

Summary
Mosaic analysis with double markers (MADM) technology enables the generation of genetic mosaic tissue in mice. MADM enables concomitant fluorescent cell labeling and introduction of a mutation of a gene of interest with single-cell resolution. This protocol highlights major steps for the generation of genetic mosaic tissue and the isolation and processing of respective tissues for downstream histological analysis.
For complete details on the use and execution of this protocol, please refer to Contreras et al. (2021).
Subject areas: Single Cell, Developmental biology, Genetics, Microscopy, Model Organisms, Molecular Biology, Neuroscience, Stem Cells, Tissue Engineering
Graphical abstract
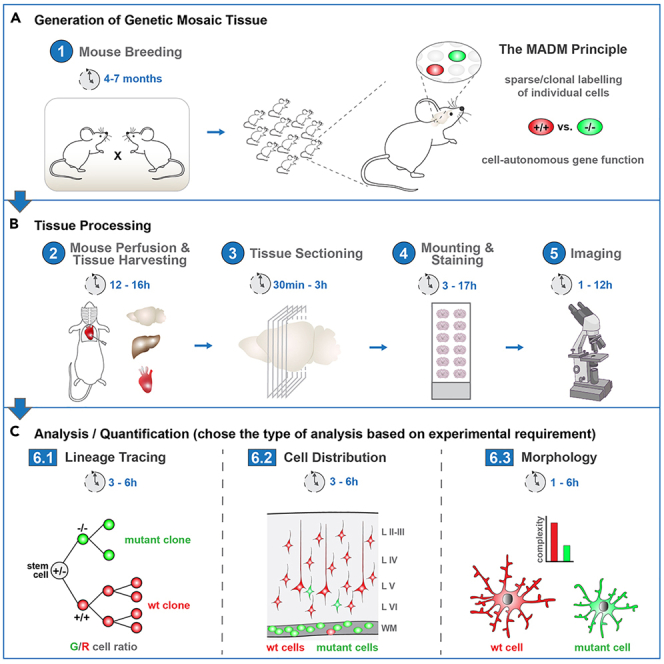
In this protocol we established an experimental pipeline for the generation of experimental MADM mice; and isolation of MADM-labeled tissue for downstream histological analysis.
Highlights
-
•
Generation of genetic mosaic mice using mosaic analysis with double markers (MADM)
-
•
Tissue harvesting from experimental MADM mice
-
•
Processing and imaging of MADM-labeled tissue
-
•
Distinct analyses to assess cell-autonomous gene function in MADM mice
Mosaic analysis with double markers (MADM) technology enables the generation of genetic mosaic tissue in mice. MADM enables concomitant fluorescent cell labeling and introduction of a mutation of a gene of interest with single-cell resolution. This protocol highlights major steps for the generation of genetic mosaic tissue and the isolation and processing of respective tissues for downstream histological analysis.
Before you begin
Background
This protocol describes the generation of genetic mosaic mice using mosaic analysis with double markers (MADM) (Zong et al., 2005; Contreras et al., 2021). A genetic mosaic individual consists of cells of distinct genotypes, for example harboring homozygous mutant cells next to wild-type cells. This phenomenon occurs naturally and is widespread across multicellular organisms. The advantages of using genetic mosaic animals for experimental studies are manifold: The controlled induction of genetic mosaicism in experimental animals allows to alter gene function at high spatiotemporal resolution and has led to many fundamental discoveries of gene function and cellular mechanisms in diverse biological contexts. Genetic mosaicism can be induced sparsely, so that individual mutant cells will be surrounded by unperturbed cells. Thus, mosaicism enables the study of cell-autonomous gene function at single cell resolution. Individual mutant cells can be studied in many ways, for example in lineage tracing, cell competition assays, morphological analysis and disease modelling (Hippenmeyer et al., 2010; Gao et al., 2014; Joo et al., 2014; Beattie et al., 2017; Tian et al., 2020; Contreras et al., 2021).
In this protocol, we present an experimental pipeline for the generation of MADM-labeled mosaic tissues and highlight a variety of downstream analysis applications. Our protocol is based on the use of distinct Cre drivers in combination with MADM cassettes targeted to all 19 mouse autosomes (Contreras et al., 2021).
Mosaic analysis with double markers (MADM) is a genetic technology that allows concomitant fluorescent labeling and introduction of a homozygous mutation of choice with unprecedented single cell resolution in a defined genetic lineage (Zong et al., 2005; Contreras et al., 2021). MADM is based on the use of split marker genes that consist of two reciprocal cassettes of partial coding sequences for green fluorescent protein (GFP) and tandem dimer Tomato (TdT), interspersed by loxP sites (Figure 1A). The two reciprocal MADM cassettes need to be maintained in two different mouse lines, the GT (N-GFP/C-TdT) line and the TG (N-TdT/C-GFP) line (Zong et al., 2005; Hippenmeyer et al., 2010).
Figure 1.

The MADM principle
(A) For MADM, two reciprocally chimeric marker genes are targeted to identical loci on homologous chromosomes. The chimeric marker genes (GT and TG alleles) consist of partial coding sequences for green (eGFP[G]) and red (tdT[T]) fluorescent proteins separated by an intron containing the loxP site. Following Cre recombinase-mediated interchromosomal recombination during mitosis, functional green and red fluorescent proteins are reconstituted.
(B) G2-X segregation of the recombinant chromosomes results in two daughter cells each expressing one of the two fluorescent proteins (recombination in G2 of the cell cycle followed by X segregation). Introduction of a mutation distal to one MADM cassette (here the TG cassette) allows the generation of genetic mosaics with single cell resolution, with wild-type daughter cells labeled in red and homozygous mutant sibling cells labelled in green in an unlabeled heterozygous environment.
(C) G2-Z segregation results in two daughter cells with no genotype alteration. One cell shows double labeling (= yellow), while the other daughter cell is unlabeled.
(D) G1 and G0 events also result in yellow cell labeling without change in genotype.
Reproduced and adapted with permission from (Contreras et al., 2021).
In order to generate genetic mosaic tissue, a modified gene of interest needs to be meiotically recombined with one of the MADM chromosomes (here shown as coupled to the TG line, resulting in an animal being heterozygous for the gene modification of interest). Additionally, one of the lines needs to carry a Cre driver of interest for the generation of experimental animals. Cells with MADM-induced mosaicism are generated by Cre/loxP-dependent mitotic interchromosomal recombination during the G2 phase in dividing progenitor/stem cells followed by X-segregation (that is the transmission of the two recombinant chromosomes to distinct daughter cells, resulting in individual green (GFP+) mutant and red (tdT+) wild-type cells) (Figures 1A and 1B). Recombination in the G2 phase of the cell cycle with segregation of both recombinant chromosomes to the same daughter cell (Z-segregation) does not alter the genotype and results in one yellow GFP+/tdT+ heterozygous cell which serves as control. At the same time, Z-Segregation results in transmission of the non-recombined chromosomes to the other sibling daughter cell and thus gives rise to an unlabeled cell (unchanged genotype, heterozygous) (Figure 1C). G1 and/or postmitotic G0 events also result in yellow cells (Figure 1D).
Importantly, the existence of sparse green mutant, red wild-type and yellow heterozygous cells in an otherwise unlabeled heterozygous environment not only allows to study gene function, but also to perform gene dosage analysis at the individual cell level within the same tissue, in a cell-type-specific manner.
Here, we demonstrate the coupling of the mutant allele of interest with the TG line, which upon successful Cre-mediated recombination will result in genetic mosaic tissue consisting of sparse genetically modified cells labeled in green, while corresponding wild-type cells will be labeled in red and all other cells (yellow and unlabeled) will be heterozygous (Figure 1).
For more details on the MADM technology, please refer to Zong et al. (2005), Beattie et al. (2020), Laukoter et al. (2020a), and Contreras et al. (2021).
For the successful generation of experimental MADM mice, a number of considerations have to be taken into account before starting with breeding (also see troubleshooting problem 1):
-
1.
Institutional and governmental permission and oversight information for the animal study should be obtained. In this study, experimental procedures were discussed and approved by the institutional ethics and animal welfare committees at IST Austria in accordance with good scientific practice guidelines and national legislation (license number: BMWF-66.018/0007-II/3b/2012 and BMWFW-66.018/0006-WF/V/3b/2017).
-
2.
For MADM to work, the presence of a Cre recombinase, active in stem or progenitor cells of your tissue of interest, is required. In other words, any Cre-driver can be used, provided Cre is expressed in dividing stem and progenitor cells, to target cell types and lineages of interest.
-
3.
MADM can be employed for both population and clonal analysis. Depending on the aim of your experiments, you are required to choose a Cre driver enabling either population analysis (constitutively active Cre) or clonal analysis (e.g., tamoxifen (TM)-inducible CreER driver).
Population analysis allows the analysis of a high number of single, sparsely labeled cells in one organ to obtain a highly quantitative read-out. However, constitutively active Cre will result in MADM events at any time point during the entire time window of Cre activity (e.g., Emx1-Cre activity starts at ∼E9.5 and is detectable up to early postnatal age). Given this relatively long activity window, the detection of labeled cells does not provide information about the spatiotemporal origin of these cells or the stem cell division mode.
In order to get to spatiotemporally controlled induction of MADM events clonal analysis, employing a TM-inducible CreER driver, can be used. Injection of one single low dose of TM allows the induction of G2-X MADM events only at the injection time point, thus generating high spatiotemporal resolution and conserving information about stem cell division modes. For more detailed information about the usage of the MADM system for clonal analysis, please refer to (Beattie et al., 2020).
-
4.
Make use of meiotic recombination in the germline to genetically link a mutant allele of your gene of interest to the corresponding MADM chromosome (Hippenmeyer et al., 2010; Laukoter et al., 2020b) (Figure 1). The probability for meiotic recombination that results in the linkage of the mutant allele with the MADM cassette can be estimated: By determining the genetic distance (centi-Morgan [cM]) between the gene locus of interest and the MADM cassette on a particular chromosome by using for example Mouse Genome Informatics (MGI) database (www.informatics.jax.org) (Figure 2A).
-
5.
Choose MADM chromosomes according to the chromosomal location of your gene of interest. For example, if your gene of interest is located on chromosome 7, you need to recombine it with a MADM cassette localized on chr.7 (MADM-7). If your gene of interest is located on chromosome 11 (as shown in our example in Figure 2), you need to recombine it with a MADM cassette localized on chr.11 (MADM-11). We have reported distinct MADM recombination frequencies for different MADM chromosomes. Consider the frequency when evaluating your results, particularly when frequency is low and results appear highly variable (also see troubleshooting problem 3). In the original manuscript corresponding to this protocol, we provide a detailed description of MADM GT and TG cassettes for all 19 mouse autosomes including primer sequences for genotyping (Contreras et al., 2021).
Figure 2.

Calculation of meiotic recombination frequency and breeding schemes for MADM mice
(A) Calculation of meiotic recombination frequency to genetically link the gene of interest (here Ndel1) to the corresponding MADM chromosome (here MADM-11).
(B) Breeding strategy 1 for maximal output efficiency in the generation of experimental MADM mice. (i) In the first breeding step, the mouse line carrying the mutation of interest is bred to the TG cassette of the corresponding MADM chromosome. (ii) In a second step, MADM cassettes are homozygosed by crossing the trans-heterozygote offspring from (i) with animals homozygous for the TG cassette. Thereby, the mutation of gene-X is genetically linked (through meiotic recombination) to the MADM TG cassette on the corresponding MADM chromosome. (iii) In parallel to steps (i) and (ii), the first breeding step for the Cre driver is performed by crossing the Cre line of choice with the GT line. (iv) In a second step, MADM cassettes are homozygosed by performing an intercross of the double heterozygous offspring from (iii). (v) Mating setup resulting in experimental MADM mice with mutant cells labelled in green and wild-type cells labelled in red. (vi) Summary of the required breeding cages to obtain 5 experimental MADM mice. The breeding strategy allows to obtain 50% of genetic mosaic MADM animals from one experimental mating.
(C) Breeding strategy 2 for maximal temporal efficiency in the generation of experimental MADM mice. (i) In the first breeding step, the mutation of gene-X is genetically linked to the MADM TG cassette on the corresponding MADM chromosome. (ii) In parallel, the Cre driver is bred to the GT line. All mice of the F1 generation will be heterozygous for MADM cassettes, gene-X and Cre, respectively. (iii) The breeding step rendering the MADM cassettes homozygous is skipped and F1 mice are used directly to set up the mating resulting in experimental MADM animals with mutant cells labelled in green and wild-type cells labelled in red. (iv) Summary of the required breeding cages to obtain 5 experimental MADM mice. The strategy allows to obtain mosaic animals at a frequency of ∼5% from ∼10 experimental matings.
We provide an example in Figure 2 using the candidate gene Ndel1, which is located on chromosome 11, and thus needs to be recombined to MADM chromosome 11. MADM-11 cassettes are ∼3 cM distal to the centromere and Ndel1 is located ∼42 cM distal to the centromere, or ∼39 cM away from the MADM cassettes. Thus, there is ∼39% probability that meiotic recombination will occur in the recombinant generating mating. However, since only half of the progeny will be of the desired genotype (TG,mutant), while the other half will be wild-type (+,+), animals with the appropriate genotype will be generated at a rate of ∼20% (39% × ½) (Figure 2A).
-
6.
The larger the genetic distance between the MADM cassette and the genomic locus of your gene of interest, the higher the recombination probability. The scale of the mating for generating recombinants using meiotic recombination needs to be adjusted according to the recombination probability. In other words, the probability of meiotic recombination determines the amount of matings you require to set up in order to successfully obtain mice where MADM cassettes and genes of interest are genetically linked. The higher the recombination probability, the less breeding cages will be needed to obtain recombinants.
Note: All information provided in this section is specific to the example gene Ndel1 in order to provide numerical calculation support to the reader. The numbers and probabilities for your specific experiments require adjustments based on your genes of interest and the required MADM chromosomes.
-
7.
The MADM targeting constructs were introduced into C57BL/6N embryonic stem cells and injected into blastocysts isolated from BALB/cRj females. The offspring were bred with C57BL/6NRj females. Further backcrossing of offspring positive for MADM cassettes was performed with CD1 mice to obtain MADM stock lines of mixed CD1-C57BL/6J genetic background. Consider variances in litter size or phenotype based on mouse background, particularly when you are usually employing pure inbred mice for your experiments.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| PFA | Sigma-Aldrich | Cat# 441244 |
| 10× PBS | Thermo Fisher Scientific | Cat# 70011 |
| Triton X 100 | Sigma-Aldrich | Cat# T878 |
| Tween 20 | Sigma-Aldrich | Cat# P9416 |
| 0.2M Tris HCL pH 8.5 | CARL ROTH | Cat# 9090 |
| NaH2PO4·2H2O | Sigma-Aldrich | Cat# 567549 |
| Na2HPO4 | Sigma-Aldrich | Cat# S3264 |
| Glycerol | Sigma-Aldrich | Cat# G5516 |
| Citric Acid | CARL ROTH | Cat# X863.1 |
| NaOH | CARL ROTH | Cat# T198.1 |
| Heat-inactivated Horse Serum (HS) | Thermo Fisher Scientific | Cat#26050088 |
| Avertin (2,2,2 triboromoethanol) | Sigma-Aldrich | Cat# T48402 |
| t-amylalcohol (2-methyl-2-butanol) | Sigma-Aldrich | Cat# 240486 |
| Ethanol | Honeywell | Cat# 24194 |
| Sucrose | Sigma-Aldrich | Cat# S8501 |
| O.C.T. Tissue Tek | Sakura | Cat# 4583 |
| Mowiol 4-88 | CARL ROTH | Cat# 0713 |
| DABCO | CARL ROTH | Cat# 0718 |
| DAPI | Molecular Probes | Cat# D1306 |
| Antibodies | ||
| Chicken anti-GFP antibody, 1:400 | Aves | RRID:AB_10000240 |
| Goat anti-tdT antibody, 1:400 | SICGEN ANTIBODIES | RRID:AB_2333092 |
| rabbit anti-PhosphoH3 Ser10, 1:800 | CellSignaling Technology | RRID:AB_1549592 |
| Donkey anti-chicken-FITC secondary antibody, 1:500 | Invitrogen | RRID:AB_923386 |
| Donkey anti-goat Alexa568 secondary antibody, 1:1000 | Molecular Probes | RRID:AB_2534104 |
| Donkey anti-rabbit Alexa647 secondary antibody, 1:1000 | Molecular Probes | RRID:AB_2762835 |
| Experimental models: Organisms/strains | ||
| Mouse: MADM-7-GT | The Jackson Laboratory | RRID:IMSR_JAX:021457 |
| Mouse: MADM-7-TG | The Jackson Laboratory | RRID:IMSR_JAX:021458 |
| Mouse: MADM-11-GT | The Jackson Laboratory | RRID:IMSR_JAX:013749 |
| Mouse: MADM-11-TG | The Jackson Laboratory | RRID:IMSR_JAX:013751 |
| Mouse: MADM-18-GT | (Contreras et al., 2021) | N/A |
| Mouse: MADM-18-TG | (Contreras et al., 2021) | N/A |
| Mouse: Emx1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:005628 |
| Mouse: Emx1-CreER | The Jackson Laboratory | RRID:IMSR_JAX:027784 |
| Mouse: Nestin-Cre | (Petersen et al., 2002) | N/A |
| Mouse: Nestin-CreER | (Imayoshi et al., 2006) | N/A |
| Mouse: Hprt-Cre | The Jackson Laboratory | RRID:IMSR_JAX:004302 |
| Mouse: Apc-flox | The Jackson Laboratory | RRID:IMSR_JAX:009045 |
| Mouse: TrkC | (Tessarollo et al., 1997) | N/A |
| Mouse: Ndel1 | The Jackson Laboratory | RRID:IMSR_JAX:026958 |
| Mouse: Lgl1-flox | (Klezovitch et al., 2004) | N/A |
| Software and algorithms | ||
| ZEN blue | ZEISS | http://www.zeiss.com/microscopy/en_us/products/microscope-software/zen.html#introduction |
| Photoshop | Adobe | adobe.com/products/photoshop |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Histosette (embedding cassette for tissue fixation) | Sakura | Cat# 4170-01 |
| Embedding molds for peripheral organs | Sakura | Cat# 4557 |
| Embedding molds for coronal brain sections | Polysciences, Inc.. | Cat# 18986-1 |
| Embedding molds for sagittal brain sections | Polysciences, Inc. | Cat# 18646A-1 |
| 6 well plates | TPP | Cat# 92406 |
| 24 well plates | TPP | Cat# 92424 |
| Flask Filters 500mL | TPP | Cat# 99505 |
| Microfuge Tubes 1.5 mL | Thermo Fisher Scientific | Cat# AM12450 |
| 50 mL Centrifuge Tubes | Sarstedt, Inc. | Cat# 62.547.254 |
| 15 mL Centrifuge Tubes | Sarstedt, Inc. | Cat# 62.554.502 |
| Syringe 60mL | Kendall | Cat# 560125 |
| Syringe 10 mL Omnifix | B.Braun | Cat# 4617100V |
| Needle 20G Sterican | B.Braun | Cat# 4657705 |
| 0.2micron filter | Nalgene | Cat# 194-2520 |
| Peristaltic Pump | Watson-Marlow | Cat# 323 S/D |
| Hydrophobic Pen | DAKO | Cat# S2002 |
| Petri dish | Thermo Fisher Scientific | Cat# NC9565080 |
| Slide Moisture Chamber black | Newcomer Supply | Cat# 68432A |
| Superfrost Glass Slides | Thermo Fisher Scientific | Cat# J1800AMNZ |
| Cover Slips 24 × 50 mm | VWR | 631–0147 |
| Dissection Tools | Freudenberg FST | Various, depending on the specific needs of the experiment |
| Fine Brush Size 1 | Ted Pella, Inc. | Cat# 11859 |
| LSM 800 Confocal | ZEISS | N/A |
| Cryostat Cryostar NX70 | Thermo Fisher Scientific | N/A |
Materials and equipment
Essential buffers and anesthesia for tissue harvesting (prepare 1 day prior to the experiment)
Timing: 60 min
1M PB buffer (pH 7.4)
| Reagent | Final concentration | Amount |
|---|---|---|
| 2M NaH2PO4. 2H2O (monobasic sodium phosphate) | 1M | 47.5 mL |
| 2M Na2HPO4 (dibasic sodium phosphate) | 1M | 202.5 mL |
| ddH2O | n/a | 250 mL |
| Total | 1M | 500 mL |
Keep at 22°C–25°C.
2N NaOH
| Reagent | Final concentration | Amount |
|---|---|---|
| NaOH | 2N | 40 g |
| ddH2O | n/a | 500 mL |
| Total | 2N | 500 mL |
Keep at 22°C–25°C.
4% PFA (in 0.1M PB buffer)—always prepare fresh PFA
| Reagent | Final concentration | Amount |
|---|---|---|
| Paraformaldehyde (PFA) | 4% | 40 g |
| 2N NaOH | 0.01N | 4 mL |
| 1M PB buffer | 0.1M | 100 mL |
| ddH2O | n/a | 850 mL |
| Total | 4% | 1,000 mL |
Details for PFA preparation: (1) Dissolve 40 g of PFA in 750 mL distilled water. (2) Add 4 mL 2N NaOH. (3) Place on a stirring heating plate at 55°C until all PFA has dissolved. (4) Add 100 mL 1M PB buffer. (5) Fill up the volume to 1000 mL with distilled water. (6) Filter through a flask filter to avoid residual undissolved PFA particles. (7) Store at 4°C for up to 1–2 days.
Anesthesia stock solution (100%)
| Reagent | Final concentration | Amount |
|---|---|---|
| Avertin | 100% | 7 g |
| t-amylalcohol | n/a | 7 mL |
| Total | 100% | 7 mL |
Details for anesthesia preparation: (1) Vortex until the solution appears homogeneous. (2) Wrap in tin foil to protect from light and store at 22°C–25°C for up to 6 months.
Anesthesia working solution (2.5%)
| Reagent | Final concentration | Amount |
|---|---|---|
| 100% anesthesia stock solution | 2N | 0.875 mL |
| 1× PBS | 1× | 34.125 mL |
| Total | 2.5% | 35 mL |
Details for anesthesia preparation: (1) Vortex until the solution appears homogeneous. (2) Filter the solution into a fresh tube using a 60 mL syringe and a 0.2micron filter. (3) Store at 4°C for up to one month.
1× PBS:
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PBS | 1× | 100 mL |
| ddH2O | n/a | 900 mL |
| Total | 1× | 1,000 mL |
Keep at 22°C–25°C.
Sucrose
| Reagent | Final concentration | Amount |
|---|---|---|
| Sucrose | 30% | 300 g |
| 1× PBS | 1× | Add up to 1,000 mL |
| Total | 30% | 1,000 mL |
We recommend to filter the sucrose solution through a 500 mL flask filter to avoid contamination with microorganisms. Store at 4°C.
Prepare perfusion equipment (on the day of experiment)
-
•
Clean all dissection tools (2 curved forceps, large and small scissors) with 70% ethanol.
-
•
Prepare a perfusion pad and whipping tissues.
-
•
Prepare a peristaltic pump or two 10 mL syringes and needles for perfusion.
-
•
Prepare 6-well plates filled with 4% PFA to collect brain tissues.
-
•
Prepare histosettes and a beaker filled with 4% PFA to collect peripheral organs.
Prepare essential buffers and reagents for histological stainings (on the day of the experiment)
Cryoprotectant
| Reagent | Final concentration | Amount |
|---|---|---|
| Glycerol | 60% | 300 mL |
| 1M PB buffer | 0.1M | 50 mL |
| ddH2O | n/a | 150 mL |
| Total | 60% | 500 mL |
Store at 4°C.
PBS with 0.5% Tween (PBS-T)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tween20 | 0.5% | 5 mL |
| 1× PBS | 1× | 995 mL |
| Total | 0.5% | 1000 mL |
Add 5 mL Tween20 to 1,000 mL 1× PBS and mix well. Store at 22°C–25°C.
Citrate buffer pH 6.0
| Reagent | Final concentration | Amount |
|---|---|---|
| Citric acid anhydrous (192M) | 1M | 1.92 g |
| Tween20 | 0.05% | 0.5 mL |
| ddH2O | n/a | 1,000 mL |
| Total | 1M | 1,000 mL |
Details for citrate buffer preparation: (1) Dissolve 1.92 g citric acid anhydrous (192M) in 1000 mL distilled water. (2) Adjust to pH 6.0 using NaOH. (3) Add 0.5 mL of Tween20. (4) Store at 4°C.
Triton-X-100 working solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Triton-X-100 | 20% | 2 mL |
| ddH2O | n/a | 8 mL |
| Total | 20% | 10 mL |
Store it in a 15 mL Falcon tube at 22°C–25°C.
Blocking buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Horse Serum | 10% | 1 mL |
| Triton-X-100 working solution | 0.5% | 0.25 mL |
| 1× PBS | 1× | 8.75 mL |
| Total | n/a | 10 mL |
Store it in a 15 mL Falcon tube at 4°C.
Staining buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Horse Serum | 5% | 0.5 mL |
| Triton-X-100 working solution | 0.5% | 0.25 mL |
| 1× PBS | 1× | 9.25 mL |
| Total | n/a | 10 mL |
Store it in a 15 mL Falcon tube at 4°C.
Note: You can prepare 1,000 ml of Blocking and Staining Buffer and aliquot in 50 mL or 15 mL tubes, which can be stored at −20°C until further usage. Once thawed, keep at 4°C and use up within 1 week.
DAPI stock solution
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 vial DAPI | 5 mg/mL | 10 mg |
| ddH2O | n/a | 2 mL |
| Total | 5 mg/mL | 2 mL |
Aliquot in 1.5 mL tubes and store at −20°C.
DAPI working solution
| Reagent | Final concentration | Amount |
|---|---|---|
| DAPI stock solution | 300nM | 1 μL |
| 1× PBS | 1× | 20 mL |
| Total | 300nM | 20 mL |
Store at 2°C–6°C up to one week, protected from light.
Embedding medium Mowiol-DABCO
| Reagent | Final concentration | Amount |
|---|---|---|
| Glycerol | n/a | 6 g |
| Mowiol | n/a | 4 g |
| ddH2O | n/a | 6 mL |
| 0.2M Tris-HCl (pH 8.5) | 0.1 | 12 mL |
| DABCO | n/a | 25 mg |
| Total | n/a | 25 mL |
Details for Mowiol-DABCO preparation: (1) Use a balance to weight 6.0 g Glycerol and 2.4 g Mowiol. Given the high viscosity of Glycerol, it is more accurate to measure its weight than its volume. (2) Mix both reagents and dissolve for 1 h on a stirring plate. (3) Add 6 mL distilled water and mix for another hour. (4) Add 12 mL 0.2M Tris-HCl (pH 8.5) and incubate for 2 h at 50°C on a stirring heating plate. (5) It is likely that not all Mowiol is going to dissolve. Thus, you can add an additional centrifugation step to remove undissolved Mowiol from the preparation by centrifuging for 15 min at 5,000 g and keeping the supernatant. (6) Add 25 mg DABCO and dissolve so that you have a clear solution. (7) Aliquot in 1.5 mL tubes and store at −20°C until further usage.
Step-by-step method details
Breeding of experimental MADM mice for the generation of genetic mosaic tissue
In order to generate genetic mosaic mice, you can either chose a breeding strategy maximizing the efficiency of experimental MADM animal output (here called “strategy 1”) or a breeding strategy maximizing the speed of obtaining experimental MADM animals (here called “strategy 2”) (Figures 2B and 2C). We provide an estimated amount of breeding mice required to obtain a realistic number of experimental MADM animals (Figures 2B and 2C). We provide step-by-step breeding guidance in the following section of this protocol (also see troubleshooting problem 1):
-
1.
For both breeding strategies, perform Breeding step 1 for gene-X of interest: In order to link the mutation of gene-X to the MADM TG cassette, cross MADMTG/TG animals with a mouse line mutant for gene-X (Figures 2B(i) and 2C(i)). The resulting F1 generation will be trans-heterozygous for the MADM TG cassette and the mutation of interest (genotype will be MADMTG/+; gene-X+/-).
-
2.
For both breeding strategies, perform Breeding step 1 for the Cre driver (Figures 2B(iii) and 2C(ii)): cross MADMGT/GT animals with a Cre driver of choice. The resulting F1 generation will be double heterozygous for the MADM GT cassette and the Cre driver specific for the cell type of interest (MADMGT/+; Cre+/-). Perform this step in parallel to the above step.
Allow F1 generation mice to grow up to 8 weeks of age. For breeding strategy 2, proceed to step 6. For breeding strategy 1, continue with steps 3, 4 and 5.
-
3.
For strategy 1, perform Breeding step 2 for the mutant by crossing MADMTG/+;gene-X+/- mice with MADMTG/TG animals in order to generate F2 offspring homozygous for the MADM TG cassette and meiotically linked gene-X (MADMTG/TG,gene-X) (Figure 2B(ii)).
-
4.
For strategy 1, perform Breeding step 2 for the Cre driver by intercrossing MADMGT/+; Cre+/- mice with MADMGT/+; Cre+/- animals in order to generate F2 offspring homozygous for the MADM GT cassette and the Cre driver. The resulting genotype will be MADMGT/GT; Cre+/+ (Figure 2B(iv)). Perform this step parallel to step 3.
Allow F2 generation mice to grow up to 8 weeks of age.
-
5.
For the generation of experimental MADM animals according to strategy 1, cross MADMTG/TG,gene-X mice with MADMGT/GT; Cre+/+ animals in order to generate experimental offspring with genetic mosaic tissue with green mutant, red wild-type and yellow heterozygous cells (Figure 2B(v)).
The genotype of the experimental MADM animals is MADMGT/TG,gene-X; Cre+/- (Figure 2B(v)).
The probability to obtain experimental MADM animals from this breeding is 50%. The probability for control-MADM animals is also 50%, i.e., animals with MADM-labelling but without mutation. With an estimated litter size of 10 pups per litter, this means that you require 1 breeding cage to obtain a total of 5 experimental MADM mice (Figure 2B(vi)).-
a.Check for the vaginal plug to monitor successful mating and to expedite the experimental planning.
-
b.Male and female offspring can be used as experimental animals.
-
c.Perform experiment at developmental stage(s) of choice.
-
a.
-
6.
For the generation of experimental MADM animals according to strategy 2, cross trans-heterozygous MADMTG/+;gene-X+/-mice directly with double heterozygous MADMGT/+; Cre+/- animals in order to generate experimental MADM offspring with genetic mosaic tissue with green mutant, red wild-type and yellow heterozygous cells (Figure 2C(iii)).
The genotype of the experimental MADM animals is MADMGT/TG,gene-X; Cre+/- (Figure 2C(iii)).
The probability to obtain experimental MADM animals from this breeding is ∼5%. With an estimated litter size of 10 pups per litter, you require ∼10 breeding cages and ∼100 pups to obtain a total of 5 experimental MADM mice (Figure 2C(iv)).-
a.Check for the vaginal plug to monitor successful mating and to expedite the experimental planning.
-
b.Male and female offspring can be used as experimental animals.
-
c.Perform experiment at developmental stage(s) of choice.Note: In the following examples described in this protocol, all tissue was analyzed at specific postnatal stages. However, experimental animals can also be used at embryonic stages, P0, and any postnatal age depending on your experimental needs.In order to provide a broad overview of applications suitable for histological analysis using MADM in this protocol, we made use of a number of different Cre drivers: (1) constitutively active Emx1-Cre and TM-inducible Emx1-CreER that primarily targets radial glia progenitors in the dorsal forebrain, which give rise to excitatory projection neurons and glial cells in the cerebral cortex (Gorski et al., 2002; Kessaris et al., 2006). (2) We include an example using constitutively active Nestin-Cre and inducible Nestin-CreER (Petersen et al., 2002; Imayoshi et al., 2006), which is a marker of stem cells across the nervous system and therefore results in MADM labeling in all neural cell types including Purkinje cells; (3) and make use of constitutively active Hprt-Cre (Tang et al., 2002) which shows ubiquitous spatiotemporal expression. The Hprt-Cre driver was utilized to investigate peripheral organs, in particular the intestine.MADM can also be applied for experiments involving flow cytometry. For details and a step-by-step protocol on the experimental workflow specific for flow cytometry experiments, we refer to (Laukoter et al., 2020a).Specific considerations when breeding experimental MADM animals are provided in the next two sections of the protocol:
-
a.
Generation of experimental MADM mice for postnatal histological analysis in brain
-
7.
For population analysis, cross MADMGT/GT; Emx1Cre/Cre or MADMGT/GT; Nestin-Cre+/+ females with MADMTG/TG;gene-X males in order to generate offspring with genetic mosaic brain tissue (Figure 2B, bottom panel) (also see troubleshooting problem 4).
For clonal analysis, cross MADMGT/GT; Emx1CreER/CreER or MADMGT/GT; Nestin-CreER+/+ females with MADMTG/TG;gene-X males in order to generate offspring with genetic mosaic brain tissue following TM injection at a time point of choice (Figure 2B, bottom panel) (also see troubleshooting problem 5).
For simplicity, the above mentioned distinct Cre/ER drivers will be abbreviated as “Cre” throughout step 7.-
a.Check for the vaginal plug to monitor successful mating and to facilitate the experimental planning.
-
b.Males and females of the resulting F1 generation can be used as experimental animals (genotype will be MADMGT/TG,gene-X; Cre+/-).Note: Upon crossing MADMGT/GT; Cre+/+ females with MADMTG/TG,gene-X males to generate MADMGT/TG,gene-X; Cre+/- mosaic animals, green cells harboring a homozygous mutation in gene-X will show uniparental paternal chromosome disomy (patUPD, PP), while red wild-type cells will show uniparental maternal chromosome disomy (matUPD, MM) (Figure 3A). We refer to this breeding scheme as ‘default breeding scheme.’Note: In order to reverse the fluorescent colors to control for a potential bias of the fluorescent reporter (color swap), couple the mutation to the MADM GT cassette and use MADMTG/TG females and MADMGT/GT,gene-X; Cre+/+ males (Laukoter et al., 2020a). Here, red cells harboring a homozygous mutation in gene-X will show patUPD (PP), while green wild-type cells will show matUPD (MM) (Figure 3B). For efficient conduction of this control experiment, we recommend to perform the color swap in the context of population analysis using a constitutively active Cre driver and not a TM-inducible CreER.Note: In order to reverse the parents to control for a potential genomic imprinting bias of the parental origin of the mutation (parent swap), use MADMTG/TG,gene-X females and MADMGT/GT; Cre+/+ males (Laukoter et al., 2020a) to generate MADMGT/TG,gene-X; Cre+/- mosaic animals. Here, green cells harboring a homozygous mutation in gene-X will show matUPD (MM), while red wild-type cells will show patUPD (PP) (Figure 3C). Given that the generation of stock lines for your experiments will yield MADMTG/TG,gene-X females and MADMGT/GT; Cre+/+ males in any case, use the opportunity of having these mice available and perform the parent swap in parallel to your ‘default breeding’ setup. For efficient conduction of this control experiment, we recommend to perform it in the context of population analysis using a constitutively active Cre driver and not a TM-inducible CreER.
-
c.Perform experiment at your desired postnatal time point, for example when generation of cell types is completed and the cells have reached a fully developed, mature state (in our examples shown in this protocol we chose time points between P1 and P21).
-
a.
Figure 3.

Breeding schemes for default setup, color swap, and parent swap
(A) Schematic showing the default breeding scheme using males where gene-X is meiotically linked to the MADM TG cassette, while females transmit the GT cassette and the Cre driver. Resulting offspring will contain red wild-type cells with uniparental maternal chromosome disomy (matUPD, MM), while green mutant cells will harbor uniparental paternal chromosome disomy (patUDP, PP).
(B) Schematic of the breeding scheme for a color swap experiment using females homozygous for the MADM TG cassette, while males transmit the Cre driver plus gene-X meiotically linked to the GT cassette. Resulting offspring will have green wild-type cells with matUPD (MM), while red mutant cells will harbor patUPD (PP).
(C) Schematic of the breeding scheme for a parent swap experiment using females where gene-X is meiotically linked to the MADM TG cassette, while males transmit the GT cassette and the Cre driver. Resulting offspring will contain red wild-type cells with patUPD (PP), while green mutant cells will harbor matUPD (MM).
Parts of the figure were reproduced and adapted with permission from (Beattie et al., 2017).
Generation of experimental MADM mice for postnatal histological analysis of genetic mosaic tissue in peripheral organs
-
8.Cross MADMGT/GT; HprtCre/+ females with MADMTG/TG,gene-X males in order to generate offspring with genetic mosaic tissues.
-
a.Check for the vaginal plug to monitor successful mating and to facilitate the experimental planning.
-
b.Males and females of the resulting F1 generation can be used as experimental animals (genotype will be MADMGT/TG,gene-X; HprtCre/+).
 CRITICAL: The Hprt-Cre transgene is located on the X chromosome, with one of the two X chromosomes undergoing random inactivation in females. Thus, Cre activity and MADM labelling are highly variable in MADMGT/TG,gene-X; HprtCre/+ females. We therefore recommend to primarily use males for your analysis in order to obtain reproducible results.Note: Upon crossing MADMGT/GT; HprtCre/+ females with MADMTG/TG,gene-X males to generate MADMGT/TG,gene-X; HprtCre/+ mosaic animals, green cells harboring a homozygous mutation in gene-X will show patUPD, while red wild-type cells will show matUPD.Note: If desired, perform color swap and parent swap (Laukoter et al., 2020a) as described in the notes for step 7 of the protocol.
CRITICAL: The Hprt-Cre transgene is located on the X chromosome, with one of the two X chromosomes undergoing random inactivation in females. Thus, Cre activity and MADM labelling are highly variable in MADMGT/TG,gene-X; HprtCre/+ females. We therefore recommend to primarily use males for your analysis in order to obtain reproducible results.Note: Upon crossing MADMGT/GT; HprtCre/+ females with MADMTG/TG,gene-X males to generate MADMGT/TG,gene-X; HprtCre/+ mosaic animals, green cells harboring a homozygous mutation in gene-X will show patUPD, while red wild-type cells will show matUPD.Note: If desired, perform color swap and parent swap (Laukoter et al., 2020a) as described in the notes for step 7 of the protocol. -
c.Perform the experiment at the desired postnatal time point, to e.g., analyze cellular phenotypes arising during development in mature tissue and/or trace disease phenotypes when a disease phenotype is evident (up to several months of age).
-
a.
Mouse perfusion and dissection for collection of MADM-labelled tissue for histological analysis
This section of the protocol documents the perfusion and dissection of postnatal mice for organ removal and further downstream histological analysis.
-
9.Arrange all solutions and tools required for the tissue collection at the dissection area
-
a.Anesthesia
-
b.1× PBS
-
c.4% PFA
-
d.Peristaltic pump or perfusion syringes
-
e.Dissection tools
-
f.6-well plates
-
a.
-
10.
Anesthetize the animal by intraperitoneal injection of anesthesia working solution at a volume of 0.16–0.24 mL/10 g mouse (equivalent to 400–600 mg/kg).
-
11.
Place the anesthetized animal in supine position on a perfusion tray and disinfect fur with 70% ethanol.
-
12.
Below the rib cage, make a medial incision with scissors and surgical forceps through the skin and the underlying muscle layer (Figure 4A). Continue to cut laterally.
-
13.
Lift the tip of the sternum and snip the diaphragm. Then trim the rib cage to expose the heart.
-
14.
Using small iris scissors make an incision to the posterior end of the right atrium for the blood to drain during the following perfusion procedure (Figure 4B).
-
15.
Insert a needle attached to a peristaltic pump or a syringe filled with 1× PBS into the lower left ventricle (Figure 4C).
-
16.
Perfuse with PBS until the liver turns pale (requires approximately 5–10 mL of PBS).
-
17.Once complete, switch the perfusion solution to 4% PFA.Note: Perfusion requirements need to be adjusted according to the age of the animals. (a) For the perfusion of pups: perfuse manually using 10 mL syringes. Prepare 2 distinct 10 mL syringes: one filled with 1× PBS and another filled with 4% PFA. Once perfusion with PBS is completed, replace PBS-containing syringe plus needle with fresh needle and syringe containing PFA through the same puncture hole in the heart. (b) For the perfusion of juvenile and adult mice, use a peristaltic pump. Place one end of the tube of the peristaltic pump into a flask or Falcon tube filled with PBS. After perfusion with PBS (speed: 5 mL at 2–4 mL/min in juvenile animals; 10 mL at 4–6 mL/min for adult mice), switch to 4% PFA.CRITICAL: briefly stop the peristaltic pump and exchange the PBS solution with ice-cold 4% PFA. This is critical to avoid bubbles in the pump tubing. Resume perfusion with PFA at the same speed.
-
18.
Remove mouse from perfusion pad and dissect the organ of interest. If organ of interest is brain, continue to follow the protocol. If organ of interest is a peripheral organ, proceed to step 27.
-
19.Dissection of brain:
-
a.Decapitate mouse.
-
b.Remove the skin over the skull (Figure 4D).
-
c.Cut the skull at the naso-frontal suture (in front of the olfactory bulbs) (Figure 4E).
-
d.Then cut along the midline (Figure 4F), followed by a cut along the parietal-interparietal sutures.
-
e.Lift the skull to the side to expose the brain (Figure 4G).
-
f.Place curved forceps or a thin spatula underneath the brain and remove it from the skull (Figure 4H).
-
a.
-
20.
Transfer the brain to a well filled with ice-cold 4%PFA in a 6-well plate (Figure 4I).
-
21.
Incubate at 4°C for minimum 10 h.
-
22.
Next day, wash 3× with 1× PBS for 5 min each.
-
23.
Transfer brain to a 15 mL Falcon tube filled with 10 mL of 30% Sucrose.
-
24.
Incubate at 4°C until the brain has sunk to the bottom of the Falcon tube.
-
25.
Remove brain from sucrose and gently remove residual sugar from the surface of the brain using lint-free tissues.
-
26.Depending on the cell type of interest and the needs of the experiment, embed brain in coronal or sagittal orientation:
-
a.For coronal orientation (Figure 4J), place brain ventral-side down on a piece of lint-free tissue and remove the cerebellum using a razor blade (Figures 4K and 4L). Place brain with the caudal side down in an embedding mold (Figure 4M), fill mold and cover tissue with O.C.T. Tissue tek and freeze. Once frozen, store it at −80°C.
-
b.For sagittal orientation (Figure 4N), place brain ventral-side down on a piece of lint-free tissue and cut it in two halves along the midline using a razor blade (Figures 4O and 4P). Place brain with the medial side down in an embedding mold (Figure 4Q), fill mold and cover tissue with O.C.T. Tissue tek and freeze. Once frozen, store it at −80°C.
 Pause point: You can keep the frozen tissue at −80°C until further usage.
Pause point: You can keep the frozen tissue at −80°C until further usage.
-
a.
-
27.Dissection of peripheral organ (this protocol is using the colon as an example):
-
28.
Incubate in a beaker filled with 4% PFA for 4 h at 22°C–25°C (Figure 4U).
-
29.
Wash 3× with 1× PBS for 5 min each.
-
30.
Incubate in 30% Sucrose at 4°C for minimum 10 h.
-
31.
Remove histosette with colon from sucrose, take out colon and gently remove residual sugar from the surface of the organ by using lint-free tissues.
-
32.
Transfer colon to an embedding mold, fill mold and cover tissue with O.C.T. Tissue Tek and freeze (Figure 4V). Once frozen, store it at −80°C.
Figure 4.

Tissue harvesting for histological analysis of experimental MADM mice
(A–C) Mouse dissection and initiation of perfusion.
(D–Q) Mouse dissection to isolate and embed brain.
(R–V) Isolation and embedding of colon.
Tissue sectioning for histological analysis
This section of the protocol lists details on sectioning MADM-labelled tissue for confocal microscopy.
-
33.
Remove tissue from −80°C and transfer to a cryostat cooled to −20°C.
-
34.
Let tissue adapt to temperature change for 20 min.
-
35.
Remove tissue block from the embedding mold.
-
36.
Attach the tissue block to the specimen disk in the cryostat by applying a drop of OCT to the disk and placing the tissue block directly into the OCT. Allow to freeze for 1–2 min.
-
37.
Insert disk with attached tissue block in the sectioning holder by placing the tissue in a correct orientation.
Note: For brain sections, insert sectioning disc with cortex facing towards the top. For peripheral organ sections, insert sectioning disc with organ of choice in an orientation that guarantees good sectioning quality for your required analysis.
-
38.
Start cutting by trimming the tissue block in 45 μm thick sections until the region of interest is reached.
-
39.
Slice the tissue in 30 μm thick sections. Either collect the slices in individual wells of a 24 well plate (= floating sections) or mount them directly onto frosted glass slides.
Tissue mounting
This part of the protocol can be skipped if the sliced tissue is directly mounted onto frosted glass slides in the course of the sectioning process. In that case, immediately proceed to step 45 of the protocol. If you prepared floating sections, continue with step 40.
-
40.
Fill a Petri dish with PBS-T.
-
41.
Place a frosted glass slide at the edge of the petri dish so that is covered up nearly to the label with PBS-T.
-
42.
Transfer all of your desired sections into the dish filled with PBS-T.
-
43.
Use a small paint brush to maneuver the sections onto the slide and arrange them side by side.
-
44.
Once all sections are placed onto the slide (for brain usually ∼12−16 sections/slide), horizontally transfer the slide to a dark slide chamber and let the sections dry completely (∼15 min) to make them adhere to the glass. Either proceed to the next steps of the staining procedure or pause the protocol at this point.
Staining
This section of the protocol explains the staining procedure for MADM-labelled cells together with an additional, cell-type specific marker staining. All steps are performed at 22°C–25°C unless otherwise stated. Please refer to troubleshooting problem 2 when observing variable signals.
-
45.
If your sections were floating sections and have been freshly mounted and dried, immediately proceed to step 46 of the protocol. If your sections were already mounted during sectioning and stored at −20°C, take the slides from the freezer and let them dry for 15 min.
-
46.
Encircle staining area with a hydrophobic pen.
-
47.
Rehydrate and wash sections 3× 5 min with 1× PBS to remove residual PBS-T.
-
48.
Meanwhile, heat up an oven to 85°C. Take the opportunity to put the antigen retrieval buffer into the oven to have the solution pre-heated. Our most commonly used antigen retrieval solution is Citrate Buffer pH 6.0.
-
49.
Incubate your sections with 1 mL pre-heated citrate buffer pH 6.0 for 25 min at 85°C in a dark and closed staining chamber (make sure it is closed. If it is open, buffer will evaporate and samples will dry out).
-
50.
Remove the staining chamber and let chill on your bench for 15 min. Open it on one corner a tiny bit so the samples can cool down more efficiently (don’t open too much, otherwise the buffer will evaporate and samples will dry out).
-
51.
Wash 3× 5 min with 1× PBS.
-
52.
Incubate with 500 μL Blocking Solution for 1 h.
-
53.
Place humidified tissues into the staining chamber to prevent your samples from dehydration during the next steps of the protocol.
-
54.
Dilute primary antibodies in Staining Solution, add 250 μL antibody mix for one sample and incubate 8–12 h at 4°C.
-
55.
Wash 3× 5 min with 1× PBS.
-
56.
Dilute secondary antibodies in Staining Solution, add 250 μL antibody mix for one sample and incubate for 2 h.
-
57.
Wash 3× 5 min with 1× PBS.
-
58.
Add 1 mL DAPI working solution for 15 min.
-
59.
Wash 1× 10 min in PBS
-
60.
Embed the slides with Mowiol and let dry at 22°C–25°C for 30 min.
Note: Instead of mounting the floating sections onto glass slides prior to the staining procedure, the whole staining protocol can be performed on floating sections in a 24well plate. Buffers are removed manually using a pasteur pipet or a P1000. We recommend the following volumes when performing stainings in 24well plates:
| Buffer volumes per well for staining of floating cryosections | |
|---|---|
| Reagent | Volume (μl) |
| PBS | 1,000 |
| Antigen retrieval buffer | 500 |
| Blocking Buffer | 500 |
| Staining Buffer (including antibody mix) | 250 |
| DAPI working solution | 500 |
Here, sections should be mounted after the final washing step following the secondary antibody staining. Mounting process is equivalent to steps 40 to 44 of the protocol. Once mounting and section drying is completed, you can proceed with DAPI staining and Mowiol embedding (steps 58 to 60 of the protocol).
Antibodies and their dilutions for MADM staining
| Antibody | Species | Dilution |
|---|---|---|
| Anti-GFP | Chicken | 1:400 |
| Anti-mCherry | Goat | 1:400 |
| Anti-chicken FITC | Donkey | 1:500 |
| Anti-goat Alexa568 | Donkey | 1:1000 |
Antibodies and their dilutions for additional marker staining, here an example for staining performed in the colon (Figure 6)
| Antibody | Species | Dilution |
|---|---|---|
| Anti-PhosphoH3 | Rabbit | 1:800 |
| Anti-rabbit Alexa647 | Donkey | 1:1000 |
Microscopy and image processing
In this section of the protocol, we describe the microscopy settings and subsequently required confocal image processing for further downstream analysis.
-
61.
For the analysis of MADM-labeled tissue we recommend to use an inverted confocal microscope, in our case Zeiss LSM800 with corresponding image acquisition software ZEN blue.
-
62.
Select the correct laser lines and filters. For MADM tissue with additional marker staining, select excitation: 405 nm (for DAPI), 488 nm (for FITC / GFP), 561 nm (for Alexa568 / tdT) and 640 nm (for Alexa647).
-
63.
Set to pinhole to 1 airy unit for optimal imaging quality.
-
64.
Select the functions “Z-stack” and “Tiles” from the acquisition dashboard.
-
65.
Select objective, scanning speed, resolution and averaging appropriate to the needs of your experiments.
-
66.
Adjust laser intensity and gain for each channel.
-
67.
Identify your area of interest and set position and imaging tiles to cover the area of interest. Adjust the z-stack so that all MADM-labeled cells in the tissue are captured.
-
68.
Assign the center position of your Z-stack to the tiles and the positions and click “Start” to initiate image acquisition.
-
69.
Save the image in .czi file format.
Note: For tissue overview images and cell number quantification, select a 10× objective and use a scanning speed pixel dwell value of 1.52–2.06 μs (values 7–8 in the ZEN blue image acquisition software) with resolution 1024 × 1024 and no averaging. For high-quality images and morphological analysis, use a high magnification objective (e.g. 63×) and use a scanning speed pixel dwell value of maximum 2.06 μs (up to value 7 in the ZEN blue image acquisition software) with resolution 1024 × 1024 and 2× or 4× averaging.
-
70.
Once image acquisition is completed, use the image processing dashboard in ZEN blue and perform maximum intensity projection using the “Orthogonal Projection” function.
-
71.
Following projection, perform “Stitching” function by fusing tiles.
-
72.
Save the projected and stitched image as a new .czi file.
-
73.
Export the projected and stitched .czi file to TIFF format. Export DAPI / GFP / tdT / Alexa647 channels individually for subsequent image analysis.
Expected outcomes
Compare your readout of green and red cells (such as cell number, cell position, morphological parameters, presence of tumors) in genetic mosaic MADM animals. Since the genotypes of red cells (wild-type) and green cells (mutant) are different, any deviation from a comparable green-to-red (G/R) read-out indicates a cell-autonomous function of gene-X in your cell type of interest.
In order to study stem cell output by counting cell numbers, determine the G/R cell ratio of your cell type of interest (Figures 5F and 5H): If G/R ratio were above 1, it indicates a proliferation increase of green mutant stem cells, whereas a G/R ratio below 1 indicates cell loss either through diminished stem cell proliferation or apoptosis of either stem cells or their descendants. Thus, gene-X has a cell-autonomous function in governing overall stem cell output.
Figure 5.

Examples and analysis of genetic mosaic MADM animals with mosaicism in the brain
(A–C) Schematic of G2-X MADM events resulting in (A) genetic mosaic tissue using (B) constitutively active Cre driver for population analysis and (C) tamoxifen-inducible CreER driver for clonal analysis.
(D–H) Analysis of central nervous system (CNS) stem cell output in Lgl1-MADM mice using Lgl1 mutant allele (Klezovitch et al., 2004). (D) schematic of Lgl1-MADM cortical tissue; (E and F) population analysis of Lgl1 using Emx1-Cre driver (Gorski et al., 2002) with (E) overview of a representative brain section at P21, (E′) insert showing the cortex, and (F) quantification of the G/R ratio of astrocytes from wild-type mice and Lgl1-MADM mice; (G and H) clonal analysis of Lgl1 using Emx1-CreER driver (Kessaris et al., 2006) with (G) reconstruction of a representative clone at P21 and (H) quantification of the number of green astrocytes in green subclones for wild-type and Lgl1 mutant clones. Scale bars: (E) 500 μm, (E′) 60 μm, (G) 70 μm). Values represent mean ± SEM. ns, nonsignificant, ∗∗∗p < 0.001. Parts of the figure and its legend were adapted and reused from (Beattie et al., 2017) with permission.
(I–N) Analysis of cortical projection neuron migration in Ndel1-MADM mice using Ndel1 knock-out allele (Sasaki et al., 2005). (I) schematic of Ndel1-MADM cortical tissue; (J–L) population analysis of Ndel1 using Emx1-Cre driver (Gorski et al., 2002) with (J) overview of a representative image of the cortex section at P1, (K) schematic indicating partitioning of the cortex into layers and layer-specific quantification of red and green neurons, and (L) quantification of the relative distribution of red wild-type and green Ndel1 mutant cells in the respective layers; (M and N) clonal analysis of Ndel1 using Nestin-CreER driver [line 1 in (Imayoshi et al., 2006)] with (M) reconstruction of a representative clone at E18.5 and (N) quantification of the relative distribution of red wild-type and green Ndel1 mutant cells in the respective layers. Scale bars: (J) 150 μm, (M) 100 μm. Values represent mean ± SEM. ns, nonsignificant; ∗p < 0.05 and ∗∗∗p < 0.001. Parts of the figure and its legend were adapted and reused from (Hippenmeyer et al., 2010) with permission.
(O–R) Analysis of Purkinje cell morphology in TrkC-MADM mice using TrkC knock-out allele (Tessarollo et al., 1997) and Nestin-Cre driver (Petersen et al., 2002). (O) schematic of TrkC-MADM neuronal tissue; (P and Q) representative high magnification images of (P) red wild-type and (Q) green TrkC mutant Purkinje cells in the cerebellum of P21 TrkC-MADM mice, (R) quantification of the height deficiency index of red wild-type and green TrkC mutant cells within the Molecular layer of the cerebellum. Scale bars: (P and Q) 50 μm. Presented morphology values are means ± SEM. ∗∗∗ p < 0.001, one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. Parts of the figure and its legend were adapted and reused from (Joo et al., 2014) with permission.
To study cell migration, measure cell position and the number of red wild-type and green mutant cells in certain positions (Figures 5L and 5N): A deviation of numbers of green cells vs. red cells in particular bins is indicative of a cell migration alteration. A large absence of green mutant neurons from cortical layers and their accumulation in the white-matter demonstrates a cell-autonomous function of gene-X in neuronal migration. Based on the sparse endogenous labeling with GFP and tdT, MADM is also well suited to study cell migration in live imaging assays and 4D tracing with high resolution (Riccio et al., 2016; Hippenmeyer et al., 2010).
Determine morphological parameters, such as the height index (Figure 5R): calculate the height index by the formula a/b (a = the distance from the pial surface to the top of the arbor of a Purkinje cell; b= the molecular layer span) and compare the values for red wild-type and green mutant cells side-by-side. If green mutant cells display higher values for height parameters than red wild-type cells, this result indicates a decreased height of the analyzed cells. As a conclusion, gene-X exerts a cell-autonomous function in cellular morphology.
In order to study a disease model such as tumorigenesis (Figure 6C), compare the numbers and area of red and green tumors in your tissue of choice. An increased number of green tumors and tumor area indicates a cell-autonomous function of gene-X in stem cell proliferation and tumor initiation, whereas a decreased number indicates a cell-autonomous disadvantage in tumor growth.
Figure 6.

Example and analysis of genetic mosaic MADM mice with mosaicism in peripheral organs
(A–C) MADM analysis of Apc tumor suppressor gene in the colon using Apc-flox allele (Cheung et al., 2010) and Hprt-Cre driver (Tang et al., 2002). (A) schematic of Apc-MADM tissue; (B) representative image of an adenoma arising from green Apc mutant stem cells in the colon of a 3month old Apc-MADM mouse. Inserts (i) and (ii) show unlabeled healthy colon epithelium, where proliferating cells (as indicated by M-Phase marker staining with P-H3 antibody) are only located in the crypt bottom, but not above the crypt region or along the differentiated epithelium. In contrast insert (iii) highlights a subepithelial region within the adenoma displaying proliferating cells. (C) Quantification of the tumor area originating from red wild-type and green Apc mutant cells. Scale bars: (B) 200 μm, (i, ii, iii) 40 μm. Values represent means ± standard deviation.
Quantification and statistical analysis
Once a stem cell undergoes a G2-X MADM event, one green labelled mutant and one red labelled wild-type daughter cell is generated in an unlabeled environment (Figure 5A). Such sparsely labelled cells can be subjected to a variety of downstream analyses, such as lineage tracing, cell number quantification, positional analysis or morphological analysis (Figure 5).
These distinct types of downstream analysis can be performed for both, population and clonal analysis. While population analysis results in sparse MADM-labeling with no spatiotemporal information (Figure 5B) clonal analysis employing a TM-inducible CreER driver can provide high spatiotemporal resolution (Figure 5C).
The following steps of the protocol provide a short description of the distinct types of histological analysis enabled by the MADM-induced genetic mosaics (also see troubleshooting problem 3):
-
1.
For the determination of stem cell output, quantify the number of red and green cells in your acquired images using the counting tool in Photoshop. To do this, load all individual channel images into one image stack to keep channels separate. Use the counting tool to manually quantify the number of distinct cell types derived from red wild-type and green mutant stem cells. Afterwards, calculate and plot the green-to-red (G/R) cell ratio. (Figures 5D–5H).
Alternatives: if photoshop is not accessible, alternative freely accessible software such as Fiji can be used for the quantification of green and red cells. In Fiji, use the tool ‘multi-points’ for quantification.
-
2.
For the assessment of cellular migration, partition the tissue into defined layers or bins. Next, quantify the number of individual red wild-type and green mutant cells within these layers or bins by using the ‘counting’ tool in photoshop. Plot the cell number per bin for red and green cells side-by-side and compare their distribution to determine if mutant cells are found in a different position than wild-type cells (Figures 5I–5N).
-
3.
In order to determine cellular morphology, import your .czi image into a software appropriate for morphological analysis, such as Imaris and use the filament tracer function. Determine parameters such as cell volume or complexity using Sholl analysis. Compare the values for red wild-type and green mutant cells to assess if the mutation causes a morphology phenotype (Figures 5O–5R). For assessment of cell height, define specific parameters, such as the distance from the pial surface to the top of the arbor of a Purkinje cell (parameter a) and the molecular layer span (parameter b) (Figure 5Q). Such parameters can be measured with the ‘ruler’ tool using ZEN software on the .czi file of your image or in photoshop or Fiji using other image formats such as .tif.
-
4.
For disease modeling such as the investigation of tumor initiation and growth, quantify the number of individual tumors using the counting tool in photoshop or determine the size of the tumor area by delineation of the tumor margin in ZEN blue or in photoshop (Figure 6).
Limitations
The genome-wide library of MADM mice allows to employ MADM to study the vast majority, nearly genome-wide (>96%), of autosomal genes in the mouse genome. Mouse autosomes have a telocentric conformation and we thus inserted the MADM cassettes as close as possible to the centromere to maximize the number of genes located distally to the MADM cassette insertion site. In order to be meiotically linked to the corresponding MADM chromosome, the gene of interest needs to be located distally to the MADM cassette. The MADM principle relies on meiotic recombination to genetically link a mutation to the MADM cassette. However, too close proximity of the gene of interest to the MADM cassette might make a recombination event difficult or impossible. In such cases, other techniques offering sparse induction of a mutation of choice could be used, such as MADR (Kim et al., 2019), Dual ifgMosaic (Pontes-Quero et al., 2017) or BATTLE (Kohara et al., 2020).
The library of MADM mice currently does not include strains with MADM cassettes inserted on the sex chromosomes, thus while the vast majority (>96%) of genes can be studied, the genes located on the X or Y chromosome are not yet covered by the MADM technique.
Troubleshooting
Problem 1
Low number of experimental mice (breeding of experimental MADM mice – steps 1–6).
Potential solution 1
Make sure the genetic background of your mice does not exhibit negative effects on their mating performance. Increase the number of breeding cages to gain a good number of F1 or F2 mice to start with.
Furthermore, control whether you have correctly followed the provided breeding strategies. Wrongly genotyped or selected parental mice can result in strongly decreased efficiency in the generation of experimental animals.
Problem 2
Weak or variable MADM signal without staining for GFP and tdT (Staining – steps 45–60).
Potential solution 2
Endogenous fluorescence intensity of GFP and tdT are pH sensitive. Make sure all of your buffers have the correct pH.
Additionally, perform staining for GFP and tdT using the antibodies indicated above.
Problem 3
Highly variable results in MADM lines with low recombination frequency (Quantification and Data Visualization – steps 74–77).
Potential solution 3
Analysis of all 19 MADM lines showed that different MADM chromosomes harbor distinct recombination frequencies [sparse, intermediate or dense, (Contreras et al., 2021)]. The required number of experimental animals and the number of tissue sections used for analysis need to be scaled up when working with a MADM chromosome that gives sparse labeling. We recommend using the entire amount of tissue sections available for one animal to avoid introduction of an analysis bias when only using a few sections from a low recombination line.
Problem 4
Low breeding performance (Generation of experimental MADM mice for histological analysis, steps 7–8).
Potential solution 4
Discuss with your veterinarian how to optimize the mating conditions (e.g., by providing special food or housing conditions).
Problem 5
High abortion rate upon Tamoxifen injection for clonal analysis (Generation of experimental MADM mice for histological analysis, step 7).
Potential solution 5
Titrate the concentration of Tamoxifen according to your experimental needs to find an optimal balance between survival and clone numbers. Additionally, recover pups by caesarian sections and perform fostering to increase the survival rate of experimental animals.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Simon Hippenmeyer (simon.hippenmeyer@ist.ac.at).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This research was supported by the Scientific Service Units (SSU) at IST Austria through resources provided by the Bioimaging (BIF) and Preclinical Facilities (PCF). We particularly thank Mohammad Goudarzi for assistance with photography of mouse perfusion and dissection. N.A. received support from FWF Firnberg-Programm (T 1031). This work was also supported by IST Austria institutional funds; FWF SFB F78 to S.H.; and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 725780 LinPro) to S.H.
Author contributions
All authors conceived the project and edited and proof-read the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
No custom code is necessary to perform this protocol.
References
- Beattie R., Postiglione M.P., Burnett L.E., Laukoter S., Streicher C., Pauler F.M., Xiao G., Klezovitch O., Vasioukhin V., Ghashghaei T.H., Hippenmeyer S. Mosaic analysis with double markers Reveals distinct sequential functions of Lgl1 in neural stem cells. Neuron. 2017;94:517–533.e3. doi: 10.1016/j.neuron.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Beattie R., Streicher C., Amberg N., Cheung G., Contreras X., Hansen A.H., Hippenmeyer S. Lineage tracing and clonal analysis in developing cerebral cortex using mosaic analysis with double markers (MADM) J. Vis. Exp. 2020 doi: 10.3791/61147. [DOI] [PubMed] [Google Scholar]
- Cheung A.F., Carter A.M., Kostova K.K., Woodruff J.F., Crowley D., Bronson R.T., Haigis K.M., Jacks T. Complete deletion of Apc results in severe polyposis in mice. Oncogene. 2010;29:1857–1864. doi: 10.1038/onc.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras X., Amberg N., Davaatseren A., Hansen A.H., Sonntag J., Andersen L., Bernthaler T., Streicher C., Heger A., Johnson R.L., et al. A genome-wide library of MADM mice for single-cell genetic mosaic analysis. Cell Rep. 2021;35:109274. doi: 10.1016/j.celrep.2021.109274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P., Postiglione M.P., Krieger T.G., Hernandez L., Wang C., Han Z., Streicher C., Papusheva E., Insolera R., Chugh K., et al. Deterministic progenitor behavior and unitary production of neurons in the neocortex. Cell. 2014;159:775–788. doi: 10.1016/j.cell.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski J.A., Talley T., Qiu M., Puelles L., Rubenstein J.L., Jones K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S., Youn Y.H., Moon H.M., Miyamichi K., Zong H., Wynshaw-Boris A., Luo L. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68:695–709. doi: 10.1016/j.neuron.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Ohtsuka T., Metzger D., Chambon P., Kageyama R. Temporal regulation of Cre recombinase activity in neural stem cells. Genesis. 2006;44:233–238. doi: 10.1002/dvg.20212. [DOI] [PubMed] [Google Scholar]
- Joo W., Hippenmeyer S., Luo L. Dendrite morphogenesis depends on relative levels of NT-3/TrkC signaling. Science. 2014;346:626–629. doi: 10.1126/science.1258996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N., Fogarty M., Iannarelli P., Grist M., Wegner M., Richardson W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.B., Rincon Fernandez Pacheco D., Saxon D., Yang A., Sabet S., Dutra-Clarke M., Levy R., Watkins A., Park H., Abbasi Akhtar A., et al. Rapid generation of somatic mouse mosaics with locus-specific, stably Integrated transgenic elements. Cell. 2019;179:251–267 e24. doi: 10.1016/j.cell.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klezovitch O., Fernandez T.E., Tapscott S.J., Vasioukhin V. Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 2004;18:559–571. doi: 10.1101/gad.1178004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K., Inoue A., Nakano Y., Hirai H., Kobayashi T., Maruyama M., Baba R., Kawashima C. BATTLE: genetically engineered strategies for split-tunable allocation of multiple transgenes in the nervous system. iScience. 2020;23:101248. doi: 10.1016/j.isci.2020.101248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoter S., Amberg N., Pauler F.M., Hippenmeyer S. Generation and isolation of single cells from mouse brain with mosaic analysis with double markers-induced uniparental chromosome disomy. STAR Protoc. 2020;1:100215. doi: 10.1016/j.xpro.2020.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoter S., Beattie R., Pauler F.M., Amberg N., Nakayama K.I., Hippenmeyer S. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat. Commun. 2020;11:195. doi: 10.1038/s41467-019-14077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen P.H., Zou K., Hwang J.K., Jan Y.N., Zhong W. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature. 2002;419:929–934. doi: 10.1038/nature01124. [DOI] [PubMed] [Google Scholar]
- Pontes-Quero S., Heredia L., Casquero-Garcia V., Fernandez-Chacon M., Luo W., Hermoso A., Bansal M., Garcia-Gonzalez I., Sanchez-Munoz M.S., Perea J.R., et al. Dual ifgMosaic: a versatile method for multispectral and combinatorial mosaic gene-function analysis. Cell. 2017;170:800–814 e18. doi: 10.1016/j.cell.2017.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio P., Cebrian C., Zong H., Hippenmeyer S., Constantini F. Ret and Etv4 Promote Directed Movements of Progenitor Cells during Renal Branching Morphogenesis. PloS Biol. 2016;14:e1002488. doi: 10.1371/journal.pbio.1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S., Mori D., Toyo-Oka K., Chen A., Garrett-Beal L., Muramatsu M., Miyagawa S., Hiraiwa N., Yoshiki A., Wynshaw-Boris A. Complete loss of Ndel1 results in neuronal migration defects and early embryonic lethality. Mol. Cell. Biol. 2005;25:7812–7827. doi: 10.1128/MCB.25.17.7812-7827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S.H.E., Silva F.J., Tsark W.M., Mann J.R. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32:199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- Tessarollo L., Tsoulfas P., Donovan M.J., Palko M.E., Blair-Flynn J., Hempstead B.L., Parada L.F. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc. Natl. Acad. Sci. U S A. 1997;94:14776–14781. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian A., Kang B., Li B., Qiu B., Jiang W., Shao F., Gao Q., Liu R., Cai C., Jing R., et al. Oncogenic state and cell identity combinatorially dictate the susceptibility of cells within glioma development hierarchy to IGF1R targeting. Adv. Sci. 2020;7:2001724. doi: 10.1002/advs.202001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H., Espinosa J.S., Su H.H., Muzumdar M.D., Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No custom code is necessary to perform this protocol.