

Abstract
Lung Cancer is the leading cause of cancer-related deaths worldwide. This is mainly due to late diagnosis and therefore advanced stage of the disease. Understanding the cell of origin of cancer and the processes that lead to its transformation will allow for earlier diagnosis and more accurate prediction of tumour type, ultimately leading to better treatments and lower patient morbidity. In this review, we focus on alveolar type 2 (AT2) cells as the cell of origin of lung adenocarcinoma (LUAD), the most common type of lung cancer. We first elaborate on the different oncogenes that are associated with LUAD and other lung cancers. After, we lay out in detail what is known about AT2 biology, to further delve into AT2 cells as cell of origin for adenocarcinoma. Understanding the precursors of LUAD and identifying the molecular changes during its progression will allow for earlier detection and better molecular targeting of the disease in early stages.
Keywords: cancer, lung adenocarcinoma
Introduction
Lung cancer is the most frequent cause of cancer-related deaths worldwide. Every year, 1.6 million people die of this disease [1]. Histologically, lung cancer has been broadly categorized into two groups: small cell lung carcinoma (SCLC) that encompasses 15% of all lung cancers, and non-small cell lung carcinoma (NSCLC) that is categorized into lung adenocarcinoma (LUAD), squamous cell carcinoma (SqCC) and large cell carcinoma (LCC). Of these types of cancer, LUAD is by far the most common form of this lung disease.
Cells from the airway and alveolar compartments are maintained by their respective resident progenitor cells which play a crucial role in lung homeostasis, repair and cancer. The lung is a complex organ in charge of the gas exchange between the air and our blood. It has two differentiated structures: airways and alveoli. These two structures have specific functions that make the respiratory system very efficient in gas exchange, whilst protecting the lung epithelia from microorganisms and particles that are inhaled on a daily basis. In the airways, basal cells can give rise to ciliated, neuroendocrine and club cells [2–5]. Basal cells are progenitor cells closely associated with the basal lamina that maintain airway homeostasis. Basal cells are considered the candidate cell of origin in lung SqCC [6]. Although LUAD sometimes exhibits features of airway basal cells, there is no evidence that these cells can be its cell of origin [7]. Ciliated cells are the predominant cell type in the lung airway. These cells clear exogenous elements out of the lung [8] by coordinated beating of their cilia [9]. Moreover, ciliated cells are also chemosensors and mechanosensors of the airways [10,11]. Ciliated cells are thought to be the most terminally differentiated cell type in the lung, with limited proliferative capacity. It is possible that ciliated cells could give rise to LUAD, first showing dysplasia and losing the cilia, becoming mucous columnar cells with enlarged nuclei that may be a precursor lesion of pulmonary LUAD [12]. They also contribute to other rare tumours such as the ciliated muconodular papillary tumour of the lung [13]. Pulmonary neuroendocrine cells (PNECs) are the only innervated airway epithelial cells [14]. They can respond to many different airway conditions such as mechanical stretch, hypoxia or hypercarbia. They also accumulate and release serotonin and other transmitters [15]. Through neuropeptides, they can activate immune responses in the lung [16]. During lung injury, PNECs may be able to give rise to club and ciliated cells, although the selective ablation of PNECs does not affect the regeneration of these cell types [17]. PNECs contribute to different types of tumours: large cell neuroendocrine carcinoma, a subtype of LCC, SCLC (10% of all lung cancers), and pulmonary carcinoid tumours. However, they are not known to contribute to LUAD [18]. Club cells are cuboidal cells that spread through the large and small airways of the lung. They secrete components that line the bronchioles and regulate the contents of secretion in the distal respiratory tract. Club cells have also a role in detoxifying the lung through cytochrome p450 enzymes in their smooth endoplasmic reticulum. They can self-renew and are precursors to ciliated, goblet and alveolar type 2 (AT2) cells [19]. Club cells are known to survive KRAS mutations and can give rise to LUAD [20]. Goblet cells secrete mucins that confer the airway surface the biophysical properties that allow for entrapment and transportation of inhaled particles and microorganisms [21]. Higher levels of specific mucins, such as MUC1, have been related to increased aggressiveness in LUAD [22]. However, LUAD composed of goblet cells is relatively rare [23, 24]. The region between the airways and the alveolar space, the bronchioalveolar duct junction (BADJ), contains bronchioalveolar stem cells (BASCs) that can give rise to both airway and alveolar cells [25, 26]. BASCs are the main source of regeneration in the distal lung after naphthalene treatment [27]. In vitro, they can give rise to bronchiolar, alveolar and bronchioalveolar organoids [28]. During KRAS-driven oncogenesis, these cells expand in the BADJ, potentially contributing to the formation of LUAD [25, 29]. In the alveolar space, we find AT2 progenitor cells that provide the alveoli with pulmonary surfactant, which is important to reduce the surface tension in the alveoli and to keep them from collapsing. AT2 cells are able to self-renew and give rise to alveolar type 1 (AT1) cells, a specialized type of epithelial cell that is part of the air–blood barrier. AT2 cells are one of the best characterized cells of origin in LUAD (Figure 1). Whilst club cells seem to need an inflammatory response to yield adenomas after oncogenic KRAS activation, AT2 cells lead to malignant adenocarcinomas regardless of their environment [30–32]. In past reviews, we covered the cells of origin of LUAD [33, 34]. However, in this review, we will focus on the genetic alterations and molecular pathways involved in the transition of AT2 cells to LUAD. We will use this example of tumour progression to understand the techniques used to address these questions.
Fig. 1.
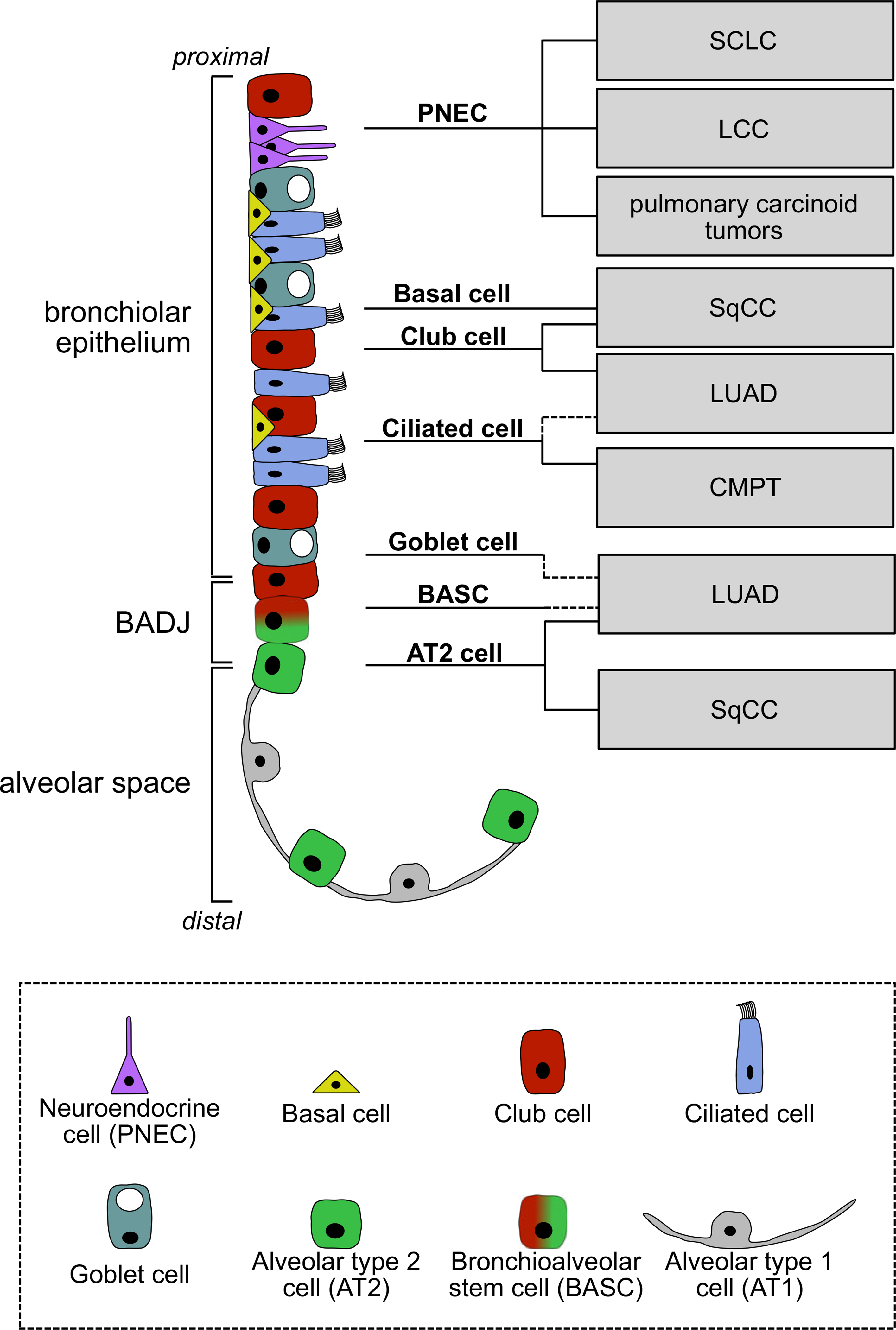
Lung epithelial cells as cells of origin of lung cancer. Different epithelial cell types line the epithelium of the different regions of the lung, the bronchiolar epithelium, the bronchioalveolar duct junction (BADJ) and the alveolar space. PNECs have been shown to be the cell of origin for small cell lung carcinoma (SCLC) and large cell lung carcinoma (LCC); basal cells, club cells and AT2 cells can give rise to squamous cell carcinoma (SqCC). AT2 cells are a confirmed cell of origin for lung adenocarcinoma (LUAD), whilst ciliated cells, goblet cells and bronchioalveolar stem cells (BASCs) are suspected to be able to give rise to LUAD in specific conditions (dotted lines). There is no evidence for AT1 cells as a cell of origin for lung cancer.
KRAS, oncogenes and adenocarcinoma
The Kirsten Rat Sarcoma viral oncogene homolog (KRAS) oncoprotein is the driver in 22% of all cancers. In LUAD, it is mutated in 33% of the patients and therefore the most mutated oncogene in this type of cancer. The most mutated tumour suppressor is TP53, present in 46% of LUAD patients. Tyrosine kinase domain of epidermal growth factor receptor (EGFR) mutations are mutually exclusive with KRAS mutations and are found in 14% of the patients, followed by BRAF mutations present in 10% of LUAD patients. Other oncogenes commonly mutated are PIK3CA (7%), MET (7%) and RIT1 (2%). Other tumour suppressor genes with mutations associated with LUAD are STK11 (17%), KEAP1 (17%), NF1 (11%), RB1 (4%) and CDKN2A (4%). Chromatin-modifying gene and RNA splicing gene mutations are also highly present in LUAD: SETD2 (9%), ARID1A (7%), SMARCA4 (6%), RBM10 (8%) and U2AF1 (3%) [1,35]. The co-occurrence of these mutations is crucial for the development of the tumour; however, not only mutations are important co-ocurrences. For instance, P53 (protein encoded by TP53) inactivation is more common in activated KRAS LUAD than the actual mutation of TP53 [36]. Co-mutations of different genes can have a considerable functional impact. STK11 mutation in a KRAS-driven LUAD mouse model causes transdifferentiation to a squamous phenotype by downregulation of the polycomb repressive complex 2 (PRC2) [37]. Furthermore, loss of KEAP1 in KRAS-G12D cells causes a change from an AT2 cell of origin to a bronchiolar cell of origin. Hence, one combination of mutations might lead to LUAD in one cell type, but not in another [38].
Specific oncogenic drivers are linked to different LUAD subtypes; nonetheless, molecular diversity of these subtypes delineate the differences within the same drivers. This intradriver molecular diversity leads to heterogeneous clinical behaviour of the tumour and its response to treatment [39]. Indeed, different mutations in the same oncogenic drivers lead to different signalling pathway activation, which translates into different clinical outcomes. An example of this is how different KRAS mutations lead to the interaction with different downstream signalling effectors: KRAS-G12C and KRAS-G12V activate RALA and RALB signalling, whilst KRAS-G12D increases PI3K-AKT and MEK-ERK activation. Therefore, different amino acid substitutions lead to different behaviour in KRAS-driven tumours. This indicates that treatment efforts should be put in different combinations of downstream inhibitors depending on the type of mutation of this oncogene [40]. Activated EGFR driven by SP-C promoter also causes adenocarcinoma in mouse models [41].
Signalling pathway alterations due to gene modifications are the drivers of tumour formation. However, the metabolism of the cell of origin has an important impact in the tumour initiation in NSCLC. Aberrant activation of the enzyme glycine decarboxylase (GLDC) is crucial for tumour initiation in NSCLC. GLDC changes the glycine/serine metabolism, which leads to changes in pyrimidine metabolism, regulating cancer cell proliferation. Aberrant activation of GLDC correlates with poor prognosis in lung cancer patients [42]. Another signalling pathway that alters the fate of LUAD is the Notch pathway. It has been observed that Notch inhibition in KRAS-G12D-positive cells inhibits adenocarcinoma formation but promotes squamous hyperplasia in the alveolar space. However, Notch activation leads to papillary adenocarcinoma formation in the bronchiolar space [43].
Alveolar cell biology
AT2 cells stabilize the epithelial barrier in the alveoli, play a role in immune defence, and maintain alveoli in homeostasis and lung injury. Moreover, they are the main source of surfactant production in the alveoli. The main role of pulmonary surfactant is the regulation of surface tension, which prevents atelectasis, the collapse of the lung and maintenance of alveolar fluid homeostasis in the alveoli. It also stabilizes alveolar size, reduces the energy necessary for breathing, and keeps the alveoli dry [44]. Surfactant is stored in lamellar bodies, and it is composed of a mix of lipids and proteins. 90% of the surfactant is made out of lipids. A lipid monolayer of dipalmitoylphosphatidylcholine covers the alveolus and is responsible for lowering the surface tension at the air-liquid interface within the alveoli, the main function of the surfactant. Another major lipid contributor to the surfactant is phosphatidylglycerol, which makes up the rest of the lipid fraction, together with phosphatidylinositol, phosphatidylethanolamine and phosphatidylserine. The surfactant proteins (SP), which make up 10% of the surfactant composition, are only four: SP-B and SP-C, small hydrophobic proteins, and SP-A and SP-D, and large hydrophilic proteins [45]. Of these, SP-C is the only protein that is exclusively present in pulmonary surfactant, and its only producing cell is the AT2 cell. Its principal characteristic is its extreme hydrophobicity [46]. The assembly of the lamellar bodies is mediated by ATP-binding cassette transporters. After their assembly in the endoplasmatic reticulum, lamellar bodies containing the surfactant fuse with the apical plasma membrane and release the content into the lumen of the alveolus in a Ca + dependent manner [47].
AT2 cells have an active role in innate immunity. They protect the lung by secreting factors that decrease bacterial growth and activate pulmonary macrophages to fight infection [48]. At the same time, macrophages can inhibit alveolar inflammation by releasing anti-inflammatory proteins such as SOCS1 and SOCS3 [49].
As progenitor cells of the alveolar region, AT2 cells have the ability to self-renew and differentiate into AT1 cells. Michael Evans et al. observed this for the first time in the 70s [50, 51], but it was not until recently that lineage tracing studies supported this hypothesis [52]. The past year, two publications uncovered a subset of AT2 cells termed alveolar epithelial progenitors (AEP) that are marked by the expression of Axin2. Besides having the same functions as Axin2-negative AT2 cells, they have been shown to regenerate both AT2 and AT1 cells after injury. These cells are regulated by Wnt signalling, which inhibits their differentiation into AT1 cells [53, 54].
AT2 origin of adenocarcinoma
Histological observations and studies with genetically engineered mouse models (GEMM) have led to the hypothesis that AT2 cells are the cell of origin of LUAD. Adenocarcinomas are typically located in peripheral regions of the lung, where the alveoli are found, and almost all tumours express SFTPC (SP-C gene). AT2 cells are not the only possible cell of origin of LUAD; however, studies in KRAS-G12D- and p53-driven GEMM have suggested that only AT2 cells progress to LUAD, whilst BASCs or club cells, in the BADJ or bronchiolar region, respectively, lead to hyperplastic lesions that rarely progress to LUAD [30–32]. In their studies, Xu et al. crossed KRAS-LSL-G12D mice that also had a heterozygous p53 deletion to CC10-CreER or SP-C-CreER mice, which enabled them to activate oncogenic KRAS in CC10 (club cells and BASCs) or SP-C (AT2 cells and BASCs) expressing cells, respectively. When Cre expression was induced in SP-C + cells, tumours occurred in the alveolar space, but no proliferation was observed at the BADJ. When Cre expression was induced in CC10 + cells, the authors observed hyperplastic lesions in the BADJ and in the alveolar space. One downside of the CC10-CreER construct is that not only club cells and BASCs were labelled, but also 10% of the AT2 cells in the alveolar space. Only SP-C + lesions in the alveolar space, and not in the BADJ, progressed to adenomas and adenocarcinomas, despite recombination throughout the whole epithelium, including the bronchioles and bronchi.
Although making a definitive statement regarding the cell of origin is difficult, the location of the tumours and the expression of SP-C imply that only AT2 cells, not club cells or BASCs progressed to LUAD upon KRAS-G12D expression in this GEMM [30]. In a similar approach, Sutherland et al. used an Adeno5–CC10–Cre or Adeno5-SP-C–Cre virus to induce KRAS-G12D in CC10 or SP-C expressing cells, respectively. In their studies, the authors observed tumours in the alveolar space when using the virus that targets SP-C + cells, and in the BADJ when using the virus that targets CC10 + cells. The BADJ contains both BASCs and club cells; both cell types were targeted with the latter virus. Interestingly, there was a higher incidence of tumours with a papillary phenotype when CC10 + cells were targeted, and a higher incidence of adenomas and adenocarcinomas when SP-C + cells were targeted, highlighting the impact of the cell of origin on tumour phenotypes [31]. In the same year, Mainardi et al. studied the cell of origin of LUAD with KRAS-G12V as the oncogenic driver. They found that several epithelial cell types had the capacity to proliferate upon KRAS-G12V expression, regardless of their location. Although transformed cell clusters were present in all lung regions, only SP-C + lesions in the alveolar space progressed to adenomas and adenocarcinomas. Lesions in the BADJ and bronchiolar epithelium only appeared in the presence of an inflammatory response and were of papillary phenotype, similar to the observations of Sutherland et al. [30]. Both studies are discussed in Rowbotham and Kim [33].
New technologies now facilitate the study of the role of AT2 cells in LUAD initiation. Recently, we used single cell RNA-sequencing (scRNA-Seq) of early LUAD lesions in the KRAS-G12D GEMM to determine the effect of oncogenic KRAS on different lung epithelial cell types. Despite ubiquitous oncogene activation and proliferation in epithelial cells, only AT2 cells formed a transcriptionally distinct cluster upon KRAS-G12D expression, indicating their role as an initiating cell for LUAD. To further examine the consequences of KRAS-G12D expression in AT2 cells, we used AT2 derived organoid cultures and induced oncogenic KRAS in vitro. Strikingly, the organoids recapitulated LUAD progression histologically and transcriptionally, and formed LUAD when transplanted orthotopically [55].
Conclusion
In this review, we have summarized the knowledge on AT2 cells as the cell of origin of LUAD. Thanks to the variety of experiments determined to trace the cell of origin of LUAD, the evidence suggests that AT2 is its main cell of origin. However, the molecular mechanisms by which transformation of AT2 cells into adenocarcinoma are many, and new regulators of this process keep surfacing. Here, we have covered gene mutations, protein inactivation, defects in signalling and metabolic enzymes. The field is focusing on detecting the first causes and modifications that lead to tumour initiation. Since AT2 cells are now a well-known source of adenocarcinoma, they have become a good target to study the earliest stages of lung cancer. Understanding the changes that occur early in the transformation process of the cell of origin will help find new early detection and treatment strategies. Factors secreted by transforming cells could potentially serve as biomarkers that can be screened for in risk populations. New early-stage targeted therapy could replace surgery as a first-line treatment and might be able to help reduce cancer cell dissemination and relapse. These are questions that once answered will eventually contribute to the generation of better treatments and diagnosis tools.
Acknowledgments
Conflict of interest
C.F.K has a sponsored research agreement from Celgene/BMS, but this funding did not support the research described in this manuscript.
References
- 1.The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rock JR, Onaitis MW, Rawlins El et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA 2009; 106: 12771–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rawlins EL, Okubo T, Xue Y et al. The role of Scgb1a1+ clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009; 4: 525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rawlins EL, Hogan BLM. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol 2008; 295: L231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014; 507: 190–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hynds RE, Janes SM. Airway basal cell heterogeneity and lung squamous cell carcinoma. Cancer Prevention Res 2017; 10: 491–3. [DOI] [PubMed] [Google Scholar]
- 7.Fukui T, Shaykhiev R, Agosto-Perez F et al. Lung adenocarcinoma subtypes based on expression of human airway basal cell genes. Eur Respir J 2013; 42: 1332–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yaghi A, Dolovich MB. Airway epithelial cell cilia and obstructive lung disease. Cells 2016; 5: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braiman A, Priel Z. Efficient mucociliary transport relies on efficient regulation of ciliary beating. Respir Physiol Neurobiol 2008; 163: 202–7. [DOI] [PubMed] [Google Scholar]
- 10.Horváth G, Sorscher EJ. Luminal fluid tonicity regulates airway ciliary beating by altering membrane stretch and intracellular calcium. Cell Motil. Cytoskeleton 2008; 65: 469–75. [DOI] [PubMed] [Google Scholar]
- 11.Shah AS, Ben-Shahar Y, Moninger TO, Kline JN, Welsh MJ. Motile Cilia of human airway epithelia are chemosensory. Science 2009; 325: 1131–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park WY, Kim MH, Shin DH et al. Ciliated adenocarcinomas of the lung: a tumor of non-terminal respiratory unit origin. Mod Pathol 2012; 25: 1265–74. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y-W, Yeh Y-C. Ciliated muconodular papillary tumors of the lung. Arch Pathol Lab Med 2019; 143: 135–9. [DOI] [PubMed] [Google Scholar]
- 14.Boers JE, den Brok JL, Koudstaal J, Arends JW, Thunnissen FB. Number and proliferation of neuroendocrine cells in normal human airway epithelium. Am J Respir Crit Care Med 1996; 154: 758–63. [DOI] [PubMed] [Google Scholar]
- 15.Cutz E, Pan J, Yeger H, Domnik NJ, Fisher JT. Recent advances and contraversies on the role of pulmonary neuroepithelial bodies as airway sensors. Semin Cell Dev Biol 2013; 24: 40–50. [DOI] [PubMed] [Google Scholar]
- 16.Branchfield K, Nantie L, Verheyden JM, Sui P, Wienhold MD, Sun X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science 2016; 351: 707–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song H, Yao E, Lin C, Gacayan R, Chen M-H, Chuang P-T. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci USA 2012; 109: 17531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fisseler-Eckhoff A, Demes M. Neuroendocrine tumors of the lung. Cancers 2012; 4: 777–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rokicki W, Rokicki M, Wojtacha J, Dżeljijli A. The role and importance of club cells (Clara cells) in the pathogenesis of some respiratory diseases. Pol J Cardio-Thorac Surg 2016; 1: 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spella M, Lilis I, Pepe MAA et al. Club cells form lung adenocarcinomas and maintain the alveoli of adult mice. eLife 2019; 8: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers DF. Airway goblet cells: Responsive and adaptable front-line defenders. Eur Respir J 1994;7:1690–706. [PubMed] [Google Scholar]
- 22.MacDermed DM, Khodarev NN, Pitroda SP et al. MUC1-associated proliferation signature predicts outcomes in lung adenocarcinoma patients. BMC Med Genomics 2010; 3: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichinokawa H, Ishii G, Nagai K et al. Clinicopathological characteristics of primary lung adenocarcinoma predominantly composed of goblet cells in surgically resected cases: Adenocarcinoma with goblet cells. Pathol Int 2011; 61: 423–29. [DOI] [PubMed] [Google Scholar]
- 24.Tajima S, Kurabe N, Okudela K et al. Extensive goblet cell metaplasia of the peripheral lung may harbor precancerous molecular changes: comparison of two cases. Pathol Int 2014; 64: 533–8. [DOI] [PubMed] [Google Scholar]
- 25.Kim CFB, Jackson Erica L, Woolfenden Amber E et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005; 121: 823–35. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q, Liu K, Cui G et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat Genet 2019; 51: 728–38. [DOI] [PubMed] [Google Scholar]
- 27.Salwig I, Spitznagel B, Vazquez-Armendariz AI et al. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J 2019; 38: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J-H, Bhang DH, Beede A et al. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 Axis. Cell 2014; 156: 440–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dovey JS, Zacharek SJ, Kim CF, Lees JA. Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem cell expansion. Proc Natl Acad Sci 2008; 105: 11857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mainardi S, Mijimolle N, Francoz S, Vicente-Duenas C, Sanchez-Garcia I, Barbacid M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc Natl Acad Sci USA 2014; 111: 255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutherland KD, Song J-Y, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras–induced mouse lung adenocarcinoma. Proc Natl Acad Sci USA 2014; 111: 4952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Rock JR, Lu Y et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci 2012; 109: 4910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowbotham SP, Kim CF. Diverse cells at the origin of lung adenocarcinoma: Table 1. Proc Natl Acad Sci USA 2014; 111: 4745–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim CF. Intersections of lung progenitor cells, lung disease and lung cancer. Eur Respir Rev 2017; 26: 170054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res 2012; 72: 2457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skoulidis F, Byers LA, Diao L et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015; 5: 860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Fillmore Brainson C, Koyama S et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun 2017; 8: 14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Best SA, Ding S, Kersbergen A et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun 2019; 10: 4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer 2019; 19: 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ihle NT, Byers LA, Kim ES et al. Effect of KRAS Oncogene substitutions on protein behavior: implications for signaling and clinical outcome. JNCI J Natl Cancer Inst 2012; 104: 228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohashi K, Kammei R, Fujiwara Y et al. Induction of lung adenocarcinoma in transgenic mice expressing activated EGFR driven by the SP-C promoter. Cancer Sci 2008; 99: 1747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang WC, Shyh-Chang N, Yang H et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012; 148: 259–72. [DOI] [PubMed] [Google Scholar]
- 43.Xu X, Huang L, Futtner C et al. The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes Dev 2014; 28: 1929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feher J The Mechanics of breathing. Quan Human Physiol 2017;1: 623–32. [Google Scholar]
- 45.Veldhuizen EJ, Haagsman HP. Role of pulmonary surfactant components in surface film formation and dynamics. Biochim Biophys Acta 2000; 1467: 255–70. [DOI] [PubMed] [Google Scholar]
- 46.Weaver TE, Whitsett JA. Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem J 1991; 273: 249–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernhard W Regulation of surfactant-associated phospholipid synthesis and secretion. Fetal Neonatal Physiol 2017;1: 813–24.e6. [Google Scholar]
- 48.Chuquimia OD, Petursdottir DH, Periolo N, Fernández C. Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect Immun 2013; 81: 381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourdonnay E, Zasłona Z, Penke LRK et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med 2015; 212: 729–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Renewal of alveolar epithelium in the rat following exposure to NO2. Am J Pathol 1973; 70: 175–98. [PMC free article] [PubMed] [Google Scholar]
- 51.Evans MJ, Cabral LJ, Stephens RJ, Freeman G. Transformation of alveolar Type 2 cells to Type 1 cells following exposure to NO2. Exp Mol Pathol 1975; 22: 142–50. [DOI] [PubMed] [Google Scholar]
- 52.Barkauskas CE, Cronce MJ, Rackley CR et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 2013; 123: 3025–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zacharias WJ, Frank DB, Zepp JA et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018; 555: 251–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018; 359: 1118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dost AFM, Moye AL, Vedaie M et al. Organoids model transcriptional hallmarks of oncogenic KRAS activation in lung epithelial progenitor cells. Cell Stem Cell 2020; 27: 663–78.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]