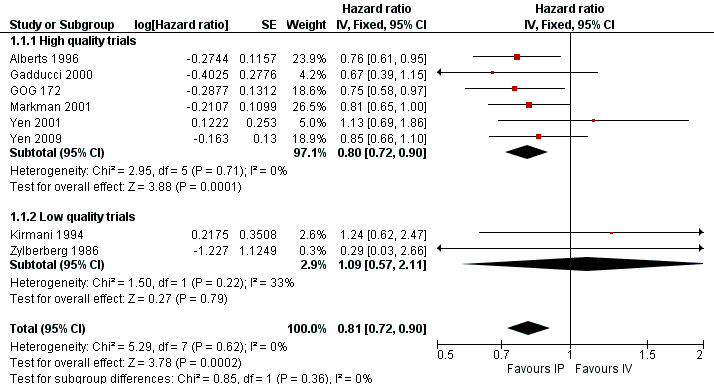
Forest plot of comparison: 1 IP component therapy versus IV therapy, outcome: 1.1 Time to death.
1.2. Analysis.
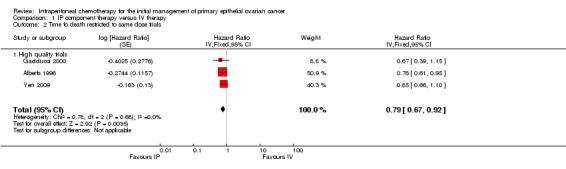
Comparison 1 IP component therapy versus IV therapy, Outcome 2 Time to death restricted to same dose trials.
Progression‐free survival (time to recurrence)
Five trials (1311 women) were included in the meta‐analysis of time to recurrence. There was a significant improvement in progression‐free survival in the IP intervention group (HR 0.78, 95% CI 0.70 to 0.86; Analysis 1.3; Figure 3). Data were homogeneous. Polyzos 1999 provided limited survival data (dichotomous) that were unusable in the meta‐analysis (33/46 = 72% of controls versus 33/44 = 75% of IP arm); time to progression was 19 (8 to 62+) and 18 months (6 to 72+) with the median survival advantage of one month in favour of the intraperitoneal arm.
1.3. Analysis.
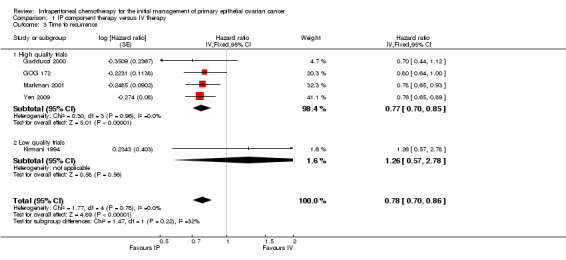
Comparison 1 IP component therapy versus IV therapy, Outcome 3 Time to recurrence.
3.
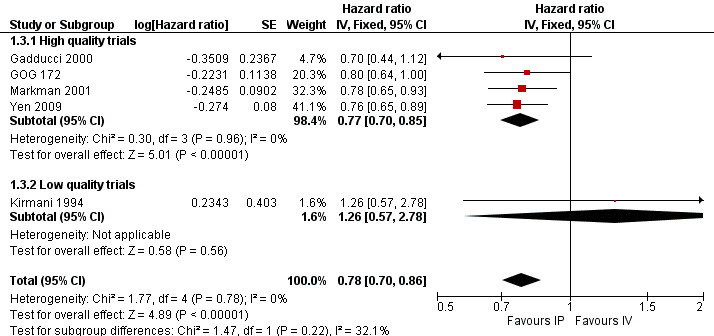
Forest plot of comparison: 1 IP component therapy versus IV therapy, outcome: 1.3 Time to recurrence.
Severe adverse effects
The following severe adverse effects (AEs grade 3/4) were more likely to occur in the IP group:
fever (five trials, 1797 women; RR 1.64, 95% CI 1.13 to 2.38; I² = 20%; Analysis 1.10; Figure 4);
fatigue (three trials, 1171 women; RR 2.32, 95% CI 1.06 to 5.07; I² = 71%; Analysis 1.11);
gastrointestinal AEs (five trials, 1339 women; RR 1.70, 95% CI 1.28 to 2.26; I² = 48%; Analysis 1.12; Figure 5). In the 'high quality trials' subgroup, I² = 0% (four trials, 1271 women; RR 1.90, 95% CI 1.57 to 2.30);
infection (three trials, 1171 women; RR 3.34, 95% CI 2.06 to 5.43; I² = 0%; Analysis 1.13; Figure 6);
metabolic AEs (two trials, 873 women; RR 4.45, 95% CI 2.72 to 7.26; I² = 0%; Analysis 1.14);
pain (three trials, 1235 women; RR 7.47; 95% CI 4.41 to 12.67; I² = 0% Analysis 1.16; Figure 7).
1.10. Analysis.
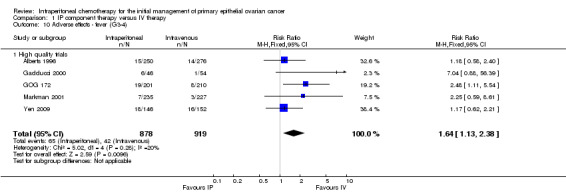
Comparison 1 IP component therapy versus IV therapy, Outcome 10 Adverse effects ‐ fever (G3‐4).
4.
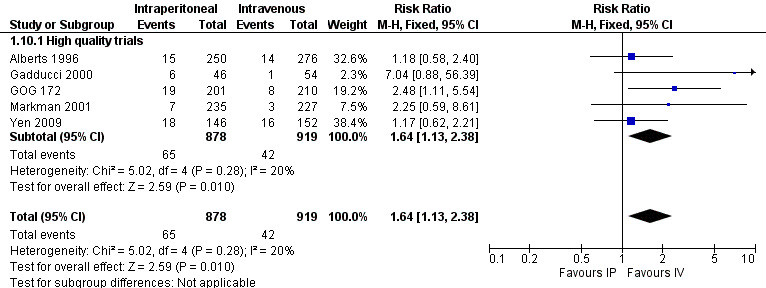
Forest plot of comparison: 1 IP component therapy versus IV therapy, outcome: 1.10 Adverse effects ‐ fever (G3‐4).
1.11. Analysis.
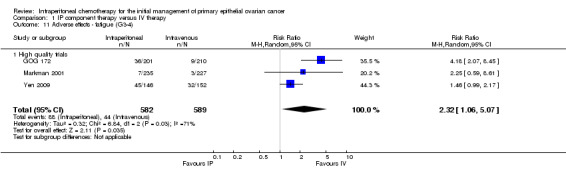
Comparison 1 IP component therapy versus IV therapy, Outcome 11 Adverse effects ‐ fatigue (G3‐4).
1.12. Analysis.
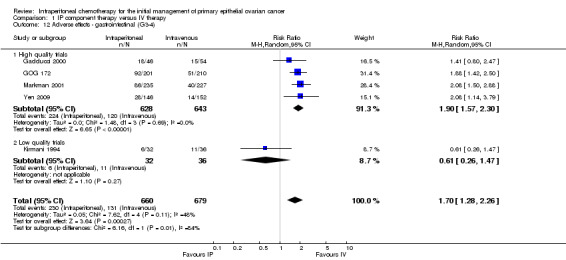
Comparison 1 IP component therapy versus IV therapy, Outcome 12 Adverse effects ‐ gastrointestinal (G3‐4).
5.
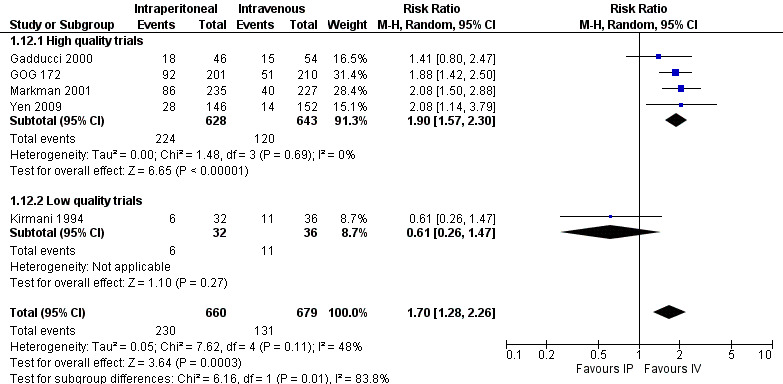
Forest plot of comparison: 1 IP component therapy versus IV therapy, outcome: 1.12 Adverse effects ‐ gastrointestinal (G3‐4).
1.13. Analysis.
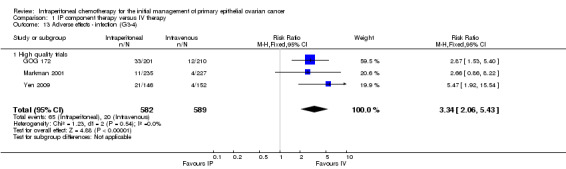
Comparison 1 IP component therapy versus IV therapy, Outcome 13 Adverse effects ‐ infection (G3‐4).
6.
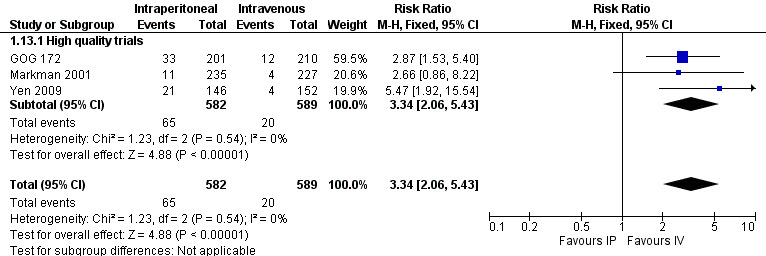
Forest plot of comparison: 1 IP component therapy versus IV therapy, outcome: 1.13 Adverse effects ‐ infection (G3‐4).
1.14. Analysis.
Comparison 1 IP component therapy versus IV therapy, Outcome 14 Adverse effects ‐ metabolic (G3‐4).
1.16. Analysis.
Comparison 1 IP component therapy versus IV therapy, Outcome 16 Adverse effects ‐ pain (G3‐4).
7.
Forest plot of comparison: 1 IP component therapy versus IV therapy, outcome: 1.16 Adverse effects ‐ pain (G3‐4).
Hearing loss was more common in the IV group (three studies, 1009 women, RR 0.67; 95% CI 0.46 to 0.99; Analysis 1.17). There were no significant differences between interventions for haematological AEs: anaemia (Analysis 1.4); thrombocytopenia (Analysis 1.5) and leukopenia (Analysis 1.6). However, substantial heterogeneity (I² > 60%) was noted in these meta‐analyses. Substantial heterogeneity also occurred in the meta‐analyses of renal, neurological and pulmonary AEs (no significant difference between interventions): Analysis 1.7, I² = 57% Analysis 1.15; I² = 69% and Analysis 1.8, I² = 79%, respectively. This could not be explained by subgroup differences.
1.17. Analysis.
Comparison 1 IP component therapy versus IV therapy, Outcome 17 Adverse effects ‐ hearing loss (G3‐4).
1.4. Analysis.
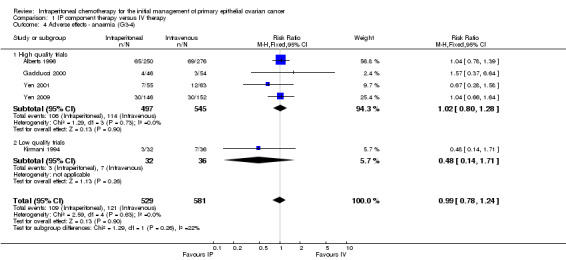
Comparison 1 IP component therapy versus IV therapy, Outcome 4 Adverse effects ‐ anaemia (G3‐4).
1.5. Analysis.
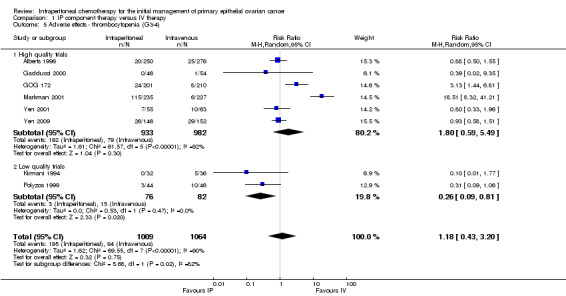
Comparison 1 IP component therapy versus IV therapy, Outcome 5 Adverse effects ‐ thrombocytopenia (G3‐4).
1.6. Analysis.
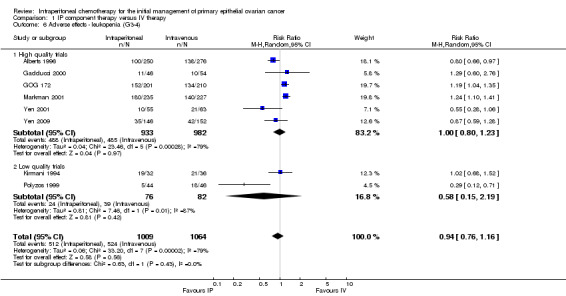
Comparison 1 IP component therapy versus IV therapy, Outcome 6 Adverse effects ‐ leukopenia (G3‐4).
1.7. Analysis.
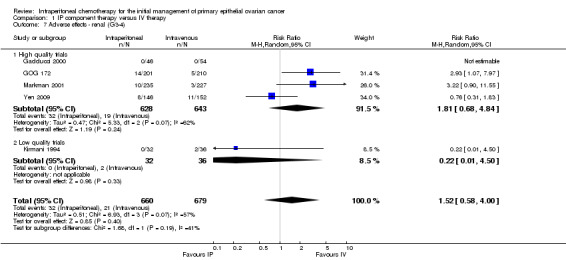
Comparison 1 IP component therapy versus IV therapy, Outcome 7 Adverse effects ‐ renal (G3‐4).
1.15. Analysis.
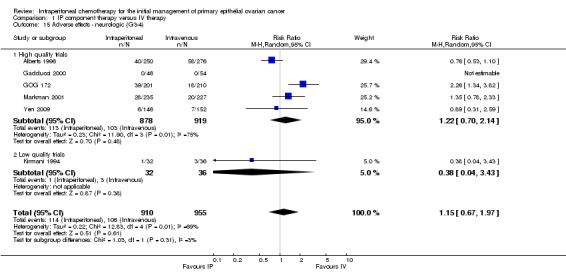
Comparison 1 IP component therapy versus IV therapy, Outcome 15 Adverse effects ‐ neurologic (G3‐4).
1.8. Analysis.
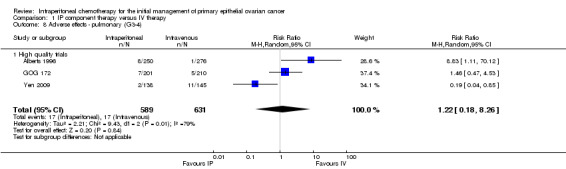
Comparison 1 IP component therapy versus IV therapy, Outcome 8 Adverse effects ‐ pulmonary (G3‐4).
Quality of life
Only the GOG 172 trial assessed QOL as an outcome measure (Wenzel 2007) and, therefore, no meta‐analysis was performed. Women who received higher dose IP therapy (paclitaxel 135 mg/m² IV, cisplatin 100 mg/m² IP, paclitaxel 60 mg/m² IP) experienced more QOL disruption when compared to those who received the more conventional dose of IV therapy (paclitaxel 135 mg/m² IV, cisplatin 75 mg/m² IV). Women in the IP arm reported worse QOL and pain prior to the fourth chemotherapy cycle (P < 0.001) and worse QOL 3 to 6 weeks post‐treatment (P = 0.009). The GOG 172 protocol exposed women in the IP arm to additional taxol and neurotoxicity was significantly worse in the IP arm 3 to 6 weeks after completing chemotherapy (P = 0.0004), and one year later (P = 0.0018). There were no significant QOL or PAIN score differences between arms one year post‐treatment. Catheter‐related complications of IP drug administration were discussed in all of the trials but data retrieved from them were insufficient to produce a meta‐analysis. Catheter blockage was described in Kirmani 1994 (10%), Gadducci 2000 (8.7%), Yen 2001 (25.5%), and GOG 172 (in 7% of those who stopped IP therapy this was the primary reason). Catheter‐related infection was reported in Yen 2001 (12.8%), Markman 2001 (severe infection noted in 4.7%) and GOG 172 (in 19.5% of those who stopped IP therapy this was the primary reason). In GOG 172, of the 205 women in the IP arm 118 (58%) did not complete 6 cycles of IP therapy. Intraperitoneal therapy was stopped in 34 (28.8%) for reasons unrelated to the peritoneal access device, but catheter failures occurred in 49 (41.5%) women. Catheter complications were the primary cause for discontinuing IP therapy in 39% participants. Descending colon or rectosigmoid colon resection appeared to decrease the number of cycles successfully delivered (P = 0.022). Also, data from the trial suggested that IP administration of paclitaxel might in itself increase complications.
Discussion
Summary of main results
We considered six of the nine studies to be of high quality. The administration of a component of chemotherapy via the intraperitoneal route, in the initial management of primary epithelial ovarian cancer, improved overall survival and disease‐free survival (see Table 1). Intraperitoneal chemotherapy was associated with significantly more pain, fever, infection, metabolic complications and gastrointestinal adverse effects than IV chemotherapy (see Table 2).
Overall completeness and applicability of evidence
Primary outcomes
Survival data were statistically homogeneous and the results are robust to the findings of the original review. Women in the IP arms of GOG 172 and Markman 2001 received a higher total chemotherapy dose initially; however, more women finished their IV course of therapy (Table 3) and very‐high‐dose‐chemotherapy intravenously did not improve clinical outcome. Women in the IP arm of GOG 172 received dose‐dense paclitaxel which may have an advantage over standard taxane administration (Katsumata 2009). When we excluded these two trials from the survival meta‐analyses for exploratory purposes, the improvement in survival in the IP group remained. Alberts 1996; Gadducci 2000; Polyzos 1999 and Yen 2009 describe trials where the chemotherapy regimes are similar in each arm and the survival outcome was similar when the analysis was restricted to these trials. Thus, we concluded that the intraperitoneal route is associated with an increased cure rate beyond the simple addition of more agents or a higher dose.
Secondary outcomes
Haematological (leukopenia, thrombocytopenia), renal, neurologic and pulmonary toxicity data were heterogeneous and so the meta‐analyses with respect to these outcomes are less reliable. This heterogeneity can be explained clinically by the different dosages, variable schedules of chemotherapy administration and different drugs used, not only between the trials, but also between arms of the same trial. It is not surprising that the Markman 2001 and GOG 172 studies revealed significantly greater haematological toxicity in the IP arm in contrast to the findings from Alberts 1996; Gadducci 2000; Polyzos 1999; Yen 2001 and Zylberberg 1986 who all used regimes which could be expected to be more balanced with respect to this adverse effect. The Markman 2001 trial used two cycles of very high dose (AUC 9) carboplatin to prime women allocated to intraperitoneal chemotherapy. The conventional IV dose is limited to AUC 6 because the additional toxicity from a higher dose does not translate into a survival advantage. This, in itself, will explain the haematologic toxicity in this group of women. The GOG 172 trial included a higher total dose of both cisplatin and paclitaxel in the IP arm and this, too, may explain the additional haematological toxicity seen here. Toxicity from IP chemotherapy including catheter‐related adverse effects and haematologic toxicity are major reasons for the lack of acceptance of this treatment option in current gynaecologic oncology practice. More attention has been focused on ameliorating this, including modification of the GOG 172 regime with lower dose IP cisplatin and slower IP infusion (Chi 2010).
Data on pulmonary toxicity, including transient dyspnoea, were also heterogeneous and included three studies (Alberts 1996; GOG 172; Yen 2009) one of which revealed a significant increase in toxicity in the IP arm (Alberts 1996). This can be explained clinically by the rapid IP injection of two litres of normal saline in this trial, resulting in compression of the base of the lungs and hence transient dyspnoea.
QOL comparison was only assessed in the GOG 172 trial. More disruption was noted in the IP arm during and shortly after treatment but quality of life improved in both arms over time. Only neurotoxicity remained significantly greater in the IP group 12 months after completion of treatment (Wenzel 2007). This difference in QOL may be due to the higher total dose of both cisplatin and paclitaxel in the IP arm rather than the method of chemotherapy administration.
When we restricted the analysis to trials that compared the same chemotherapy regimes and differed only by route of administration, we found that the IP catheter causes pain but there are no other detectable differences that are statistically significant.
Treatment regimens
The trials in this review studied very different regimens, most of them including cisplatin in the IP arm. However, current standard therapy for the primary treatment of ovarian cancer is carboplatin with or without a taxane (du Bois 2003; Ozols 2003). Carboplatin has been used with good effect and it remains in the peritoneal cavity for longer than if it was given by the intravenous route. Twenty‐four hour platinum AUC in the peritoneal cavity is approximately 17 times higher when carboplatin is administered IP compared with IV administration, whilst 24 hour serum concentrations are the same.
Multiple studies have addressed the potential benefits of carboplatin over cisplatin for intraperitoneal use with reports of better tolerance, less discontinuation of therapy, less impact on quality of life, but with greater haematologic toxicity (Fujiwara 2008; Fujiwara 2009; Fujiwara 2012; Gray 2010; Zeimet 2009). Hence, IP carboplatin has been included in all three ongoing inter‐group trials: GOG 0252, NCIC CTG OV.21 and GOTIC‐001/JGOG‐3019 (see Table 4 and Characteristics of ongoing studies).
2. Currently recruiting GCIG RCTs.
| GOG 0252 | |
| Arm 1 | Carboplatin AUC 6 IV for 6 cycles, given every 21 days + paclitaxel 80 mg/m2 IV on days 1, 8, and 15 + bevacizumab 15 mg/kg IV on day 1 in cycles 2‐6, then bevacizumab alone for cycles 7‐22 |
| Arm 2 | Carboplatin AUC 6 IP for 6 cycles, given every 21 days + paclitaxel 80 mg/m2 IV on days 1, 8, and 15 + bevacizumab 15 mg/kg IV on day 1 in cycles 2‐6, then bevacizumab alone for cycles 7‐22 |
| Arm 3 | Cisplatin 75 mg/m2 IP on day 2 for 6 cycles, given every 21 days + paclitaxel 135 mg/m2 IV on day 1 + paclitaxel 60 mg/m2 IP on day 8 + bevacizumab 15 mg/kg IV on day 1 in cycles 2‐6, then bevacizumab alone for cycles 7‐22 |
|
NCIC CTG OV21 3 ‐ 4 cycles of neoadjuvant platinum‐based chemotherapy after histologically confirmed initial diagnosis of advanced epithelial ovarian cancer, serous type peritoneal or fallopian tube cancer, followed by optimal cytoreduction (< 1 cm residual) then randomisation | |
| Phase II portion of trial evaluating two IP regimes and one IV regime | |
| Arm 1 | Carboplatin AUC 5‐6 IV for 3 cycles, given every 21 days + paclitaxel 135 mg/m2 IV on day 1, and 60 mg/m2 on day 8 |
| Arm 2 | Cisplatin 75 mg/m2 IP for 3 cycles, given every 21 days + paclitaxel 135 mg/m2 IV on day 1, and 60 mg/m2 on day 8 |
| Arm 3 | Carboplatin AUC 5‐6 IP for 3 cycles, given every 21 days + paclitaxel 135 mg/m2 IV on day 1, and 60 mg/m2 on day 8 |
| Phase III portion of trial comparing Arm 1 to either Arm 2 or Arm 3 | |
| GOTIC‐001/JGOG 3019 | |
| Arm 1 | Carboplatin AUC 6 IV for 6‐8 cycles, given every 21 days + paclitaxel 80 mg/m2 IV on days 1, 8, and 15 |
| Arm 2 | Carboplatin AUC 6 IP for 6‐8 cycles, given every 21 days + paclitaxel 80 mg/m2 IV on days 1, 8, and 15 |
GOG 0252 is an ongoing phase III trial comparing two different IP regimens to IV chemotherapy, all given with bevacizumab during chemotherapy and as consolidation. They also incorporate dose‐dense paclitaxel in all three arms, based on the results of the JGOG study reporting a significant improvement in survival associated with dose‐dense paclitaxel (Katsumata 2009). The NCIC CTG OV.21 phase III trial is comparing IP/IV against IV chemotherapy after three to four courses of neoadjuvant chemotherapy. The Japanese GOG and Gynecologic Oncology Trial and Investigations Consortium (GOTIC) are currently recruiting to a phase III trial comparing IP carboplatin versus IV carboplatin, both given with weekly dose‐dense IV paclitaxel (GOTIC‐001/JGOG‐3019). Results of these trials should supplement the evidence supporting intraperitoneal chemotherapy for advanced ovarian cancer.
Women likely to benefit most from IP chemotherapy
Evidence from GOG 172; Makhija 2001 and Yen 2009 suggests that distal colonic resections predispose to catheter complications and reduce the number of successful cycles delivered. We consider upper abdominal bowel surgery to be a relative contraindication as bowel injury from a catheter may be avoided by intermittent intraperitoneal injections. Current opinion, however, favours preferentially selecting IP therapy for women with minimal residual disease, who have not required upper abdominal bowel surgery, and have chemosensitive tumours such as serous papillary morphology. This group is most likely to benefit and to gain an exaggerated survival advantage.
Mathematical modelling of the data reported by Yen 2009 shows that one, two or three cycles of IP therapy were all associated with a median survival of about 44 months, compared with 53 months if five or more cycles were given. This implies that we should aim for five cycles of IP chemotherapy. The modelling also suggests that the prognosis is so poor for elderly women with clear cell tumours, especially those who have not been optimally debulked, that the added toxicity and side effects from IP therapy over IV treatment exceeds the benefits.
Authors' conclusions
Implications for practice.
The results of this review support the use of IP chemotherapy. IP chemotherapy increases both overall survival and progression‐free survival in women with advanced ovarian cancer. The potential for catheter‐related complications and complications relating to toxicity need to be taken into consideration in dialogue between the clinician and patient when deciding on the most appropriate form of treatment for the individual. However, this meta‐analysis provides the current most reliable estimates of the survival benefits associated with IP therapy and should be used as part of this decision making process.
It is difficult to translate these data into real advantages. A HR of 0.81 and 0.78 for time to death and time to recurrence reflects a significant survival advantage. However, one woman out of 13 is likely to suffer fever, one in nine will get an infection, one in six will experience pain from the catheter that she would not have experienced had she received intravenous chemotherapy alone, and one in three might suffer serious (grade 3 or 4) gastrointestinal toxicity to gain this survival advantage. These complications might be reduced by selecting a less toxic regime or better administration technique. Until new data are available, we can tell women that the trials show that the IP route has significant difficulties but there is a survival advantage to the IP route of administration.
Implications for research.
This meta‐analysis cannot determine if the taxane component should be given intraperitoneally. The theoretical disadvantage of intraperitoneal taxane is that it is associated with a significant risk of catheter adhesions and the theoretical therapeutic advantages are limited. Taxanes are complex molecules that are not well absorbed from the peritoneal cavity and consequently very high concentrations are found intraperitoneally after intravenous administration. The role of intraperitoneal paclitaxel is being addressed specifically in the NCIC CTG OV.21 trial.
The three ongoing GCIG trials will add data to answer the outstanding questions around the optimal IP drug, dose, combination and number of courses of IP chemotherapy. There has been some progress made on elucidating which subgroups may benefit from IP chemotherapy (Yen 2009). Yen 2009 modelling also showed that we should aim for 5 cycles of IP therapy, but more can be done in this area.
GOG 0252 stratifies the participants by stage and size of residual disease which should help to determine whether IP chemotherapy is beneficial for more advanced disease. IP catheter complications include blockage, catheter leakage, infection, diarrhoea, bowel perforation and fistula formation. These problems might be reduced by a single‐use catheter rather than the semi‐permanent subcutaneous implantable port and catheter system, like the Tenckhoff, Port‐A‐Cath or BardPort systems; this is also in need of evaluation.
To date, there are few QOL data from trials using similar chemotherapy regimes and more data on this outcome, when the ongoing GCIG trials mature, will be invaluable. More trials comparing different IP chemotherapy administration techniques may be worthwhile. The main focus must be to compare IP carboplatin with cisplatin. VEGF inhibitors are effective at reducing ascites and have a significant progression‐free survival advantage (Zhao 2013) but their role in IP treatment is unknown and, to date, there are no data on their use with IP chemotherapy. Similarly, there is limited evidence evaluating heated IP therapy but a small Italian phase II trial has recently been published with promising results in terms of survival with comparable morbidity (Deraco 2011), and a Dutch trial is ongoing (Hyperthermic IP therapy).
What's new
| Date | Event | Description |
|---|---|---|
| 13 January 2016 | Amended | Acknowledgements amended |
History
Protocol first published: Issue 3, 2005 Review first published: Issue 1, 2006
| Date | Event | Description |
|---|---|---|
| 17 December 2015 | New citation required but conclusions have not changed | No new studies identified for inclusion. |
| 17 December 2015 | New search has been performed | Searches updated. |
| 26 September 2011 | New search has been performed | Search updated: one new trial (Yen 2009), five additional publications related to the GOG 172 trial and three ongoing trials (GOG 0252; GOTIC‐001/JGOG‐3019; NCIC CTG OV.21) were identified. |
| 26 September 2011 | New citation required but conclusions have not changed | Review content updated: one new trial and three ongoing studies added. New author added. |
| 17 August 2010 | New search has been performed | Search updated |
| 9 September 2008 | Amended | Converted to new review format. |
| 1 May 2007 | New search has been performed | Minor update: 01/05/07 The literature search as described in the search strategy sections was updated on 23 February 2007. No new relevant studies were identified. |
| 15 November 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We gratefully acknowledge the help and advice of Chris Williams, Heather Dickinson, Gail Quinn, Jill Porthouse, Anne Oestmann and Clare Jess, all of the Cochrane Gynaecological Cancer Review Group. We are also grateful to Dr Deborah Armstrong of the Gynecologic Oncology Group for providing data from the GOG 172 randomised controlled trial prior to publication and Dr CM Juang for providing unpublished data from Yen 2009. Finally, we would like to thank all those women who took part in the trials and contributed to this research.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Gynaecological, Neuro‐oncology and Orphan Cancer Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. MEDLINE Ovid Search strategy
1 exp Ovarian Neoplasms/ 2 (ovar* adj5 (cancer* or neoplasm* or malignan* or tumor* or tumour* or carcinom*)).mp. 3 1 or 2˜ 4 exp Infusions, Parenteral/ 5 Injections, Intraperitoneal/ 6 intraperitoneal.mp. 7 intra‐peritoneal.mp. 8 peritone*.mp. 9 regional.mp. 10 parenteral.mp. 11 4 or 5 or 6 or 7 or 8 or 9 or 10 12 drug therapy.fs. 13 exp Antineoplastic Agents/ 14 exp Antineoplastic Combined Chemotherapy Protocols/ 15 exp Chemotherapy, Adjuvant/ 16 chemotherap*.mp. 17 cisplatin.mp. 18 carboplatin.mp. 19 cyclophosphamide.mp. 20 etoposide.mp. 21 paclitaxel.mp. 22 doxorubicin.mp. 23 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 24 3 and 11 and 23 25 randomized controlled trial.pt. 26 controlled clinical trial.pt. 27 randomized.ab. 28 placebo.ab. 29 drug therapy.fs. 30 randomly.ab. 31 trial.ab. 32 25 or 26 or 27 or 28 or 29 or 30 or 31 33 24 and 32
key: mp = title, original title, abstract, name of substance word, subject heading word, unique identifier pt = publication type ab = abstract fs = floating subheading
Appendix 2. EMBASE Ovid search strategy
1 exp ovary tumor/ 2 (ovar* adj5 (cancer* or neoplasm* or malignan* or tumor* to tumour* or carcinom*)).mp. 3 1 or 2 4 exp infusion/ 5 intraperitoneal drug administration/ 6 intraperitoneal.mp. 7 intra‐peritoneal.mp. 8 peritone*.mp. 9 regional.mp. 10 parenteral.mp. 11 4 or 5 or 6 or 7 or 8 or 9 or 10 12 dt.fs. 13 exp antineoplastic agent/ 14 exp chemotherapy/ 15 chemotherap*.mp. 16 cisplatin.mp. 17 carboplatin.mp. 18 cyclophosphamide.mp. 19 etoposide.mp. 20 paclitaxel.mp. 21 doxorubicin.mp. 22 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 23 3 and 11 and 22 24 crossover procedure/ 25 double blind procedure/ 26 randomized controlled trial/ 27 single blind procedure/ 28 random*.mp. 29 factorial*.mp. 30 crossover*.mp. 31 cross over*.mp. 32 cross‐over*.mp. 33 placebo*.mp. 34 (double* adj blind*).mp. 35 (singl* adj blind*).mp. 36 assign*.mp. 37 allocat*.mp. 38 volunteer*.mp. 39 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 40 23 and 39
key: mp = title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer
Appendix 3. CENTRAL search strategy
#1 MeSH descriptor Ovarian Neoplasms explode all trees #2 ovar* near/5 (cancer* or neoplasm* or malignan* or tumor* or tumour* or carcinom*) #3 (#1 OR #2) #4 MeSH descriptor Infusions, Parenteral explode all trees #5 MeSH descriptor Injections, Intraperitoneal explode all trees #6 intraperitoneal #7 intra‐peritoneal #8 peritone* #9 regional #10 parenteral #11 (#4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10) #12 Any MeSH descriptor with qualifier: DT #13 MeSH descriptor Antineoplastic Agents explode all trees #14 MeSH descriptor Antineoplastic Combined Chemotherapy Protocols explode all trees #15 MeSH descriptor Chemotherapy, Adjuvant explode all trees #16 chemotherap*.mp. #17 cisplatin #18 carboplatin #19 cyclophosphamide #20 etoposide #21 paclitaxel #22 doxorubicin #23 (#12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22) #24 (#3 AND #11 AND #23)
Appendix 4. Methodological differences of this review with previous versions
Assessment of risk of bias
The following methods of assessment of bias were used in earlier versions of this review. Where the method of allocation was truly random e.g. computer generated random numbers, coin tossed etc, this was coded as:
(a) Adequate, e.g. women allocated to treatment arms using a sequence of random (or pseudo‐random) numbers; (b) Inadequate randomisation, e.g. women allocated to treatment arms on basis of clinic id, surname, month, day of the week; (c) Unclear.
Where there was proper concealment of allocation; this was coded as:
(a) Adequate, e.g. randomisation carried out on a central site and transmitted to treatment providers by telephone, fax, or sealed opaque envelope; (b) Inadequate; (c) Unclear.
Where blinding of the outcome assessor was performed, this was recorded as:
(a) Blinding of the outcome assessor was performed; (b) Blinding of the outcome assessor was not performed; (c) Unclear.
Where loss to follow‐up was accounted for, numbers lost to follow‐up in each treatment arm were noted.
It was noted whether intention‐to‐treat analysis (understood to mean that participants were analysed in the groups to which they were initially assigned, regardless of which treatment they actually received) was performed. This was recorded as:
(a) Intention‐to‐treat analysis was performed; (b) Intention‐to‐treat analysis was not performed; (c) Unclear.
An overall description of the quality of trials is included in the report. Trials were defined as being of low quality if they were of poor quality in at least three of the above areas (Table 1).
Data and analyses
Comparison 1. IP component therapy versus IV therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Time to death | 8 | Hazard ratio (Fixed, 95% CI) | 0.81 [0.72, 0.90] | |
| 1.1 High quality trials | 6 | Hazard ratio (Fixed, 95% CI) | 0.80 [0.72, 0.90] | |
| 1.2 Low quality trials | 2 | Hazard ratio (Fixed, 95% CI) | 1.09 [0.57, 2.11] | |
| 2 Time to death restricted to same dose trials | 3 | Hazard Ratio (Fixed, 95% CI) | 0.79 [0.67, 0.92] | |
| 2.1 High quality trials | 3 | Hazard Ratio (Fixed, 95% CI) | 0.79 [0.67, 0.92] | |
| 3 Time to recurrence | 5 | Hazard ratio (Fixed, 95% CI) | 0.78 [0.70, 0.86] | |
| 3.1 High quality trials | 4 | Hazard ratio (Fixed, 95% CI) | 0.77 [0.70, 0.85] | |
| 3.2 Low quality trials | 1 | Hazard ratio (Fixed, 95% CI) | 1.26 [0.57, 2.78] | |
| 4 Adverse effects ‐ anaemia (G3‐4) | 5 | 1110 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.78, 1.24] |
| 4.1 High quality trials | 4 | 1042 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.80, 1.28] |
| 4.2 Low quality trials | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.14, 1.71] |
| 5 Adverse effects ‐ thrombocytopenia (G3‐4) | 8 | 2073 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.43, 3.20] |
| 5.1 High quality trials | 6 | 1915 | Risk Ratio (M‐H, Random, 95% CI) | 1.80 [0.59, 5.49] |
| 5.2 Low quality trials | 2 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.09, 0.81] |
| 6 Adverse effects ‐ leukopenia (G3‐4) | 8 | 2073 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.76, 1.16] |
| 6.1 High quality trials | 6 | 1915 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.80, 1.23] |
| 6.2 Low quality trials | 2 | 158 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.15, 2.19] |
| 7 Adverse effects ‐ renal (G3‐4) | 5 | 1339 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.58, 4.00] |
| 7.1 High quality trials | 4 | 1271 | Risk Ratio (M‐H, Random, 95% CI) | 1.81 [0.68, 4.84] |
| 7.2 Low quality trials | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.01, 4.50] |
| 8 Adverse effects ‐ pulmonary (G3‐4) | 3 | 1220 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.18, 8.26] |
| 8.1 High quality trials | 3 | 1220 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.18, 8.26] |
| 9 Adverse effects ‐ cardiovascular (G3‐4) | 3 | 1152 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.85, 2.45] |
| 9.1 High quality trials | 3 | 1152 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.85, 2.45] |
| 10 Adverse effects ‐ fever (G3‐4) | 5 | 1797 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [1.13, 2.38] |
| 10.1 High quality trials | 5 | 1797 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [1.13, 2.38] |
| 11 Adverse effects ‐ fatigue (G3‐4) | 3 | 1171 | Risk Ratio (M‐H, Random, 95% CI) | 2.32 [1.06, 5.07] |
| 11.1 High quality trials | 3 | 1171 | Risk Ratio (M‐H, Random, 95% CI) | 2.32 [1.06, 5.07] |
| 12 Adverse effects ‐ gastrointestinal (G3‐4) | 5 | 1339 | Risk Ratio (M‐H, Random, 95% CI) | 1.70 [1.28, 2.26] |
| 12.1 High quality trials | 4 | 1271 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [1.57, 2.30] |
| 12.2 Low quality trials | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.26, 1.47] |
| 13 Adverse effects ‐ infection (G3‐4) | 3 | 1171 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.34 [2.06, 5.43] |
| 13.1 High quality trials | 3 | 1171 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.34 [2.06, 5.43] |
| 14 Adverse effects ‐ metabolic (G3‐4) | 2 | 873 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.45 [2.72, 7.26] |
| 14.1 High quality trials | 2 | 873 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.45 [2.72, 7.26] |
| 15 Adverse effects ‐ neurologic (G3‐4) | 6 | 1865 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.67, 1.97] |
| 15.1 High quality trials | 5 | 1797 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.70, 2.14] |
| 15.2 Low quality trials | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.04, 3.43] |
| 16 Adverse effects ‐ pain (G3‐4) | 3 | 1235 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.47 [4.41, 12.67] |
| 16.1 High quality trials | 3 | 1235 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.47 [4.41, 12.67] |
| 17 Adverse effects ‐ hearing loss (G3‐4) | 3 | 1009 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.46, 0.99] |
| 17.1 High quality trials | 2 | 941 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.44, 1.00] |
| 17.2 Low quality trials | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.23, 2.42] |
1.9. Analysis.
Comparison 1 IP component therapy versus IV therapy, Outcome 9 Adverse effects ‐ cardiovascular (G3‐4).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alberts 1996.
| Methods | Study duration: 06/1986 ‐ 07/1992 Type of trial: Multicentre RCT, intention‐to‐treat basis Number ineligible: 108 | |
| Participants | Stage III Residual disease status ≤ 2cm Performance status: SWOG 0‐2 Number randomised: 654 Number evaluable: 546 | |
| Interventions | Arm 1: IV cyclophosphamide (600 mg/m²) + IV cisplatin (100 mg/m²). Repeated every three weeks for a total of six cycles Arm 2: IV cyclophosphamide (600 mg/m²) + IP cisplatin (100 mg/m²). Repeated every three weeks for a total of six cycles | |
| Outcomes | Overall survival Pathological CR Adverse effects: Anaemia, leukopenia, thrombocytopenia, abdominal pain, fever, tinnitus, hearing loss, neuromuscular effects, pulmonary effects | |
| Notes | Survival = time from randomisation to death (any cause) Protocol change in Jan 1991, extending accrual for an additional year, to achieve large enough sample ≤ 0.5cm Methodological quality = high | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Multicentre RCT |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessor blinding performed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low attrition |
| Selective reporting (reporting bias) | Low risk | Analysis by ITT; all expected outcomes reported |
| Other bias | Low risk | High quality trial |
Gadducci 2000.
| Methods | Study duration: 04/1989 ‐ 12/1996 Type of trial: multicentre RCT, intention‐to‐treat Number ineligible: 5 Co‐interventions and other potential confounders: 7 clear cell cancer patients randomly allocated to systemic treatment were excluded from analyses | |
| Participants | Stage: II ‐ IV Residual disease status: < 2cm Performance status: ECOG < 2 Number randomised: 113 Number evaluable: 100 | |
| Interventions | Arm 1: IV epidox 60 mg/m² + IV CTX 600 mg/m² + IV cisplatin 50 mg/m². Repeated every four weeks for a total of six cycles Arm 2: IV epidox 60 mg/m² + IV CTX 600 mg/m² + IP cisplatin 50 mg/m². Repeated every four weeks for a total of six cycles | |
| Outcomes | Overall survival Disease‐free survival Adverse effects | |
| Notes | Survival = time from randomisation to death (any cause) or Sept 1998 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Multicentre RCT |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | < 20% attrition |
| Selective reporting (reporting bias) | Low risk | Analysis by ITT; all expected outcomes reported |
| Other bias | Low risk | Methodological quality = high |
GOG 172.
| Methods | Study duration: 03/1998 ‐ 01/2001 Type of trial: Multicentre RCT, intention‐to‐treat basis Number ineligible: 14 | |
| Participants | Stage: III Residual disease status: ≤ 1cm Performance status: GOG 0‐2 Number randomised: 429 Number evaluable: 415 | |
| Interventions | Arm 1: IV paclitaxel 135 mg/m² over 24 h (day 1) + IV cisplatin 75 mg/m² (day 2). Repeated every three weeks for a total of six cycles Arm 2: IV paclitaxel 135 mg/m² over 24 h (day 1) + IP cisplatin 100 mg/m² (day 2) + IP paclitaxel 60 mg/m² (day 8). Repeated every three weeks for a total of six cycles | |
| Outcomes | Overall survival
Disease‐free survival
Adverse effects: Leukopenia, thrombocytopenia, GI, GU/renal, pulmonary, cardiovascular, neurologic, fever, alopecia, fatigue, infection, metabolic, pain, lymphatic, hepatic, renal, tinnitus
QOL: Functional assessment of cancer therapy Biochemical measures: serum CA 125 |
|
| Notes | Methodological quality = high | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Multicentre RCT |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not strictly blinded but pathology reports, operative notes and eligibility information were collected before registration |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 415/429 evaluated; baseline characteristics similar; 11/205 vs 5/210 lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | Analysis by ITT; all expected outcomes reported |
| Other bias | Low risk | High quality trial |
Kirmani 1994.
| Methods | Study duration: 01/1988 ‐ 02/1992 Includes data to 15 Sept 1993 Type of trial: Single centre RCT Number ineligible: 10 (pathology review) + 7 (refused assigned treatment, 4 IP, 3 IV) + 8 (not evaluable) = 25 | |
| Participants | Stage: IIC to IV Residual disease status: ≤ 1 cm or > 1 cm Performance status: ECOG 1‐2 Number randomised: 87 Number evaluable: 62 evaluable for survival data, 67 evaluable for toxicity | |
| Interventions | Arm 1: IV cisplatin 100 mg/m² + IV cyclophosphamide 600 mg/m². Repeated every three weeks for a total of six cycles Arm 2: IP cisplatin 200 mg/m² + IP etoposide 350 mg/m². Repeated every four weeks for a total of six cycles | |
| Outcomes | Overall survival Disease‐free survival Adverse effects: leukopenia, GI, neurologic, renal, IP catheter‐related complications | |
| Notes | Survival = time from randomisation to death (any cause) or last follow‐up Methodological quality = low | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | > 20% incomplete data for disease‐free survival and response to treatment |
| Selective reporting (reporting bias) | Unclear risk | Expected outcomes reported; ITT not described |
| Other bias | Unclear risk | More women in the IV group had ECOG performance status of 0 (14 vs 5) |
Markman 2001.
| Methods | Study duration: 08/1992 ‐ 04/1995 Type of trial: multicentre RCT, intention‐to‐treat Number ineligible: 61 | |
| Participants | Stage: III Residual disease status: ≤ 1 cm Time from surgery to chemotherapy: 6 weeks Performance status: GOG < 3 Number randomised: 523 Number evaluable: 462 | |
| Interventions | Arm 1: IV paclitaxel 135 mg/m² over 24 hours (day 1) + IV cisplatin 75 mg/m² (day 2). Repeated every three weeks for a total of six cycles Arm 2: IV carboplatin (AUC 9) for two courses every 28 days, followed 4 weeks later by IV paclitaxel 135 mg/m2 over 24 hours (day 1) + IP cisplatin 100 mg/m2 (day 2). Repeated every three weeks for a total of six cycles. | |
| Outcomes | Overall survival Progression‐free survival Adverse effects: Leukopenia, GI, creatinine clearance, cardiovascular, neurologic, fever, allergic, fatigue, infection, metabolic | |
| Notes | Survival = time from randomisation to death (any cause) or date of last contact PFS = time from randomisation to date of appearance of disease (clinically or radiologically detected) 2 deaths in each arm from chemo toxicity Methodological quality = high | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Multicentre RCT |
| Allocation concealment (selection bias) | Low risk | Central allocation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessor blinding performed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | < 20% attrition |
| Selective reporting (reporting bias) | Low risk | Analyses by ITT; all expected outcomes reported |
| Other bias | Low risk | High quality trial |
Polyzos 1999.
| Methods | Study duration: 1990 ‐ 1996 Type of trial: single centre RCT | |
| Participants | Stage: III Residual disease status: < or > 2cm Time from surgery to chemotherapy: unclear Performance status: ECOG < 4 Number randomised: 90 Number evaluable: 90 (46 IV and 44 IP) | |
| Interventions | Arm 1: IV carboplatin 350 mg/m² + IV cyclophosphamide 600 mg/m². Repeated every three to four weeks for a total of six cycles Arm 2: IP carboplatin 350 mg/m² + IV cyclophosphamide 600 mg/m². Repeated every three to four weeks for a total of six cycles | |
| Outcomes | Overall survival Disease‐free survival Adverse effects: Leukopenia, thrombocytopenia, IP administration complications | |
| Notes | No salvage chemotherapy offered to any patient after recurrence Methodological quality = low | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information for assessment |
| Selective reporting (reporting bias) | Unclear risk | Expected outcomes reported; ITT not described |
| Other bias | Unclear risk | Low quality trial |
Yen 2001.
| Methods | Study duration: 04/1990 ‐ 03/1995 Type of trial: single centre RCT; intention‐to‐treat Number ineligible: 14 | |
| Participants | Stage: III Residual disease status: ≤ 1 cm Performance status: SWOG < 3 Number randomised: 132 Number evaluable: 118 | |
| Interventions | Arm 1: IV cyclophosphamide (500 mg/m²) over 1 hour (day 1)+ IV adriamycin or epirubicin (50 mg/m²) over 60‐min (day 1) + IV cisplatin (50 mg/m²) over 1 hour (day 1). Repeated every three weeks for a total of six cycles Arm 2: IV cyclophosphamide (500 mg/m²) over 1 hour (day 1) + IV adriamycin or epirubicin (50 mg/m²) over 60‐min (day 1) + IP cisplatin (100 mg/m²) (day 1). Repeated every 3 weeks for a total of six cycles | |
| Outcomes | Overall survival Adverse effects: Leukopenia, thrombocytopenia, anaemia, IP catheter‐related complications | |
| Notes | Methodological quality = high | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number tables |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low attrition |
| Selective reporting (reporting bias) | Low risk | Analyses by ITT; all expected outcomes reported |
| Other bias | Low risk | High quality trial |
Yen 2009.
| Methods | RCT conducted in Taiwan. Patients randomized postoperatively between Jan 2001 and Dec 2007. Overall survival duration was calculated from cytoreductive surgery to disease‐specific death or last follow‐up. | |
| Participants | 367 women with Stage III epithelial ovarian cancer following cytoreductive surgery. | |
| Interventions | IP versus IV chemotherapy following standard cytoreductive surgery. Chemotherapy consisted of paclitaxel on day 1 combined with cisplatin or carboplatin administered IP or IV on day 2. This was repeated every 3 weeks for 6 cycles provided serum creatinine was < or equal to 2 mg/dl, WCC > 3000/mm3, and platelet count was > 80,000/mm3. IP therapy was discontinued and shifted to IV chemo if tube problems occurred. | |
| Outcomes | Nomogram construction/model for survival using age, CA‐125, IV/IP, Stage, histology, upper abdominal metastases | |
| Notes | Patients who had a sub‐optimal cytoreductive status (tumour greater than 1 cm) or incomplete clinicopathological data or were operated on by a non‐sub specialist were excluded from the analysis. Patients lost to follow‐up were 'censored'. 298 included in final analyses (152 in IV group and 146 in IP group). Baseline characteristics similar. Analyzed the impact of delivered IP cycles on survival and found that 5 or more IP cycles were needed to achieve better survival. Authors provided additional unpublished data (including survival data and adverse effect data) and methodological details. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated random numbers (additional unpublished information) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not possible |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Group sizes were similar, as were baseline characteristics. Unpublished data was complete for survival outcomes and adverse effects. |
| Selective reporting (reporting bias) | Low risk | Objective was to construct a nomogram for survival. Analyses by ITT (unpublished information). |
| Other bias | Low risk | Considered a high quality trial |
Zylberberg 1986.
| Methods | Study duration: 01/1980 ‐ 03/1984 Type of trial: single centre RCT | |
| Participants | Stage: III Residual disease status: as little as possible Number randomised: 20 Number evaluable: 20 | |
| Interventions | Arm 1: IV adriamycin 35 mg x 2 + fluorouracil 750 mg x 2 + bleomycin 15 mg + cisplatin 100 mg + vincaleucoblastine 10 mg + ifosfamide 1 g x 2 Arm 2: IV adriamycin 20 mg x 2 + fluorouracil 500 mg x 2 + cisplatin 50 mg + vincaleucoblastine 10 mg + ifosfamide 1 g x 2 + IP bleomycin 15 mg + cisplatin 50 mg + fluorouracil 500 mg + adriamycin 30 mg | |
| Outcomes | Overall survival Disease‐free survival | |
| Notes | Study translated from French Methodological quality = low | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information for assessment |
| Selective reporting (reporting bias) | Unclear risk | ITT not described; did not report adverse events/toxicity |
| Other bias | Unclear risk | Small study, low quality trial |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Piccart 2003 | This study specifically included ovarian cancer patients with a pathologically complete remission after platinum‐based intravenous chemotherapy. This was not considered the initial management of ovarian cancer but rather consolidation treatment. |
Characteristics of ongoing studies [ordered by study ID]
GOG 0252.
| Trial name or title | GOG 0252 |
| Methods | Study duration: projected to complete in 2016 Type of trial: US multicentre RCT Planned follow‐up duration: 5 years Patients are stratified according to disease stage and size of residual disease after initial staging/debulking surgery |
| Participants | Patients with FIGO stage II ‐ IV (no gross residual disease vs gross residual disease with lesion ≤ 1 cm vs any gross residual disease with lesion > 1 cm). Number to be randomised: 1500 |
| Interventions | Patients are randomized to 1 of 3 treatment arms. See Table 4. |
| Outcomes | PFS, OS, adverse events and QOL |
| Starting date | Aug 2009 |
| Contact information | Joan Walker USA 405‐271‐8707 |
| Notes |
GOTIC‐001/JGOG‐3019.
| Trial name or title | GOTIC‐001/JGOG‐3019 |
| Methods | Study duration: projected to take 3 years to complete accrual Type of trial: multicentre RCT Planned follow‐up duration of 3 years |
| Participants | Stage: II‐IV newly diagnosed ovarian, fallopian tube and primary peritoneal cancer Residual disease status: Includes women with optimal and sub‐optimal residual disease Performance status: 0‐2 Number to be randomised: 746 Number evaluable: (to be determined) |
| Interventions | Arm 1: IV dose‐dense paclitaxel plus IP carboplatin Arm 2: IV dose‐dense paclitaxel plus IV carboplatin IV paclitaxel will be infused on days 1, 8 and 15 at 80 mg/m². Carboplatin at AUC 6 given on day 1, either IP or IV. Repeated every three weeks for six cycles |
| Outcomes | PFS, OS, QOL and cost/benefit |
| Starting date | June 2010 |
| Contact information | fujiwara@saitama‐med.ac.jp |
| Notes | The first 120 will be evaluated for the feasibility of the intraperitoneal arm |
Hyperthermic IP therapy.
| Trial name or title | Secondary Debulking Surgery +/‐ Hyperthermic Intraperitoneal Chemotherapy in Stage III Ovarian Cancer (NCT00426257) |
| Methods | This phase III randomised, open‐label trial compares a single intraperitoneal perfusion of chemotherapy under hyperthermic conditions immediately after interval debulking surgery (OVHIPEC) versus no intraperitoneal therapy |
| Participants | Women with stage III ovarian, peritoneal or tubal carcinoma who are eligible for interval debulking surgery either following primary chemotherapy or following incomplete primary debulking and chemotherapy. Participants must be between 18 and 76 yr old and not have had relevant competing previous malignancies. Estimated enrolment = 280 women |
| Interventions | Arm 1: Secondary debulking surgery with hyperthermic intraperitoneal chemotherapy (cisplatin 100 mg/m2 at the end of surgery) Arm 2: Secondary debulking surgery |
| Outcomes | Primary: duration of recurrence free survival following completion of treatment. Secondary: toxicity and morbidity, quality of life, tumour response following treatment and overall survival |
| Starting date | Feb 2007 |
| Contact information | Willemien J van Driel, MD Ph: +31 20 512 2975 Email: w.v.driel@nki.nl |
| Notes | Estimated completion date 2013 |
NCIC CTG OV.21.
| Trial name or title | NCIC CTG OV.21 |
| Methods | Study duration: Projected to take 4.5 years to complete accrual Type of trial: Multicentre RCT with collaborating institutions in Canada, UK, Spain and the USA No. ineligible: (to be determined) |
| Participants | Women with EOC and optimal tumour debulking after 3‐4 cycles of neoadjuvant chemotherapy Stage: IIb‐IIIa Residual disease status: ≤ 1 cm Performance status: ≤ 2 Number to be randomised: 150 (Phase II trial) and 630 (Phase III trial) Number evaluable: (to be determined) |
| Interventions | Arm 1: IV paclitaxel 135 mg/m² plus IV carboplatin on day 1; IV paclitaxel 60 mg/m² on day 8 Arm 2: IV paclitaxel 135 mg/m² plus IP carboplatin or cisplatin* on day 1; IP paclitaxel on day 8 Repeated every 21 days for 3 cycles |
| Outcomes | PFS, OS, toxic effects, QOL, cost/benefit and correlative biology studies |
| Starting date | 2011 |
| Contact information | Helen.Mackay@uhn.on.ca |
| Notes | Phase III trial to be preceded by a Phase II trial with three arms: IV only, cisplatin IP component therapy and carboplatin IP component therapy. 50 women will be recruited to each arm. *Arm 2 IP chemo agent to be determined by the outcome of the Phase II part of the trial |
Differences between protocol and review
The methodology of the current version of this review was updated in May 2011. See Appendix 4 for original methods.
Contributions of authors
Ken Jaaback: protocol and review development, methodological quality assessment, data extraction, analysis and content. Nick Johnson: protocol and review development, data analysis and verification. Tess Lawrie: study selection, data extraction, data analyses and editing of the updated review.
Declarations of interest
There is no conflict of interest amongst the authors of this review.
Edited (no change to conclusions)
References
References to studies included in this review
Alberts 1996 {published data only}
- Alberts DS, Liu PY, Hannigan EV, O'Toole R, Williams SD, Young JA, et al. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. New England Journal of Medicine 1996;335(26):1950‐5. [DOI] [PubMed] [Google Scholar]
Gadducci 2000 {published data only}
- Gadducci A, Carnino F, Chiara S, Brunetti I, Tanganelli L, Romanini A, et al. Intraperitoneal versus intravenous cisplatin in combination with intravenous cyclophosphamide and epidoxorubicin in optimally cytoreduced advanced epithelial ovarian cancer: a randomised trial of the Gruppo Oncologico Nord‐Ovest. Gynecologic Oncology 2000;76(2):157‐62. [DOI] [PubMed] [Google Scholar]
GOG 172 {published and unpublished data}
- Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. The New England Journal of Medicine 2006;354(1):34‐43. [DOI] [PubMed] [Google Scholar]
- Armstrong DK, Bundy BN, Baergen R, Lele SB, Copeland LJ, Walker, et al. Randomized phase III study of intravenous (IV) paclitaxel and cisplatin versus IV paclitaxel, intraperitoneal (IP) cisplatin and IP paclitaxel in optimal stage III epithelial ovarian cancer (OC): a Gynecologic Oncology Group trial (GOG 172). Proceedings of the American Society of Clinical Oncology. 2002; Vol. 21:201a.
- Krivak TC, Tian C, Rose GS, Armstrong DK, Maxwell GL. A Gynecologic Oncology Group Study of serum CA‐125 levels in patients with stage III optimally debulked ovarian cancer treated with intraperitoneal compared to intravenous chemotherapy: an analysis of patients enrolled in GOG 172. Gynecologic Oncology 2009;115(1):81‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnock J, Darcy K, Tian C, Gallion H, DeLoia J, Armstrong D, et al. Association between reduced BRCA1 expression and survival in patients with sporadic epithelial ovarian cancer treated with intraperitoneal versus intravenous cisplatin and paclitaxel: A Gynecologic Oncology Group study. Gynecologic Oncology 2010;116(Conference Publication: 3 Suppl.1):S3‐S4. [Google Scholar]
- Walker JL, Armstrong DK, Huang HQ, Fowler J, Webster K, Burger RA, et al. Intraperitoneal catheter outcomes in a phase III trial of intravenous versus intraperitoneal chemotherapy in optimal stage III ovarian and primary peritoneal cancer: a Gynecologic Oncology Group Study. Gynecologic Oncology 2006;100(1):27‐32. [DOI] [PubMed] [Google Scholar]
- Wenzel LB, Huang HQ, Armstrong DK, Walker JL, Cella D, and Gynecologic Oncology Group. Health‐related quality of life during and after intraperitoneal versus intravenous chemotherapy for optimally debulked ovarian cancer: a Gynecologic Oncology Group Study. Journal of Clinical Oncology 2007;25(4):437‐43. [DOI] [PubMed] [Google Scholar]
Kirmani 1994 {published data only}
- Kirmani S, Braly PS, McClay EF, Saltzstein SL, Plaxe SC, Kim S, et al. A comparison of intravenous versus intraperitoneal chemotherapy for the initial treatment of ovarian cancer. Gynecologic Oncology 1994;54(3):338‐44. [DOI] [PubMed] [Google Scholar]
Markman 2001 {published data only}
- Markman M, Bundy BN, Alberts DS, Fowler JM, Clark‐Pearson DL, Carson LF, et al. Phase III trial of standard‐dose intravenous cisplatin plus paclitaxel versus moderately high‐dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small‐volume stage III ovarian carcinoma: an intergroup study of the Gynecologic Oncology Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. Journal of Clinical Oncology 2001;19(4):1001‐7. [DOI] [PubMed] [Google Scholar]
Polyzos 1999 {published data only}
- Polyzos A, Tsavaris N, Kosmas C, Giannikos L, Katsikas M, Kalahanis N, et al. A comparative study of intraperitoneal carboplatin versus intravenous carboplatin with intravenous cyclophosphamide in both arms as initial chemotherapy for stage III ovarian cancer. Oncology 1999;56(4):291‐6. [DOI] [PubMed] [Google Scholar]
Yen 2001 {published data only}
- Yen MS, Juang CM, Lai CR, Chao GC, Ng HT, Yuan CC. Intraperitoneal cisplatin‐based chemotherapy vs. intravenous cisplatin‐based chemotherapy for stage III optimally cytoreduced epithelial ovarian cancer. International Journal of Gynecology and Obstetrics 2001;72(1):55‐60. [DOI] [PubMed] [Google Scholar]
Yen 2009 {published and unpublished data}
- Yen MS, Twu NF, Lai CR, Horng HC, Chao KC, Juang CM. Importance of delivered cycles and nomogram for intraperitoneal chemotherapy in ovarian cancer. Gynecologic Oncology 2009;114(3):415‐9. [DOI] [PubMed] [Google Scholar]
Zylberberg 1986 {published data only}
- Zylberberg B, Ravina JH, Salat‐Baroux J, Dormont D, Lipp B, Guillet JL. Polychemotherapy of ovarian cancer via combined intravenous and intraperitoneal routes. Technic and preliminary results [Polychimiotherapie des cancers de l'ovaire par voie mixte intraveineuse et intraperitoneale. Technique et resultats preliminaires]. Journal de Gynécologie Obstétrique et Biologie de la Reproduction 1986;15(5):671‐6. [PubMed] [Google Scholar]
References to studies excluded from this review
Piccart 2003 {published data only}
- Piccart MJ, Floquet A, Scarfone G, Willemse PH, Emerich J, Vergote I, et al. Intraperitoneal cisplatin versus no further treatment: 8‐year results of EORTC 55875, a randomised phase III study in ovarian cancer patients with a pathologically complete remission after platinum‐based intravenous chemotherapy. International Journal of Gynecological Cancer 2003;13 Suppl 2:196‐203. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
GOG 0252 {unpublished data only}
- GOG‐0252. Bevacizumab and Intravenous or Intraperitoneal Chemotherapy in Treating Patients With Stage II, Stage III, or Stage IV Ovarian Epithelial Cancer, Fallopian Tube Cancer, or Primary Peritoneal Cancer. ClinicalTrials.gov Identifier: NCT00951496.
GOTIC‐001/JGOG‐3019 {published and unpublished data}
- Aotani E, Kukino S, Nonaka M, Nagao S, Fujiwara K. First attempt of large phase III oncology trial using Japanese new trial evaluation system, the evaluation system of investigational medical care. Japanese Pharmacology and Therapeutics 2010;38(Suppl.1):S59‐S64. [Google Scholar]
- Fujiwara K, Aotani E, Hamano T, Nagao S, Yoshikawa H, Sugiyama T, et al. A randomized phase II/III trial of 3 weekly intraperitoneal versus intravenous carboplatin in combination with intravenous weekly dose‐dense paclitaxel for newly diagnosed ovarian, fallopian tube and primary peritoneal cancer. Japanese Journal of Clinical Oncology 2011;41(2):278‐82. [DOI] [PubMed] [Google Scholar]
Hyperthermic IP therapy {unpublished data only}
- Secondary debulking surgery +/‐ hyperthermic intraperitoneal chemotherapy in stage III ovarian cancer. http://www.bioportfolio.com/resources/trial/102235/Secondary‐Debulking‐Surgery‐Hyperthermic‐Intraperitoneal‐Chemotherapy‐In‐Stage‐Iii‐Ovarian‐Cancer.html.
- Secondary debulking surgery +/‐ hyperthermic intraperitoneal chemotherapy in stage III ovarian cancer. http://www.cancer.gov/clinicaltrials/search/view?cdrid=533399&version=HealthProfessional&protocolsearchid=9267648.
NCIC CTG OV.21 {published and unpublished data}
- Mackay HJ, Provencheur D, Heywood M, Tu D, Eisenhauer EA, Oza AM. Phase II/III study of intraperitoneal chemotherapy after neoadjuvant chemotherapy for ovarian cancer: NCIC CTG OV.21. Current Oncology 2011;18(2):84‐90. [NCT00993655] [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
ACS 2005 [Computer program]
- American Cancer Society. Detailed Guide: Ovarian Cancer. What are the key statistics about ovarian cancer?. American Cancer Society, 2005.
Armstrong 2006
- Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. The New England Journal of Medicine 2006;354(1):34‐43. [DOI] [PubMed] [Google Scholar]
Bristow 2002
- Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta‐analysis. Journal of Clinical Oncology 2002;20:1248‐59. [DOI] [PubMed] [Google Scholar]
Burghardt 1991
- Burghardt E, Girardi F, Lahousen M, Tamussino K, Stettner H. Patterns of pelvic and paraaortic lymph node involvement in ovarian cancer. Gynecologic Oncology 1991;40 (2):103‐6. [DOI] [PubMed] [Google Scholar]
Chi 2010
- Chi D, Dao F, Ferguson S, Sabbatini P, Hensley M, Konner J, et al. Analysis of progression‐free survival, tolerability and toxicity in patients with stage III ovarian, tubal and peritoneal carcinoma treated with an outpatient regimen of primary intravenous/ intraperitoneal paclitaxel and intraperitoneal cisplatin. Gynecologic Oncology 2010;Conference Publication: 116(3 Suppl 1):S123. [Google Scholar]
Deraco 2011
- Deraco M, Kusamura S, Virzì S, Puccio F, Macrì A, Famulari C, et al. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy as upfront therapy for advanced epithelial ovarian cancer: Multi‐institutional phase‐II trial. Gynecologic Oncology 2011;122(2):215‐20. [DOI] [PubMed] [Google Scholar]
du Bois 2003
- du Bois A, Luck HJ, Meier W, Adams HP, Mobus V, Costa S, et al. A randomised clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first‐line treatment of ovarian cancer. Journal of the National Cancer Institute 2003;95(17):1320‐9. [DOI] [PubMed] [Google Scholar]
Fujiwara 2008
- Fujiwara 2008. Can carboplatin replace cisplatin for intraperitoneal use?. International Journal of Gynecological Cancer 2008;18(Suppl 1):29‐32. [DOI] [PubMed] [Google Scholar]
Fujiwara 2009
- Fujiwara K, Nagao S, Kigawa J, Noma J, Akamatsu N, Miyagi Y, et al. Phase II study of intraperitoneal carboplatin with intravenous paclitaxel in patients with suboptimal residual epithelial ovarian or primary peritoneal cancer: A Sankai Gynecology Cancer Study Group Study. International Journal of Gynecological Cancer 19;5:834‐7. [DOI] [PubMed] [Google Scholar]
Fujiwara 2012
- Fujiwara K. Three ongoing intraperitoneal chemotherapy trials in ovarian cancer. Journal of Gynecologic Oncology 2012;23(2):75‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gray 2010
- Gray HJ, Shah CA, Swensen RE, Tamimi HK, Goff BA. Alternative intraperitoneal chemotherapy regimens for optimally debulked ovarian cancer. Gynecologic Oncology 2010;116(3):340‐4. [DOI] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2009. Available from www.cochrane‐handbook.org.
Howell 1982
- Howell SB, Pfeifle CL, Wung WE, Olshen RA, Lucas WE, Yon JL, et al. Intraperitoneal cisplatin with systemic thiosulfate protection. Annals of Internal Medicine 1982;97:845‐51. [DOI] [PubMed] [Google Scholar]
Jones 1978
- Jones RB, Myers CE, Guarino AM, Dedrick RL, Hubbard SM, DeVita VT. High volume intraperitoneal chemotherapy ("belly bath") for ovarian cancer. Pharmacologic basis and early results. Cancer Chemotherapy and Pharmacology 1978;1(3):161‐6. [DOI] [PubMed] [Google Scholar]
Katsumata 2009
- Katsumata N, Yasuda M, Takahashi F, Isonishi S, Jobo T, Aoki D, et al. Dose‐dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open‐label, randomised controlled trial. Lancet 2009;374:1331‐8. [DOI] [PubMed] [Google Scholar]
Lopez 1985
- Lopez JA, Krikorian JG, Reich SD, Smyth RD, Lee FH, Issell BF. Clinical pharmacology of intraperitoneal cisplatin. Gynecologic Oncology 1985;20(1):1‐9. [DOI] [PubMed] [Google Scholar]
Los 1990
- Los G, Mutsaers PH, Lenglet WJ, Baldew GS, McVie JG. Platinum distribution in intraperitoneal tumors after intraperitoneal cisplatin treatment. Cancer Chemotherapy and Pharmacology 1990;25(6):389‐94. [DOI] [PubMed] [Google Scholar]
Makhija 2001
- Makhija S, Leitao M, Sabbatini P, Bellin N, Almadrones L, Leon L, et al. Complications associated with intraperitoneal chemotherapy catheters. Gynecologic Oncology 2001;81(1):77‐81. [DOI] [PubMed] [Google Scholar]
Markman 1999
- Markman M. IP chemotherapy. Critical Reviews in Oncology/ Hematology 1999;31:239‐46. [DOI] [PubMed] [Google Scholar]
Ozols 2003
- Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke‐Pearson D, Burger RA, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. Journal of Clinical Oncology 2003;21(17):3194‐200. [DOI] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815‐34. [DOI] [PubMed] [Google Scholar]
RevMan 2008 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Weisberger 1955
- Weisberger AS, Levine B, Storaasli JP. Use of nitrogen mustard in treatment of serous effusions of neoplastic origin. JAMA 1955;159(18):1704‐7. [DOI] [PubMed] [Google Scholar]
Wenzel 2004
- Wenzel LB, Huang H, Armstrong D, Walker J, Cella D. Quality of life (QOL) results of a randomised study of intravenous (IV) paclitaxel and cisplatin vs IV paclitaxel, intraperitoneal (IP) cisplatin and IP paclitaxel in optimal stage III epithelial ovarian cancer (OC): A Gynecologic Oncology Group trial. Journal of Clinical Oncology 2004;22(14S):5026. [Google Scholar]
Wenzel 2007
- Wenzel LB, Huang HQ, Armstrong DK, Walker JL, Cella D, and Gynecologic Oncology Group. Health‐related quality of life during and after intraperitoneal versus intravenous chemotherapy for optimally debulked ovarian cancer: a Gynecologic Oncology Group Study. Journal of Clinical Oncology 2007;25(4):437‐43. [DOI] [PubMed] [Google Scholar]
Zeimet 2009
- Zeimet AG, Reimer D, Radl AC, Reinthaller A, Schauer C, Petru E, et al. Review: Pros and cons of intraperitoneal chemotherapy in the treatment of epithelial ovarian cancer. Anticancer Research 2009;29(7):2803‐8. [PubMed] [Google Scholar]
Zhao 2013
- Zhao H, Du N, Fu Y, Wang H, Fan Z. Clinical study of intraperitoneal injection bevacizumab (BV) combined with intraperitoneal hyperthermic perfusion chemotherapy (CT) in treatment of malignant ascites of ovarian cancer (OC). Journal of Clinical Oncology. Annual Meeting of the American Society of Clinical Oncology, ASCO Chicago, IL United States. 2013; Vol. 31:15 Suppl. 1.
References to other published versions of this review
Jaaback 2007
- Jaaback K, Johnson N. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD005340.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaaback 2011
- Jaaback K, Johnson N, Lawrie TA. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD005340.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]