

Abstract
The major brain abnormality underlying cerebral palsy in premature infants is periventricular leukomalacia (PVL), a lesion of the immature cerebral white matter. Oligodendrocyte precursors (pre-OLs; O4+O1–) predominate in human cerebral white matter during the peak time frame for PVL (24–32 gestational weeks) and are vulnerable to excitotoxicity. We hypothesize that PVL reflects, in part, excitotoxicity to pre-OLs resulting from cerebral ischemia/reperfusion. Reversal of glutamate transport in the setting of energy failure is a major source of pathologic accumulation of extracellular glutamate. Here, we identify and localize the glutamate transporters in human cerebral white matter during the age range of PVL. In situ hybridization was performed with digoxigenin-labeled probes directed against the full-length coding regions of EAAT1, EAAT2, and EAAT3. EAAT2 mRNA was abundant in human fetal white matter during the period of peak incidence of PVL and virtually disappeared by 2 postnatal months. Its developmental profile differed significantly from that of both EAAT1 and EAAT3 mRNA. Immunoblotting demonstrated that EAAT2 protein was highly expressed in early development relative to adult values. Double-label immunocytochemistry detected EAAT2 in OLs but not astrocytes or axons in the human fetal white matter. We conclude that transient expression of EAAT2 occurs during the window of peak vulnerability for PVL, suggesting that this developmentally up-regulated transporter may be a major source of extracellular glutamate in ischemic injury to the cerebral white matter of the preterm infant. J. Comp. Neurol. 501:879–890, 2007.
Keywords: cerebral palsy, excitoxicity, oligodendrocyte, hypoxic-ischemic injury, myelination, periventricular leukomalacia
Approximately 10–15% of babies born with birth weights less than 1,500 g develop cerebral palsy (Volpe, 2003). The major brain abnormality underlying cerebral palsy is periventricular leukomalacia (PVL), a disorder of the immature cerebral white matter hypothesized to result, at least in part, from cerebral ischemia/reperfusion. It is characterized by injury to premyelinating oligodendrocytes (pre-OLs) that appears to result in delayed or hypomyelination. The major risk period for PVL is at 24–32 gestational weeks, when pre-OLs predominate in human white matter (Back et al., 2001). Animal and cell culture data implicate excitotoxicity in injury to premyelinating OLs and thus have obvious relevance to understanding the cellular and molecular mechanisms underlying PVL (Deng et al., 2003; Follett et al., 2000; Itoh et al., 2002; Rosenberg et al., 2003).
Although excitotoxicity appears to be an important pathway of cell death in hypoxic-ischemic injury to white matter in the perinatal period, the source of the excess glutamate remains in question. During energy failure, glutamate transporters, which are physiologically responsible for clearing extracellular glutamate, operate in reverse and thereby release glutamate into the surrounding tissue (Deng et al., 2003; Fern and Möller, 2000; Roettger and Lipton, 1996; Rossi et al., 2000; Seki et al., 1999). The glutamate transporter family has five subtypes, known as EAAT1–5 in humans; EAAT1–3 are known as GLAST (EAAT1), GLT1 (EAAT2), and EAAC1 (EAAT3) in animals, rat or rabbit, in which they were originally discovered (Arriza et al., 1994, 1997; Fairman et al., 1995; Pines et al., 1992; Storck et al., 1992). In adult mammalian brains, GLAST is expressed mainly in astrocytes and EAAC1 in neurons. The GLT1 transporter, previously thought to be expressed exclusively in astrocytes, is also expressed in neurons in at least two variant forms, GLT1a and GLT1b (Chen et al., 2002, 2004; Schmitt et al., 2002). In experimental animals, GLT1a protein is transiently expressed in growing axons in rat white matter, mouse spinal cord, and sheep corpus callosum, before the establishment of expression in astrocytes postnatally (Furuta et al., 1997; Northington et al., 1999; Yamada et al., 1998). Although studies of human adult brains have shown GLT1a expression in astrocytes and most recently in OLs (Bar-Peled et al., 1997; Milton et al., 1997; Pitt et al., 2000), there is no information about glutamate transporter expression in developing human cerebral white matter.
We hypothesized, in light of the developmental expression of GLT1 in central white matter axons in animals, that the human homologue EAAT2 is similarly elevated in early life, particularly during the peak period of vulnerability to PVL, and declines to adult levels thereafter. A transient expression of GLT could account, at least in part, for the vulnerability of immature cerebral white matter to ischemic injury. To test this hypothesis, we characterized glutamate transporter expression in the developing human cerebral white matter of autopsy cases without significant white matter pathology by using in situ hybridization, quantitative immunoblot analysis, and immunocytochemistry.
MATERIALS AND METHODS
Clinical database
Tissue was accrued from the cerebra of human fetuses and infants from a standardized level, i.e., parietooccipital lobe at the level of the occipital horn of the lateral ventricle, and from the body of the corpus callosum (level of posterior frontal white matter). Tissue samples were obtained through the Departments of Pathology, Brigham and Women’s Hospital and Children’s Hospital (Boston, MA), with parental and institutional approval. Parietooccipital white matter was also obtained from adult patients without neurological disease (21–75 years), as an index of maturation for comparison. The age of the infant cases is expressed in postconceptional (gestational plus postnatal) weeks (Table 1). Only samples with a post-mortem interval of less than 24 hours was used.
TABLE 1.
Summary of Human Cases Used for In Situ Hyridization, Immunoblotting, and Immunocytochemistry1
| Case No. | Age | PMI | In situ | Blot | ICC |
|---|---|---|---|---|---|
| 1 | 19 Weeks | 4 Hours | × | ||
| 2 | 20 Weeks | 4 Hours | × | ||
| 3 | 21 Weeks | 24 Hours | × | ||
| 4 | 22 Weeks | 4 Hours | × | ||
| 5 | 23 Weeks | 4 Hours | × | × | × |
| 6 | 23 Weeks | Uk | |||
| 7 | 24 Weeks | 26 Hours | × | ||
| 8 | 25 Weeks | Uk | × | ||
| 9 | 26 Weeks | 62 Hours | × | ||
| 10 | 30 Weeks | 24 Hours | × | ||
| 11 | 31 Weeks | 6 Hours | |||
| 12 | 31 Weeks | 4 Hours | × | × | |
| 13 | 32–33 Weeks | 22 Hours | × | × | |
| 14 | 34 Weeks | 17 Hours | × | ||
| 15 | 34 Weeks | 10 Hours | × | ||
| 5 | 38 Weeks | 16.5 Hours | × | ||
| 6 | 38 Weeks | 5 Hours | × | × | × |
| 7 | 40 Weeks | 24 Hours | × | × | |
| 8 | 42 Weeks | 41 Hours | × | ||
| 9 | 45 Weeks | 7 Hours | × | × | |
| 10 | 49 Weeks | 12 Hours | × | × | |
| 11 | 53 Weeks | 12 Hours | × | × | |
| 12 | 54 Weeks | 16 Hours | × | ||
| 13 | 55 Weeks | Uk | × | ||
| 14 | 74 Weeks | 20 Hours | × | × | |
| 15 | 106 Weeks | 21 Hours | × | ||
| 16 | 114 Weeks | 27 Hours | × | ||
| 17 | 3 Years | Uk | |||
| 18 | 3 Years | 20 Hours | × | ||
| 19 | 3.3 Years | Uk | × | ||
| 20 | 21 Years | 13 Hours | × | × | |
| 21 | 38 Years | 5 Years | × | × | |
| 22 | 43 Years | 21 Hours | × | × | |
| 23 | 61 Years | Uk | × |
Age, gestational + postconceptional age; PMI, post-mortem interval; Uk, unknown.
At autopsy, the parietooccipital poles were removed from cases and snap frozen immediately in blocks at –70°C for in situ hybridization (19 cases) and immunoblot assay (21 cases), or were immersed in freshly prepared 4% paraformaldehyde (Sigma, St. Louis, MO) in phosphate-buffered saline for immunocytochemistry (seven cases). Adjacent blocks were paraffin embedded for histopathologic assessment. Standard neuropathologic evaluations of cerebral cortex, white matter, diencephalon, hippocampus, brainstem, and cerebellum were performed in each case.
In situ hybridization
Frozen brain tissue was sectioned at 15 mm on a Leitz cryostat and mounted on glass slides. Plasmids (gifts of Dr. Susan Amara) containing the cDNAs of EAAT1, EAAT2, and EAAT3 (GenBank accession Nos. NM_004172, NM_004171, and NM_004170, respectively) were used to amplify by PCR the sense and antisense full-length coding regions of each transporter with primers that contained RNA polymerase initiation sites. Analyses of EAAT1 and EAAT3 mRNA were performed to determine the differential patterns of expression of the other transporters relative to EAAT2, the focus of this study. The amplified cDNA was used for transcription to generate digoxigenin (DIG)-labeled cRNA probes for each transporter. After transcription, all probes were alkali hydrolyzed to an average length of 500 base pairs. Nonisotopic in situ hybridization was performed according to a previously described method (Berger et al., 2005; Berger and Hediger, 1998; Chen et al., 2004), which is an improved method based on a previously published protocol (Schaeren-Wiemers and Gerfin-Moser, 1993). Briefly, in situ hybridization probes were labeled with DIG. After hybridization, sections were incubated with a sheep anti-DIG-Fab antibody fragment conjugated to alkaline phosphatase and developed with 5-bromo-4 chloro-indoylphosphate and nitro blue tetrazolium (NBT/BCIP). As a negative control, adjacent sections were stained with a noncomplementary sense probe. A regression analysis of post-mortem interval (PMI) on EAAT1, EAAT2, and EAAT3 expression in control cases showed no statistically significant effect of PMI.
Cell counting from tissue sections labeled by using in situ hybridization
Counting of transporter-positive cells/high-power field (hpf) in single-labeled tissue sections was performed. Positive cells per hpf at “400 magnification (0.173 mm2) were counted in an area representative of all fields. The number of positive cells containing mRNA per hpf (cell/hpf) are reported in Figure 3 after being subjected to the post hoc correction factor for cell size used by Abercrombie (1946) and Guillery (2002). The thickness of all sections was 15 μm. The diameter of 20 cells from two different human cases for each probe was measured with a Nikon E800 digital microscope with Spot camera software. The average cell diameter in tissue stained for each probe was 11.7 ± 0.65 μm (EAAT1), 11.6 ± 0.87 μm (EAAT2), and 11.3 ± 1.0 μm (EAAT3), giving a correction factor of 56–57% for all probes. Two observers scored each case together, without knowledge of case diagnosis or age.
Fig. 3.

EAAT1, EAAT2, and EAAT3 expression as a function of age. A regression analysis of the effect of age on mRNA expression for each glutamate transporter is indicated by the curve drawn through each plot of individual data points. These data show that EAAT1 increases quadratically with age. EAAT2 changes quadratically with age, first increasing, then decreasing. EAAT3 increases with age. Regression analysis did not include ages more than 1 postnatal year, because the large variation in ages of older cases would influence the regression analysis.
Northern blot analysis of RNA probes on fetal brain lysates
The specificity of the in situ probes was tested by Northern blot analysis based on a method described previously (Sambrook and Russell, 2001). Briefly, fetal human forebrain total RNA (Stratagene, La Jolla, CA) at 10 μg/lane was separated by electrophoresis on a 0.9% agarose-formaldehyde gel, electroblotted onto a charged cellulose membrane, and hybridized to 32P-labeled RNA probes generated against the full-length coding regions of EAAT1, EAAT2, and EAAT3. 32P-UTP-labeled probes were generated with a Maxiscript kit from Ambion (Austin, TX). The sizes of the glutamate transporter transcripts were determined by using a standard curve prepared from the 18S and 28S ribosomal RNA bands and RNA molecular weight markers (Invitrogen, Carlsbad, CA).
Antibodies for Western blot analysis and immunocytochemistry
A polyclonal antibody against the N-terminus of GLT1 (anti-nGLT1), which detects both variant forms of GLT1, GLT1a and GLT1b, was generated in New Zealand white rabbits (Research Genetics, Huntsville, AL) and has been previously characterized (Chen et al., 2002, 2004). This antibody was generated based on the published sequences for rat GLT1a and GLT1b (aa 1–15; GenBank accession No. AF451299), which are identical to each other and to the human sequence, and thus was used to detect EAAT2a and EAAT2b in human tissue in this study. To determine the specific cell types in which glutamate transporters are located, the following antibodies were used for double-labeling immunocytochemistry: anti-GFAP for astrocytes (Sternberger Monoclonals, Lutherville, MD), antivimentin (Chemicon, Temecula, CA) for radial glia, A2B5 monoclonal antibody for glial progenitors [grown from cells provided by the American Type Culture Collection (ATCC)], O4 monoclonal antibody for precursor OLs, O1 monoclonal antibody for immature OLs, antimyelin basic protein (MBP) for mature OLs (Sternberger Monoclonals), antiNeuN (Chemicon) for neuronal cell bodies, SMI-312 (Sternberger Monoclonals), and GAP-43 for axons. Hybridoma cells producing O4 and O1 monoclonal antibodies were generously provided by Dr. Stephen Pfeiffer. A summary of antibodies used in this study is included in Table 2.
TABLE 2.
Antibodies Used For Single- and Double-Label Immunocytochemistry and Western Blot1
| Antibody | Source | Host and type | Antigen | Specificity | Dilution |
|---|---|---|---|---|---|
| Neurofilament (SMI312) | Sternberger Monoclonals, Lutherville, MD | Mouse mono IgG1 (mixture) | Homogenized hypothalami from Fisher 344 rats | Detects neruofilaments on axons in human fetal brain2 | 1:500 |
| Vimentin (MAB 3400) | Chemicon International | Mouse mono IgG | Purified porcine vimentin | Detects a 57-kDa band specific for vimentin on immunoblots of human glioma cell lines3 | 1:1,000 |
| GFAP (SMI 22) | Sternberger Monoclonals, Lutherville, MD | Mouse mono IgG (mixture) | Purified bovine GFAP | All components (clones 1B4, 2E1, 4A11) are specific for GFAP by radioimmunoassay, Western blot and immunocytochemistry4 | 1:500 |
| O1 | Gift from Dr S. Pfeiffer, Farmington, CT | Mouse mono IgM | Bovine corpus callosum | Recognizes specifically GalC and MG expressed on OL surface on lipid immunodot blots5,6 | 1:500 |
| O4 | Gift from Dr. S. Pfeiffer | Mouse mono IgM | Bovine corpus callosum | Reacts selectively with OL surface antigens sulfatide and seminolipid on lipid immunodot blots5,6 | 1:500 |
| MBP (SMI 99) | Sternberger Monoclonals | Mouse mono IgG | Human MBP peptide containing aa 131-136 (–A–S–D–Y–K–S–) | Detects four bands between 14 and 21 kDa, corresponding to four MBP isoforms on immunoblots of rat cerebellum7 | 1:1,000 |
| NeuN (MAB 377) | Chemicon International | Mouse mono IgG | Purified cell nuclei from mouse brain | Recognizes two bands on Western blot of mouse brain, corresponding to the 46- and 48-kDa NeuN isoforms8 | 1:100 |
| nGLT1 | Rosenberg Lab | Rabbit polyIgG | Rat peptide containing aa 1-15 (M–A–S–T–E–G–A–N–N–M–P–K–Q–V–E) | Recognizes one band on immunoblot of rat brain (67 kDa); does not stain GLT1 knockout using immunocytochemistry and immunoblotting9,10 | 1:500 |
Poly, polyclonal; mono, monoclonal; aa, amino acids; GFAP, glial fibrillary acidic protein; O4, O1, OL progenitor surface markers; MBP, myelin basic protein; NeuN, neuronal nuclei; nGLT1, rat glutamate transporter homologue/EAAT2 in human.
Ulfig N, et al. 1998. Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res 291:433–443.
Osborn M, et al. 1984. Monoclonal antibodies specific for vimentin. Eur J Cell Biol 34:137–143.
McLendon RE, et al. 1986. The immunohistochemical application of three anti-GFAP monoclonal antibodies to formalin-fixed, paraffin-embedded, normal and neoplastic brain tissues. J Neuropathol Exp Neurol 45:692–703.
Sommer I, Schachner M. 1981. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol 83:311–327.
Bansal R, et al. 1989. Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res 24:548–557.
Dyer CA, Matthieu J-M. 1994. Antibodies to myelin/oligodendrocyte-specific protein and myelin/oligodendrocyte glycoprotein signal distinct changes in the organization of cultured oligodendroglial membrane sheets. J Neurochem 62:777–787.
Lind D, et al. 2005.Characterization of the neuronal marker NeuN as a multiply phosphorylated antigen with discrete subcellular localization. J Neurosci Res 79:295–302.
Chen W, et al. 2004. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24:1136–1148.
Chen W, et al. 2002. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22:2142–2152.
Immunoblot analysis of human fetal brain lysates
Lysates were made from human brain tissue. Briefly, tissue was homogenized in a frosted glass homogenizer in 1% sodium dodecyl sulfate (SDS) containing a protease inhibitor cocktail with EDTA (Roche). After homogenization, samples were dispersed in an ultrasonic bath for approximately 15 minutes until the solution was clear. Protein concentration was measured using the DC Protein Assay (Bio-Rad, Hercules, CA). Samples (40 μg/lane) were run on an 8–18% polyacrylamide gel and electroblotted onto a polyvinylidene fluoride (PVDF) membrane (Perkin Elmer, Wellesley, MA). PVDF membranes were incubated with anti-nGLT1 at 1 μg/ml overnight at 4°C in TBST buffer (50 mM Tris, 150 mM NaCl, 0.01% Triton, pH 7.4) containing 5% nonfat milk. Blots were then washed three times in TBST buffer, followed by a 1-hour incubation with horseradish peroxidase (HRP)-conjugated goat antirabbit IgG (Amersham Life Science, Piscataway, NJ). For protein detection, membranes were incubated in Western Lightning Chemiluminescence Reagent (Perkin Elmer) and exposed on X-Omat Blue XB-1 film (Kodak). Densitometric analysis was performed on film using the MCID Elite version 7.0 software published by Imaging Research (Ontario, Canada). A human adult standard lysate containing parietal white matter from three pooled cases (ages 55, 65, and 75 years) was run as a control on each blot. Density of the individual bands obtained from the human developmental series of cerebral white matter lysates was calculated and plotted as a percentage of the human adult standard. A regression analysis of PMI on EAAT2 expression in controls and in controls and PVL cases combined showed no statistically significant effect of PMI.
Immunocytochemistry
Immunocytochemical studies were performed on 4% paraformaldehyde-fixed tissue that was cryoprotected with 30% sucrose. Tissue was sectioned on a freezing vibratome and prepared as 50-μm floating sections. Briefly, sections were washed in phosphate-buffered saline (PBS; 10 mM sodium phosphate, 2 mM potassium phosphate, 2.7 mM potassium chloride, 137 mM sodium chloride, pH 7.4) with 0.1% Triton X-100 (except when using O4 and O1 monoclonal antibodies against surface markers that do not require permeabilization) before blocking with 5% goat serum. Sections were incubated with primary antibodies overnight, followed by washing in PBS. Primary antibody labeling of tissue sections was detected with immunofluorescent secondary antibodies: Alexa 594 (red) and Oregon 488 (green; Molecular Probes, Eugene, OR). Sections were mounted with Fluoromount-G (Southern Biotechnology, Birmingham, AL) with added bisbenzamide to identify cell nuclei. Slides were observed with a Nikon E800 microscope, and photographs were taken with a Spot digital camera.
Statistical analysis
To assess the relationship between the mRNA and protein expression of each transporter and age, linear and quadratic regression analyses were performed. To determine whether levels differed before and after term birth (40 weeks), Wilcoxon rank sum tests were utilized. To test the EAAT2 protein expression (as percentage of adult standard) against the adult standard, representing 100% expression, a one-sample t-test was used. The measure of dispersion used throughout Results is the standard error of the mean.
RESULTS
Clinicopathological data set
Table 1 shows the cases used for the ensuing studies: 19 cases for in situ hybridization, 21 for immunoblot analysis, and seven for immunocytochemical analysis. They had died of a variety of disorders, but standard neuropathologic and histopathogic examination in each case did not reveal diagnostic cerebral white matter damage or other CNS abnormalities.
Specificity of glutamate transporter mRNA probes for in situ hybridization
The specificity of glutamate transporter mRNA probes used for in situ hybridization was tested by Northern blot analysis. The probes were transcribed from the same full-length coding regions of the transporters used for in situ and were labeled with 32P. Each mRNA probe detected a distinct mRNA transcript for its respective glutamate transporter subtype in human fetal brain total RNA. The EAAT2 probe hybridized to a 11.7-kb band, the EAAT1 probe hybridized to a 4.0-kb band, and the EAAT3 probe hybridized to a 3.7-kb band (Fig. 1). Similar results were obtained with cDNA probes used to detect glutamate transporters in total brain RNA prepared from human cases ranging in age from 16 to 75 years (Nakayama et al., 1996).
Fig. 1.

Northern blot analysis of glutamate transporter mRNA expression in total RNA extracted from human fetal brain tissue. Ten micrograms of fetal total RNA were loaded per lane. 32P-labeled RNA probes were generated against the full-length coding regions of EAAT1, EAAT2, and EAAT3 and hybridized to RNA at 4.0, 11.7, and 3.7 kb, respectively. 28s RNA is also observed at 4.5 kDa.
In situ hybridization of EAAT1, EAAT2, and EAAT3 mRNA in developing human cerebral white matter
Analysis of mRNA expression was performed via in situ hybridization in 19 baseline cases, without neurological disease or significant brain histopathology, with probes against the full-length coding region of EAAT1, EAAT2, and EAAT3. Tissue was available from seven fetuses, seven infants, one child, and four adults (for comparison), although tissue was not available from all cases for each probe (Table 1). The sense cRNA probes were used as control probes for all three transporters, and did not yield any staining. The developmental profiles for the different transporters varied from one another substantially (Figs. 2, 3). EAAT2 mRNA expression in the parietooccipital white matter was low at midgestation; increased to its highest level of expression at 25 and 32 gestational weeks, i.e., the peak age window of PVL (Fig. 2C); and declined after 50 postconceptional weeks, i.e., 1.5 postnatal months to reach low adult levels between 55 and 74 postconceptional weeks (Figs. 2D, 3B). EAAT1 expression increased across the last half of gestation between 25 and 32 postconceptional weeks (Fig. 2A), the earliest time points examined, until approximately 50 postconceptional weeks, i.e., 1.5 postnatal months, and remained level at high adult values (Figs. 2B, 3A). EAAT3 mRNA expression was low at midgestation (Fig. 2E) and increased linearly between 34 and 40 postconceptional weeks to an intermediate level as seen in adults compared with EAAT2 and EAAT3 (Figs. 2F, 3C). We also considered the glutamate transporter mRNA levels relative to term birth (40 gestational weeks), because the peak age of vulnerability of PVL is in the mid- to late fetal period of pregnancy (Fig. 4). The differences before and after term birth were significant for all three transporters, giving the following P values determined from Wilcoxon tests: EAAT1 (0.003), EAAT2 (0.014), and EAAT3 (0.003). EAAT1 and EAAT3 mRNA levels were higher after than before birth; in contrast, EAAT2 mRNA levels were higher before than after birth (Fig. 4).
Fig. 2.

Nonisotopic in situ hybridization of EAAT2/GLT1, EAAT1/GLAST, and EAAT3/EAAC1 mRNA in human fetal cerebral white matter. C shows a section of white matter (WM) and cortex (CTX) of a 32-week-old subject, demonstrating a prominent amount of EAAT2 mRNA in the white matter. In contrast, a 7-month-old subject (D) expresses almost no EAAT2 mRNA in the white matter. Cortex serves as a positive control, showing EAAT2 labeling in both subjects. EAAT1 (A,B) and EAAT3 (E,F) expression was present and increased with development, in contrast to EAAT2. Scale bars &50 μm.
Fig. 4.

Comparison of preterm (≤40 weeks) and postterm (>40 weeks) expression of EAAT1, EAAT2, and EAAT3 mRNA. EAAT1 and EAAT3 levels were significantly higher after term, whereas EAAT2 levels were significantly lower after birth. Statistical analysis of preterm and postterm expression using the Wilcoxon test gives P values of 0.003 (EAAT1), 0.014 (EAAT2), and 0.003 (EAAT3). *P % 0.05, **P % 0.01.
Evaluation of EAAT2 protein expression in the human fetus and infant
Because the in situ results (Figs. 2, 3) demonstrated that EAAT2 is developmentally up-regulated during the peak of vulnerability for premature infants to PVL, we analyzed the expression of EAAT2 at the protein level by using an antibody raised against the N terminus of GLT1/EAAT2, known to be shared by the two known variant forms of GLT1/EAAT2. To test the specificity of the antinGLT1 antibody in human brain tissue, immunoblot analysis was performed on lysates prepared from normal human motor cortex (Fig. 5). A dense band appeared at 67 kDa (Fig. 5, lane 2). A light band was also observed above the 120-kDa marker (Fig. 5, lane 2), probably representing dimers of the transporter. These data are consistent with results obtained with this antibody in rodent brain lysates (Chen et al., 2002, 2004). To demonstrate the specificity of the immune response to the antigenic N-terminus GLT1 peptide, the immunoreactivity of purified anti-nGLT1 antibody was compared with that of the preimmune serum (Fig. 5, lane 1). In addition, the ability of the peptide against which the antibody was generated to block immunoreactivity was tested (Fig. 5, lane 3). Preimmune serum was not immunoreactive (Fig. 5, lane 1). Antigenic peptide nearly completely blocked the immunoreactivity of the anti-nGLT1 antibody against human brain lysate (Fig. 5, lane 3). Thus, the anti-nGLT1 antibody specifically detected protein containing the epitope against which it was generated in human lysates. Previous studies have shown that the immunreactivity produced by this antibody is not present in brain lysates or in tissue sections from GLT1 knockout mice (Chen et al., 2004).
Fig. 5.

Western blot analysis of the specificity of the anti-nGLT1 antibody in human brain tissue. To characterize the anti-nGLT1 antibody, it was tested on lysates prepared from normal human motor cortex and appears as a band at 67 kDa (lane 2). A lighter band is also observed above the 120-kDa marker (lane 2). To demonstrate the specificity of the immune response to the nGLT1 peptide, the immunoreactivity of the anti-nGLT1 antibody was compared with that of the preimmune serum (lane 1). In addition, the ability of the peptide against which the antibody was generated to block immunoreactivity was tested (lane 3).
To assess whether the transient expression of EAAT2 mRNA occurs at the protein level, an immunoblot using anti-nGLT1 antibody in parietooccipital cerebral white matter lysates was performed (Fig. 6). Lane 1 (Fig. 6) contains adult human standard cerebral white matter lysate (see Materials and Methods). Lane 2 (Fig. 6) contains white matter from a 30-week-old preterm infant. Comparison of lanes 1 and 2 (Fig. 6) shows a substantially increased expression of EAAT2 protein at 30 weeks compared with the adult. Lane 3 (Fig. 6) contains cerebral gray matter from the same 30-week-old preterm infant and shows that at this age the expression of EAAT2 in the cerebral white matter is comparable to the expression in the cerebral gray matter. Another example of cerebral white matter from a 30-week-old preterm infant is shown in lane 4 (Fig. 6), demonstrating similar expression to the case shown in lane 2 (Fig. 6).
Fig. 6.

A representative immunoblot of anti-nGLT1 antibody in human cerebral white matter lysates. Lane 1 contains an adult human cerebral white matter standard made from three brains 55, 65, and 75 years old. Lanes 2 and 3 contain cerebral white matter (2) and cerebral gray matter (3) from the same 30-week-old preterm infant. Lane 4 contains another case of cerebral white matter from a 30-weekold preterm infant. The molecular weight of GLT1 is 67 kDa.
To quantitate EAAT2 protein expression across human development, using densitometric analysis, lysates taken from parietooccipital white matter at different postconceptional ages were immunoblotted with anti-nGLT1 antibody. Data are expressed as percentage of adult human standard, run on each blot. White matter samples were obtained from 20 fetuses and infants and from one adult, aged 21 years (Table 1). Comparison of protein expression of EAAT2 at different ages is shown in Figure 7 and grouped according to biologically relevant epoch; 19–34 weeks, preterm; 38–41 weeks, term; 1–6 months, infant; 1–3 year, child; 21–75 years, adult. A linear regression showed no statistically significant change of EAAT2 protein expression across age. For the group of cases at 19–41 weeks, however, the mean densitometric value was 614% ± 37% of the 100% adult (standard) level. This value was significantly higher than the adult standard value (onesample t-test; P % 0.001).
Fig. 7.

Comparison of protein expression of EAAT2 at different ages. Densitometric values were grouped as shown. A one-sample t-test was used to compare each sample group with adult standard (21–75 years), giving a significant P value of 0.001 for 19–41 weeks.
Immunocytochemistry of EAAT2 in fetal human white matter
To determine the cellular localization of EAAT2 expression in the developing human white matter, we performed single- and double-label immunocytochemistry on seven human cases (Table 1) Relatively little EAAT2 staining was present in the cerebral white matter at 3 years of age compared with a 31-week-old case (data not shown). Cerebral cortex (CTX) staining was present in both cases and served as a positive internal control. To identify cellular elements expressing EAAT2, double-labeling experiments with anti-nGLT1 antibody and cell-specific markers were performed. Fluorescence microscopy of the parietooccipital white matter (Fig. 8) from a representative case at 31 gestational weeks demonstrated anti-GFAP labeling of astrocytes (Fig. 8A), vimentin labeling of radial glia (Fig. 8C), and neurofilament labeling of axons (Fig. 8E). Double labeling showed no colocalization of EAAT2 with GFAP (Fig. 8B), vimentin (Fig. 8D), or neurofilament (Fig. 8F). In contrast, in pre-OLs in a representative 23-week case (midgestation) colocalization was seen with the antinGLT1 antibody (Fig. 9A) and the O4 monoclonal antibody (Fig. 9B). Coincidence of labeling with the anti-GLT1 and O4 antibodies is indicated by the appearance of yellow labeling of the overlying images (Fig. 9C).
Fig. 8.
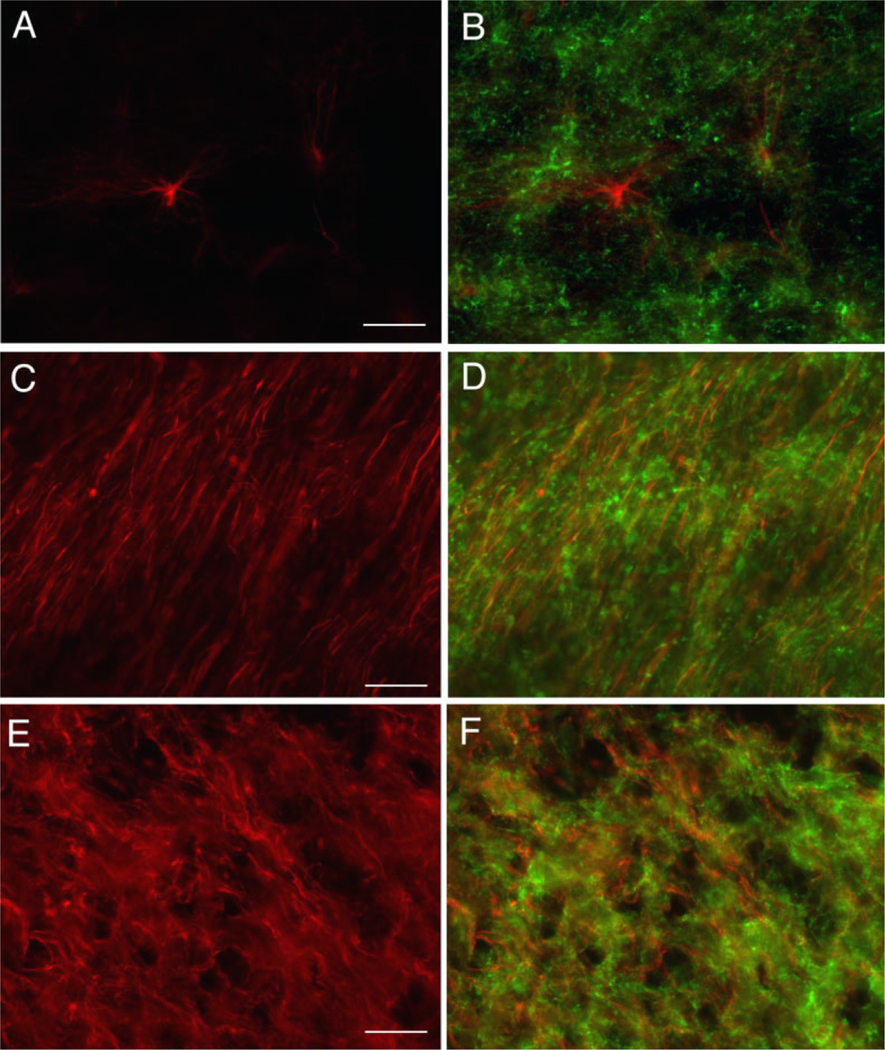
Immunocytochemistry of EAAT2 and cell-specific markers in human cerebral white matter. Fluorescence microscopy (×20) of 31-week-old human cerebral white matter labeled with GFAP (A), vimentin (C), and neurofilament (E). Double labeling of GFAP, vimentin, and neurofilament was performed with anti-GLT1 antibody, and the overlay shows no colocalization of GLT1 with these markers in B, D, and F, respectively. Scale bars = 50 μm.
Fig. 9.

Immunofluorescent double labeling of EAAT2 and O4+ OLs in human developing cerebral white matter. Fluorescence confocal microscopy (×40) of cerebral white matter from a 31-week-old preterm infant labeled with anti-nGLT1 polyclonal antibody (A) counterstained with the O4 monoclonal antibody (B). Overlay of A and B shows EAAT2 labeling in O4+ OLs (C). Scale bars = 50 μm.
DISCUSSION
In this study, we identify and localize glutamate transporters in the cerebral white matter in the human fetus and infant. Our main finding is that EAAT2 mRNA, as demonstrated by in situ hybridization, is abundant in human fetal white matter during the period of peak incidence of PVL and is substantially down-regulated at a time when myelination is initiated postnatally. The developmental profile of EAAT2 differed considerably from that of both EAAT1, which increased significantly during development and remained highly expressed through adulthood, and EAAT3, which was expressed at relatively low levels during pre- and postnatal development. Immunoblotting demonstrated that EAAT2 protein was highly expressed in early human development relative to adult values, and double-label immunocytochemistry detected EAAT2 in O4+ OLs. The transient expression of EAAT2 coincides with the window of vulnerability for PVL, suggesting that reversal of this transporter might be a major source of glutamate in hypoxic-ischemic injury to the cerebral white matter of the preterm infant.
Pathophysiological implications of upregulation of EAAT2 in developing cerebral white matter
The role of glutamate excitotoxicity in hypoxic-ischemic injury to the neonate has been suggested clinically by the elevation of glutamate in the cerebrospinal fluid of term infants after perinatal hypoxia-ischemia (Hagberg, 1992) and experimentally by in vivo microdialysis in a perinatal rat model of ischemia (Benveniste et al., 1984; Silverstein et al., 1991) and, more recently, in a fetal sheep model (Loeliger et al., 2003). In culture, pre-OLs (O4+O1–MBP– and O4+O1+MBP–) are sensitive to AMPA/kainate receptor-mediated excitotoxicity (Matute et al., 1997; McDonald et al., 1998; Rosenberg et al., 2003; Yoshioka et al., 1996). However, mature OLs (O4+O1+MBP+) are resistant to excitotoxicity (Rosenberg et al., 2003) because of down-regulation of glutamate receptors in this stage (Itoh et al., 2002; Rosenberg et al., 2003). The relevance of the in vitro studies is suggested by in vivo studies in which an AMPA receptor antagonist was protective against cerebral white matter damage in a perinatal rat model of selective white matter injury (Follett et al., 2000, 2004), and calcium-permeable AMPA receptors were shown to be upregulated in cerebral white matter during the period of peak vulnerability to PVL (Follett et al., 2003, 2004; Talos et al., 2005a,b). Furthermore, recent data have shown that NMDA receptors are also expressed in developing oligodendrocyte processes and mediate injury (Karadottir et al., 2005; Micu et al., 2006; Salter and Fern, 2005). Although excitotoxicity may be a fundamental mechanism in the pathogenesis of PVL, the source of the excess extracellular glutamate remains in question. The results presented herein suggest that part of the basis of the maturation-dependent vulnerability of white matter is due to the transient up-regulation of EAAT2.
Glutamate transporters provide the only known mechanism for maintaining extracellular glutamate homeostasis (Danbolt, 2001; Tanaka et al., 1997). Control of extracellular glutamate concentration is disrupted under conditions leading to excitotoxic injury (Lipton and Rosenberg, 1994; Meldrum and Garthwaite, 1990). When there is a dissipation of electrochemical gradients across the plasma membrane, such as occurs during hypoxiaischemia, glutamate transporters effectively act in reverse to release glutamate (Nicholls and Attwell, 1990; Szatkowski et al., 1990). In addition, OLs subjected to oxygen-glucose deprivation in vitro have been shown to release glutamate by reversal of GLT1, insofar as dihydrokainate, a specific blocker of GLT1, is highly protective (Deng et al., 2003; Fern and Möller, 2000). GLT1 knockout mice are more vulnerable to neuronal loss in the hippocampus and attain higher extracellular glutamate levels in response to a 5-minute episode of ischemia than wild-type mice. However, after 20 minutes of ischemia, wild-type mice are more vulnerable to neuronal death and attain higher glutamate levels than knockout mice (Mitani and Tanaka, 2003). Thus, whether expression of EAAT2 in developing human white matter is helpful or harmful may depend on the duration of the ischemic episode. Presumably, in the Mitani and Tanaka experiments, prolonged ischemia compromised energy metabolism sufficiently to induce release of glutamate from cells through their glutamate transporters. Taken together, these data suggest that the upregulation of EAAT2 in developing OLs may be the source of excessive extracellular glutamate (by reverse transport) that overstimulates glutamate receptors, also present on OLs at this time, leading to excitotoxicity. Other possible sources of excessive extracellular glutamate accumulation, besides glutamate transporters, are synaptic release of glutamate from neurons; release through swellingactivated anion channels in astrocytes (Kimelberg et al., 1990; Takano et al., 2005) or hemichannels in activated microglia (Takeuchi et al., 2006); and exocytotic release from astrocytes mediated by chemokines (Bezzi et al., 2001), prostaglandins (Bezzi et al., 1998), and neuropeptides (Parpura et al., 1994). Although there are several possible sources of extracellular glutamate, glutamate transport is the only known clearance mechanism. Therefore, any disturbance in glutamate transport, such as down-regulation or blockage of uptake by cytokines (Fine et al., 1996; Hu et al., 2000; Liao and Chen, 2001; Ye and Sontheimer, 1996), reactive oxygen species (Volterra et al., 1994), or reactive nitrogen species (Trotti et al., 1996), will impair glutamate homeostasis.
Glutamate uptake in OLs may be critical to glutamate homeostasis in developing cerebral white matter
The present data show that astrocytes in human cerebral white matter during the peak age of vulnerability for PVL do not express EAAT2. Previously, no astrocytic expression of EAAT2 protein was found in the developing human CNS (Furuta et al., 2005) or in human cultured glial progenitors (Maragakis et al., 2004). Astrocytic expression of GLT1 was also not observed in rat white matter, mouse spinal cord, and sheep corpus callosum until postnatal development (Furuta et al., 1997; Northington et al., 1999; Yamada et al., 1998). Our data show that EAAT2 is abundant in developing human white matter and is located on pre-OLs when the white matter is most vulnerable to PVL. It has been assumed that astrocytes have the major role in maintaining glutamate homeostasis (Bergles et al., 1999; Danbolt, 2001; Rosenberg and Aizenman, 1989; Tanaka et al., 1997). In fetal human white matter, oligodendroctyes appear to have an important role in glutamate homeostasis, in that pre-OLs and not astrocytes are the site of expression of EAAT2.
We did not observe the localization of GLT1/EAAT2 protein in growing axons that has been observed in studies of developing rodent and sheep brain. One explanation for this observation is that EAAT2 is transiently expressed in human axons during the fetal period at an earlier time (>23 weeks) than was included in our clinical database. This is suggested by one study that showed partial coexpression of EAAT2 and (-tubulin in the area of corticofugal bundles adjacent to radial fibers in the intermediate zone only at 20 GW in the human fetus (Furuta et al., 2005). The absence of EAAT2 from axons and astrocytes during the period of maximal vulnerability to PVL and the presence of EAAT2 in pre-OLs during this period suggest that pre-OLs may make a significant contribution to glutamate homeostasis during development.
Physiological implications of up-regulation of EAAT2 in the cerebral white matter of the human fetus and infant
A key physiological question concerns the significance of the developmental up-regulation of EAAT2 in the human white matter in early life. This up-regulation occurs at a time when pre-OLs express glutamate receptors, and the coincidence of these two phenomena suggests that glutamate receptor activation is important in the development and function of these cells. The expression of both glutamate receptors and transporters in pre-OLs also strongly suggests that there is a source for glutamate in developing white matter that causes activation of the OL glutamate receptors. Possible sources include astrocytes (Bezzi et al., 1998, 2001; Kimelberg et al., 1990; Parpura et al., 1994), microglia (Nakamura et al., 2003), and growing axons (Li and Stys, 2001; Lin et al., 2005). The functional consequences of glutamate receptor activation may be regulation of OL proliferation and differentiation, migration, and/or myelination (Gallo et al., 1996; Yuan et al., 1998). Glutamatergic synapses on oligodendroglial progenitors have been shown in the hippocampus (Bergles et al., 2000). It has become increasingly evident that developing oligodendrocytes are a target for glutamate signaling; therefore, further study is warranted focusing on the regulation of glutamate transporter expression and function in OLs.
ACKNOWLEDGMENTS
The authors are grateful to Drs. Pamela Follett, Robin L. Haynes, and Delia Talos for helpful discussions and to Dr. Rebecca D. Folkerth for help with neuropathological review of cases.
Grant sponsor: National Institutes of Health; Grant number: NS41883; Grant number: NS40753; Grant number: NS07473; Grant number: NS38475; Grant number: HD18655; Grant sponsor: Hearst Fund Award; Grant sponsor: United Cerebral Palsy Foundation.
LITERATURE CITED
- Abercrombie M. 1946. Estimation of nuclear population from microtome sections. Anat Rec 94:239–247. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. 1994. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14:5559–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. 1997. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A 94:4155–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. 2001. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21:1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled O, Ben-Hur H, Biegon A, Groner Y, Dewhurst S, Furuta A, Rothstein JD. 1997. Distribution of glutamate transporter subtypes during human brain development. J Neurochem 69:2571–2580. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. 1984. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43:1369–1374. [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. 1998. Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol 198:13–30. [DOI] [PubMed] [Google Scholar]
- Berger UV, Desilva TM, Chen W, Rosenberg PA. 2005. Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol 492:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE. 1999. Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol 9:293–298. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Roberts JDB, Somogyi P, Jahr CE. 2000. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405:187–191. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. 1998. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391:281–285. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. 2001. CXCR4activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–710. [DOI] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. 2002. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci 22:2142–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. 2004. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24:1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. 2001. Glutamate uptake. Prog Neurobiol 65:1–105. [DOI] [PubMed] [Google Scholar]
- Deng W, Rosenberg PA, Volpe JJ, Jensen FE. 2003. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci U S A 100:6801–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. 1995. An excitatory amino-acid transporter with properties of a ligandgated chloride channel. Nature 375:599–603. [DOI] [PubMed] [Google Scholar]
- Fern R, Möller T. 2000. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci 20:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA. 1996. Tumor necrosis factor ) inhibits glutamate uptake by primary human astrocytes—implications for pathogenesis of HIV-1 dementia. J Biol Chem 271:15303–15306. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. 2000. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci 20:9235–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Talos DM, Volpe JJ, Jensen FE. 2003. AMPA receptor subunit expression in human white matter during window of susceptibility to periventricular leukomalacia. Ann Neurol 54(Suppl 3):S103.12891660 [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. 2004. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci 24:4412–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. 1997. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17:8363–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Takashima S, Yokoo H, Rothstein JD, Wada K, Iwaki T. 2005. Expression of glutamate transporter subtypes during normal human corticogenesis and type II lissencephaly. Brain Res Dev Brain Res 155:155–164. [DOI] [PubMed] [Google Scholar]
- Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. 1996. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci 16:2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW. 2002. On counting and counting errors. J Comp Neurol 447:1–7. [DOI] [PubMed] [Google Scholar]
- Hagberg H. 1992. Hypoxic-ischemic damage in the neonatal brain: excitatory amino acids. Dev Pharmacol Ther 18:139–144. [PubMed] [Google Scholar]
- Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. 2000. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7:153–159. [DOI] [PubMed] [Google Scholar]
- Itoh T, Beesley J, Itoh A, Cohen AS, Kavanaugh B, Coulter DA, Grinspan JB, Pleasure D. 2002. AMPA glutamate receptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J Neurochem 81:390–402. [DOI] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, Attwell D. 2005. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438:1162–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. 1990. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci 10:1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Stys PK. 2001. Na+-K+-ATPase inhibition and depolarization induce glutamate release via reverse Na+-dependent transport in spinal cord white matter. Neuroscience 107:675–683. [DOI] [PubMed] [Google Scholar]
- Liao SL, Chen CJ. 2001. Differential effects of cytokines and redox potential on glutamate uptake in rat cortical glial cultures. Neurosci Lett 299:113–116. [DOI] [PubMed] [Google Scholar]
- Lin SC, Huck JH, Roberts JD, Macklin WB, Somogyi P, Bergles DE. 2005. Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron 46:773–785. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. 1994. Mechanisms of disease: excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622. [DOI] [PubMed] [Google Scholar]
- Loeliger M, Watson CS, Reynolds JD, Penning DH, Harding R, Bocking AD, Rees SM. 2003. Extracellular glutamate levels and neuropathology in cerebral white matter following repeated umbilical cord occlusion in the near term fetal sheep. Neuroscience 116:705–714. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Dietrich J, Wong V, Xue H, Mayer-Proschel M, Rao MS, Rothstein JD. 2004. Glutamate transporter expression and function in human glial progenitors. Glia 45:133–143. [DOI] [PubMed] [Google Scholar]
- Matute C, Sánchez-Gómez MV, Martíınez-Millán L, Miledi R. 1997. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci U S A 94:8830–8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. 1998. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med 4:291–297. [DOI] [PubMed] [Google Scholar]
- Meldrum B, Garthwaite J. 1990. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 11:379–387. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. 2006. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439:988–992. [DOI] [PubMed] [Google Scholar]
- Milton ID, Banner SJ, Ince PG, Piggott NH, Fray AE, Thatcher N, Horne CH, Shaw PJ. 1997. Expression of the glial glutamate transporter EAAT2 in the human CNS: an immunohistochemical study. Brain Res Mol Brain Res 52:17–31. [DOI] [PubMed] [Google Scholar]
- Mitani A, Tanaka K. 2003. Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. J Neurosci 23:7176–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Ohmaki M, Murakami K, Yoneda Y. 2003. Involvement of protein kinase C in glutamate release from cultured microglia. Brain Res 962:122–128. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Kawakami H, Tanaka K, Nakamura S. 1996. Expression of three glutamate transporter subtype mRNAs in human brain regions and peripheral tissues. Brain Res Mol Brain Res 36:189–192. [DOI] [PubMed] [Google Scholar]
- Nicholls D, Attwell D. 1990. The release and uptake of excitatory amino acids. Trends Pharmacol Sci 11:462–468. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Traystman RJ, Koehler RC, Martin LJ. 1999. GLT1, glial glutamate transporter, is transiently expressed in neurons and develops astrocyte specificity only after midgestation in the ovine fetal brain. J Neurobiol 39:515–526. [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. 1994. Glutamate-mediated astrocyte-neuron signalling. Nature 369:744–747. [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. 1992. Cloning and expression of a rat brain L-glutamate transporter. Nature 360:464–467. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. 2000. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 6:67–70. [DOI] [PubMed] [Google Scholar]
- Roettger V, Lipton P. 1996. Mechanism of glutamate release from rat hippocampal slices during in vitro ischemia. Neuroscience 75:677–685. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. 1989. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett 103:162–168. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Dai WM, Gan XD, Ali S, Fu J, Back SA, Sanchez RM, Segal MM, Follett PL, Jensen FE, Volpe JJ. 2003. Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res 71:237–245. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. 2000. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403:316–321. [DOI] [PubMed] [Google Scholar]
- Salter MG, Fern R. 2005. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438:1167–1171. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. 2001. Northern Hybridization. Molecular Cloning. Cold Spring Harbor: CSHL Press p. 7.42–7.44. [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. 1993. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100:431–440. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Lesch K-P, Kugler P. 2002. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience 109:45–61. [DOI] [PubMed] [Google Scholar]
- Seki Y, Feustel PJ, Keller RW Jr, Tranmer BI, Kimelberg HK. 1999. Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30:433–440. [DOI] [PubMed] [Google Scholar]
- Silverstein FS, Naik B, Simpson J. 1991. Hypoxia-ischemia stimulates hippocampal glutamate efflux in perinatal rat brain: an in vivo microdialysis study. Pediatr Res 30:587–590. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. 1992. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A 89:10955–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkowski M, Barbour B, Attwell D. 1990. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature 348:443–446. [DOI] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, Li Y, Yang J, Dienel G, Zielke HR, Nedergaard M. 2005. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci U S A 102:16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. 2002. Tumor necrosis factor-) induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 281:21362–21368. [DOI] [PubMed] [Google Scholar]
- Talos DM, Fishman RE, Park H-K, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. 2005a. Developmental regulation of AMPA receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischcemic injury: part I. Rodent cerebral white matter and cortex. J Comp Neurol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, Jensen FE. 2005b. Developmental regulation of AMPA receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury: part II. Human cerebral white matter and cortex. J Comp Neurol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. 1997. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A. 1996. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem 271:5976–5979. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. 2003. Cerebral white matter injury of the premature infant— more common than you think. Pediatrics 112:176–180. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Floridi S, Racagni G. 1994. Reactive oxygen species inhibit high-affinity glutamate uptake: molecular mechanism and neuropathological implications. Ann N Y Acad Sci 738:153–162. [DOI] [PubMed] [Google Scholar]
- Yamada K, Watanabe M, Shibata T, Nagashima M, Tanaka K, Inoue Y. 1998. Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. J Neurosci 18:5706–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. 1996. Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport 7:2181–2185. [DOI] [PubMed] [Google Scholar]
- Yoshioka A, Ikegaki N, Williams M, Pleasure D. 1996. Expression of N-methyl-D-aspartate (NMDA) and non-NMDA glutamate receptor genes in neuroblastoma, medulloblastoma, and other cell lines. J Neurosci Res 46:164–172. [DOI] [PubMed] [Google Scholar]
- Yuan X, Eisen AM, McBain CJ, Gallo V. 1998. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 125:2901–2914. [DOI] [PubMed] [Google Scholar]