

Abstract
The study of cell senescence is a burgeoning field. Senescent cells can modify the cellular microenvironment through the secretion of a plethora of biologically active products referred to as the senescence‐associated secretory phenotype (SASP). The consequences of these paracrine signals can be either beneficial for tissue homeostasis, if senescent cells are properly cleared and SASP activation is transient, or result in organ dysfunction, when senescent cells accumulate within the tissues and SASP activation is persistent. Several studies have provided evidence for the role of senescence and SASP in promoting age‐related diseases or driving organismal ageing. The hype about senescence has been further amplified by the fact that a group of drugs, named senolytics, have been used to successfully ameliorate the burden of age‐related diseases and increase health and life span in mice. Ablation of senescent cells in the brain prevents disease progression and improves cognition in murine models of neurodegenerative conditions. The role of senescence in cancer has been more thoroughly investigated, and it is now accepted that senescence is a double‐edged sword that can paradoxically prevent or promote tumourigenesis in a context‐dependent manner. In addition, senescence induction followed by senolytic treatment is starting to emerge as a novel therapeutic avenue that could improve current anti‐cancer therapies and reduce tumour recurrence. In this review, we discuss recent findings supporting the role of cell senescence in the pathogenesis of neurodegenerative diseases and in brain tumours. A better understanding of senescence is likely to result in the development of novel and efficacious anti‐senescence therapies against these brain pathologies.
Keywords: Alzheimer's disease, cell senescence, craniopharyngioma, diffuse midline glioma, glioblastoma multiforme, low‐grade glioma, medulloblastoma, multiple sclerosis, Parkinson's disease, SASP, senolytic
This review discusses the role of senescent cells in brain pathologies, in particular age‐associated neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease and multiple sclerosis, as well as brain tumours, specifically craniopharyngioma, low‐grade glioma, glioblastoma multiforme, medulloblastoma and diffuse midline glioma. We present evidence accumulated from in vitro and in vivo studies in both mice and humans. The translational implications of such studies will also be discussed.

INTRODUCTION
Since the initial discovery and description of senescent cells by Hayflick and Moorhead in 1961 [1], the field of senescence has evolved and expanded immensely. Hayflick originally observed that primary human fibroblast cell cultures had a finite proliferative capacity in vitro. This finite proliferative capacity is now termed replicative senescence and is due to the gradual attrition of telomeres over serial passages [2]. It was initially proposed that replicative senescence was the driver of organismal ageing due to the possible lack of cell replacement and repair [3]. A few decades later, it is now clear that senescent cells are present in many living organisms, from mice to humans and their presence can either be beneficial or detrimental depending on the biological context [4, 5 Figure 1.
FIGURE 1.
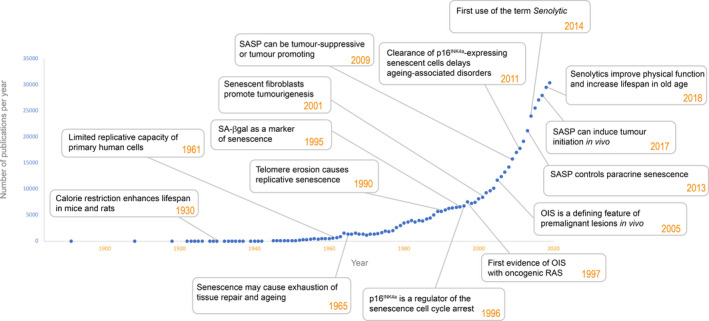
Timeline of research on senescence indicating the number of published papers and milestones
Cellular senescence is a survival programme that can be induced by a range of damaging stress signals such as radiation, chemotherapy, replicative stress and oncogenic signalling. Senescent cells are characterised by the stable and irreversible cell‐cycle arrest whilst maintaining metabolic activity and viability [4, 6. This is distinct from cellular quiescence, which is defined as a reversible proliferative arrest, such as adult stem cells, which can be stimulated to re‐enter the cell‐cycle by mitogenic signals [9]. In contrast, senescent cells do not proliferate in respond to these signals. However, they can re‐enter the cell cycle, mostly in the context of developing cancers, whereby accumulation of genetic or epigenetic alterations results in the disruption of the key molecular pathways maintaining cell‐cycle arrest [10, 11, 12, 13.
A hallmark of senescent cells is the activation of a senescence‐associated secretory phenotype (SASP), characterised by the synthesis and secretion of a plethora of biologically active molecules (e.g. inflammatory mediators, growth factors, extracellular matrix proteins) [14, 15, 16. The SASP underpins the paracrine functions of senescent cells. Senescent cells are involved in essential physiological processes such as embryonic development, immune modulation, tissue regeneration, cell plasticity and reprogramming [17, 18, 19. In these contexts, senescent cells are present only transiently to be subsequently eliminated by the immune system [20, 21. In contrast, persistence of senescent cells within tissues results in the deterioration of organ function, which can lead to disease. For instance, age‐related conditions such as osteoarthritis, atherosclerosis, fibrosis of the lungs, kidney and heart, sarcopenia, glaucoma, cataracts and type 2 diabetes are all associated with increased numbers of senescent cells [22, 23, 24. The repertoire of senescence‐associated pathologies has recently been expanded to include neurodegenerative diseases such as Alzheimer's, Parkinson's and multiple sclerosis [25, 26, 27. Moreover, in addition to their role in age‐related diseases, evidence is mounting that accumulation of senescent cells within tissues may be driving organismal ageing itself [8, 28.
Senescence and SASP play a critical role in cancer [36, 37. An early landmark study revealed that the expression of oncogenic RAS, induces premature senescence in primary cell cultures, a process now known as oncogene‐induced senescence (OIS) [38]. This has been further corroborated in different in vivo studies, where oncogenic mutations in different contexts lead to the accumulation of senescent cells [36, 39. Activation of a senescence phenotype constitutes an excellent cell autonomous barrier against development of cancer, by preventing the proliferation of cells harbouring DNA mutations Figure 2A. Cancer progression is understood to require the inactivation of cell‐cycle inhibitors (e.g. p53 and p16INK4a) resulting in senescence escape, cell‐cycle re‐entry and tumour cell proliferation [13, 44 Figure 2B. However, this view has been challenged,it has been shown that loss of p53 may not be sufficient for senescence escape, but p53‐defficient cells are able to bypass the establishment of a senescence programme when targeted to express oncogenic mutations [36, 45 Figure 2C. Beyond the cell autonomous role of senescence in cancer prevention and progression through senescence escape or bypass, the paracrine activities of senescent cells can be pro‐tumourigenic Figure 2D. Evidence from in vitro and in vivo studies has demonstrated a critical role of SASP in tumour initiation, progression to malignancy and metastasis [37, 47. In addition to these cell autonomous and non‐autonomous roles of senescent cells in cancer pathogenesis, the senescence response is relevant in the context of cancer therapy. Several standard anti‐cancer treatments such as DNA‐damaging chemotherapy, radiotherapy and even specific targeted therapies against critical pathogenic cancer drivers can trigger therapy‐induced senescence (TIS) in cancer cells or in the microenvironment [51, 52 Figure 2E. New compounds capable of selectively killing senescent cells, termed senolytics, have been identified [5]. These chemical drugs inhibit pathways that are essential for survival of senescent cells but are dispensable in non‐senescent cellular states. Exploiting these new vulnerabilities in cancer is a very exciting area of research, which is likely to lead to novel, efficacious anti‐cancer therapies. Finally, senescent cells can remodel the tumour microenvironment through the SASP creating a permissive setting that allows tumours to progress Figure 2F [37, 53.
FIGURE 2.
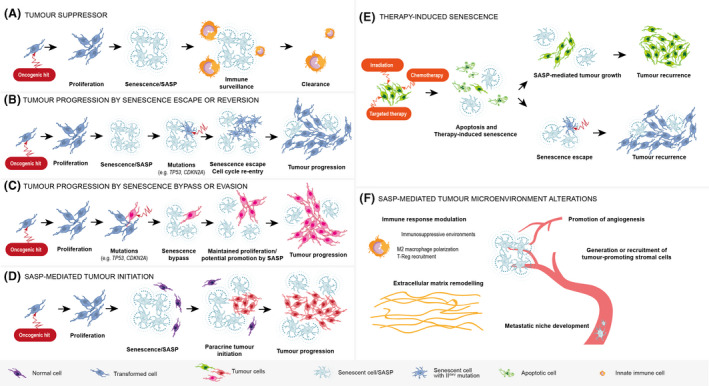
Proposed roles of senescence and SASP in tumourigenesis. (A) Tumour suppressor mechanism. Oncogenic signalling leads to transient proliferation followed by senescence induction (oncogene‐induced senescence). Senescent cells activate SASP and attract immune cells that clear them from the tissues, thus preventing subsequent tumour development. (B) Tumour progression by senescence escape or reversion. Following senescence induction, one cell accumulates further mutations (e.g. TP53, encoding p53, or CDKN2A, encoding p16INK4a) resulting in senescence escape, cell‐cycle re‐entry, proliferation and tumour formation. (C) Tumour progression by senescence bypass or evasion. Upon oncogenic signalling and initial proliferation burst, most of the cells become senescent but one cell continues proliferating due to pre‐existing mutations in key senescence regulators (e.g. p53 or p16) that prevents the establishment of a senescence response. (D) SASP‐mediated tumour initiation. Senescent cells, through the SASP, create a pro‐tumourigenic microenvironment that leads to cell transformation of a non‐tumour cell and tumour formation. (E) Therapy‐induced senescence. Following irradiation, chemotherapy and targeted therapy, most of the cells in the tumour bed are killed (e.g. by apoptosis) or induced into senescence with some cells being unaffected (green cell). Senescent cells will eventually contribute to tumour recurrence either in a paracrine manner through SASP‐mediated tumour growth, or by senescence escape or bypass. (F) SASP‐mediated tumour microenvironment alterations. Senescent cells, through the SASP, can remodel the tumour microenvironment. For instance, by (i) modulating the immune response to create immunosuppressive environment (e.g. M2 macrophage polarisation, T‐reg recruitment), (ii) driving extracellular matrix remodelling and tumour vascularisation and (iii) promoting the development of metastatic niches
In this review we will discuss the role of senescent cells in brain pathologies, in particular age‐associated neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease and multiple sclerosis, as well as brain tumours, specifically craniopharyngioma, low‐grade glioma, glioblastoma multiforme, medulloblastoma and diffuse midline glioma. We will present evidence accumulated from in vitro and in vivo studies in both mice and humans. The translational implications of such studies will also be discussed. For further reading, we recommend other reviews in the field [4, 5, 37, 54.
CELLULAR SENESCENCE AND SASP
The permanent cell‐cycle arrest of senescent cells is mediated principally by the p16INK4a/Rb and p21Cip1/p53 pathways in response to stress stimuli [56, 57, 58, 59. Expression of p53 due to cellular stress signals activates a multitude of responses, including cell‐cycle arrest, which is mediated by the p53 target p21Cip1. p16INK4a mediates cell‐cycle arrest by inhibiting CDK4/6 leading to hypo‐phosphorylation of RB and inhibition of cell‐cycle progression into S phase. p16INK4 has been termed the master regulator of cell‐cycle arrest in senescent cells [57, 58. Activation of the senescence programme leads to further molecular changes: (i) chromatin remodelling (e.g. presence of senescence‐associated chromatin foci), (ii) activation of a DNA damage response (e.g. expression of γH2AX), (iii) enlargement of the lysosomal compartment (e.g. increased expression of GLB1 and lipofuscin accumulation); (iv) macromolecular damage (e.g. telomere attrition); (v) deregulated metabolism (e.g. a shift from oxidative phosphorylation to glycolysis); (vi) anti‐apoptotic response (e.g. increased expression of BCL‐2 family proteins and inhibition of caspase 3) [4, 6. Resistance to apoptotic death is primarily mediated through the stress‐induced p53 pathway, which upregulates the expression of anti‐apoptotic BCL‐2 proteins [60, 61. Additionally, the p53‐transcriptional target p21Cip1 has been shown to be able to directly inhibit caspase 3, hence contributing to apoptotic resistance [62].
A main feature of senescent cells is the activation of the SASP [63, 64, 65. SASP factors include cytokines and chemokines (e.g. IL1A, IL1B, IL6, L8, CCL2, CCL5, CCL20 TGFβ and TNFα), growth factors (e.g. EGF, bFGF, HGF, VEGF), proteases (e.g. MMP‐1, −3, −10, −12, −13, −14), extra cellular matrix components (e.g. fibronectin, collagens, laminin) and non‐protein components such as lipids and prostaglandins [36, 63, 66. The SASP is dependent on several signalling pathways such as NFκB, p38MAPK, mTOR, NOTCH, C/EBPβ and activation of the cGAS‐STING pathway [68, 69, 70, 71, 72, 73, 74. There is no unique SASP, on the contrary the composition and intensity of the SASP depends on the cell type, the senescence‐inducing stimulus and the timing after SASP activation [75]. SASP factors reinforce the senescent phenotype by autocrine or paracrine signalling, but can also affect the microenvironment via paracrine signalling [53, 64. Senescent cells can also affect their neighbouring cells via juxtacrine NOTCH signalling or by the transfer of cellular cargo by either cytoplasmic bridges or exosome production [14]. The consequences of SASP activation can be either beneficial or detrimental depending on the biological context. For example, senescent cells in skin secrete the SASP factor PDGF‐AA upon wounding, which is important for optimal healing [76]. Furthermore, senescent cells and the expression of SASP can contribute to the dedifferentiation and reprogramming on non‐senescent adjacent cells [19]. Dysregulated and chronic SASP is detrimental and can cause age‐related diseases, organismal ageing and cancer[37].
The identification of senescence in vivo is difficult, and no single marker can unambiguously be used to define senescent cells. Initially the expression of β‐D galactosidase and its detection in a colorimetric enzymatic assay at acidic pH (SA‐β‐Gal) was used to define senescent cells. However, this staining can be unreliable in vivo. Therefore, a consensus has been published, where a combination of markers is recommended to assess cellular senescence [5] Table 1.
TABLE 1.
Detection of senescence: senescence hallmarks, markers, biological consequences and limitations
| Senescence hallmarks | Markers | Limitations | Biological consequences |
|---|---|---|---|
| Cell‐cycle withdrawal | Ki67 negativity, phopho‐histone H3 negativity, EdU exclusion | Also in quiescence | Irreversible cell‐cycle arrest |
| p21Cip1 positivity, CDKN1A upregulation | Also in quiescence | CDK2 inhibitor p21Cip1 accumulation | |
| p16INK4a positivity, CDKN2A upregulation | Express by non‐senescent cells (macrophages), not express by all senescent cells | CDK4/6 inhibitor p16INK4A accumulation | |
| p15INK4b positivity, CDK2B upregulation | Other cyclin inhibitors accumulation | ||
| Cyclin‐A2 (CCNA2) and Cyclin‐E2 (CCNE2) downregulation | Decreased expression of cyclins | ||
| Persistent activation of RB family proteins (e.g phosphorylation of RB1, p107, p130) | Also in quiescence | Stability of the senescent state | |
| Heterochromatinization of E2F target genes | |||
| Macromolecular damage | Telomere shortening | DNA Damage | |
| Persistent nuclear DNA damage foci | |||
| Expression of γ‐H2AX and PARP‐1 | |||
| Phosphorylation of DNA‐PKcs and ATM | |||
| Ubiquitin proteasome system active | Protein Damage | ||
| Autophagy | |||
| Fatty acids and free cholesterol increase/phospholipids and cholesteryl decrease | High variability of the senescence‐associated lipid profile | Lipid Damage | |
| Secretory phenotype (SASP) | NF‐κB, C/EBPβ, GATA4, mTOR and p38MAPK signaling pathways (phosphorylation of IkBa, p38,…) | High Variability: duration, cell type, inducer stimuli, and cell‐to‐cell variability | Activation of transcription factors |
| IL6; IL7; IL1; IL1B; IL13; IL15 ; TGFβ; GM‐CSE; G‐CSE; IFN‐γ; BLC; MIF | Pro‐inflammatory cytokines release | ||
| IL8; GRO‐a, ‐b, ‐g; MCP‐2; MCP‐4; MIP‐1a; MIP‐3a; HCC‐4; eotaxin; eotaxin‐3; TECK; ENA‐78; I‐309; I‐TAC | Chemokines production | ||
| Amphiregulin; epiregulin; heregulin; EGF; bFGF; HGF; KGF (FGF7); VEGF; angiogenin; SCF; CXCL12; PIGF; NGF; IGFBP2, IGFBP3, IGFBP4, IGFBP6, IGFBP7 | Growth modulators, angiogenic factors | ||
| MMP‐1, ‐3, ‐10, ‐12, ‐13, ‐14; TIMP‐1; TIMP‐2; PAI‐1, ‐2; tPA; uPA; cathepsin B | Proteases, matrix metalloproteinases | ||
| ICAM‐1, ‐3; OPG; sTNFRI; sTNFRII; TRAIL‐R3; Fas; uPAR; SGP130; EGF‐R ; Fibronectin; collagens; laminin | Secretion of other factors | ||
| Deregulated metabolism | Increase number, decreased membrane potential, increased proton leak | Less functional mitochondria | |
| PML nuclear bodies (isoform IV) | Also during apoptosis | Reactive oxygen species (ROS) production | |
| Senescence‐associated beta‐galactosidasde (SA‐β‐gal) activity | Not required for the senescent phenotype | Lysosomes increase in number and size | |
| Galactosidase, beta 1 (GLB1) upregulation | |||
| LAMP1, LAMP2, Lysozime C upregulation | |||
| Senescence‐associated epigenetic | H4K16ac, H3.3, H4K20me3 and H3K9me3 | Global increase in chromatin accessibility | |
| Senescence‐associated heterochromatin foci (SAHFs) | |||
| Global loss of linker histone H1 | |||
| Lamin B1 (LMNB1) loss and reduced nuclear integrity | |||
| Upregulation of specific miRNAs (e.g. miR‐504, miR‐605) | Change in miRNAs expression | ||
| Resistance to apoptosis | Increased expression of BCL‐2 family members | Anti‐apoptotic protein upregulation | |
| TRAIL‐Decoy Receptor DcR2 over expression | Markers not present in mice | Hiding from Immune system | |
| NKG2D ligands over expression |
ANTI‐SENESCENCE AND ANTI‐SASP STRATEGIES
After the discovery of the detrimental role that senescent cells play in ageing and in numerous pathologies, it soon became relevant to develop specific targeted strategies. The first proof‐of‐concept that the ablation of senescent cells was beneficial and reduced ageing‐associated disorders was published in 2011 [28, 77. This was followed by studies showing that the selective killing of senescent cells using chemical compounds improves organ function in ageing mice [33, 78. A non‐exhaustive list of current and promising strategies is presented in Table 2. There are four main approaches of anti‐senescence and anti‐SASP strategies currently being investigated.
TABLE 2.
Anti‐senescence and anti‐SASP strategies: target proteins, agents and clinical development status
| Candidate senotherapy | Target class/target | Agent | Class | Development status | ||
|---|---|---|---|---|---|---|
| In vitro inducer | Preclinical models | Clinical trials | ||||
| Senolytics | Pan‐kinase | Quercetin | Small inhibitor | Replicative Senescence | Phase II Chronic Kidney Disease NCT02848131 | |
| Pan‐receptor tyrosine kinase | Dasatinib | Small inhibitor | Replicative Senescence | Phase II Skeletal Health in aging NCT04313634 | ||
|
Pro‐survival proteins BCL‐2, BCL‐XL, BCL‐W |
ABT‐737 | Small inhibitor | Replicative Senescence Oncogene Induced Senescence Irradiation, Cytostatic drugs | |||
| A1155463 | Small inhibitor | |||||
| A‐1331852 | Small inhibitor | |||||
| ABT‐263 (Navitoclax) | Small inhibitor | |||||
| MDM2/p53 protein interaction | UBX0101 | Small inhibitor | Replicative Senescence | Osteoarthritis mouse model | Phase II Knee Osteoarthritis NCT04129944 | |
| P5091, P22077 | USP7 inhibitor | Replicative Senescence Irradiation, Cytostatic drugs | Doxorubicin‐treated mice | |||
| FOXO4 | FOXO4 peptide | Peptide blocking interaction with p53 | Replicative Senescence Irradiation, Cytostatic drugs | XpdTTD/TTD mice, p16‐3MR aged mice | ||
| HSP90 | Alvespimycine (17‐DMAG) | Small inhibitor, antibiotic | Replicative Senescence Irradiation, Oxidative stress | Ercc1 ‐/∆ progeroid mice | ||
| Ganetespib | Small inhibitor | |||||
| Unknown | Fisetin | Flavonoid polyphenol | Replicative Senescence | Ercc1 ‐/∆ progeroid and old INK‐ATTAC mice | Phase II Frail Elderly Syndrome NCT03675724, NCT03430037 | |
| Luteolin | Flavonoid polyphenol | Replicative Senescence | Phase I/II Osteoarthritis NCT04210986 | |||
| Phase II Chronic Kidney Diseases, Diabetes NCT03325322 | ||||||
| Phase II Skeletal Health in aging NCT04313634 | ||||||
| Na+/K+ ATPase | Ouabain | Cardiac Glycosides | Replicative Senescence Irradiation, Cytostatic drugs. | Lung adenocarcinoma, melanoma xenograft | ||
| Liver cancer, ACP mouse models | ||||||
| Digoxin | Irradiated mice | |||||
| Ageing mice | ||||||
| Mitochondria | MitoTam | Mitochondria‐targeted tamoxifen | Replicative Senescence, Cytostatic drugs | Ageing mice | ||
| Vectorisation | Encapsulation of drugs with galacto‐oligosaccharides | Mesoporous Silica Nanoparticles | Oncogene Induced Senescence, Cytostatic drugs | Melanoma xenograft | ||
| Multiple cargo: doxorubicin, navitoclax, | Triple negative breast cancer xenograft | |||||
| Fibrotic lungs mouse model | ||||||
| Galactose‐modified prodrug | Duocarmycin derivatives | Replicative Senescence, Oncogene Induced Senescence Irradiation Cytostatic drugs | ACP mouse model | |||
| Irradiated mice | ||||||
| Navitoclax | Oncogene Induced Senescence Irradiation, Cytostatic drugs | NSCLC mouse models | ||||
| SASP prouction modulators | IKK, NF‐κB and DICER | Metformin | Small inhibitor | Replicative Senescence, Oncogene Induced Senescence | Approved for Diabetes | |
| mTOR | Rapamycin | Small inhibitor | Replicative Senescence, Irradiation, Cytostatic drugs | Ageing mice | Approved immunosuppressor | |
| JAK‐STAT | Ruxolitinib | Small inhibitor | Replicative Senescence | Ageing mice | Approved immunosuppressor | |
| Glucocorticoids Receptor | Corticosterone, Cortisol | Glucocorticoids | Irradiation | Approved immunosuppressor | ||
| p38MAPK | Small inhibitor | |||||
| SASP blocking antibodies | IL1A, IL1B, IL1Receptor | Rilonacept, Canakinumab, Anakinra | Clinicaly approved immunosuppressors | |||
| TNF | Etanercept, Infliximab | |||||
| IL6, IL6R | Siltuximab, Tocilizumab | |||||
| Immunotherapies | Antibody‐dependent cell‐mediated cytotoxicity with NK cells | DPP4 antibody | Antibody | Replicative Senescence | ||
| NKG2D CAR‐T cells | ||||||
| Calories restriction mimetics | 2‐deoxy‐D‐glucose | Glucose analogue | Replicative Senescence Oncogene Induced Senescence | |||
| Resveratrol | Polyphenol | Replicative Senescence Oncogene Induced Senescence | Ageing mice | |||
Prevention of senescent cell accumulation
The chronic reduction of total calorie intake has been reported to counteract several age‐associated alterations, through molecular and physiological effects, including prevention of senescent cells accumulation [80, 81, 82. As a consequence, caloric restriction mimetics are studied in the context of ageing, particularly among them the modulation of glucose metabolism by 2‐deoxy‐D‐glucose, which has been shown to reduce degeneration of dopaminergic neurons in a Parkinson's disease mouse model [83]. Resveratrol and other polyphenols are also able to suppress the formation of reactive oxygen species (ROS) and to limit cellular senescence in neurons [84, 85. Cells treated with caloric restriction mimetics express molecular pathways similar to cells affected by long‐term calorie restriction or short‐term fasting, including the autophagy pathway. The crosstalk between autophagy and SASP production is an important element to investigate to better understand the regulation of cell senescence by these drugs.
Ablation of senescent cells: senolytics
Among the senescence hallmarks, the anti‐apoptotic programme is not only required for senescent cell survival, but also the easiest to target. Thus, the first senolytic drugs that have been reported are inhibitors of the anti‐apoptotic B cell lymphoma 2 (BCL‐2) protein family [33, 78. Two of these promising drugs, ABT‐263 and ABT‐737, have been shown to be capable of selective elimination of senescent cells and causing therapeutic benefits in several physiological and disease contexts (e.g. regeneration [86], cancer [87], type 1 diabetes [88], and atherosclerosis [89]. Other anti‐apoptotic pathways have been investigated, in particular the inhibition of the MDM2/p53 interaction (e.g. UBX0101 [90] and USP7 inhibitor [91]. In mouse models, UBX0101 is able to attenuate the development of osteoarthritis by selective clearance of senescent cells, however a phase II trial did not replicate these results (NCT04129944) [92]. Recently, high throughput drug screenings have uncovered the senolytic activity of cardiac glycosides, through a process mediated by induction of the pro‐apoptotic BCL2‐family protein NOXA [93, 94. Another class of senolytics take advantage of the high lysosomal β‐galactosidase activity of senescent cells to deliver more specifically cytotoxic drugs to senescent cells and reduce the toxic side effects [95, 96.
Making senescent cells harmless: SASP‐modulating drugs
An additional approach to interfere with the detrimental effects of senescent cells is the modulation of their secretome, either by disrupting the overall production, maturation or secretion of SASP factors, or by the selective blockade of specific components. The NF‐κB inhibitor Metformin, the mTOR inhibitor Rapamycin and the JAK/STAT inhibitor Ruxolitinib have been shown to suppress SASP activation by inhibition of critical pro‐SASP cascade signalling [71, 97. The blockade of specific SASP factors (e.g. IL1B and IL6) or their receptors has the potential to reduce off‐target effects, and anti‐inflammatory drugs inhibiting these factors have already been clinically approved (e.g. Tocilizumab against the IL6 receptor, Anakinra against IL1B). However, the redundancy and pleiotropic functions of SASP factors may make it difficult to target SASP therapeutically and no trials have yet been successfully conducted using this strategy.
Enhancing organismal anti‐senescence systems: immune clearance
Senescence activates the innate and adaptive immune responses, which result in elimination of senescent cells in physiological contexts (i.e. immune clearance). However, decline of immune system surveillance associated with ageing or immunosuppressive microenvironments results in immune evasion of senescent cells leading to tissue accumulation and subsequent deterioration of organ function [20, 99. Boosting immune surveillance through the use of biotherapeutics, such as engineered immune cells (e.g. chimeric antigen receptor T (CarT) cells or natural killer cells) has been successfully used for the treatment of human cancer [102]. Similar approaches could be effective against membrane‐bound proteins that are present in senescent cells (e.g. uPAR [103] and DPP4) [104], although toxicity may hamper the potential therapeutic use of this strategy.
More research is needed to identify safe and efficacious anti‐senescence therapies able to counteract the detrimental effects of ageing and cancer. As previously mentioned, senescent cells are highly heterogeneous and the activation of specific transcriptomic programmes is dependent on cell type, stress inducers and duration of senescent induction [75, 105. Therefore, the identification of the best anti‐senescence approach may need to be tailored to the specific cellular context, whether ageing, specific degenerative disease or cancer.
SENESCENCE IN NEURODEGENERATIVE DISEASES
Age is the most common risk factor for neurodegenerative diseases [106]. The incidence of conditions such as Alzheimer's and Parkinson's disease, which are characterised by cognitive decline and loss of neurons and synaptic connections, increases with age [107]. Age is also a risk factor for inflammatory diseases such as multiple sclerosis, which show loss of axons, dendrites, and neurons [108]. Senescent cells have been identified in different cell types of the nervous system, including neural stem cells, neurons, astrocytes, oligodendrocytes and microglia [109, 110, 111, 112, 113, 114, 115. Although neurons are characterised by permanent exit of the cell cycle, they have been shown to accumulate DNA damage and acquire additional features that typify senescence, including SASP activation [113]. These senescent cell types have been implicated in the pathogenesis of Alzheimer's disease, Parkinson's disease, multiple sclerosis, frontotemporal dementia and ischaemia/stroke. Cellular senescence may contribute to the initiation and/or progression of neurodegenerative diseases by promoting chronic inflammation, causing loss of regenerative properties and enhancing age‐related decline in the blood‐brain barrier and micro‐vasculature [116].
Alzheimer's disease
Alzheimer's disease (AD) is the most common neurogenerative disease with an incidence of 11.08 per 1000, doubling every 5 years after 65 years of age. AD affects more than 35 million people worldwide. The main pathological features that define AD are an accumulation of Aβ peptide amyloid plaques and neurofibrillary tangles (NFTs) of hyperphosphorylated tau proteins in the hippocampus and cerebral cortex [117]. Additional features include loss of neurons and synapses in the dentate gyrus of the hippocampus and cerebral cortex resulting in progressive cognitive decline and memory loss [118]. Senescent cells have been identified in both AD human samples and mouse models opening the possibility that these cells contribute to the pathogenesis. Supporting this statement, expression of p16INK4a and p53 are elevated in post‐mortem human AD samples compared with age‐matched control brains [119, 120.
Progressive loss of neurons and neural stem cells in the dentate gyrus of an ageing hippocampus may contribute to the aetiology of AD. Neural stem cell senescence could explain the loss of neural progenitor proliferation that is observed in both, mouse models of AD and premature ageing, as well as in human brains from old individuals [121, 122, 123, 124. Recent experiments in vitro have demonstrated the presence of senescent neural stem cells in AD. The formation of Aβ oligomers has been shown to induce senescence in hippocampal neural stem cells of the APP/PS1 AD mouse model [125]. In this study, Aβ fibrils can accelerate neural stem cell senescence via activation of the MAPK pathway, ultimately leading to loss of neurogenesis.
Higher levels of pro‐inflammatory SASP factors has been reported in aged human and mouse brains compared with younger controls [126]. Inflammation is a key feature which contributes to the initiation, severity and progression of most neurodegenerative diseases including AD [127]. Expression of SASP factors, e.g. IL6, IL1B, TGFβ, TNFα and MMP‐1, −3 and −10 and activation of the p38MAPK pathway are upregulated in human AD samples and murine models [72, 128.
Microglia, the resident macrophages of the central nervous system (CNS) whose functions are tightly regulated by their microenvironment, can secrete SASP factors [133]. Ageing or neurodegenerative accumulation of misfolded protein induces microglia proliferation and promotes an activated state. This state is known as microglia priming and initiates the reactive defence program characterised by phagocytosis and increased release of cytokines, tumour necrosis factor (TNF) and nitric oxide [134]. Primed microglia are also prone to be stimulated by secondary sources of inflammation, triggering an exaggerated and chronic inflammatory response in the CNS [135]. Both aged and AD brain samples show microglia priming and an increase of their pro‐inflammatory response [134].
Ex vivo and in vitro studies have revealed that aged microglia secrete higher levels of SASP factors such as IL6 and TNFα compared with young microglia. Aged microglia lose their ability to phagocytose Aβ fibrils and undergo replicative senescence due to telomere shortening [110, 136. Evidence linking age‐related senescence and AD pathogenesis has been provided by a study in which aged rat microglia were isolated and treated with Aβ oligomers in vitro. Upon treatment, these activated microglia become senescent, shown by SA‐βgal staining and production of IL1B, TNFα and MMP2 [137]. This suggests that age‐related senescence in AD microglia may play a role in disease progression.
Primed microglia and neuroinflammation are considered to play key roles in the initiation and progression of AD. An increase in numbers of primed microglia correlates with AD disease progression in humans [138], however, the mechanisms by which these cells could detrimentally affect AD pathogenesis are not yet fully elucidated. Primed microglia release IL1B and IL18 [139], and in a study on human AD, it has been shown that IL1B induces the secretion of TNFα, promoting the formation of amyloid plaques [140].
Another heterogenous cell population implicated in AD are astrocytes [141]. These cells have diverse homeostatic roles in the CNS including neurotransmitter uptake/recycling, synaptic activity, maintenance of the blood brain barrier and inflammation [142]. Single cell sequencing from wild type and AD mouse models has identified disease‐specific astrocytes that are apparent before the onset of neurological phenotypes and are increased with disease progression [138]. These astrocytes express an inflammatory and neurotoxic gene profile that is analogous to that observed in aged wild‐type astrocytes (i.e. upregulation of genes involved in development and differentiation, metabolic pathways of lipid and cholesterol, response to toxic compounds and inflammatory signalling, including NfκB signalling and ROS). Furthermore, these upregulated genes have been identified in aged human brain samples from AD post‐mortem samples, confirming previous studies in which overexpression of IL6 in murine astrocytes results in the appearance of AD‐like neurological symptoms [143] and in the formation of amyloid plaques that are similar to those observed in human AD patients [144, 145. These studies provide evidence that neuroinflammation, caused by the secretion of chemokines and cytokines commonly found in the SASP, contributes to the initiation and progression of AD.
Senolytic therapy in AD
A recent report has identified senescent oligodendrocyte precursors (OPCs) (a subset of glial cells in the brain) in human AD and in the APP/PS1 AD murine model [26]. These senescent cells are associated with Aβ plaques in both species and the study suggests that Aβ fibrils trigger OPC senescence. The senescent cells in vivo are positive for SA‐β gal staining, co‐express OPC markers together with p16INK4a and p21Cip1, and show increase levels of CDKN2A (encoding p16INK4a) mRNA. Acute oral administration of Dasatinib and Quercetin (a senolytic drug combination) selectively kills senescent OPCs in the Aβ plaques, consequently reducing IL6 levels. The selective killing of these senescent cells not only reduces neuroinflammation but decreases Aβ loads and ameliorates the AD cognitive defects [26]. A phase II clinical trial has been initiated in patients with AD using this senolytic combination [146].
In a study using micro‐dissected post‐mortem human AD, a senescent transcriptomic profile has been identified in neurons containing neurofibrillary tangles (NFTs) of aggregated tau protein [147]. NFT‐accumulating neurons in different AD murine models display a senescent phenotype, evidenced by expression of CDKN2A mRNA. Treatment with Dasatinib and Quercetin can kill these senescent cells resulting in reduction of both NFT density and neuronal loss.
Collectively, these findings indicate a strong association between the presence of cellular senescence in the brain and neurodegeneration, which is supported by mechanistic studies in murine models. Potential therapeutic avenues that selectively kill senescent cells could revolutionise AD treatments.
Parkinson's disease
Parkinson's disease is the second most common neurodegenerative disease after AD, affecting about 7–10 million people worldwide over the age of 65. It is characterised by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta of the midbrain, leading to progressive motor degeneration. A key pathological feature is the presence of Lewy bodies, composed of aggregates of α‐synuclein, a protein involved in DNA damage repair [148]. PD symptoms manifest once 80% of the dopaminergic neurons are lost [149]. Currently there are no chemical treatments that can prevent disease progression.
Evidence of senescence in PD has been shown in various studies. Higher expression levels of p16INK4a, p21Cip1 and inflammatory markers (such as IL6) have been identified in PD patients compared with healthy controls. Increased expression of these factors is associated with faster cognitive decline in the patients [27]. A recent study has revealed that a DNA binding protein, STAB1, which is associated with PD, prevents cellular senescence in dopaminergic neurons in vivo [150]. Loss of STAB1 in human stem cell‐derived dopaminergic neurons causes activation of a senescence programme in vitro. Inhibition of STAB1 in human PD brain slice cultures or loss of STAB1 in mice also activates a senescence programme. STAB1 directly represses CDKN1A (encoding p21Cip1) in DA neurons and activates a senescence and SASP programme. Expression levels of p21Cip1 in DA neurons in the substantia nigra are elevated compared with age‐matched controls. Increased p21Cip1 protein levels, potentially suggesting senescence, have also been observed in neural stem cells in the Parkin‐deficient (Prkn −/−) PD mouse model. Parkin is an E3 ubiquitin ligase needed for targeted protein degradation, which is essential for neurogenesis. Protein ubiquitination plays a key role in neural stem cell renewal and differentiation and impaired neurogenesis is found in PD [151].
A common feature of PD is the presence of activated microglia and astrocytes, which could contribute to chronic neuroinflammation. Elevated levels of inflammatory factors have been detected in the cerebrospinal fluid (e.g. IL1B) [152] as well as in DA neurons (e.g. IL1B and IL6) [153] in PD patients. Post‐mortem examination of PD samples has revealed the expression of senescence and SASP markers such as p16INK4a, IL6, IL1A, IL8 and MMP3. Dysfunctional lysosomes and increased SA‐βgal staining has also been identified in human PD samples [154, 155.
Senescent astrocytes have been observed in both human PD samples and a PD murine model [115]. Sporadic PD in humans has been associated with exposure to the herbicide paraquat (PQ), and PQ administration to mice is sufficient to induce PD‐like phenotypes. Human astrocytes cultured in vitro with PQ show positive SA‐βgal staining and reduced proliferation. Conditioned medium from these senescent astrocytes reduces human DA neurone viability and decreases neural stem cell proliferation [115]. The genetic ablation of p16INK4a‐expressing senescent cells in the context of a PQ‐induced PD mouse model is sufficient to abrogate PD‐associated motor deficits and neuropathology. Together, there is evidence supporting the presence of disease‐relevant senescent cells with an activated SASP in human and murine PD, providing a rationale for the therapeutic targeting of these cells.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease causing severe physical incapacitation and neurological damage, affecting around 2.5 million people worldwide [156]. The debilitating causes of MS are due to CNS demyelination and neurodegeneration with limited remyelination. Infiltrating macrophages and lymphocytes cause multifocal inflammation and oligodendrocyte cell death, which lead to demyelination, neuronal and axonal loss, and generation of CNS plaques that contain inflammatory cells and demyelinated axons. The aetiology of MS remains unknown, but certain genetic and environmental factors might influence the likelihood of developing MS [108]. Several immunosuppressive and immunomodulatory treatments are available,however, disease progression is still common.
The presence of senescent cells with activated SASP has been observed in mouse models and human MS samples. Using a gliotoxin‐induced demyelination MS murine model, it has been reported that aged mice show slower rates of remyelination than younger mice, suggesting that age‐related senescence could play a role in the onset and progression of this disease [157]. In another mouse model, in which demyelination is induced by feeding the mice cuprizone, increased numbers of SA‐βgal‐positive glial cells have been identified in demyelinating fibres of the corpus callosum [158]. In comparison with age‐matched control tissue, demyelinated human MS lesions show increased numbers of SOX2+ cells (a marker of neural progenitors) co‐expressing p16INK4a, suggesting the presence of senescent progenitor cells in progressive MS [159]. This finding has been corroborated in in vitro differentiation experiments of iPSC lines derived from either MS patients or age‐matched controls. For instance, expression of senescent markers (e.g. SA‐βgal staining, p16INK4a and p53) is elevated in iPSC‐derived neuronal progenitor cells (NPCs) from MS patients relative to healthy controls. Interestingly, the inhibition of the mTOR pathway with rapamycin reverses the senescent phenotype and results in reduced SA‐βgal staining and p16INK4a expression levels in iPSC‐derived NPC from MS patients. Further evidence that SASP activities may be involved in MS comes from in vitro studies assessing the capacity of senescent NPCs to promote differentiation of oligodendrocyte progenitor cells (OPCs) into myelinating oligodendrocytes (MOs). Conditioned medium from healthy control‐derived NPCs can induce differentiation of OPCs into MOs in vitro, whilst MS‐patient‐derived NPCs conditioned medium fails to induce differentiation. However, rapamycin treatment of NPCs from MS patient‐derived NPCs yields conditioned medium with comparable OPC to MO differentiation potential to that of healthy control‐derived NPCs. These findings suggest that paracrine signals from MS‐patient‐derived NPCs inhibit OPC to MO differentiation, which can be reversed by inhibition of mTOR, a critical pathway maintaining SASP activation. Proteomic analysis of conditioned medium from MS‐patient‐derived NPCs has shown the presence of secreted proteins previously associated with cellular senescence, such as heat shock proteins 90 and 60, DJ‐1, and HMGB1 and a similar molecular profile to aged NPCs derived from healthy controls.
Currently there is no cure for MS, treatments so far rely on immunomodulation. However, the identification of senescent cells and SASP factors in MS patients and murine models supports the possibility that senescence may play a role in the pathogenesis of MS, hence providing a rationale for the specific targeting of these cells using senolytics.
SENESCENCE IN BRAIN TUMOURS
Senescent cells play an important role in tumourigenesis and can act as a double‐edged sword. On one hand, senescence limits proliferation of cells bearing DNA damage in a cell autonomous manner, thus preventing tumour progression, which can occur only by senescence bypass or escape. Paradoxically, senescent cells through the SASP can generate a pro‐tumourigenic microenvironment that fuels tumour initiation and progression, including senescence escape or bypass. Additionally, standard anti‐cancer treatments (e.g. chemo‐, radio‐ and targeted therapies) can effectively trigger therapy‐induced senescence (TIS) to create new vulnerabilities in tumour cells that could be exploited using senolytics and/or SASP modulators. Here, we will discuss the role of senescent cells in brain tumours.
Craniopharyngiomas
Craniopharyngiomas (CPs) are benign epithelial tumours (WHO grade 1) of the sellar region (an anatomical region between the hypothalamus and the cranial base where the pituitary gland is located). There are two types: (i) adamantinomatous (ACP), which carry mutations in CTNNB1 (encoding β‐catenin) resulting in the activation of the WNT/β‐catenin pathway; (ii) papillary (PCP), associated with mutations in BRAF‐V600E leading to the activation of the MAPK/ERK pathway. Although these tumours are associated with high survival (over 90% 5 years survival), they cause significant morbidity and poor quality of life for the patients, in particular ACPs due to their tendency to invade the hypothalamus and optic chiasm [160, 161.
Senescence has not been characterised in PCP; however, a bulk of research has demonstrated the existence of senescent cells in human ACP. Despite the presence of CTNNB1‐activating mutations in all the tumour cells, [162] accumulation of nucleocytoplasmic β‐catenin and activation of the WNT pathway occurs in sporadic cells, most of which form groups of cells referred to as clusters. These cell clusters are not present in PCP or any other pituitary tumours. Immunohistochemistry has demonstrated that these cluster cells show the hallmarks of senescence: they are Ki‐67‐ve, express cell‐cycle inhibitors (e.g. p21Cip1), show DNA damage (e.g. γ‐H2aX staining), activate the DNA damage response (e.g. phospho‐DNA‐PKc staining), exhibit enlargement of the lysosomal compartment (e.g. GLB1 expression) and turn on the NFκB pathway (phospho‐IκB staining) [48, 163. Laser capture microdissection followed by transcriptomic analysis has confirmed that human cluster cells are senescent and activate a SASP resulting in the expression of numerous inflammatory mediators and growth factors [48, 162, 164. The location of the clusters within the finger‐like protrusion invading the brain strongly suggests that the paracrine activities of the cluster cells may play a role in tumour epithelium remodelling, proliferation and invasion [165] Figure 3.
FIGURE 3.
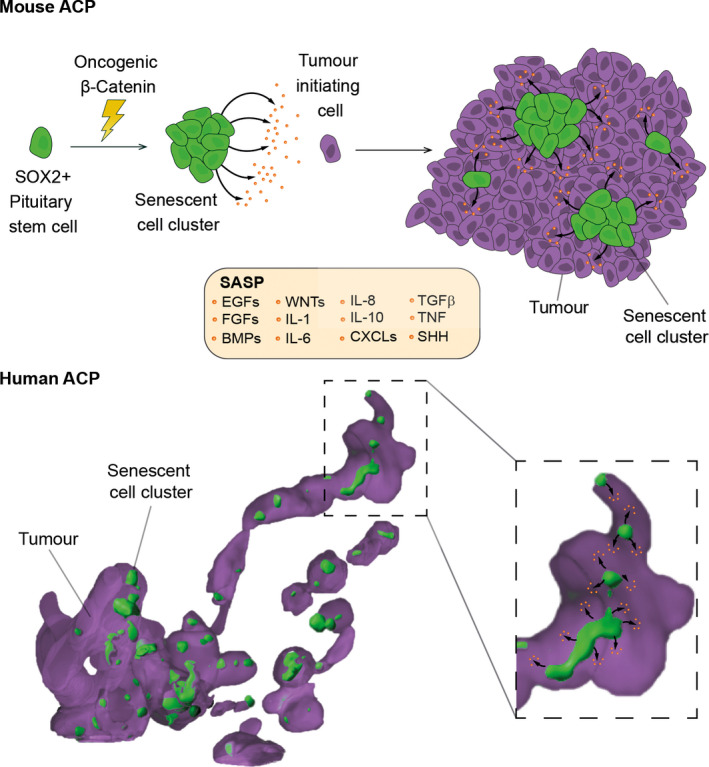
Schematic showing a working model for the role of the β‐catenin‐accumulating cell clusters in mouse and human ACP. (A) Expression of oncogenic β‐catenin in SOX2+ pituitary stem cells (both embryonic and postnatal) results in the formation of β‐catenin‐accumulating cell clusters, which contain senescent cells (oncogene‐induced senescence). Senescent cluster cells activate a senescence‐associated secretory phenotype (SASP), which leads to the synthesis and secretion of a plethora of active peptides, some of which are included in the box. The persistent activity of the SASP factors on surrounding cells eventually causes cell transformation of a cell not of the SOX2 cell lineage (purple cell) and subsequent tumour development in a paracrine manner. (B). The human tumour depicted in the schematic derives from a three‐dimensional reconstruction of a micro‐CT‐imaged human ACP sample, in which the glial reactive tissue has not been rendered. Purple indicates the stellate reticulum and cells of the palisading epithelium, and green represents the β‐catenin‐accumulating cell clusters. Note the presence of finger‐like protrusions of tumour cells, which project away from a tumour epithelium mass, containing a string of clusters inside. These human clusters are molecularly analogous to the mouse clusters and share a signature of senescence and SASP. The model proposes that the SASP activities underlie tumour growth and invasive behaviour by promoting epithelial remodelling and proliferation. Reproduced with permission from S. Karger AG, Basel (Martinez‐Barbera J. P. and Andoniadou C. L., Biological Behaviour of Craniopharyngiomas. Neuroendocrinology 2020. https://doi.org/10.1159/000506904)
These findings in human ACP have been confirmed in ACP mouse models. Genetically modified mouse models of ACP have been generated by expression of a functionally equivalent mutant form of β‐catenin in SOX2+ve stem cells of the pituitary gland during embryonic development and adulthood [166, 167. Like human ACP, the mouse tumours contain cell clusters expressing senescence markers. Interestingly, genetic tracing in the ACP mouse models has revealed that the clusters derive from SOX2+ve stem cells expressing oncogenic β‐catenin. In contrast, the tumours are derived from a different cell lineage and do not express oncogenic β‐catenin [167]. This initial study led to propose a paracrine model of tumourigenesis, whereby oncogenic SOX2+ stem cells give rise to senescent clusters that induce tumour formation in a cell non‐autonomous manner [167, 168. More recently, a mechanism for this paracrine model has been postulated Figure 3. Gene profiling has revealed that mouse and human clusters are molecularly analogous and share a signature of senescence with activated SASP [48, 163. In agreement, cluster cells are sensitive to several senolytic agents [48, 95. Providing functional relevance, the attenuation of the senescence/SASP response in mice has been shown to result in a significant reduction in the tumour‐inducing potential of the cluster cells [48].
Together, this research has clearly demonstrated the presence of senescent cells in mouse and human ACP. A model is starting to emerge in which senescent cluster cells through the SASP play a critical role in tumour initiation in mouse ACP and tumour invasion in human ACP.
Low‐grade gliomas
Low‐grade gliomas (LGGs) are a diverse group of benign brain tumours (WHO grade I and II). Symptoms are variable and largely attributable to mass effect from invasion into surrounding parenchyma (such as seizures, headache, cognitive or behavioural changes). LGGs are characterised by slow growth without invasive properties and generally low Ki‐67 proliferative index. Clinical management includes surgical resection if possible, radiation, chemotherapy and specific targeted therapies [169, 170, 171.
Senescence has been more thoroughly investigated in paediatric than adult LGG. Pilocytic astrocytoma (PA; WHO grade I) is the most prevalent paediatric LGG and the most frequent paediatric brain tumour in children. Constitutive activation of MAPK pathway, by genetic mutations, is detectable in nearly all cases [172], which leads to oncogene‐induced senescence, as shown by β‐galactosidase activity and induction of p16INK4a expression in up to 90% of primary PA samples [173]. SASP factors (e.g. IL1B and IL6) are found to be upregulated in primary human tumours as well as in a PA mouse model [174]. SASP expression in PA tumours is associated with favourable prognosis whereas anti‐inflammatory treatment with dexamethasone inhibits the SASP and induces regrowth of senescent cells. These results highlight the importance of paracrine propagation and maintenance of senescence in paediatric LGG. Of relevance, senescent PA cells can be ablated using senolytics (i.e. ABT‐263 and ABT‐737), paving the way to a new type of treatment for these patients.
Homozygous deletion of CDKN2A (encoding p16INK4a) can be observed with low frequency in paediatric LGG [175], but is more common in higher‐grade tumours, such as pleomorphic xanthoastrocytoma and anaplastic astrocytoma with piloid features, suggesting that it probably acts as a second oncogenic hit, promoting senescence escape and facilitating transformation into high‐grade glioma [176, 177. Secondary alterations involving homozygous or hemizygous losses of CDKN2A and TP53 are more characteristic in adult LGG. Increased survival has been associated with absence of mutations in CDKN2A and TP53, suggesting that senescence escape may promote tumour progression [178, 179.
Together, this research area has highlighted the presence of a large number of senescent cells in LGG. These cells through the SASP seem to play a critical role in tumour control, preventing the progression of the tumour to a more aggressive cancer. However, senescent cells in LGG are susceptible to a second oncogenic hit, promoting senescence escape and tumour progression.
Glioblastoma multiforme
Glioblastoma multiforme (GBM; WHO grade IV) is one of the most common and aggressive primary brain tumours accounting for 60% of brain tumours in adults. They are highly infiltrative and have an average survival of less than 25% after two years due to the high recurrence rate [180]. GBM tumours contain heterogenous populations of cells characterised by various different genetic aberrations with a tendency to occur in any location in the brain. GBM tumours commonly show inhibition of the p53 and RB signalling pathways, or activation of RAS, PI3K and receptor tyrosine kinase pathways. Current standard of care therapy consists of surgical resection, adjuvant chemo/radiotherapy and administration of Temozolomide (TMZ) [181]. Even with this radical treatment regime, progression and recurrence rates are high and no other chemical treatments have shown great promise.
Evidence of therapy‐induced senescence in GBM has been shown following TMZ treatment and radiotherapy [182, 183. Culture of GBM cell lines in the presence of TMZ induces senescence through a DNA damage response pathway and expression of p21Cip1. Subsequently, the NF‐κB pathway is activated, accompanied by the production of the SASP components IL6 and IL8 [183, 184. Confirming the in vitro data, orthotopic transplantation of GBM cell lines into immunodeficient mice followed by oral administration of TMZ, leads to a senescence response evidenced by p21Cip1 expression and NF‐κB pathway activation in the tumour.
It is thought that radiotherapy in GBM leads to increased recurrence rates due to the induction of a tumour‐promoting microenvironment [182, 185. The DNA damage caused by irradiation results in the induction of senescence and SASP in both tumour cells and/or non‐tumour cells in the microenvironment, which as previously discussed can be pro‐tumourigenic and lead to recurrence [7, 15, 52. It has been shown that irradiation of GBM primary cells results in cellular senescence and SASP in vitro, as shown by morphological cellular changes, positive SA‐βgal staining, cell‐cycle arrest and p21Cip1 expression [51]. Upon irradiation, SASP factors such as IL6, IL1A and IL1B are induced and the NfκB pathway is activated. Furthermore, co‐injection of irradiated, senescent primary GBM cells with non‐irradiated GBM cells results in larger more aggressive tumours compared with the injection of only non‐irradiated cells in xenograft mouse models [188].
A recent study has demonstrated that GBM cell lines can be driven into senescence, by either TMZ treatment or irradiation, to subsequently be selectively ablated with Navitoclax (ABT‐263) as a senolytic [189]. Since the induction of senescence and SASP, caused by TMZ treatment or radiotherapy, can cause GBM recurrence, i.e. by creating a pro‐tumourigenic microenvironment favouring senescent escape or bypass, the ablation of GBM senescent cells could be expected to reduce tumour relapse and improve survival of the patients.
Medulloblastoma
Medulloblastoma (MB; WHO grade IV)) is an embryonal tumour of the cerebellum originating from different neuronal progenitor cell populations. MB most commonly affects children and young adults with an average age of diagnosis of 6–8 years. MB is the most common high‐grade paediatric brain tumour. Gene expression analysis has subdivided this tumour into four major subgroups: WNT, SHH, Group 3 and Group 4 (Groups 3 and 4 have recently been subdivided into eight different types) [190, 191. These groups differ not only in their gene expression but also their methylation patterns, histology, clinical characteristics, metastatic potential, incidence and rate of recurrence. Despite the extensive clinical treatment stratification, outcomes of therapy can still be poor due to recurrence [192].
An initial study using a mouse model of SHH (sonic hedgehog) MB has identified p16INK4a and p21Cip1 expressing senescent cells in the pre‐neoplastic MB lesions [46]. These senescent cells are not detectable in advanced tumours, which are characterised by the presence of spontaneous p53 mutations, suggesting that senescence escape underlies tumour progression. This research has also shown that human SHH MB samples exhibit CDKN2A (encoding p16INK4a) promoter methylation, supporting a senescence evasion mechanism. Additional evidence of senescence has been proposed from in vitro studies using the cell lines DAOY and ONS‐76 [193]. Knockdown of citron kinase protein (CITK), which is required for normal proliferation and survival of neural progenitors, induces senescence and apoptosis via p53. In an MB mouse model, CITK deletion results in decreased tumour growth and increased overall survival, which is associated with increased expression of senescence markers such as p21Cip1, p27Kip1 and p16INK4a in the tumours. There is a need to understand better the role of senescence in MB, not only as a tumour‐suppressive mechanism, but also the contribution of senescent cell‐mediated paracrine signalling to tumourigenesis.
Diffuse Midline Glioma
Diffuse Midline Glioma (DMG) represents an incurable Grade IV group of paediatric tumours and accounts for 10% of all brain tumours in children. Found in the brainstem and midline structures including the thalamus, DMG are characterised by carrying mutations in histone‐encoding genes, i.e. histone H3 gene H3F3A (encoding H3.3) or in the related HIST1H3B (encoding H3.1) gene, often associated with loss of TP53 [194]. Expression of senescence markers, such as p16INK4a, is very low in this tumour type, and this is probably due to the oncogenic driver mutations’ ability to represses the CDKN2A locus [195]. In contrast to the tumour cells, p16INK4a+ve cells are often found in the tumour microenvironment (up to 80% of tumours [196], suggesting that these potentially senescent cells could have a role in tumourigenesis and/or treatment resistance.
Conventional clinical management by radiotherapy or new targeted therapies could be used to trigger TIS in DMG tumours, as suggested by in vitro studies on patient‐derived DMG cell lines. The combination of radiation and the mTOR inhibitor AZD2014 has been shown to result in a strong synergistic antitumour activity preclinically [197], suggesting that the use of senolytics or SASP modulators could be of therapeutic relevance. Likewise, a recent study has proposed a new model where senescence is induced in DMG tumour cells by inhibition of BMI1. In vivo, the clearance of these treatment‐induced senescent cells with ABT‐263 attenuates tumour growth and prolongs animal survival [198].
CONCLUDING REMARKS AND PERSPECTIVES
There is sufficient evidence to support the idea that senescent cells play a critical role in the pathogenesis of neurodegenerative conditions and brain tumours. The ablation experiments using genetic and chemical approaches have fuelled the interest in anti‐senescence therapies as potential treatments against these pathologies. However, several questions still remain that should be addressed to support further the development of senotherapies.
The function of senescent cells is highly context‐dependent. This has been better demonstrated in the cancer field, whereby senescent cells can be either anti‐ or pro‐ tumourigenic. This raises the possibility that senescent cells may also elicit beneficial or detrimental functions not just in cancer but also in other disease contexts. For instance, although a body of research has demonstrated the beneficial effects of ablating senescent cells with senolytics (e.g. by clearing organ‐resident senescent macrophages), a recent study using a new mouse model has revealed that the genetic ablation of vascular endothelial senescent cells in liver sinusoids, which express high levels of p16INK4a, results in premature death due to hepatic dysfunction [199]. This study highlights that the balance between the beneficial and detrimental functions of senescence must be thoroughly understood.
The ablation experiments in neurodegeneration mouse models suggest that senescent cells are not just bystanders, but they contribute to disease progression and cognitive loss. It will be interesting to assess whether such a role is preserved in humans. Another cerebral disease highly associated with old age is ischaemia/stroke and aneurysms [200]. During the acute phase of ischaemia in humans, pathogenic processes such as neuroinflammation (cytokines and chemokines) and oxidative stress have been shown to be upregulated. Furthermore, aged murine models have demonstrated a higher inflammatory response during the acute phase of ischaemia, which results in increased cerebral injury compared to young animals [200]. It will be important to define whether senotherapies are only able to prevent disease progression, or in addition, senolytics can improve cognitive decline and restore brain function in patients with advanced disease. These questions can be addressed in human trials, as those already running to test the efficacy of senotherapies against other human conditions.
Senescence is postulated to be a cell autonomous barrier against cancer that maintains potential cancer‐initiating cells in a benign, non‐proliferative state. Only through senescence bypass or senescence escape, caused by genetic or epigenetic alterations, can those benign lesion progress to give rise to malignant tumours. This model of cancer is supported mostly by in vitro studies and the fact that senescent cells are usually abundant in benign tumours while rare in malignant cancers [39, 40, 41, 43. Further studies using mouse models capable of genetic tracing the fate of senescent cells may clarify whether this model is universal and provide mechanistic insights. This is particularly important in view of data suggesting that senescent cells through paracrine signalling can, not only promote tumour progression to malignancy and metastasis, but also initiate tumour formation in a cell non‐autonomous manner [37].
One of the main problems with current anti‐cancer therapies is tumour recurrence. It is thought that senescent cells within the tumour bed are therapy‐resistant and will eventually re‐enter the cell cycle and give rise to a relapsed tumour. It has been shown that passing through a senescent state, even if transiently, can bestow features of stemness upon tumour cells making them more aggressive and malignant [12]. Therefore, there is a strong rationale to use senotherapies as adjuvant treatments to eliminate senescent cells prior to tumour recurrence. This is a promising approach, whereby current effective senescence‐inducing treatments, such as cytostatic chemotherapy, radiotherapy or specific targeted therapies, could be combined with senolytics in order to ablate the senescent cells prior to senescence escape and progression to recurrence. Preclinical research using suitable models of brain tumours will facilitate the development of clinical trials to test these combination therapies.
Despite the clear advantages of selectively eliminating senescent cells in mouse models in a variety of different human conditions, challenges remain to be addressed before senotherapies can be safely applied in clinical practice. Possibly, the most important one is to fully understand the mechanism involved in ‘good’ vs ‘bad’ senescence (i.e. beneficial vs detrimental effects). Such understanding requires a better characterisation of the different senescent cell populations within different organs in both normal physiology and disease contexts (e.g. specific diseases, ageing or cancer). This knowledge will provide a rationale for the use of senotherapies against certain human conditions and inform on the potential side effects, senolytic dosing regime, length of treatment and other important factors when designing clinical trials. Single cell profiling approaches and mouse models specifically designed to study senescence in vivo are ideal strategies to further our knowledge on the heterogeneity of senescent cells and reveal their functions. An additional problem is that there are very few drugs with proven senolytic activity and, in most cases, the mechanisms underlying such function remain poorly elucidated. There is an urgent need to discover novel senolytics and characterise their mechanisms of action through well‐designed senolytic screens.
We wonder whether Hayflick thought that his initial observations would ever become the catalyst that fuelled a vast research field, which potentially could improve clinical outcomes for the most relevant human diseases or even prolong a healthy life span. Despite current limitations and unknowns, it is difficult to not be affected by an encouraging optimism towards the translational potential of anti‐senescence therapies against brain pathologies and cancer. Future research will reveal key mechanistic insights into how senescent cells contribute to human disease paving the path to novel anti‐senescence treatments.
ETHICS STATEMENT
Ethics approval was not required since this paper does not concern animal experimentation or the use of human volunteers.
CONFLICT OF INTEREST
G.C. and R.G. have no conflicts of interest to declare. J.P.M.‐B. is an executive editor of Neuropathology and Applied Neurobiology. The Editors of Neuropathology and Applied Neurobiology are committed to peer‐review integrity and upholding the highest standards of review. As such, this article was peer‐reviewed by independent, anonymous expert referees and J.P.M.‐B. had no role in either the editorial decision or the handling of the paper.
AUTHOR CONTRIBUTIONS
This paper was written, reviewed and approved by all three authors.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12689.
ACKNOWLEDGEMENTS
The authors wish to thank Prof. T. Jacques and Prof. Rick Livesey for their comments and suggestions. We also thank Dr J. Grey for his feedback.
Funding Information
J.P.M.‐B. laboratory is funded by Cancer Research UK, Children's Cancer and Leukaemia Group, Children with Cancer UK, The Brain Tumour Charity (SIGNAL and EVEREST), Great Ormond Street Hospital Children's Charity, the Morgan Adams Foundation and National Institute of Health Research Biomedical Research Centre at the Great Ormond Street Hospital for Children NHS Foundation Trust and the University College London. J.P.M.‐B. is a Great Ormond Street Hospital for Children's Charity Principal Investigator.
Contributor Information
Gabriela Carreno, Email: g.carreno@ucl.ac.uk.
Romain Guiho, Email: r.guiho@ucl.ac.uk.
Juan Pedro Martinez‐Barbera, Email: j.martinez-barbera@ucl.ac.uk.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during this study.
REFERENCES
- 1. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25(3):585‐621. 10.1016/0014-4827(61)90192-6 [DOI] [PubMed] [Google Scholar]
- 2. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019;20(5):299‐309. 10.1038/s41576-019-0099-1 [DOI] [PubMed] [Google Scholar]
- 3. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37(3):614‐636. 10.1016/0014-4827(65)90211-9 [DOI] [PubMed] [Google Scholar]
- 4. Hernandez‐Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28(6):436‐453. 10.1016/j.tcb.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 5. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813‐827. 10.1016/j.cell.2019.10.005 [DOI] [PubMed] [Google Scholar]
- 6. Muñoz‐Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482‐496. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 7. He S, Sharpless NE. Senescence in health and disease. Cell. 2017; 169(6):1000‐1011. 10.1016/j.cell.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192(4):547‐556. 10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mohammad K, Dakik P, Medkour Y, Mitrofanova D, Titorenko VI. Quiescence entry, maintenance, and exit in adult stem cells. Int J Mol Sci. 2019;20(9):2158. 10.3390/ijms20092158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature. 2006;444(7119):638‐642. 10.1038/nature05327 [DOI] [PubMed] [Google Scholar]
- 11. Kuilman T, Michaloglou C, Vredeveld LCW, et al. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell. 2008;133(6):1019‐1031. 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- 12. Milanovic M, Fan DNY, Belenki D, et al. Senescence‐associated reprogramming promotes cancer stemness. Nature. 2018; 553(7686):96‐100. 10.1038/nature25167 [DOI] [PubMed] [Google Scholar]
- 13. Saleh T, Tyutyunyk‐Massey L, Gewirtz DA. Tumor cell escape from therapy‐induced senescence as a model of disease recurrence after dormancy. Cancer Res. 2019; 79(6):1044‐1046. 10.1158/0008-5472.CAN-18-3437 [DOI] [PubMed] [Google Scholar]
- 14. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128(4):1238‐1246. 10.1172/JCI95148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coppé J‐P, Desprez P‐Y, Krtolica A, Campisi J. The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol Mech Dis. 2010;5(1):99‐118. 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966‐972. 10.1172/JCI64098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muñoz‐Espín D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104‐1118. 10.1016/j.cell.2013.10.019 [DOI] [PubMed] [Google Scholar]
- 18. Lujambio A, Akkari L, Simon J, et al. Non‐cell‐autonomous tumor suppression by p53. Cell. 2013;153(2):449‐460. 10.1016/j.cell.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mosteiro L, Pantoja C, de Martino A, Serrano M. Senescence promotes in vivo reprogramming through p16 INK4a and IL‐6. Aging Cell. 2018;17(2). 10.1111/acel.12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ovadya Y, Landsberger T, Leins H, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun. 2018; 9(1):1‐15. 10.1038/s41467-018-07825-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kale A, Sharma A, Stolzing A, et al. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing. 2020;17(1). 10.1186/s12979-020-00187-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pignolo RJ, Passos JF, Khosla S, Tchkonia T, Kirkland JL. Reducing senescent cell burden in aging and disease. Trends Mol Med. 2020;26(7):630‐638. 10.1016/j.molmed.2020.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. 2020;16(5):263‐275. 10.1038/s41574-020-0335-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paez‐Ribes M, González‐Gualda E, Doherty GJ, Muñoz‐Espín D. Targeting senescent cells in translational medicine. EMBO Mol Med. 2019;11(12). 10.15252/emmm.201810234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oost W, Talma N, Meilof JF, Laman JD. Targeting senescence to delay progression of multiple sclerosis. J Mol Med. 2018;96(11):1153‐1166. 10.1007/s00109-018-1686-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ‐associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22(5):719‐728. 10.1038/s41593-019-0372-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin‐Ruiz C, Williams‐Gray CH, Yarnall AJ, et al. Senescence and inflammatory markers for predicting clinical progression in parkinson’s disease: the ICICLE‐PD study. J Parkinsons Dis. 2020;10(1):193‐206. 10.3233/JPD-191724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16 Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature. 2011;479(7372):232‐236. 10.1038/nature10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baker DJ, Perez‐Terzic C, Jin F, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10(7):825‐836. 10.1038/ncb1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92(20):9363‐9367. 10.1073/pnas.92.20.9363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16 Ink4a‐positive cells shorten healthy lifespan. Nature. 2016;530(7589):184‐189. 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age‐related bone loss in mice. Nat Med. 2017;23(9):1072‐1079. 10.1038/nm.4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78‐83. 10.1038/nm.4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973‐977. 10.1111/acel.12458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246‐1256. 10.1038/s41591-018-0092-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. 2019;21(1):94‐101. 10.1038/s41556-018-0249-2 [DOI] [PubMed] [Google Scholar]
- 37. Gonzalez‐Meljem JM, Apps JR, Fraser HC, Martinez‐Barbera JP. Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer. 2018;118(10):1283‐1288. 10.1038/s41416-018-0066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell. 1997;88(5):593‐602. 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- 39. Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. 10.1038/436642a [DOI] [PubMed] [Google Scholar]
- 40. Braig M, Lee S, Loddenkemper C, et al. Oncogene‐induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660‐665. 10.1038/nature03841 [DOI] [PubMed] [Google Scholar]
- 41. Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature. 2005;436(7051):725‐730. 10.1038/nature03918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Michaloglou C, Vredeveld LCW, Soengas MS, et al. BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature. 2005;436(7051):720‐724. 10.1038/nature03890 [DOI] [PubMed] [Google Scholar]
- 43. Denchi EL, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence‐like features in the mouse pituitary gland. Mol Cell Biol. 2005; 25(7):2660‐2672. 10.1128/mcb.25.7.2660-2672.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roberson RS, Kussick SJ, Vallieres E, Chen SYJ, Wu DY. Escape from therapy‐induced accelerated cellular senescence in p53‐null lung cancer cells and in human lung cancers. Cancer Res. 2005; 65(7):2795‐2803. 10.1158/0008-5472.CAN-04-1270 [DOI] [PubMed] [Google Scholar]
- 45. Garnett S, Dutchak KL, McDonough RV, Dankort D. P53 loss does not permit escape from Braf V600E ‐induced senescence in a mouse model of lung cancer. Oncogene. 2017; 36(45):6325‐6335. 10.1038/onc.2017.235 [DOI] [PubMed] [Google Scholar]
- 46. Tamayo‐Orrego L, Wu CL, Bouchard N, et al. Evasion of cell senescence leads to medulloblastoma progression. Cell Rep. 2016;14(12):2925‐2937. 10.1016/j.celrep.2016.02.061 [DOI] [PubMed] [Google Scholar]
- 47. Kim YH, Choi YW, Lee J, Soh EY, Kim JH, Park TJ. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun. 2017;8(1):1‐14. 10.1038/ncomms15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mario Gonzalez‐Meljem J, Haston S, Carreno G, et al. Stem cell senescence drives age‐attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun. 2017;8(1). 10.1038/s41467-017-01992-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoshimoto S, Loo TM, Atarashi K, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97‐101. 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- 50. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98(21):12072‐12077. 10.1073/pnas.211053698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jeon HY, Kim JK, Ham SW, et al. Irradiation induces glioblastoma cell senescence and senescence‐associated secretory phenotype. Tumor Biol. 2016;37(5):5857‐5867. 10.1007/s13277-015-4439-2 [DOI] [PubMed] [Google Scholar]
- 52. Demaria M, O’Leary MN, Chang J, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7(2):165‐176. 10.1158/2159-8290.CD-16-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context‐dependent effects of SASP in cancer. Nat Rev Cancer. 2019;19(8):439‐453. 10.1038/s41568-019-0156-2 [DOI] [PubMed] [Google Scholar]
- 54. Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128(4):1208‐1216. 10.1172/JCI95145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martínez‐Cué C, Rueda N. Cellular senescence in neurodegenerative diseases. Front Cell Neurosci. 2020;14. 10.3389/fncel.2020.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF). Cell. 1997;91(5):649‐659. 10.1016/S0092-8674(00)80452-3 [DOI] [PubMed] [Google Scholar]
- 57. Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of genetic and epigenetic alterations of CDKN2A (p16INK4a) in cancer. EBioMedicine. 2016;8:30‐39. 10.1016/j.ebiom.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell‐cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366(6456):704‐707. 10.1038/366704a0 [DOI] [PubMed] [Google Scholar]
- 59. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014; 28(2):99‐114. 10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thomas A, Giesler T, White E. p53 mediates Bcl‐2 phosphorylation and apoptosis via activation of the Cdc42/JNK1 pathway. Oncogene. 2000;19(46):5259‐5269. 10.1038/sj.onc.1203895 [DOI] [PubMed] [Google Scholar]
- 61. Hemann MT, Lowe SW. The p53‐Bcl‐2 connection. Cell Death Differ. 2006;13(8):1256‐1259. 10.1038/sj.cdd.4401962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sohn D, Essmann F, Schulze‐Osthoff K, Jänicke RU. p21 blocks irradiation‐induced apoptosis downstream of mitochondria by inhibition of cyclin‐dependent kinase‐mediated caspase‐9 activation. Cancer Res. 2006;66(23):11254‐11262. 10.1158/0008-5472.CAN-06-1569 [DOI] [PubMed] [Google Scholar]
- 63. Özcan S, Alessio N, Acar MB, et al. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging (Albany NY). 2016;8(7):1316‐1329. 10.18632/aging.100971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978‐990. 10.1038/ncb2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hoare M, Narita M. Transmitting senescence to the cell neighbourhood. Nat Cell Biol. 2013;15(8):887‐889. 10.1038/ncb2811 [DOI] [PubMed] [Google Scholar]
- 66. Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006‐1018. 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- 67. Kang C, Xu Q, Martin TD, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612. 10.1126/science.aaa5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang H, Wang H, Ren U, Chen Q, Chena ZJ. CGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114(23):E4612‐E4620. 10.1073/pnas.1705499114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hoare M, Ito Y, Kang TW, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol. 2016;18(9):979‐992. 10.1038/ncb3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Herranz N, Gallage S, Mellone M, et al. mTOR regulates MAPKAPK2 translation to control the senescence‐associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205‐1217. 10.1038/ncb3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049‐1061. 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J. 2011;30(8):1536‐1548. 10.1038/emboj.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Glück S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061‐1070. 10.1038/ncb3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen Y, Wang L, Jin J, et al. p38 inhibition provides anti‐DNA virus immunity by regulation of USP21 phosphorylation and STI NG activation. J Exp Med. 2017;214(4):991‐1010. 10.1084/jem.20161387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ito Y, Hoare M, Narita M. Spatial and temporal control of senescence. Trends Cell Biol. 2017; 27(11):820‐832. 10.1016/j.tcb.2017.07.004 [DOI] [PubMed] [Google Scholar]
- 76. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell. 2014;31(6):722‐733. 10.1016/j.devcel.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gil J. Cellular senescence causes ageing. Nat Rev Mol Cell Biol. 2019; 20(7):388. 10.1038/s41580-019-0128-0 [DOI] [PubMed] [Google Scholar]
- 78. Zhu Y, Tchkonia T, Fuhrmann‐Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl‐2 family of anti‐apoptotic factors. Aging Cell. 2016; 15(3):428‐435. 10.1111/acel.12445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yosef R, Pilpel N, Tokarsky‐Amiel R, et al. Directed elimination of senescent cells by inhibition of BCL‐W and BCL‐XL. Nat Commun. 2016;7. 10.1038/ncomms11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Madeo F, Carmona‐Gutierrez D, Hofer SJ, Kroemer G. Caloric restriction mimetics against age‐associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 2019;29(3):592‐610. 10.1016/j.cmet.2019.01.018 [DOI] [PubMed] [Google Scholar]
- 81. Fontana L, Mitchell SE, Wang B, et al. The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell. 2018;17(3):e12746. 10.1111/acel.12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fontana L, Nehme J, Demaria M. Caloric restriction and cellular senescence. Mech Ageing Dev. 2018;176:19‐23. 10.1016/j.mad.2018.10.005 [DOI] [PubMed] [Google Scholar]
- 83. Duan W, Mattson MP. Dietary restriction and 2‐deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J Neurosci Res. 1999;57(2):15–206. . [DOI] [PubMed] [Google Scholar]
- 84. Lin KL, Lin KJ, Wang PW, et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic Res. 2018;52(11‐12):1371‐1386. 10.1080/10715762.2018.1489128 [DOI] [PubMed] [Google Scholar]
- 85. Yessenkyzy A, Saliev T, Zhanaliyeva M, et al. Polyphenols as caloric‐restriction mimetics and autophagy inducers in aging research. Nutrients. 2020;12(5):1344. 10.3390/nu12051344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ritschka B, Knauer‐Meyer T, Gonçalves DS, et al. The senotherapeutic drug ABT‐737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. 2020;34:489‐494. 10.1101/gad.332643.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Saleh T, Carpenter VJ, Tyutyunyk‐Massey L, et al. Clearance of therapy‐induced senescent tumor cells by the senolytic ABT‐263 via interference with BCL‐X L ‐BAX interaction. Mol Oncol. 2020. 2504–2519. 10.1002/1878-0261.12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. 2019;29(5):1045‐1060.e10. 10.1016/j.cmet.2019.01.021 [DOI] [PubMed] [Google Scholar]
- 89. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, Van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472‐477. 10.1126/science.aaf6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Safety T Pharmacokinetics, and Clinical Outcomes Following Single‐Dose IA Administration of UBX0101, a Senolytic MDM2/p53 Interaction Inhibitor, in Patients with Knee OA ‐ ACR Meeting Abstracts. https://acrabstracts.org/abstract/safety‐tolerability‐pharmacokinetics‐and‐clinical‐outcomes‐following‐single‐dose‐ia‐administration‐of‐ubx0101‐a‐senolytic‐mdm2‐p53‐interaction‐inhibitor‐in‐patients‐with‐knee‐oa/. Accessed July 16, 2020.
- 91. He Y, Li W, Lv D, et al. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell. 2020;19(3). 10.1111/acel.13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jeon OH, Kim C, Laberge RM, et al. Local clearance of senescent cells attenuates the development of post‐traumatic osteoarthritis and creates a pro‐regenerative environment. Nat Med. 2017; 23(6):775‐781. 10.1038/nm.4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Triana‐Martínez F, Picallos‐Rabina P, Da Silva‐Álvarez S, et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun. 2019;10(1). 10.1038/s41467-019-12888-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Guerrero A, Herranz N, Sun B, et al. Cardiac glycosides are broad‐spectrum senolytics. Nat Metab. 2019;1(11):1074‐1088. 10.1038/s42255-019-0122-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guerrero A, Guiho R, Herranz N, et al. Galactose‐modified duocarmycin prodrugs as senolytics. Aging Cell. 2020;19(4). 10.1111/acel.13133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. González‐Gualda E, Pàez‐Ribes M, Lozano‐Torres B, et al. Galacto‐conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell. 2020;19(4). 10.1111/acel.13142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xu M, Tchkonia T, Ding H, et al. JAK inhibition alleviates the cellular senescence‐associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. 2015;112(46):E6301‐E6310. 10.1073/pnas.1515386112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Moiseeva O, Deschênes‐Simard X, St‐Germain E, et al. Metformin inhibits the senescence‐associated secretory phenotype by interfering with IKK/NF‐κB activation. Aging Cell. 2013;12(3):489‐498. 10.1111/acel.12075 [DOI] [PubMed] [Google Scholar]
- 99. Burton DGA, Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res Rev. 2018;43:17‐25. 10.1016/j.arr.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 100. Ruhland MK, Loza AJ, Capietto AH, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7. 10.1038/ncomms11762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656‐660. 10.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ghorashian S, Kramer AM, Onuoha S, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low‐affinity CD19 CAR. Nat Med. 2019;25(9):1408‐1414. 10.1038/s41591-019-0549-5 [DOI] [PubMed] [Google Scholar]
- 103. Amor C, Feucht J, Leibold J, et al. Senolytic CAR T cells reverse senescence‐associated pathologies. Nature. 2020;583(7814):127‐132. 10.1038/s41586-020-2403-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kim KM, Noh JH, Bodogai M, et al. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017;31(15):1529‐1534. 10.1101/gad.302570.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hernandez‐Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol. 2017;27(17):2652‐2660.e4. 10.1016/j.cub.2017.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Johnson IP. Age‐related neurodegenerative disease research needs aging models. Front Aging Neurosci. 2015;7:168. 10.3389/fnagi.2015.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rocca WA, Petersen RC, Knopman DS, et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimer’s Dement. 2011;7(1):80‐93. 10.1016/j.jalz.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Scalfari A, Neuhaus A, Daumer M, Ebers GC, Muraro PA. Age and disability accumulation in multiple sclerosis. Neurology. 2011;77(13):1246‐1252. 10.1212/WNL.0b013e318230a17d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence‐associated secretory phenotype. Eur J Neurosci. 2011;34(1):3‐11. 10.1111/j.1460-9568.2011.07738.x [DOI] [PubMed] [Google Scholar]
- 110. Flanary BE, Streit WJ. Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia. 2004;45(1):75‐88. 10.1002/glia.10301 [DOI] [PubMed] [Google Scholar]
- 111. Kujuro Y, Suzuki N, Kondo T. Esophageal cancer‐related gene 4 is a secreted inducer of cell senescence expressed by aged CNS precursor cells. Proc Natl Acad Sci USA. 2010;107(18):8259‐8264. 10.1073/pnas.0911446107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Al‐Mashhadi S, Simpson JE, Heath PR, et al. Oxidative glial cell damage associated with white matter lesions in the aging human brain. Brain Pathol. 2015;25(5):565‐574. 10.1111/bpa.12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21‐dependent senescence‐like phenotype driven by a DNA damage response. Aging Cell. 2012;11(6):996‐1004. 10.1111/j.1474-9726.2012.00870.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double‐strand breaks. Nat Cell Biol. 2004;6(2):168‐170. 10.1038/ncb1095 [DOI] [PubMed] [Google Scholar]
- 115. Chinta SJ, Woods G, Demaria M, et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018;22(4):930‐940. 10.1016/j.celrep.2017.12.092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Han X, Zhang T, Liu H, Mi Y, Gou X. Astrocyte senescence and Alzheimer’s disease. A review. Front Aging Neurosci. 2020;12:148–153. 10.3389/fnagi.2020.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Deture MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14(1):1‐18. 10.1186/s13024-019-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8):a006239. 10.1101/cshperspect.a006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Arendt T, Holzer M, Gärtner U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J Neural Transm. 1998;105(8‐9):949‐960. 10.1007/s007020050104 [DOI] [PubMed] [Google Scholar]
- 120. De La Monte SM, Sohn YK, Wands JR. Correlates of p53‐ and Fas (CD95)‐mediated apoptosis in Alzheimer’s disease. J Neurol Sci. 1997;152(1):73‐83. 10.1016/S0022-510X(97)00131-7 [DOI] [PubMed] [Google Scholar]
- 121. Yang Z, Jun H, Choi CI, et al. Age‐related decline in BubR1 impairs adult hippocampal neurogenesis. Aging Cell. 2017;16(3):598‐601. 10.1111/acel.12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mathews KJ, Allen KM, Boerrigter D, Ball H, Shannon Weickert C, Double KL. Evidence for reduced neurogenesis in the aging human hippocampus despite stable stem cell markers. Aging Cell. 2017;16(5):1195‐1199. 10.1111/acel.12641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hollands C, Tobin MK, Hsu M, et al. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol Neurodegener. 2017;12(1). 10.1186/s13024-017-0207-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lee IS, Jung K, Kim IS, et al. Human neural stem cells alleviate Alzheimer‐like pathology in a mouse model. Mol Neurodegener. 2015;10(1):38. 10.1186/s13024-015-0035-6. http://sfx.library.uu.nl/utrecht?sid=EMBASE&issn=17501326&id=10.1186%2Fs13024‐015‐0035‐6&atitle=Human+neural+stem+cells+alleviate+Alzheimer‐like+pathology+in+a+mouse+model&stitle=Mol.+Neurodegeneration&title=Molecular+Neurodegeneration&volume=10&issue=1&spage=&epage=&aulast=Lee&aufirst=Il‐Shin&auinit=I.‐S.&aufull=Lee+I.‐S.&coden=&isbn=&pages=‐&date=2015&auinit1=I&auinitm=‐S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ. Amyloid‐β1‐42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013;4(11). 10.1038/cddis.2013.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kevin Howcroft T, Campisi J, Louis GB, et al. The role of inflammation in age‐related disease. Aging (Albany NY). 2013;5(1):84‐93. 10.18632/aging.100531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Giunta B, Fernandez F, Nikolic WV, et al. Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation. 2008;5(1):51. 10.1186/1742-2094-5-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lai KSP, Liu CS, Rau A, et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta‐analysis of 175 studies. J Neurol Neurosurg Psychiatry. 2017;88(10):876‐882. 10.1136/jnnp-2017-316201 [DOI] [PubMed] [Google Scholar]
- 129. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Denis Alexander H, Ross OA. Age and age‐related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9(APR):586–592. 10.3389/fimmu.2018.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cacabelos R, Alvarez XA, Fernandez‐Novoa L, et al. Brain interleukin‐1beta in Alzheimer’s disease and vascular dementia. Methods Find Exp Clin Pharmacol. 1994;16(2):141‐151. [PubMed] [Google Scholar]
- 131. Swardfager W, Lanctt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta‐analysis of cytokines in Alzheimer’s disease. Biol Psychiatry. 2010;68(10):930‐941. 10.1016/j.biopsych.2010.06.012 [DOI] [PubMed] [Google Scholar]
- 132. Yoshiyama Y, Asahina M, Hattori T. Selective distribution of matrix metalloproteinase‐3 (MMP‐3) in Alzheimer’s disease brain. Acta Neuropathol. 2000;99(2):91‐95. 10.1007/PL00007428 [DOI] [PubMed] [Google Scholar]
- 133. Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133‐139. 10.1002/glia.10154 [DOI] [PubMed] [Google Scholar]
- 134. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10(4):217‐224. 10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- 135. Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39(1):19‐34. 10.1111/j.1365-2990.2012.01306.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Floden AM, Combs CK. Microglia demonstrate age‐dependent interaction with amyloid‐β fibrils. J Alzheimer’s Dis. 2011;25(2):279‐293. 10.3233/JAD-2011-101014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Njie MG, Boelen E, Stassen FR, Steinbusch HWM, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age‐related changes in microglial function. Neurobiol Aging. 2012;33(1):195.e1‐195.e12. 10.1016/j.neurobiolaging.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Habib N, McCabe C, Medina S, et al. Disease‐associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23(6):701‐706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wang WY, Tan MS, Yu JT, Tan L. Role of pro‐inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3(10):136–142. 10.3978/j.issn.2305-5839.2015.03.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Griffin WST, Stanley LC, Ling C, et al. Brain interleukin 1 and S‐100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86(19):7611‐7615. 10.1073/pnas.86.19.7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chun H, Lee CJ. Reactive astrocytes in Alzheimer’s disease: a double‐edged sword. Neurosci Res. 2018;126:44‐52. 10.1016/j.neures.2017.11.012 [DOI] [PubMed] [Google Scholar]
- 142. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7‐35. 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Campbell IL, Abraham CR, Masliah E, et al. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci USA. 1993;90(21):10061‐10065. 10.1073/pnas.90.21.10061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bauer J, Strauss S, Schreiter‐Gasser U, et al. Interleukin‐6 and α‐2‐macroglobulin indicate an acute‐phase state in Alzheimer’s disease cortices. FEBS Lett. 1991;285(1):111‐114. 10.1016/0014-5793(91)80737-N [DOI] [PubMed] [Google Scholar]
- 145. Huell M, Strauss S, Volk B, Berger M, Bauer J. Interleukin‐6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol. 1995;89(6):544‐551. 10.1007/BF00571510 [DOI] [PubMed] [Google Scholar]
- 146. Senolytic therapy to modulate progression of Alzheimer’s disease ‐ full text view ‐ ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04063124. Accessed June 1, 2020.
- 147. Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6):e12840. 10.1111/acel.12840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9(1):13‐24. 10.1038/nrneurol.2012.242 [DOI] [PubMed] [Google Scholar]
- 149. Nikonova EV, Xiong Y, Tanis KQ, et al. Transcriptional responses to loss or gain of function of the leucine‐rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Hum Mol Genet. 2012;21(1):163‐174. 10.1093/hmg/ddr451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Riessland M, Kolisnyk B, Kim TW, et al. Loss of SATB1 induces a p21 dependent cellular senescence phenotype in dopaminergic neurons. bioRxiv. 2018;514–530. 10.1101/452243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Park MH, Lee HJ, Lee HL, et al. Parkin knockout inhibits neuronal development via regulation of proteasomal degradation of p21. Theranostics. 2017;7(7):2033‐2045. 10.7150/thno.19824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Tan FCC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15(6):643‐660. 10.1007/s10522-014-9532-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Mogi M, Harada M, Kondo T, et al. Interleukin‐1β, interleukin‐6, epidermal growth factor and transforming growth factor‐α are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180(2):147‐150. 10.1016/0304-3940(94)90508-8 [DOI] [PubMed] [Google Scholar]
- 154. van Dijk KD, Persichetti E, Chiasserini D, et al. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov Disord. 2013;28(6):747‐754. 10.1002/mds.25495 [DOI] [PubMed] [Google Scholar]
- 155. Choi DH, Kim YJ, Kim YG, Joh TH, Beal MF, Kim YS. Role of matrix metalloproteinase 3‐mediated α‐synuclein cleavage in dopaminergic cell death. J Biol Chem. 2011;286(16):14168‐14177. 10.1074/jbc.M111.222430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Ghasemi N, Razavi S, Nikzad E. Multiple sclerosis: pathogenesis, symptoms, diagnoses and cell‐based therapy. Cell J. 2017;19(1):1‐10. 10.22074/cellj.2016.4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Shields SA, Gilson JM, Blakemore WF, Franklin RJM. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin‐induced CNS demyelination. Glia. 1999;28(1):77‐83. [DOI] [PubMed] [Google Scholar]
- 158. Karamita M, Nicholas R, Kokoti L, et al. Cellular senescence correlates with demyelination, brain atrophy and motor impairment in a model of multiple sclerosis (P2.405). Neurology. 2018;90(15 Supplement). [Google Scholar]
- 159. Nicaise AM, Wagstaff LJ, Willis CM, et al. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc Natl Acad Sci USA. 2019;116(18):9030‐9039. 10.1073/pnas.1818348116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Müller HL, Merchant TE, Warmuth‐Metz M, Martinez‐Barbera JP, Puget S. Craniopharyngioma. Nat Rev Dis Prim. 2019;5:1. 10.1038/s41572-019-0125-9 [DOI] [PubMed] [Google Scholar]
- 161. Martinez‐Barbera JP, Andoniadou CL. Biological behaviour of craniopharyngiomas. Neuroendocrinology. 2020;110:797‐804. 10.1159/000506904 [DOI] [PubMed] [Google Scholar]
- 162. Apps JR, Carreno G, Gonzalez‐Meljem JM, et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol. 2018;135(5):757‐777. 10.1007/s00401-018-1830-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Gonzalez‐Meljem JM, Martinez‐Barbera JP. Senescence drives non‐cell autonomous tumorigenesis in the pituitary gland. Mol Cell Oncol. 2018;5(3):e1435180. 10.1080/23723556.2018.1435180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Andoniadou CL, Gaston‐Massuet C, Reddy R, et al. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012;124(2):259‐271. 10.1007/s00401-012-0957-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Apps JR, Hutchinson JC, Arthurs OJ, et al. Imaging Invasion: micro‐CT imaging of adamantinomatous craniopharyngioma highlights cell type specific spatial relationships of tissue invasion. Acta Neuropathol Commun. 2016;4(1):57. 10.1186/s40478-016-0321-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Gaston‐Massuet C, Andoniadou CLCL, Signore M, et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci USA. 2011;108(28):11482‐11487. 10.1073/pnas.1101553108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Andoniadou CL, Matsushima D, Mousavy‐gharavy SN, et al. The Sox2 + population of the adult murine pituitary includes progenitor/stem cells with tumour‐inducing potential. Cell Stem Cell. 2013;419–432. [DOI] [PubMed] [Google Scholar]
- 168. Apps JR, Martinez‐Barbera JP. Genetically engineered mouse models of craniopharyngioma: an opportunity for therapy development and understanding of tumor biology. Brain Pathol. 2017;27(3):364‐369. 10.1111/bpa.12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Ryall S, Tabori U, Hawkins C. Pediatric low‐grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun. 2020;8(1). 10.1186/s40478-020-00902-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Schiff D, Brown PD, Giannini C. Outcome in adult low‐grade glioma: the impact of prognostic factors and treatment. Neurology. 2007;69(13):1366‐1373. 10.1212/01.wnl.0000277271.47601.a1 [DOI] [PubMed] [Google Scholar]
- 171. Jones DTW, Bandopadhayay P, Jabado N. The power of human cancer genetics as revealed by low‐grade gliomas. Annu Rev Genet. 2019;53(1):483‐503. 10.1146/annurev-genet-120417-031642 [DOI] [PubMed] [Google Scholar]
- 172. Jones DTW, Hutter B, Jäger N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927‐932. 10.1038/ng.2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Jacob K, Quang‐Khuong DA, Jones DTW, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene‐induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17(14):4650‐4660. 10.1158/1078-0432.CCR-11-0127 [DOI] [PubMed] [Google Scholar]
- 174. Buhl JL, Selt F, Hielscher T, et al. The senescence‐associated secretory phenotype mediates oncogene‐induced senescence in pediatric pilocytic astrocytoma. Clin Cancer Res. 2019;25(6):1851‐1866. 10.1158/1078-0432.CCR-18-1965 [DOI] [PubMed] [Google Scholar]
- 175. Bax DA, Mackay A, Little SE, et al. A distinct spectrum of copy number aberrations in pediatric high‐grade gliomas. Clin Cancer Res. 2010;16(13):3368‐3377. 10.1158/1078-0432.CCR-10-0438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Schiffman JD, Hodgson JG, Vandenberg SR, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70(2):512‐519. 10.1158/0008-5472.CAN-09-1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Reinhardt A, Stichel D, Schrimpf D, et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol. 2018;136(2):273‐291. 10.1007/s00401-018-1837-8 [DOI] [PubMed] [Google Scholar]
- 178. Reis GF, Pekmezci M, Hansen HM, et al. CDKN2A loss is associated with shortened overall survival in lower‐grade (World Health Organization Grades II‐III) astrocytomas. J Neuropathol Exp Neurol. 2015;74(5):442‐452. 10.1097/NEN.0000000000000188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Brat DJ, Aldape K, Colman H, et al. cIMPACT‐NOW update 5: recommended grading criteria and terminologies for IDH‐mutant astrocytomas. Acta Neuropathol. 2020;139(3):603‐608. 10.1007/s00401-020-02127-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Taylor OG, Brzozowski JS, Skelding KA. Glioblastoma multiforme: an overview of emerging therapeutic targets. Front Oncol. 2019;9:963. 10.3389/fonc.2019.00963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Aasland D, Gotzinger L, Hauck L, et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR‐CHK1, p21, and NF‐kB. Cancer Res. 2019;79(1):99–113. 10.1158/0008-5472.CAN-18-1733 [DOI] [PubMed] [Google Scholar]
- 182. Gupta K, Burns TC. Radiation‐induced alterations in the recurrent glioblastoma microenvironment: therapeutic implications. Front Oncol. 2018;8:503. 10.3389/fonc.2018.00503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. He Y, Kaina B. Are there thresholds in glioblastoma cell death responses triggered by temozolomide? Int J Mol Sci. 2019;20(7):1562. 10.3390/ijms20071562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Tomicic MT, Christmann M. Targeting anticancer drug‐induced senescence in glioblastoma therapy. Oncotarget. 2018;9(101):37466‐37467. 10.18632/oncotarget.26502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Duan C, Yang R, Yuan L, et al. Late effects of radiation prime the brain microenvironment for accelerated tumor growth. Int J Radiat Oncol Biol Phys. 2019;103(1):190‐194. 10.1016/j.ijrobp.2018.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild‐type p53 glioblastoma multiforme cells. J Neurosurg. 2006;105(1):111‐118. 10.3171/jns.2006.105.1.111 [DOI] [PubMed] [Google Scholar]
- 187. Ghorai A, Mahaddalkar T, Thorat R, Dutt S. Sustained inhibition of PARP‐1 activity delays glioblastoma recurrence by enhancing radiation‐induced senescence. Cancer Lett. 2020;490:44‐53. 10.1016/j.canlet.2020.06.023 [DOI] [PubMed] [Google Scholar]
- 188. Halliday J, Helmy K, Pattwell SS, et al. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural‐mesenchymal shift. Proc Natl Acad Sci. 2014;111(14):5248‐5253. 10.1073/pnas.1321014111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Rahman M, Olson IE, Mansour M, et al. Selective vulnerability of senescent glioblastoma cells to Bcl‐XL inhibition. bioRxiv. 2020. 10.1101/2020.06.03.132712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Sharma T, Schwalbe EC, Williamson D, et al. Second‐generation molecular subgrouping of medulloblastoma: an international meta‐analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. 2019;138(2):309‐326. 10.1007/s00401-019-02020-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465‐472. 10.1007/s00401-011-0922-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Northcott PA, Robinson GW, Kratz CP, et al. Medulloblastoma. Nat Rev Dis Prim. 2019;5(1):1‐20. 10.1038/s41572-019-0063-6 [DOI] [PubMed] [Google Scholar]
- 193. Pallavicini G, Sgro F, Garello F, et al. Inactivation of citron kinase inhibits medulloblastoma progression by inducing apoptosis and cell senescence. Cancer Res. 2018;78(16):4599‐4612. 10.1158/0008-5472.CAN-17-4060 [DOI] [PubMed] [Google Scholar]
- 194. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803‐820. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 195. Cordero FJ, Huang Z, Grenier C, et al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res. 2017;15(9):1243‐1254. 10.1158/1541-7786.MCR-16-0389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Ballester LY, Wang Z, Shandilya S, et al. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol. 2013;37(9):1357‐1364. 10.1097/PAS.0b013e318294e817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Flannery PC, DeSisto JA, Amani V, et al. Preclinical analysis of MTOR complex 1/2 inhibition in diffuse intrinsic pontine glioma. Oncol Rep. 2018;39(2):455‐464. 10.3892/or.2017.6122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Balakrishnan I, Danis E, Pierce A, et al. Senescence induced by BMI1 inhibition is a therapeutic vulnerability in H3K27M‐mutant DIPG. Cell Rep. 2020;33(3):108286. 10.1016/j.celrep.2020.108286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Grosse L, Wagner N, Emelyanov A, et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020;32(1):87‐99.e6. 10.1016/j.cmet.2020.05.002 [DOI] [PubMed] [Google Scholar]
- 200. Di Napoli M, Shah IM. Neuroinflammation and cerebrovascular disease in old age: a translational medicine perspective. J Aging Res. 2011;2011:1‐18. 10.4061/2011/857484 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during this study.