

Abstract
Background and Aim:
HEV is a significant cause of acute hepatitis globally. Some genotypes establish persistent infection when immunity is impaired. Adaptive immune mechanisms that mediate resolution of infection have not been identified. Here, the requirement for CD8+ T cells to control HEV infection was assessed in rhesus macaques, a model of acute and persistent HEV infection in humans.
Methods:
Rhesus macaques were untreated or treated with depleting anti-CD8α monoclonal antibodies before challenge with an HEV gt3 isolate derived from a chronically infected human subject. HEV replication, ALT, anti-capsid antibody and HEV-specific CD4+ and CD8+ T cell responses were assessed after infection.
Results:
HEV control in untreated macaques coincided with the onset of a neutralizing IgG response against the ORF2 capsid and liver infiltration of functional HEV-specific CD4+ and CD8+ T cells. Virus control was delayed by 1 week in CD8+ T cell depleted macaques. Infection resolved with onset of a neutralizing IgG antibody response and a much more robust expansion of CD4+ T cells with antiviral effector function.
Conclusions:
Liver infiltration of functional CD8+ T cells coincident with HEV clearance in untreated RM, and a 1 week delay in HEV clearance in CD8+ T cell depleted RM, support a role for this subset in timely control of virus replication. Resolution of infection in the absence of CD8+ T cells nonetheless indicates that neutralizing antibodies and/or CD4+ T cells may act autonomously to inhibit HEV replication. HEV susceptibility to multiple adaptive effector mechanisms may explain why persistence occurs only with generalized immune suppression. The findings also suggest that neutralizing antibodies and/or CD4+ T cells should be considered as a component of immunotherapy for chronic infection.
Keywords: Hepatitis E Virus, CD4+ and CD8+ T lymphocyte, depletion, rhesus macaque
Lay Abstract.
The hepatitis E virus is a major cause of liver disease globally. Some genetic types (genotypes) of HEV persist in the body if immunity is impaired. Our objective was to identify immune responses that promote clearance of HEV. Findings indicate that HEV may be susceptible to multiple arms of the immune response that can act independently to terminate infection. They also provide a pathway to assess immune therapies for chronic HEV infection.
Graphical Abstract
Introduction.
The hepatitis E virus (HEV) infects approximately 20 million humans each year and is a major cause of acute enteric hepatitis globally[1, 2]. Human infections are most commonly caused by genotypes within the HEV Orthohepevirus A species. Infections are typically sub-clinical and resolve spontaneously in individuals with normal immune function[2, 3]. Adaptive immunity is considered essential for termination of acute infections. An early IgM response against the open reading frame 2 (ORF2) capsid is diagnostic of acute primary infection[1] and rapidly transitions to an IgG response as virus replication is controlled[1]. Serum ORF2-specific IgG and IgM antibodies can neutralize HEV infectivity[4–6]. T cell responses against the 3 HEV open reading frames (ORFs) have been described in immune competent humans with acute and resolved infections[7]. Some Orthohepevirus species A genotypes (gt3, gt4, and gt7) [3] and at least one species C genotype (gt1)[8] can cause chronic infection and liver injury, but only when immunity is compromised by treatment with immune suppressive drugs, HIV co-infection, or hematological malignancies[1, 2]. HEV-specific antibody, CD4+ and CD8+ T cell responses are weak or undetectable in persistent infection[9–11].
Adaptive responses required for resolution of acute HEV infection have not been identified. Delayed clearance but no persistence of an avian Orthohepevirus species B virus was described in chickens treated simultaneously with cyclosporin A and an antibody against the chicken CD8α protein[12]. Interpretation of this study was limited by challenges in measuring chicken HEV-specific T cell responses[12]. Rhesus macaques (RM) are susceptible to infection with human HEV strains[13], including the Orthohepevirus species A gt3 viruses that persist when immunity is suppressed[14]. Here, we compared the course of HEV gt3 infection in RM that were untreated or treated with a depleting anti-CD8 monoclonal antibody (mAb) to more precisely define the role of CD8+ T cells in control of virus replication.
Experimental Procedures.
Rhesus macaques and HEV challenge.
RM (Macaca mulatta) of Indian origin selected for study were HEV capsid antibody negative. Expression of Mamu class I and II alleles was determined by PCR with sequence-specific primers [15, 16]. Studies were performed at the Abigail Wexner Research Institute at Nationwide Children’s Hospital, an AAALAC accredited Institution, in accordance with protocols approved by the Institutional Animal Care and Use Committee. An RM passaged inoculum was derived from a gt3 HEV strain (Kernow; see CTAT Methods). In brief, a fecal suspension from a chronically infected human[17] was clarified by centrifugation, filtered (0.45 μM) and diluted in PBS with HEV antibody negative RM serum (2% v/v). To generate sufficient virus stock for infection studies, an RM was challenged with 2×106 genome equivalents (GE) of the fecal suspension. Feces with a high HEV RNA titer were processed as described above, and 2×106 GE were delivered intravenously to RM in this study. Fecal HEV RNA titers were determined by qPCR as described in Supplementary Materials and Methods and Supplementary Table 1.
M-T807R1 treatment.
M-T807R1 is an IgG1 mouse/rhesus CDR-grafted form of the depleting anti-CD8α antibody M-T807[18] obtained from the Non-Human Primate Reagent Resource (described in CTAT Methods). Four RM were treated with M-T807R1 (50 mg/Kg of body weight, i.v.) 1 day before and at 4 weekly intervals after HEV challenge. CD8+ T cell depletion in blood and liver was monitored with anti-CD8α mAb DK25 (Dako) that recognizes a CD8α epitope distinct the one that binds M-T807R1.
Mamu multimers and antibodies.
HEV and rhesus CMV (rhCMV) epitopes were mapped using virus-specific CD4+ and CD8+ T cell lines derived from RM infected with these viruses (Supplementary Materials and Methods and Supplementary Table 2). Mamu-A*0101, A*0801, and DRB*w201 restriction was determined as described[19]. Tetramers containing Mamu-A*0801 class I epitopes (HEV ORF21923 and rhCMV pp65906) and a Mamu-DRB*w201 class II epitope (HEV ORF21913) were produced by the NIH Tetramer Core Facility at Emory University. Dextramers containing Mamu-A*0101 epitopes (HEV X917, Pol1724, ORF22088) were produced by Immudex ApS (Denmark). Specificity of class I and II multimer binding (Supplementary Fig. 1), details of fluorophore-conjugated mAb (Supplementary Table 3 and CTAT Methods) and gating strategies (Supplementary Fig. 2) are provided. Class II multimer frequencies in untreated and M-T807R1 treated RM were determined by gating on CD3+CD4+ T cells. Depletion of CD8+ T cells from the blood and liver of M-T807R1 treated RM was profound. Gating on CD3+CD8+ T cells was therefore not possible for class I multimer analysis. Frequencies of class I multimer-positive cells in M-T807R1 treated and untreated RM were therefore assessed by gating on CD3+CD4- T cells (Supplementary Materials and Methods).
T cell analyses.
ELISpot and ICS assays are described in Supplementary Materials and Methods and elsewhere[20]. For both assays, responses were assessed against 14 pools of synthetic overlapping peptides (18 amino acids overlapping by 11 residues) spanning HEV gt3 Kernow ORF1, ORF2, and ORF3 proteins, based on Genbank sequence HQ709170.1, with glutamine (Q) replacing any amino acid (X) at position 1058 (Supplementary Table 4). ELISpot background was <10 spot forming colonies per 106PBMCs without HEV peptide stimulation (Supplementary Fig. 3). For flow-based assays, cells were analyzed using LSRII or Fortessa instruments (BD). Data was analyzed using FlowJo (v.9.8, FlowJo LLC). Between 500,000 and 1,500,000 events were acquired for cytokine analysis. A response was scored as positive when the number of peptide-stimulated cells that produced a cytokine was more than twice that of unstimulated cells, and the value after background subtraction was at least 0.003%. For multimer analysis, between 1,000,000 and 2,000,000 events were acquired. Background multimer staining was <0.001%.
HEV ORF2 antibody ELISA.
Serum HEV IgM and IgG titers were determined by ELISA using the manufacturers protocol (Xpress Biotech International, Frederick, MD). A WHO IgG Reference Reagent for HEV (NIBSC code: 95/584) provided a standard to determine international units of anti-ORF2 IgG antibodies in RM serum.
Neutralization assays.
HEV neutralization has been described[21]. In brief, serum collected before and at 1 week intervals after HEV challenge was untreated or pre-treated with dithiothreitol (DTT, 0.5 mM at 37° C for 1 hour). Six serial 3-fold dilutions of serum in DMEM, starting at a 1:200 dilution, were incubated (37°C for 1 h) with gradient purified naked HEV particles (3×107 GE of Kernow C1/p6) before inoculation onto HepG2-shMAVS cells (see CTAT Methods). DTT treatment denatures pentameric IgM antibodies so that the contribution of IgM versus IgG to neutralization can be determined[22]. HEV antigen was quantified in the culture medium 5 days later using the WANTAI HEV-Ag ELISA kit (Beijing Wantai Biological Pharmacy, Beijing, China). Neutralization was calculated as percentage of inhibition in virus infection.
To determine antibody inhibition of HEV spread, HepG2-shMAVS cells were infected with purified HEV particles (1× 107 GE). On days 5 and 7 post infection, 10 μL of RM serum was added to the culture medium (100 μL). On day 10 post infection, fixed cells were immunostained with a mouse anti-ORF2 FITC-labelled antibody (mAb #4)[21]. Images were acquired using a Zeiss LSM 510 meta inverted confocal microscope. The number of infected cells identified by immune fluorescence assay was quantified with the Image J software (NIH).
Results.
Circulating HEV-specific T cell responses, serum ALT, and fecal HEV shedding were first assessed in 2 groups of 4 RM that were either untreated or treated with M-T807R1, a depleting anti-CD8α mAb, before HEV challenge. High levels of HEV fecal shedding were measured in 4 untreated RM at weeks 1 and 2 before dropping below the detection threshold at week 3 (Fig. 1A). An initial dose of M-T807R1 mAb at study day −1 reduced circulating CD8+ T cells by ~99% 24 hours later when the 4 RM were challenged with HEV (Supplementary Fig. 4A). Virus load did not differ between untreated and M-T807R1 treated RM at week 2 (Figs. 1A and 1B). However, HEV fecal shedding was detectable until week 4 in M-T807R1 treated RM (Fig. 1B), 1 week longer than untreated RM (Fig.1A). Serum ALT remained within normal limits for both groups (Fig. 1A and 1B).
Figure 1. HEV infection and T cell immunity in untreated and M-T807R1 treated RM.

Fecal HEV titers, serum ALT, and blood T cell responses for (A) 4 untreated and (B) 4 M-T807R1 treated RM. Arrows indicate dosing with M-T807R1 (B). IFN-γ ELISpot, sum of all spot forming units (SFU) against ORF1, ORF2, and ORF3 peptide pools. Circulating CD4+ and CD8+ T cells were visualized with class II (ORF21913) and class I (HEV ORF21923 and rhCMV pp65906) multimers. The Y axis represents the percent of CD3+CD4+ PBMC from 4 untreated and 3 M-T807R1 treated RM that were positive for ORF21913 class II multimer binding, and the percent of CD3+CD4- positive PBMC from 4 treated and untreated RM that were positive for ORF21923 and rhCMV pp65906 binding. Data are presented as the mean+/−SD for RM in each group.
T cell responses in both RM groups were first compared by IFN-γ ELISpot assay with ORF1, ORF2 and ORF3 peptide pools. The T cell response peaked in untreated RM at week 2, immediately before control of virus replication at week 3 (Fig. 1A). The response was delayed in M-T807R1 treated RM, peaking at week 4 before contracting slowly through week 8 (Fig. 1B). T cell responses were next evaluated by multimer visualization. The 8 RM in this first experiment were selected for expression of the Mamu-A*0801 class I (4/4 untreated and M-T807R1 treated RM) and the class II Mamu-DRBw*0201 alleles (4/4 untreated and 3/4 M-T807R1 treated RM). This combination of class I and II alleles facilitated visualization of circulating ORF2-specific CD8+ T cells (ORF21923) and CD4+ T cells (ORF21913) at multiple time points within the same individual. In untreated RM, an ORF21913-specific CD4+ T cell response was detected in blood at week 2 (Fig. 1A). CD4+ T cells contracted sharply at week 3 when an ORF21923-specific CD8+ T cell population peaked. Rapid CD8+ T cell contraction followed (Fig. 1A). Circulating ORF21913-specific CD4+ T cells were also visualized in M-T807R1 treated RM at week 2 post-infection, but with a different kinetic (Fig. 1B). They expanded through week 5, and peaked at a higher frequency when compared with the week 2 peak in untreated RM (Figs. 1A and 1B). HEV-specific Mamu-A*0801 ORF21923-specific CD8+ T cells were not visualized in the blood of M-T807R1 treated RM after infection (Fig. 1B). Of note, all RM in both groups were naturally infected with rhCMV. CD8+ T cells against a Mamu-A*0801 restricted rhCMV pp65 epitope were present in all RM at the baseline timepoint. Frequencies remained stable in the 4 untreated RM after HEV infection (Fig.1A). One dose of M-T807R1 resulted in loss of rhCMV pp65-specific CD8+ T cells from blood that was sustained through week 8 (Fig. 1B). M-T807R1 treatment therefore caused highly efficient depletion of circulating CD8+ T cells that was sustained until HEV infection resolved.
Intrahepatic CD8+ T cell responses.
A profound loss of intrahepatic CD8+ T cells was observed in M-T807R1 treated RM when compared with baseline values (Supplementary Fig. 4B). At week 3, the reduction in CD3+CD8+ T cells was ~95% in all 4 treated RM (Fig. 2A). At week 4, CD8+ T cells remained undetectable in 2 RM and partially recovered in the other 2 (Fig. 2A). ORF21923-specific T cells were not detected in liver of any RM at week 3 by multimer analysis (Fig. 2A). A CD8-negative ORF21923-positive population was visualized at low frequency (0.24%) in only 1 RM at week 4, the time point when all 4 animals in the group resolved the infection (Fig. 2A). Sampling permitted a direct ex vivo IFN-γ ICS assay on 2 RM at week 3 and 2 RM at week 4. Functional HEV-specific CD3+CD4- T cells were not detected in any of the RM, including the only animal with an ORF21923-positive, CD8-negative T cell population visualized by multimer staining at week 4 (Fig. 2B). Infection therefore resolved with no consistent pattern of CD8+ T cell recovery, or detection of HEV-specific CD8+ T cells by multimer visualization or effector function.
Figure 2. Intrahepatic HEV-specific CD8+ T cell responses.

(A) Staining of liver mononuclear cells (LMC) from M-T807R1 treated RM with anti-CD8 antibodies and the ORF21923 multimer. Dot plots (gated on CD3+CD4- LMC) are from 4 individual RM sampled at week 3 (top row) or week 4 (bottom row). (B) IFN-γ ICS analysis of LMC from M-T807R1 treated RM against the indicated HEV peptide pool at week 3 or week 4 post-infection (2 RM per time point). Peptide pools and HEV protein designations are described in Supplementary Table 4. (C) Direct visualization of HEV-specific CD8+ T cells in liver of 7 untreated RM at week 3. Each dot plot represents staining of CD3+CD4- LMC from 4 RM that expressed Mamu-A*0101 (epitopes ORF1 X917, Pol1724 and ORF22088) and 5 RM that expressed (or co-expressed) Mamu-A*0801 (ORF21923). (D) Intracellular IFN-γ production by week 3 LMC from 7 untreated RM.
Intrahepatic CD8+ T cell responses were also assessed in 7 untreated RM selected for expression of Mamu-A*0101 (2 RM), Mamu-A*0801 (3 RM), or both class I alleles (2 RM), for a total of 4 RM that expressed Mamu-A*0101 and 5 that expressed (or co-expressed) Mamu-A*0801. Inclusion of Mamu-A*0101 positive RM facilitated an expansion of the liver multimer analysis to 2 ORF1 epitopes (X917 and Pol1724) and an additional ORF2 epitope (ORF22088) (Supplementary Table 2). HEV replication patterns were remarkably consistent in all 7 untreated RM (Supplementary Fig. 5), with high levels of fecal shedding at weeks 1 and 2 followed by a sharp decline at week 3 post-infection. At week 3, CD8+ T cells were visualized in liver of all 4 RM that expressed the Mamu-A*0101 allele (ORF1 X917, Pol1724, ORF22088)and 5 RM that expressed (or co-expressed) Mamu-A*0801 (ORF21923) (Fig. 2C). A broad functional CD8+ T cell response was predicted based on multimer visualization of intrahepatic ORF1 and/or ORF2-specific populations. However, functional ORF1-specific CD8+ T cells were absent or present at very low frequency by IFN-γ ICS assay (Fig. 2D). Strong ORF2-specific responses were detected in all 7 untreated RM, including 2 sub-pools that contained the same dominant ORF22088 epitope in the overlapping region between the last (pool B) and first (pool C) peptide in each set (Fig. 2D). Differences in the magnitude of the CD8+ T cell response against these pools likely reflects variability in processing efficiency of the ORF22088 epitope from each peptide, or additive effects of unidentified class I epitopes in Pool B that stimulated a stronger response. Multifunctionality of CD8+ T cells was assessed in 5/7 untreated RM. IFN-γ production and cytotoxic degranulation, as measured by CD107a expression, were the dominant effector functions of ORF2-specific CD8+ T cells (Supplementary Fig. 6). Together, these observations indicate that ORF1 and ORF2 specific CD8+ T cells expanded in liver when virus replication was controlled, but antiviral effector function was apparent only for those targeting ORF2.
Intrahepatic CD4+ T cell responses.
An IFN-γ CD4+ T cell response against multiple HEV proteins was observed in the liver of 7 untreated (Fig. 3A) and 4 M-T807R1 treated (Fig. 3B), with notable differences between the groups. Specifically, liver CD4+ T cell frequencies against all proteins were higher in the M-T807R1 treated RM (Fig. 3A versus 3B), consistent with the stronger and more sustained circulating CD4+ T cell response (Fig. 1A versus 1B). Intrahepatic HEV-specific CD4+ T cells were assessed by ICS for IFN-γ, TNF-α, IL-2 and IL-21 production in 4 M-T807R1 treated and 5 of 7 untreated RM. Representative dot plots from 1 RM in each group are shown (Fig. 3C). CD4+ T cells from both groups were multifunctional. Overall, only a minor difference in the number of CD4+ T cells producing 2 or more cytokines was observed in the untreated (~60%) versus treated (~50%) groups (Fig. 3D). Notably, the difference is attributable to a reduced frequency of IL-2 and IL-21 producing CD4+ T cells in M-T807R1 treated RM. The frequency of IL-21 positive CD4+ T cells was ~30% in M-T807R1 treated RM versus ~60% in untreated RM (P value .0013 by unpaired t test, GraphPad Prism)(Fig. 3D,E). The fraction of CD4+ T cells producing IFN-γ or TNF-α was not significantly different between the groups (Fig. 3D,E).
Figure. 3. Intrahepatic CD4+ T cell responses.

IFN-γ production by liver CD3+CD4+ T cells from (A) 7 untreated RM (week 3) and (B) 4 M-T807R1 treated RM (2 each at weeks 3 or 4) after stimulation with the indicated peptide pool (see Supplementary Table 4). (C) Representative dot plots of IFN-γ (X axis) versus TNF-α, IL-2 and IL-21 (Y axis) production by week 3 liver CD4+ T cells from one untreated and one M-T807R1 treated RM. (D)Pie charts indicate the percentage of liver HEV-specific CD4+ T cells from 5 untreated and 4 M-T807R1 treated RM. CD4+ T cell production of 1, 2, 3, or 4 cytokines, as indicated in the legend. Arcs indicate the cytokines produced within each pie segment. (E). Percentage of cytokine CD4+ T cells positive for the indicated cytokine in 5 untreated and 4 M-T807R1 treated RM (mean +/− SD) compared by an unpaired t test (Graph Pad Prism).
Antibody responses.
ORF2 ELISA IgM and IgG antibody responses were remarkably consistent in untreated RM. IgM antibodies first appeared at week 2 and peaked at week 3 (Fig. 4A). IgM titers declined at week 4 with transition to an anti-ORF2 IgG antibody response (Fig. 4A). Notably, the ORF2 IgM response was delayed until week 3 in M-T807R1 treated RM, and peak titers were reduced when compared with untreated RM (Fig. 4A). The anti-ORF2 IgG ELISA response was lower at week 3 in treated versus untreated RM, but titers equalized by week 4 (Fig. 4B).
Figure 4. ORF2 capsid antibody response.

HEV fecal shedding and serum IgM and IgG antibody titers were measured by ORF2 capsid ELISA for (A) 4 untreated and (B) 4 M-T807R1 treated RM (mean +/−SD). Serum neutralizing antibodies in (C) 4 untreated and (D) 4 M-T807R1 treated RM (mean +/−SD) were measured at the indicated time points. Fecal virus titers for the RM are also shown. Each bar represents mean percent neutralization for 6 serial 3-fold dilutions of serum from 4 animals in each group, beginning at a 1:200 dilution. Neutralization titer 50 (NT50) was calculated from the dilution series. Percent neutralization at each dilution with (black bars) or without (open bars) DTT treatment of serum is shown. Serum from (E) 4 untreated and (F) 4 M-T807R1 treated RM was added to culture at days 5 and 7 after HEV infection at a final dilution of 1:10 and immunostained at day 10 with FITC-labelled anti-ORF2 antibodies. Representative images of infected cells cultured in the presence of serum collected at the indicated week post challenge from 2 untreated RM (E, U1 and U2) and 2 M-T807R1 treated RM (F, T1 and T2) are shown. Percent inhibition (mean+/−SD) of virus spread in cell culture and mean HEV RNA titer in fecal samples for 4 animals in each group are shown in right panels at the indicated week post infection.
Antibody neutralization was measured after co-incubating naked HEV particles with 6 serial 3-fold dilutions of serum from 4 untreated and 4 M-T807R1 treated RM, starting at a 1:200 dilution. Strong neutralization of HEV infectivity in untreated RM at week 2 was mediated by IgM as the activity was ablated by treatment of serum with DTT, a reducing agent that disrupts IgM structure (Fig. 4C). A decline in fecal shedding below the detection threshold at week 3 was associated with appearance of DTT resistant IgG neutralizing antibodies (Fig. 4C). At week 2, IgM neutralization was weak in M-T807R1 versus untreated RM (Fig. 4C versus 4D), consistent with delayed onset of this response by ELISA (Fig. 4B). Serum neutralization increased substantially at week 3 but was mediated almost entirely by DTT-sensitive IgM antibodies (Fig. 4D). Control of fecal shedding at week 4 was associated with a sharp increase in IgG-mediated neutralization (Fig. 4D).
The capacity of serum antibodies to block spread of quasi-enveloped HEV between hepatocytes was also assessed. Serum collected at week 2 from untreated RM (Fig. 4E) and week 3 from M-T807R1 treated RM (Fig. 4F) did not prevent HEV spread in culture despite high titers of IgM neutralizing antibodies. Transition to an IgG response was associated with significant inhibition of virus spread in culture by week 3 serum from untreated RM (Fig. 4E) and week 4 serum from M-T807R1 treated RM (Fig. 4F). In summary, serum IgM and IgG antibodies neutralized infectivity of naked HEV virions. Inhibition of quasi-enveloped HEV spread in cultured hepatocytes was mediated predominately by serum IgG antibodies that appeared coincident with the sharp decline in fecal virus shedding.
Discussion
The RM model of acute HEV infection facilitated detailed mapping of temporal relationships between virus replication and onset of adaptive immune responses. Control of HEV replication in untreated RM was kinetically associated with (i) an anti-ORF2 neutralizing IgG antibody response that inhibited virus spread in a cell culture model, and (ii) liver infiltration of virus-specific CD4+ and CD8+ T cells. Intrahepatic CD8+ T cells targeting ORF-2 produced IFN-γ but only a small subset co-produced TNF-α. Production of these cytokines and cytotoxic degranulation upon antigen stimulation is consistent with a role for ORF2-specific CD8+ T cells in control of acute infection. ORF1-specific CD8+ T cells were also visualized in liver but their contribution to infection control in this model is less certain because of an apparent deficit in effector function. This defect was selective for ORF1-specific CD8+ T cells as CD4+ T cells targeting this non-structural domain were multifunctional. Functional ORF1-specific CD8+ T cell have been identified by ICS assay in the blood of human subjects with acute and resolved HEV infections[10, 11]. Whether poor effector function by intrahepatic CD8+ T cells targeting ORF1 is specific to the RM model, or reflects conditions unique to the liver, remains to be determined. The selective effector function of ORF2-specific CD8+ T cells is also unexplained. HEV encodes a secreted, glycosylated form of the ORF2 capsid protein[1, 21] that could potentially elicit a more functional CD8+ T cell response.
Antibody-mediated depletion of CD8+ T cells prolonged HEV infection by 1 week when compared with untreated RM, an observation that is also consistent with a contribution of this T cell subset to timely virus control. Nonetheless, M-T807R1 treated RM resolved infection with no or only partial recovery of the liver CD8+ T cell compartment. A the time of resolution, a T cell population targeting class I epitope ORF21923 that was dominant in all untreated RM was detected in only 1/4 M-T807R1 treated RM. A functional HEV-specific CD8+ T cells were not detected against any HEV protein, including the ORF2 capsid that was targeted by IFN-γ producing CD8+ T cells in untreated RM. These observations indicate that CD8+ T cells are not necessarily required for control of acute HEV infection when other arms of the adaptive immune system are intact.
ALT values remained within normal limits in both groups, despite detection of ORF2-specific CD8+ T cells that underwent cytotoxic degranulation in the liver of untreated RM. The absence of overt hepatocellular injury is consistent with mild to inapparent acute HEV gt3 infection in most humans[3]. This may indicate that non-cytotoxic effector mechanisms, perhaps mediated by CD4+ T cells or antibodies, had a dominant role in HEV clearance from the liver of both RM groups. In support of this possibility, the CD4+ T cell response was remarkably broad and multifunctional, consistent with observations of circulating HEV-specific CD4+ T cells in human subjects[10]. HEV-specific CD4+ T cells may have been particularly important in termination of infection in M-T807R1 treated RM. CD4+ T cell frequencies were higher and the response was prolonged by several weeks in M-T807R1 treated RM, perhaps due to homeostatic proliferation [23] or depletion of CD8α+ NK cells that may modulate CD4+ T cell responses[24]. The proportion of CD4+ T cells that produced IL-21 was significantly lower in M-T807R1 treated versus untreated RM. Reasons for this reduction are not clear, but may have contributed to a slower ORF2-specific neutralizing antibody response in M-T807R1 treated RM given the essential role of IL-21 in B cell activation and germinal center development[25]. CD4+ T cells from both groups also produced IFN-γ and TNF-α. TNF-α production may be significant because this cytokine suppresses HEV replication in cell culture models[26, 27]. In a patient with chronic hepatitis E, infusion of anti-TNF-α antibodies to treat an autoimmune condition enhanced HEV replication and worsened liver disease[27]. Whether TNF-α production by CD4+ T cells contributed directly to inhibition of HEV replication merits further study.
Anti-ORF2 antibody responses may also have contributed to virus control in untreated and M-T807R1 treated RM, but in an isotype specific manner. While both antibody isotypes neutralized infectivity of naked HEV particles found in feces, only IgG inhibited spread of the virus between hepatocytes. This is most likely explained by the quasi-enveloped nature of HEV particles. The quasi-envelope, a lipid bilayer acquired upon release of HEV from the basolateral surface of hepatocytes, provides resistance to antibody-mediated neutralization [6, 28]. Loss of the membrane in endolysosomes during cellular entry enhances HEV susceptibility to IgG antibody-mediated neutralization [28]. The pentameric structure of IgM is thought to be too large or unstable to neutralize HEV in this environment. Of note, quasi-enveloped HAV is similarly susceptible to IgG neutralizing antibodies [29] that can provide effective post-exposure therapy for infection[7]. This concept has not been rigorously assessed for HEV infection. If successful, it could be of particular value in management of persistent virus replication.
Why HEV gt3 infection did not persist in M-T807R1 treated RM remains unknown. This outcome was most likely prevented by robust CD4+ T cell and neutralizing IgG antibody responses. HEV may therefore be susceptible to multiple adaptive effector mechanisms that can act autonomously to control virus replication. A deficit in one arm of the adaptive response (for instance, CD8+ T cells in this study) can therefore be compensated by other arms, most likely neutralizing antibodies and/or CD4+ T cells that have antiviral effector function. Such a multi-layered defense could explain the inability of HEV gt3 viruses to persist in humans[3] or macaques[14] in the absence of generalized immune suppression affecting all arms of the adaptive response.
Finally, findings from this study provide a conceptual and practical approach towards immune therapy for chronic HEV infection. Passive transfer of neutralizing anti-ORF2 IgG antibodies is an option, although it may be challenging given high levels of soluble ORF2 protein in chronic infection[30, 31]. HEV antigen levels in chronically infected humans are inversely correlated with circulating HEV-specific CD8+ and CD4+ T cell frequencies[11]. Liver infiltration of functional HEV-specific CD8+ T cells with clearance of acute infection in RM also suggests that this response contributes to virus control. Reconstitution of this response, perhaps by transfer of multifunctional HEV T cell receptor (TcR) redirected CD8+ T cells[32], has been proposed to treat persistent HEV infection. Our observations from CD8+ T cell depleted RM suggest that consideration should also be given to transfer of multifunctional CD4+ T cells. The potential for multifunctional chimeric antigen receptor (CAR) CD4+ T cells to control virus replication was recently demonstrated in a small animal model of HIV infection[33]. Transfer of CD4+ T cells with a protective cytokine profile that includes IL-21 and TNF-α could provide therapeutic benefit in persistent HEV infections that are refractory to a reduction in immune suppression or ribavirin treatment. Detailed mapping of functional CD4+ and CD8+ T cell responses in an animal model that is highly relevant to persistent human HEV infection[14] should facilitate a direct test of cellular therapeutic strategies.
Supplementary Material
Highlights.
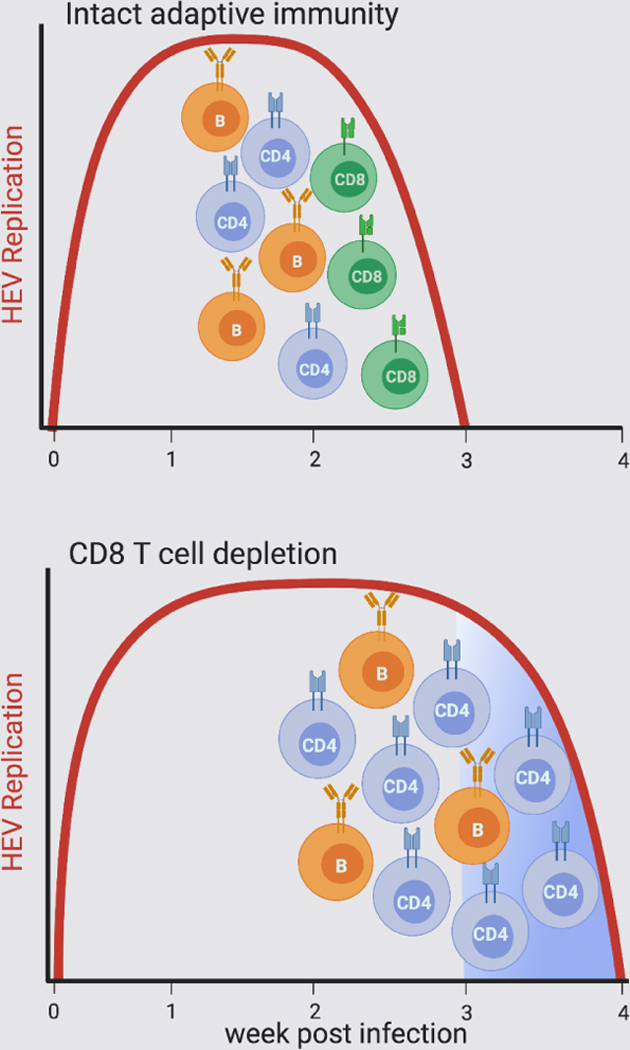
HEV genotype 3 infection in rhesus macaques cleared with onset of a neutralizing IgG antibody response and liver infiltration of antiviral CD4+ and CD8+ T cells.
Antibody-mediated depletion of CD8+ T cells before HEV challenge prolonged infection by 1 week.
In the absence of CD8+ T cells, infection resolved with onset of IgG neutralizing antibody and enhanced CD4+ T cell responses.
CD4+ T cells produced IFN-γ and TNF-α, cytokines with the ability to suppress HEV replication.
HEV is susceptible to multiple adaptive immune mechanisms that can act autonomously and perhaps be adapted to cure persistent infection.
Acknowledgements.
The authors gratefully acknowledge Dr. S. Emerson for providing the HEV gt3 Kernow inoculum and key reagents used in these studies. We also thank the NIH Tetramer Core Facility for production of class I and II tetramers and the Non-Human Primate Reagent Resource for provision of M-T807R1. We are grateful to Dr. Laurie Goodchild, DVM, and staff of the Nationwide Children’s Animal Resource Core for expert veterinary care and technical assistance.
Financial support. Research reported in this manuscript was funded in part by the National Institutes of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI139511 and R21AI137912.
Abbreviations.
- RM
Rhesus macaque
- gt
genotype
- ORF
open reading frame
- mAb
monoclonal antibody
- GE
genome equivalents
Footnotes
Conflicts of interest: None.
Data availability: Data are available upon request.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lhomme S, Marion O, Abravanel F, Izopet J, Kamar N. Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. J Clin Med 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Horvatits T, Schulze Zur Wiesch J, Lutgehetmann M, Lohse AW, Pischke S. The Clinical Perspective on Hepatitis E. Viruses 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kamar N, Pischke S. Acute and Persistent Hepatitis E Virus Genotype 3 and 4 Infection: Clinical Features, Pathogenesis, and Treatment. Cold Spring Harb Perspect Med 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Emerson SU, Clemente-Casares P, Moiduddin N, Arankalle VA, Torian U, Purcell RH. Putative neutralization epitopes and broad cross-genotype neutralization of Hepatitis E virus confirmed by a quantitative cell-culture assay. J Gen Virol 2006;87:697–704. [DOI] [PubMed] [Google Scholar]
- [5].Tanaka T, Takahashi M, Kusano E, Okamoto H. Development and evaluation of an efficient cell-culture system for Hepatitis E virus. J Gen Virol 2007;88:903–911. [DOI] [PubMed] [Google Scholar]
- [6].Takahashi M, Yamada K, Hoshino Y, Takahashi H, Ichiyama K, Tanaka T, et al. Monoclonal antibodies raised against the ORF3 protein of hepatitis E virus (HEV) can capture HEV particles in culture supernatant and serum but not those in feces. Arch Virol 2008;153:1703–1713. [DOI] [PubMed] [Google Scholar]
- [7].Walker CM. Adaptive Immune Responses in Hepatitis A Virus and Hepatitis E Virus Infections. Cold Spring Harb Perspect Med 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sridhar S, Yip CC, Wu S, Chew NF, Leung KH, Chan JF, et al. Transmission of Rat Hepatitis E Virus Infection to Humans in Hong Kong: A Clinical and Epidemiological Analysis. Hepatology 2020. [DOI] [PubMed] [Google Scholar]
- [9].Suneetha PV, Pischke S, Schlaphoff V, Grabowski J, Fytili P, Gronert A, et al. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology 2012;55:695–708. [DOI] [PubMed] [Google Scholar]
- [10].Brown A, Halliday JS, Swadling L, Madden RG, Bendall R, Hunter JG, et al. Characterization of the Specificity, Functionality, and Durability of Host T-Cell Responses Against the Full-Length Hepatitis E Virus. Hepatology 2016;64:1934–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Al-Ayoubi J, Behrendt P, Bremer B, Suneetha PV, Gisa A, Rinker F, et al. Hepatitis E virus ORF 1 induces proliferative and functional T-cell responses in patients with ongoing and resolved hepatitis E. Liver Int 2018;38:266–277. [DOI] [PubMed] [Google Scholar]
- [12].Rogers E, Todd SM, Pierson FW, Kenney SP, Heffron CL, Yugo DM, et al. CD8(+) lymphocytes but not B lymphocytes are required for protection against chronic hepatitis E virus infection in chickens. J Med Virol 2019;91:1960–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Corneillie L, Banda DH, Meuleman P. Animal Models for Hepatitis E virus. Viruses 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gardinali NR, Guimaraes JR, Melgaco JG, Kevorkian YB, Bottino FO, Vieira YR, et al. Cynomolgus monkeys are successfully and persistently infected with hepatitis E virus genotype 3 (HEV-3) after long-term immunosuppressive therapy. PLoS One 2017;12:e0174070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lobashevsky A, Smith JP, Kasten-Jolly J, Horton H, Knapp L, Bontrop RE, et al. Identification of DRB alleles in rhesus monkeys using polymerase chain reaction-sequence-specific primers (PCR-SSP) amplification. Tissue Antigens 1999;54:254–263. [DOI] [PubMed] [Google Scholar]
- [16].Kaizu M, Borchardt GJ, Glidden CE, Fisk DL, Loffredo JT, Watkins DI, et al. Molecular typing of major histocompatibility complex class I alleles in the Indian rhesus macaque which restrict SIV CD8+ T cell epitopes. Immunogenetics 2007;59:693–703. [DOI] [PubMed] [Google Scholar]
- [17].Dalton HR, Bendall RP, Keane FE, Tedder RS, Ijaz S. Persistent carriage of hepatitis E virus in patients with HIV infection. N Engl J Med 2009;361:1025–1027. [DOI] [PubMed] [Google Scholar]
- [18].Schmitz JE, Simon MA, Kuroda MJ, Lifton MA, Ollert MW, Vogel CW, et al. A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. The American journal of pathology 1999;154:1923–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Furchner M, Erickson AL, Allen T, Watkins DI, Sette A, Johnson PR, et al. The simian immunodeficiency virus envelope glycoprotein contains two epitopes presented by the Mamu-A*01 class I molecule. J Virol 1999;73:8035–8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou Y, Callendret B, Xu D, Brasky KM, Feng Z, Hensley LL, et al. Dominance of the CD4(+) T helper cell response during acute resolving hepatitis A virus infection. The Journal of experimental medicine 2012;209:1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yin X, Ying D, Lhomme S, Tang Z, Walker CM, Xia N, et al. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc Natl Acad Sci U S A 2018;115:4773–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Okuno T, Kondelis N. Evaluation of dithiothreitol (DTT) for inactivation of IgM antibodies. J Clin Pathol 1978;31:1152–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Okoye A, Park H, Rohankhedkar M, Coyne-Johnson L, Lum R, Walker JM, et al. Profound CD4+/CCR5+ T cell expansion is induced by CD8+ lymphocyte depletion but does not account for accelerated SIV pathogenesis. The Journal of experimental medicine 2009;206:1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2011;481:394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tangye SG, Ma CS. Regulation of the germinal center and humoral immunity by interleukin-21. The Journal of experimental medicine 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Todt D, Francois C, Anggakusuma, Behrendt P, Engelmann M, Knegendorf L, et al. Antiviral Activities of Different Interferon Types and Subtypes against Hepatitis E Virus Replication. Antimicrob Agents Chemother 2016;60:2132–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Behrendt P, Lüth S, Dammermann W, Drave S, Brown RJ, Todt D, et al. Exacerbation of hepatitis E virus infection during anti-TNFα treatment. Joint bone spine 2017;84:217–219. [DOI] [PubMed] [Google Scholar]
- [28].Yin X, Ambardekar C, Lu Y, Feng Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J Virol 2016;90:4232–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013;496:367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ankcorn M, Gallacher J, Ijaz S, Taha Y, Harvala H, Maclennan S, et al. Convalescent plasma therapy for persistent hepatitis E virus infection. J Hepatol 2019;71:434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Behrendt P, Bremer B, Todt D, Brown RJ, Heim A, Manns MP, et al. Hepatitis E Virus (HEV) ORF2 Antigen Levels Differentiate Between Acute and Chronic HEV Infection. J Infect Dis 2016;214:361–368. [DOI] [PubMed] [Google Scholar]
- [32].Soon CF, Behrendt P, Todt D, Manns MP, Wedemeyer H, Sallberg Chen M, et al. Defining virus-specific CD8+ TCR repertoires for therapeutic regeneration of T cells against chronic hepatitis E. J Hepatol 2019;71:673–684. [DOI] [PubMed] [Google Scholar]
- [33].Maldini CR, Claiborne DT, Okawa K, Chen T, Dopkin DL, Shan X, et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat Med 2020;26:1776–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.