

SUMMARY
In tumors, a subset of CD8+ T cells expressing the transcription factor TCF-1 drives the response to immune checkpoint blockade. We examined the mechanisms that maintain these cells in an autochthonous model of lung adenocarcinoma. Longitudinal sampling and single-cell sequencing of tumor-antigen specific TCF-1+ CD8+ T cells revealed that whereas intratumoral TCF-1+ CD8+ T cells acquired dysfunctional features and decreased in number as tumors progressed, TCF-1+ CD8+ T cell frequency in the tumor draining LN (dLN) remained stable. Two discrete intratumoral TCF-1+ CD8+ T cell subsets developed over time – a proliferative SlamF6+ subset and a non-cycling SlamF6- subset; blocking dLN egress decreased the frequency of intratumoral SlamF6+ TCF-1+ CD8+ T cells. Conventional type I dendritic cell (cDC1) in dLN decreased in number with tumor progression, and Flt3L+anti-CD40 treatment recovered SlamF6+ T cell frequencies and decreased tumor burden. Thus, cDC1s in tumor dLN maintain a reservoir of TCF-1+ CD8+ T cells and their decrease contributes to failed anti-tumor immunity.
Graphical Abstract
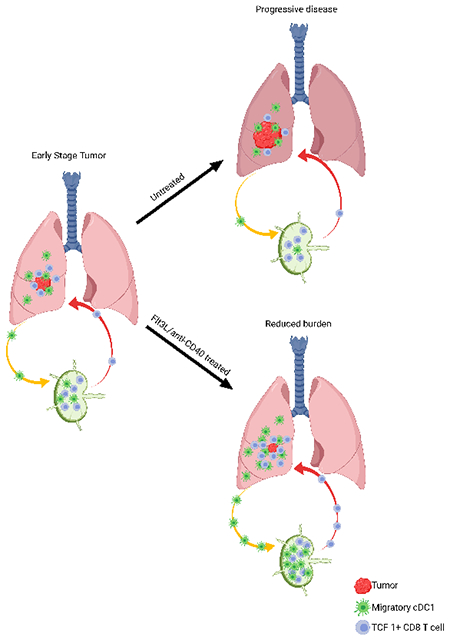
eTOC:
In tumors, TCF-1+ CD8+ T cells drive the response to immune checkpoint blockade. Schenkel, Herbst et al. reveal that, in lung adenocarcinoma, a reservoir of TCF-1+ CD8+ T cells is maintained in tumor draining lymph nodes by conventional type I dendritic cell (cDC1). Decrease of these cDC1 as the tumor progresses contributes to failed anti-tumor immunity.
INTRODUCTION
After arriving in a tissue, effector CD8+ T cells kill cells that present cognate peptide bound to major histocompatibility class I (MHC-I (Masopust and Schenkel, 2013; Mueller et al., 2013). When pathogens or tumors persist, however, CD8+ T cells become dysfunctional, increasing and sustaining expression of inhibitory receptors (IRs) (e.g., PD-1, Lag-3, Tim-3), and losing the ability to proliferate, secrete cytokines, and kill cells (Blank et al., 2019; Wherry and Kurachi, 2015). The acquisition of T cell dysfunction is progressive, developing over the course of weeks to months (Angelosanto et al., 2012; Pauken et al., 2016; Penaloza-MacMaster et al., 2015; Philip et al., 2017; Schietinger et al., 2016). Importantly, although dysfunctional T cells are not as protective as bona fide effector or memory T cells, they are not inert (Kallies et al., 2020; Pauken and Wherry, 2015). Dysfunctional T cells represent a significant therapeutic opportunity, as restoring effector potential to these T cells can provide potent anti-tumor immunity (Blank et al., 2019; Kallies et al., 2020; Pauken and Wherry, 2015). While early successes have provided proof of concept that immunotherapy, particularly checkpoint blockade, can drive productive T cell responses against tumors, most cancer patients do not show durable clinical responses (Ribas and Wolchok, 2018; Sharpe and Pauken, 2017; Wei et al., 2018). Thus, a better understanding of CD8+ T cell dysfunction during tumor development and of the barriers that prevent the successful harnessing of responding T cells is paramount to improving outcomes for more cancer patients.
CD8+ T cells arising during chronic infection or in tumors can be epigenetically sustained in a state of dysfunction (Pauken et al., 2016; Philip et al., 2017; Sen et al., 2016). Moreover, studies have revealed two subsets within chronically stimulated CD8+ T cells (He et al., 2016; Im et al., 2016; Miller et al., 2019; Paley et al., 2012; Siddiqui et al., 2019; Utzschneider et al., 2016). One subset expresses the IR Tim-3, has low proliferative potential and poor cytokine production, but mediates contact-dependent cytotoxicity. The other expresses the transcription factor TCF-1, and is less dysfunctional, maintaining some degree of proliferative potential and cytokine function. TCF-1+ CD8+ T cells are critical for maintaining the Tim-3+ population (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016), and the TCF-1+ subset is preferentially reinvigorated after checkpoint blockade (He et al., 2016; Im et al., 2016; Kurtulus et al., 2019; Miller et al., 2019; Sade-Feldman et al., 2018; Siddiqui et al., 2019). These studies have demonstrated residual activity and complementary functions within a heterogenous population of responding CD8+ T cells, with implications for the efficacy of checkpoint blockade.
Most preclinical models of anti-tumor CD8+ T cells use transplantable cancer cells that are injected subcutaneously into mice. Such models have short experimental timescales of a few weeks, which limits their utility. Genetically engineered mouse models (GEMMs), where the tumor is induced in vivo, represent a complementary approach. One such model is the mutant Kras, loss of p53 (KP) GEMM of lung adenocarcinoma (DuPage et al., 2011; DuPage et al., 2012; Jackson et al., 2005). Addition of two CD8+ T cells antigens to the C-terminus of luciferase, SIINFEKL and SIYRRGYL (LucOS), results in a trackable anti-tumor CD8+ T cell response (DuPage et al., 2011; DuPage et al., 2012). Importantly, tumor cell expression of the SIINFEKL and SIYRRGYL antigens is maintained throughout tumor progression (DuPage et al., 2011). While CD8+ T cells are initially able to slow growth and progression of KP lung tumors, the response ultimately falters, and LucOS tumors reach similar sizes and grades as tumors that do not express strong T cell antigens in late-stage disease (DuPage et al., 2011; DuPage et al., 2012).
The KP GEMM of lung adenocarcinoma is an excellent model for interrogating the immune response to lung cancer because it (1) recapitulates human disease in the native lung environment, (2) follows a stereotypical progression from hyperplasia to adenocarcinoma, and (3) has a prolonged course, developing over several months (DuPage et al., 2011; DuPage et al., 2012; Jackson et al., 2005; Li et al., 2019). Here, we interrogated how CD8+ T cell dysfunction changed in KP tumors over several months. In early stage adenomas, we observed a bifurcation of the tumor-specific CD8+ T cell response into a TCF-1+ population a TIM-3+ dysfunctional population, consistent with previous studies (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016). However, the TCF-1+ CD8+ T cell population numerically and functionally eroded over time. Single cell RNA sequencing revealed discrete subpopulations of TCF-1+ CD8+ T population - a cycling SlamF6+ cells and a non-cycling SlamF6− cells, with the latter population being dominant in later stage disease. Further, the tumor-draining lymph node (dLN) acted as a reservoir for the more functional SlamF6+ TCF-1+ CD8+ T cell population. Concurrent with the erosion of the anti-tumor CD8+ T cell response in the lung, the number and stimulatory phenotype of migratory cDC1s entering the dLN decreased over time. Therapeutically boosting the migratory cDC1 population with a combination of Flt3L and agonistic CD40 antibody resulted in improved stimulation of the SlamF6+ TCF-1+ CD8+ in the dLN, which resulted in improved T cell trafficking into the tumor and reduced tumor burden. Collectively, these data highlight the dynamic nature of the TCF-1+ CD8+ T cell population in slowly progressing tumors, and point to the therapeutic potential of targeting the migratory cDC1 axis to mobilize the reservoir of SlamF6+ TCF-1+ CD8+ T cells in the dLN.
RESULTS
Tumor-specific CD8+ T cells become dysfunctional during progression of lung adenocarcinoma
To determine the kinetics of the T cell response in the KP model of lung adenocarcinoma, we intratracheally injected mice with lentivirus expressing LucOS to induce tumors (DuPage et al., 2011), and assessed tumor-specific CD8+ T cells using SIINFEKL peptide-MHC class I (pMHC-I) tetramers (Figure 1A). We found tumor-specific CD8+ T cells in tumor-bearing lungs by flow cytometry, and immunofluorescence microscopy showed these cells localize within tumors (Figure 1B). Both the number and proportion of tumor-specific CD8+ T cells peaked at 5 weeks and remained elevated until week 8, after which it declined (Figure 1C). In parallel to the contraction of the CD8+ T cell response, tumors grew, progressing from ~2.5% of total lung area at 5 weeks post-initiation to ~22% of total lung area by 16 weeks post-initiation (Figure S1A).
Figure 1. Tumor-specific CD8+ T cells become dysfunctional during progression of lung adenocarcinoma.
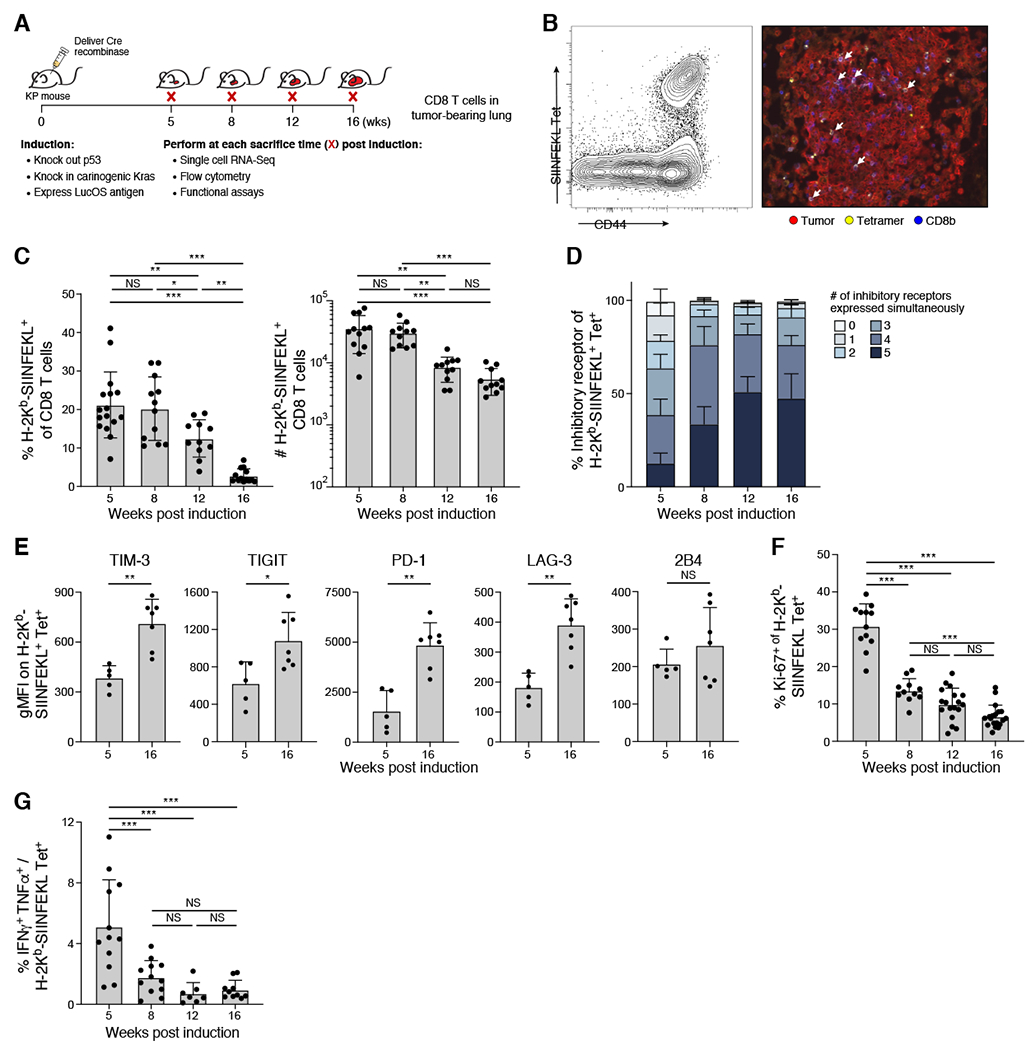
(A) Experimental schematic. Leukocytes were isolated from lungs. (B) Tumor-specific CD8+ T cells infiltrated tumors. Representative H-2Kb/SIINFEKL tetramer staining by flow cytometry (left, gated on CD8+ T cells) and by immunofluorescence microscopy (right). (C) Percent (left) and number (right) of tumor specific CD8+ T cells of total CD8+ T cells throughout KP tumor development. (D-E) tumor specific CD8+ T cells upregulated number (D) and level (E) of IR expression over time. (D) Percent of tumor-specific CD8+ T cells expressing combinations (indicated by color) of IR by flow cytometry (TIM-3, TIGIT, PD-1, LAG-3, 2B4). (E) IR geometric MFI at 5 and 16 weeks on tumor specific CD8+ T cells from 1 experiment, representative of 2 experiments (dot: one mouse), Mann-Whitney test. (F) Percent Ki-67+ of tumor specific CD8+ T cells at indicated weeks. (G)Ratio of IFNγ+ TNFα+ to H-2Kb/SIINFEKL tetramer+ of CD8+ T cells after in-vitro peptide stimulation. *p<0.05, **p<0.01, ***p<0.001, ns=not significant (p>0.05). Tukey’s multiple comparisons test unless otherwise noted. See also Supplemental Figure 1.
We hypothesized that the tumor-specific CD8+ T cell population would become more dysfunctional with tumor progression. Supporting this hypothesis, the portion of tumor specific CD8+ T cells co-expressing IRs increased with time (Figure 1D) (Blank et al., 2019; Wherry and Kurachi, 2015). The proportion of tumor-specific CD8+ T cells co-expressing PD-1, Lag-3, Tim-3, TIGIT and 2B4, increased from 10% at 5 weeks post initiation, to ~50% by week 12 (Figure 1D and S1B, Two way ANOVA, p<.001). The expression of different IRs was higher at week 16 vs. week 5 (Figure 1E and S1C, Mann Whitney, PD-1 p<0.01, Lag-3 p<0.01, Tim-3 p<0.01, TIGIT p<0.05, 2B4 not significant). Additionally, tumor-specific CD8+ T cells developed canonical features of dysfunction by 8 weeks, including decreases in proliferation and cytokine production, which persisted thereafter. The proportion of cycling tumor-specific CD8+ T cells was decreased 4.7 fold in 16 vs. 5 weeks (~30% to ~6.5%, Figure 1F and S1D, One way ANOVA, p<0.001). Additionally, following ex vivo simulation with SIINFEKL peptide, a minority of CD8+ T cells produced TNFα and IFNγ at 5 weeks, which decreased and remained low at 8, 12 and 16 weeks (Figure 1G and S1E). Collectively, these data suggest that tumor-specific CD8+ T cells become progressively dysfunctional in the KP tumor model.
Single cell RNA-seq highlights distinct effector and dysfunction programs that arise during tumor progression in tumor-specific CD8+ T cells
To identify gene programs associated with the dysfunctional states we observed in tumor-specific CD8+ T cells in KP tumors, we performed full-length, plate-based single-cell RNA-Seq (sc-RNAseq) at 5, 8, 12 and 20 weeks post-initiation on sorted H-2Kb SIINFEKL Tetramer+ and Tetramer− CD8+ T cells (Figure 2A–C, STAR Methods). While CD8+ T cells did segregate by tetramer binding (Figure 2A and 2B), the cells did not segregate into discrete clusters but instead occupied transcriptional states across a continuum. To identify relevant cellular states and transcriptional programs associated with tumor reactivity over the course of tumor progression, we performed topic modeling using latent dirichlet allocations (Dey et al., 2017; Pritchard et al., 2000).
Figure 2. Single cell RNA-seq highlights distinct effector and dysfunction programs that arise during tumor progression in tumor-specific CD8+ T cells.
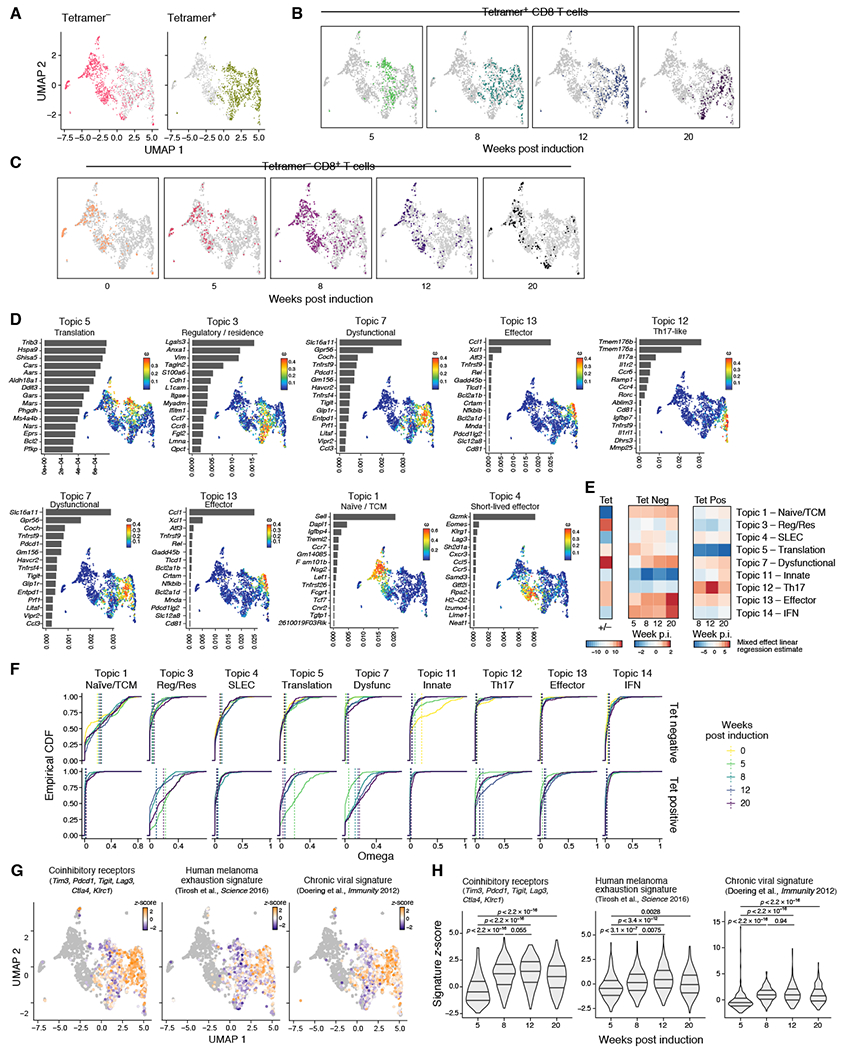
Data showing results for plate-based scRNA-seq data. (A) UMAP embedding of all CD8+ T cells colored by tetramer binding. (B&C) UMAP embedding of all CD8+ T cells where (B) tetramer+ and (C) tetramer− CD8+ T cells are colored and faceted by weeks p.i. (D) Topic modeling of tumor specific-CD8+ T cells profiled by plate based scRNA-seq. Shown is a bar plot of topic scores for top ranked genes (left), and UMAP of the cell profiles (as in A) colored by topic’s weight per cell (right). (E) Heatmap showing the estimate of a linear mixed effect model on the topic (rows) weights by tetramer binding (left), by timepoint for tetramer− (left) and tetramer+ (right). (F) Empirical cumulative distribution functions (ECDFs) of topic weights across timepoints (color) and tetramer staining (rows). Average topic weights by timepoint are indicated with vertical dotted lines. TCM – Central Memory, SLEC – Short lived effector, Dysfunc – Dysfunctional, Res - Resident, Reg – Regulatory. (G) UMAP embedding where tumor-specific CD8+ T cells are colored by respective signature (as indicated) z-score and tetramer− CD8+ T cells in grey. (H) Quantification of signature z-scores as shown in Figure 2G. Violin plot of z-scores across time points. Wilcox-test.
An effector-like Topic (13) and a dysfunctional-like Topic (7) were specifically expressed in tumor-specific CD8+ T cells, where they emerged at 8 weeks post-initiation (Figure 2D–F). Moreover, the expression of other known signatures of T cell dysfunction was increased in tumor specific CD8+ T cells as early as 8 weeks post-induction (Figure 2G and 2H, Table S1). Taken together, the sustained expression of IRs, loss of cell cycle, decreased ability to secrete cytokines, and transcriptional programs were consistent with the acquisition of dysfunction during tumor progression.
TCF-1+ tumor-specific CD8+ T cells become increasingly dysfunctional over time
Because phenotypic, functional, and transcriptional features of dysfunction developed in the tumor-specific CD8+ T cell population between 5 and 8 weeks, we investigated whether a gradual program mediated these alterations during early stages of tumor growth. We generated a higher resolution scRNA-Seq map of tumor-specific CD8+ T cells at 6, 7, 8 and 12 weeks post-initiation using droplet-based scRNA-seq (Figure 3A). Topic modeling revealed several different effector and dysfunctional programs (Figure S2A, Table S1), whose expression in cells changed over time (Figure S2B–C), many consistent with topics from our plate-based scRNA-seq (Figure S2D).
Figure 3. TCF-1+ tumor-specific CD8 T cells become increasingly dysfunctional over time.
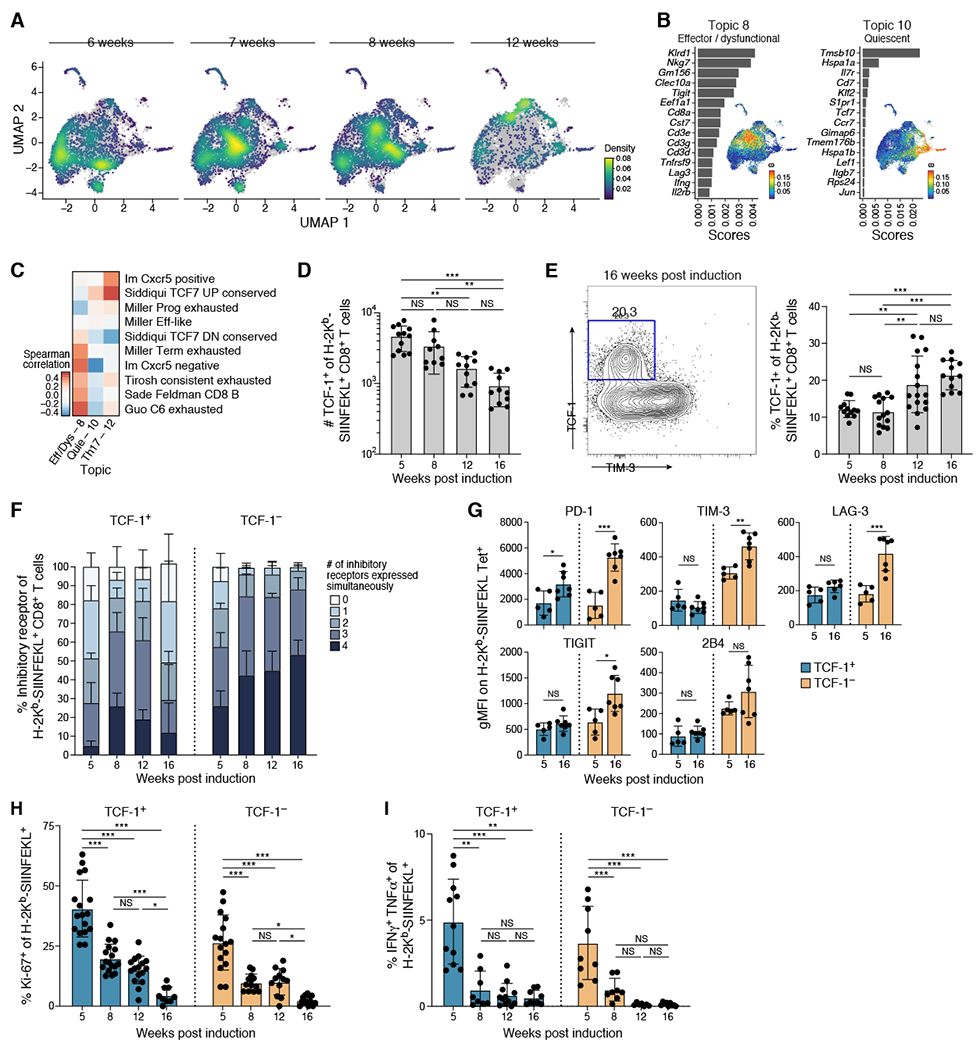
(A) UMAP embedding of tumor specific CD8+ T cells (Bodhankar et al.) profiled with droplet-based scRNA-seq colored by cell density and faceted by weeks. (B) Topic modeling identified a precursor-like (Topic 10) and dysfunctional (Topic 8) transcriptional program of tumor-specific CD8+ T cells. Shown is a bar plot of topic scores for top ranked genes (left), and UMAP of the cell profiles (as in A) colored by topic’s weight per cell (right). (C) Topic expression correlated with signatures associated with different functional states. Spearman correlation coefficient (color bar) between topic (columns) weights and signature z-score across cells for published signatures (rows) for droplet-based scRNA-seq data. Quie – Quiescent, Eff – Effector, Dys – Dysfunctional. (D-E) TCF-1+ tumor specific CD8+ T cells decreased in absolute numbers (D) but increased in fraction (E) over tumor development. (D) Number of TCF-1+ tumor specific CD8+ T cells at indicated timepoints. (E) Representative flow cytometry staining for TCF-1 and Tim-3 on tumor-specific CD8+ T cells (left) and % TCF1+ of tumor specific CD8+ T cells at indicated timepoints (right). (F-G) TCF1− tumor-specific CD8+ T cells upregulated number (F) and level (G) of IR expression over time unlike TCF1+ tumor-specific CD8+ T cells. (F) Percent of tumor-specific CD8+ T cells expressing combinations of IR by flow cytometry (TIGIT, PD-1, LAG-3, 2B4). (G) IR geometric MFI at 5 and 16 weeks on tumor-specific CD8+ T cells from 1 experiment, representative of 2 experiments (dot: one mouse), Mann-Whitney test. (H-I) Both TCF1+/− tumor-specific CD8+ T cells lost ability to proliferate (H) and produce cytokine (I) with time. (H) Percent Ki-67+ of TCF1+/− tumor-specific CD8+ T cells at indicated weeks. (I) Ratio of IFNγ+ TNFα+ to H-2Kb/SIINFEKL tetramer+ of CD8+ T cells. Data from at least 3 experiments unless otherwise noted (dot: one mouse), Error bars: SD. *p<0.05, **p<0.01, ***p<0.001, Tukey’s multiple comparisons test unless otherwise noted. NS: non-significant (p>0.05). See also Supplemental Figures 2 and 3.
Among the key programs were a canonical dysfunctional-like program (Topic 8), including Tigit and Lag3, and a quiescent-like program (Topic 10), including Tcf7 (Figure 3B and S2E). TCF-1+ cells were previously described as “progenitor” dysfunctional CD8+ T cells which maintain residual proliferative capacity, cytokine secretion, and responsiveness to checkpoint blockade immunotherapy despite chronic activation (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016). The scores of the quiescent-like program (Topic 10) and a Th17-like program (Topic 12) across our cells correlated with those of previous signatures associated with the Tcf7+ progenitor-like population, while those of an effector/dysfunctional program (Topic 8), including multiple IR genes, correlated with those of previous programs defined for terminally dysfunctional-like Havcr2+ cells, including from human tumors (Figure 3C and S2F, Table S1). Moreover, longitudinal analysis revealed that progenitor-like Topics 10 and 12, together with a Tcf7+ signature, increased with time, while the more dysfunctional-like Topic 8 peaked at week 8 and then decreased by week 12, suggesting that these cell states are differentially modulated as tumors progressed (Figure S2C and S2G). Thus, the tumor-specific CD8+ T cells diversity in the KP-LucOS model grossly encompassed the transcriptional programs of T cells in previous models and human tumors.
Next, we confirmed the presence of TCF-1+ tumor-specific CD8+ T cells by flow cytometry, showing that while their numbers steadily decreased between weeks 5 and 16 (Figure 3D), their fraction among tumor-specific CD8+ T cells increased over time (Figure 3E). Consistent with previous reports, fewer TCF-1+ tumor-specific CD8+ T cells expressed four IRs than the TCF-1− counterparts (Figure 3F, Two way ANOVA, 5, 8, 12, and 16 weeks p<0.001), and followed a different temporal pattern. IR expression increased from week 5 to 8, but gradually decreased by week 16, while in TCF-1− cells the number and level of IRs increased throughout the course of progression (Figure 3G and S3A). Collectively, these data suggest a more limited functional change to the TCF-1+ of tumor specific CD8+ T cells as tumors grow out.
TCF-1+ dysfunctional CD8+ T cells maintain some residual self-renewal and cytokine-secreting capacities, while TCF-1− counterparts have greater cytotoxic potential (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016). Consistent with these observations, TCF-1+ tumor-specific CD8+ T cells had a higher initial proportion of Ki-67+ cells at 5 weeks compared to TCF-1− counterparts (1.72 fold, One way ANOVA, p<0.001) (Figure 3H and S3B). However, the fraction of cycling TCF-1+ tumor-specific CD8+ T cells decreased ~7.5 fold between weeks 5 and 16 (Figure 3H). Moreover, the fraction of cycling TCF-1+ CD8+ T cells was not different than that of cycling TCF-1− CD8+ T cells at 16 weeks (p=0.55), suggesting that the higher residual proliferative capacity of TCF-1+ CD8+ T cells was not sustained long term. Further, while both TCF-1+ and TCF-1− CD8+ T cells could produce IFNγ and TNFα after in-vitro stimulation with SIINFEKL peptide at 5 weeks to a similar extent (Figure 3I and S3C–D, one way ANOVA, not significant), this decreased in both subsets by week 8, and remained low thereafter (Figure 3I). Nevertheless, TCF-1− tumor-specific CD8+ T cells expressed more granzyme B than their TCF-1+ counterparts, consistent with greater cytotoxicity (Figure S3E). Taken together, these data suggested that the residual functionality of TCF-1+ CD8+ T cells eroded over time despite sustained TCF-1 expression, increasingly resembling their TCF-1− counterparts.
TCF-1+ tumor-specific CD8+ T cells are heterogeneous and appear more quiescent over the course of tumor progression
The erosion in functionality in TCF-1+ tumor-specific CD8+ T cells can arise due to either a homogenous shift towards a dysfunctional, cell-intrinsic program or a change in the proportion of different subsets within TCF-1+ cells with different functional capacities. To determine if the composition of TCF-1+ tumor-specific CD8+ T cells changes during tumor progression, we analyzed the subset of cells in the sc-RNAseq dataset that expressed Tcf7 (Figure 4A), or that were highly weighted for Topic 10 (quiescent) and 12 (Th-17 like) (Figure S2B), and found several patterns of transcriptional changes with time (Figure 4B,C and S4A,B, Table S1). In particular, effector-like Topic 12 (with chemotactic and inflammatory cytokines and co-stimulatory molecules) decreased with time, while quiescent-like Topic 3 increased (DuPage et al., 2011). Other programs, including the regulatory-like Topic 9, which contained galectins and the anti-inflammatory cytokine Fgl2, changed less consistently during progression (Henderson and Sethi, 2009; Sundblad et al., 2017). Taken together, the changes reflect fundamental alterations in T cell state during tumor progression.
Figure 4. TCF-1+ tumor-specific CD8 T cells are transcriptionally diverse and shift over the course of tumor progression.
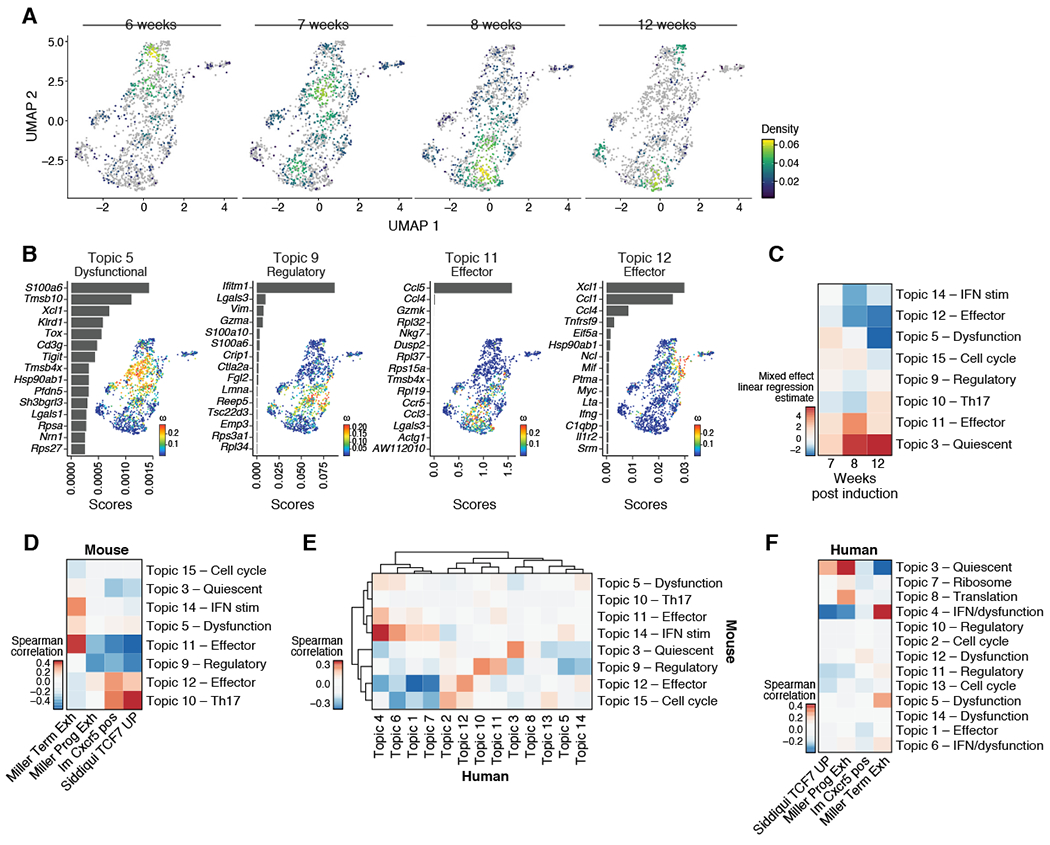
(A) UMAP embedding of all Tcf7+ tumor-specific CD8+ T cells (Bodhankar et al.) colored by cell density and faceted by time point. (B) Topic modeling of Tcf7+ tumor-specific CD8+ T cells. Shown is a bar plot of topic scores for top ranked genes (left), and UMAP of the cell profiles (as in A) colored by topic’s weight per cell (right). (C) Heatmap showing the estimates per time points (columns) relative to 6 weeks p.i. of a linear mixed effect model on the topic (rows) weights. (D) Only two topics of Tcf7+ tumor-specific CD8+ T cells correlated with reported signatures of precursor-exhausted Tcf7+ CD8+ T cells. Spearman correlation coefficient (color bar) between topic (rows) weights and signature (columns) z-score across cells for published signatures of Tcf7+ tumor-specific CD8+ T cells. (E-F) Data taken from Sade-Feldman et al., 2018. (E) Topics identified in Tcf7+ tumor-specific CD8+ T cells of this manuscript correlated with topics identified in human melanoma patients. Heatmap shows the spearman correlation coefficient (color bar) between human inferred topic (columns) weights and signature z-score when taking top 50 feature genes of mouse topics (rows) across TCF7+ CD8+ T cells of human melanoma patients. (F) Few topics of TCF7+ CD8+ T cells of human melanoma patients correlated with reported signatures of precursor-exhausted Tcf7+ CD8+ T cells in mice. Spearman correlation coefficient (color bar) between topic (rows) weights and signature (columns) z-scores across cells for published signatures for human Tcf7+ tumor specific CD8+ T cells. See also Supplemental Figure 4.
We next examined how the heterogeneity within TCF-1+ tumor-specific CD8+ T cells in KP-LucOS tumors related to previous bulk and single cell RNA-seq derived signatures of Tcf7+ CD8+ T cells from other mouse tumor models or chronic infection models. Comparing the Topics to these signatures derived from more traditional “hard clustering” methods allowed for more precise overlap assessment between the different Tcf7+ subpopulations identified in our model with progenitor-like populations identified in the literature. Compared to published signatures from Tcf7+ CD8+ T cells from mouse models (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019), only the cell scores for Th17-like Topic 10 and effector-like Topic 12 correlated well with previous Tcf7 signatures from the literature, even though all cells expressed Tcf7 (Figure 4D). Conversely, the cell scores for Topic 5 (dysfunctional-like), 11 (effector-like) and 14 (interferon stimulated) correlated better with scores for a signature derived from terminally dysfunctional Havcr2+ CD8+ T cells (Figure 4D). These data were consistent with the notion that the Tcf7+ subpopulation in the KP LucOS model were diverse and dynamic, and acquired gene programs that more closely resembled terminally dysfunctional populations.
In order to understand how Tcf7+ Topics related to Tcf7+ populations identified in human tumors, we performed Topic modeling on scRNA-seq data from Tcf7+ CD8+ TILs from patients with advanced melanoma (Sade-Feldman et al., 2018; Tirosh et al., 2016), and performed a Topic by Topic comparison. Here, we found that the variation we observed in mouse tumors corresponded to Topics of Tcf7+ CD8+ TILs in human tumors (Sade-Feldman et al., 2018; Tirosh et al., 2016). Programs identified by topic modeling of Tcf7+ CD8+ T cell from human melanoma included effector-like (Topic 12), quiescent-like (Topic 3), and interferon stimulated-like topics (Topics 4 and 6), all correlated with those identified in mouse Tcf7+ tumor-specific CD8+ T cells (Figure 4E, S4C–E, Table S1). Further, the human programs largely did not correspond to previous bulk Tcf7+ signatures from mouse models (Figure 4F and S4F), highlighting the utility of using Topic modeling to more finely dissect the transcriptional programs contributing to these CD8+ T cell subpopulations. Collectively, these data support a model where the Tcf7+ CD8+ TIL population can be highly dynamic and diverse – While some of these cells may display a “progenitor-like” or “quiescent” transcriptional program, others may be more effector-like or dysfunctional. Moreover, these data highlights the utility of defining the active transcriptional state within a cell, as expression of Tcf7 in our dataset is not synonymous with quiescence or a progenitor-like state.
SlamF6 expression distinguishes two functionally distinct populations within the TCF-1+ population
Because previous Tcf7 signatures failed to adequately recapitulate the heterogeneity encompassed within the Tcf7 compartment in our dataset, we next interrogated whether this lack of concordance was due to variable expression of signature genes. This analysis revealed that 34 out of 206 Tcf7+ signature genes detected in our data were expressed with higher relative variance across our Tcf7+ tumor-specific CD8+ T cells (Figure S5A, Methods). Slamf6, a member of the CD2 immunoglobulin superfamily, was one of those 34 variable genes (Figure S5A) and its expression decreased over time (Figure 5A and S5B). We validated the progressive loss of SlamF6 protein on TCF-1+ tumor specific CD8+ T cells by flow cytometry (~35% decrease from week 5 to week 8, which continued into week 16) (Figure 5B). Several topics were differentially weighted as a function of Slamf6 expression when accounting for cell complexity differences (UMI) (Methods, Figure S5C). In particular, Topic 10 (Th17-like) was higher and Topics 9 (regulatory-like) and 11 (effector-like) were lower weighted as a function of Slamf6 expression, especially early, suggesting functional differences between Slamf6+ and Slamf6− cells. To further explore functional distinctions of Tcf7+ tumor-specific CD8+ T cells expressing Slamf6, we identified genes that correlated with Slamf6 expression across the cells (Figure S5D). Lower Slamf6 expression was correlated with higher expression of the genes encoding the chemokines Ccl3, Ccl4, and Ccl5 (Figure S5D,E), an inhibitory ligand for Lag-3, Lgals3, and the immunosuppressive cytokine Fgl2 (Figure S5D,E). We confirmed this at the protein level for CCL5 and Galectin 3 (encoded by Lgals3) (Figure 5C,D and S5F). Overall, these data suggest that SlamF6+ vs. SlamF6− TCF-1+ tumor-specific CD8+ T cells may play different roles in modulating immune cells at the tumor site. We next determined whether these putative functional differences in the Slamf6+ and Slamf6− cells could be due to differences in their TCRs. Because Ccl5 was anti-correlated with Slamf6 expression, we used Ccl5 expression to identify bona-fide Slamf6− cells. We found that Slamf6+ and Ccl5+ cells, by and large, shared clonotypes, suggesting that differences in their TCR could not account for the transcriptional differences seen and that the cells were derived from similar progenitors (Figure S5G).
Figure 5. SlamF6 expression distinguishes two functionally distinct populations within the TCF-1+ population.
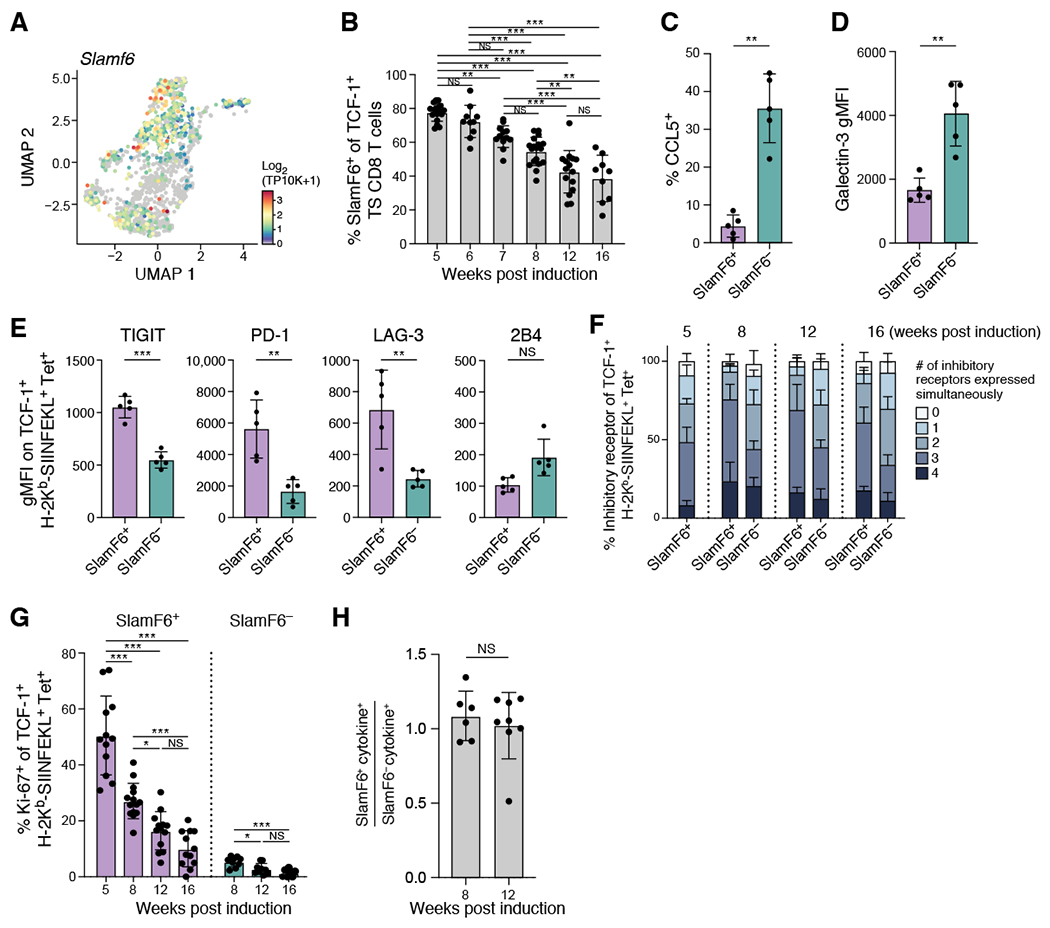
(A) Slamf6 was variably expressed among Tcf7+ tumor-specific CD8+ T cells. UMAP embedding of all Tcf7+ tumor specific CD8+ T cells colored by Slamf6 expression. (B) Percent SlamF6+ of TCF-1+ tumor specific CD8+ T cells decreased steadily with tumor development. Percent shown by flow cytometry at the indicated week. (C) Percent CCL5+ of SlamF6+ or SlamF6− TCF-1+ tumor-specific CD8+ T cells at 8 weeks post-initiation. (D) Geometric MFI of Galectin-3+ of SlamF6+ or SlamF6− TCF-1+ tumor-specific CD8+ T cells at 8 weeks post-initiation. (E-F) SlamF6+ expressed IR at higher levels (E) and more combinations (F) compared to SlamF6− early. (E) IR geometric MFI at 8 weeks on SlamF6+ and SlamF6− TCF-1+ tumor specific CD8+ T cells from 1 experiment, representative of at least 3 experiments (dot: one mouse), Mann-Whitney test. (F) Percent of tumor-specific SlamF6+ or SlamF6− TCF-1+ CD8+ T cells expressing combinations of IR by flow cytometry (TIGIT, PD-1, LAG-3, 2B4) at indicated week post induction. (G) Percent Ki-67+ of SlamF6+ or SlamF6− TCF-1+ tumor-specific CD8+ T cells at indicated week post induction. (H) Ratio of IFNγ+ TNFα+ SlamF6+to SlamF6− TCF-1+ CD8+ T cells. Data from at least 2 experiments unless otherwise noted (dot: one mouse), Error bars: SD. *p<0.05, **p<0.01, ***p<0.001, Tukey’s multiple comparisons test. NS: non-significant (p>0.05). See also Supplemental Figures 5 and 6.
To test the hypothesis that SlamF6+ and SlamF6− populations were functionally distinct, we next examined IR expression and found that SlamF6+ TCF-1+ CD8+ T cells expressed 3 or more IRs more frequently and at higher levels than SlamF6− TCF-1+ CD8+ T cells (Figure 5E,F, Wilcoxon test, week 8 p<0.01, week 12 p<0.01, week 16 p<0.05; and S6A–D, paired t test, PD-1 p<0.01, Lag-3 p<0.01, TIGIT p<0.001, 2B4 not significant). Only 2B4 was more highly expressed in SlamF6− cells than SlamF6+ cells. While SlamF6− cells expressed fewer of several IRs and at lower levels than SlamF6+ cells, they did express IRs such as PD-1, Lag-3 and TIGIT at higher levels than naïve CD8+ T cells (Figure S6B,C, one-way ANOVA, PD-1 p<0.01, Lag-3 p<0.01, TIGIT p<0.01). Additionally, SlamF6+ TCF-1+ CD8+ T cells were more proliferative than SlamF6− counterparts at all time points (Figure 5G and S6E), but their proliferation decreased with tumor progression. Conversely, while SlamF6+ and SlamF6− populations had comparable ability to secrete IFNγ and TNFα (Figure 5H), these levels were low (~1%) at both 8 and 12 weeks (Figure S6F). As expected, neither population expressed significant levels of granzyme B (Figure S6G). Taken together, the SlamF6+ and SlamF6− TCF-1+ tumor-specific CD8+ T cell populations appear phenotypically and functionally distinct, and may have different roles in promoting and maintaining the anti-tumor response.
LN-derived TCF-1+ tumor-specific CD8+ T cells are continuously recruited to the tumor
Considering the phenotypic, functional and quantitative differences between SlamF6+ and SlamF6− TCF-1+ CD8+ T cells, we were interested in whether this heterogeneity originated in the tumor microevironment, or whether the diversification of the TCF-1 subset occurried earlier during CD8+ T cell differentiation in the dLN. Previous studies have shown that TCF-1+ CD8+ T cells are more numerically abundant in secondary lymphoid organs, while more dysfunctional CD8+ T cell populations predominate in non-lymphoid tissues and tumors (Paley et al., 2012; Siddiqui et al.). Additionally, the dLN is critical in the efficacy of checkpoint blockade, and responding anti-tumor CD8+ T cells can be identified in peripheral blood after treatment (Fransen et al., 2018; Valpione et al., 2020; Wu et al., 2020; Yost et al., 2019). Considering these findings, we hypothesized that the dLN could serve as a reservoir for the more functional TCF-1+ SlamF6+ CD8+ T cell subset.
We found that the number of tumor-specific CD8+ T cells in the dLN, and the proportion of TCF-1+ cells among them (~75%) did not change during tumor progression (Figure 6A and 6B, one way ANOVA, no significant differences). Moreover, the vast majority (>90%) of TCF-1+ tumor-specific CD8+ T cells expressed SlamF6 throughout (Figure S7A,B one way ANOVA, no significant differences). The percentage of dLN tumor-specific CD8+ T cells that expressed 3 or more IRs increased 20-30% from week 5 to week 8, but remained stable there after (Figure 6C, S7C). Additionally, the ability of dLN tumor-specific CD8+ T cells to secrete cytokines after restimulation (~10% of tumor-specific CD8+ T cells; Figure 6D) remained constant throughout. Interestingly, however, there was a decrease in the portion of proliferative (Ki-67+) tumor-specific CD8+ T cells in the dLN, from ~36% at 5 weeks to ~8% by 8 weeks and beyond (Figure 6E). The fraction of cycling tumor-specific CD8+ T cells in peripheral blood followed the same temporal pattern (Figure 6F). Consistently, the proportion of SlamF6+ cells among blood TCF-1+ tumor-specific CD8+ T cells steadily decreased over several weeks (Figure 6G), in a pattern that roughly matched the decrease in SlamF6+ CD8+ T cells in tumor-bearing lungs (Figure 5B). This suggested that the dLN may be an important reservoir of SlamF6+ tumor specific CD8+ T cells.
Figure 6. dLN-derived TCF-1+ tumor-specific CD8+ T cells are continuously recruited to the tumor microenvironment.
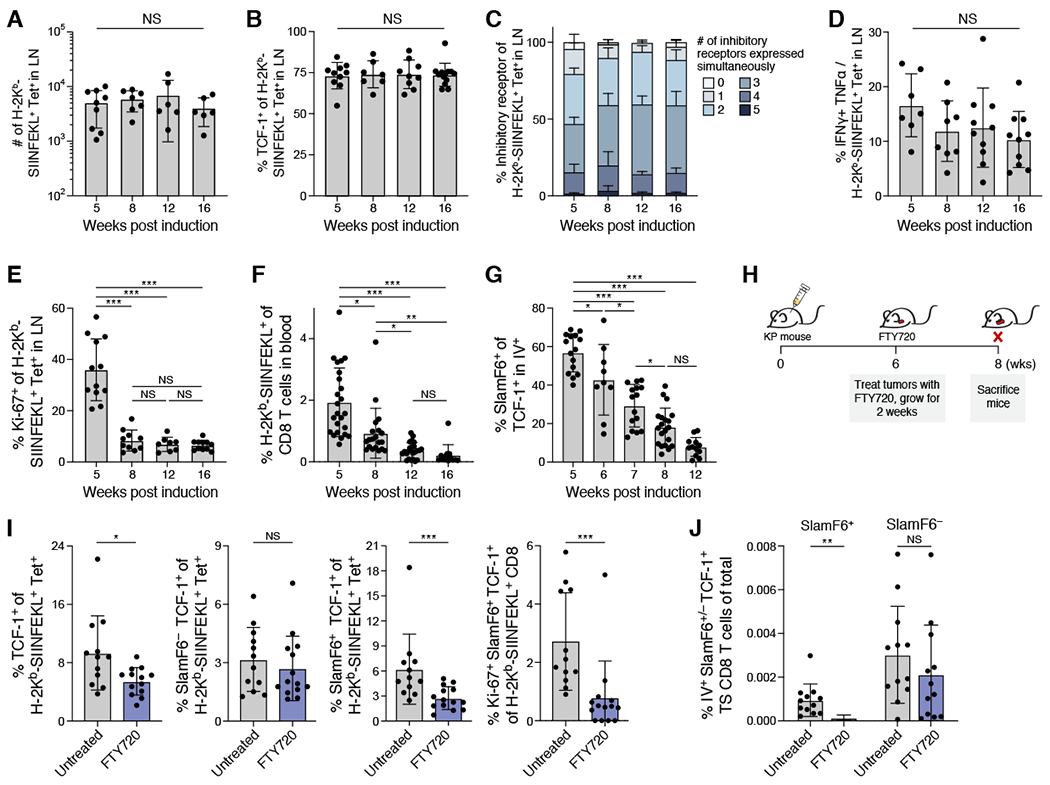
(A) Number of tumor-specific CD8+ T cells in the dLN remained stable over time. (B) Percent TCF-1+ of tumor specific CD8+ T cells in the dLN by flow cytometry over time. (C) Percent of tumor-specific CD8+ T cells expressing combinations of IRs (color bar) by flow cytometry (TIGIT, PD-1, LAG-3, 2B4) in the dLN. (D) Ratio of IFNγ+ TNFβ+ to H-2Kb/SIINFEKL tetramer of CD8+ T cells over time. (E) % Ki-67+ of tumor-specific CD8+ T cells in dLN at indicated timepoints. (F) % tumor-specific CD8+ T cells of total CD8+ T cells in blood at the indicated timepoint. (G) % SlamF6+ of TCF-1+ tumor-specific CD8+ T cells in blood. (H) Blocking egress from dLN; FTY720 experimental schematic. (I) Treating mice with FTY720 significantly reduced fraction of SlamF6+ cells, in particular Ki-67+ SlamF6+, but not SlamF6− cells. From left to right; % TCF-1+ of tumor specific CD8+ T cells, % SlamF6− TCF-1+ of tumor specific CD8+ T cells, % SlamF6+ TCF-1+ of tumor-specific CD8+ T cells, and % Ki-67+ of SlamF6+ TCF-1+ tumor-specific CD8+ T cells isolated from lungs of untreated or FTY720 treated mice. (J) % SlamF6+ or SlamF6− TCF-1+ tumor-specific CD8+ T cells of total isolated cells in blood of untreated and FTY720 treated mice. Student t-test for I and J. Data from at least 3 experiments unless otherwise noted (dot: one mouse), Error bars: SD. *p<0.05, **p<0.01, ***p<0.001, Tukey’s multiple comparisons test. NS: non-significant (p>0.05). See also Supplemental Figure 7.
To determine if SlamF6+ tumor-specific CD8+ T cells migrate from the dLN into the lungs, we blocked LN egress by treating mice with the sphingosine-1 phosphate receptor agonist FTY720 (Schwab and Cyster, 2007) (Figure 6H). We observed the expected decrease in circulating naive CD8+ T cells (Figure S7D), and an increase in tumor-specific CD8+ T cells in the dLN (Figure S7E,F). In the lungs, the fraction of TCF-1+ cells decreased among tumor-specific CD8+ T cells (Figure 6I), which was almost exclusively accounted for by a decrease of SlamF6+ TCF-1+ tumor-specific CD8+ T cells, whereas the proportion of SlamF6− TCF-1+ did not change in either tumor bearing lungs or blood (Figure 6I,J and S7G,H). The total number of TCF-1− Tim-3+ cells, like the SlamF6+ TCF-1+ CD8+ T cells, also decreased with FTY720, consistent with previous reports (Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019) that SlamF6+ TCF-1+ CD8+ T cells give rise to Tim-3+ CD8+ T cells (Figure S7I). Furthermore, the proportion of Ki-67+ SlamF6+ TCF-1+ cells among tumor-specific CD8+ T cells decreased in tumor-bearing lungs from FTY720-treated mice (Figure 6I and S7J), suggesting that recently recruited SlamF6+ tumor-specific CD8+ T cells have greater proliferative capacity than SlamF6+ tumor-specific CD8+ T cells already present within the tumor. Taken together, these data suggest that SlamF6+ TCF-1+ T cells migrate from the dLN to the lungs, particularly during early stage disease, and that the tumor-specific CD8+ T cells in the dLN are a vital source of SlamF6+ TCF-1+ tumor-specific CD8+ T cells in the tumor.
Recent thymic CD8+ T cell emigrants (CD8+ RTE) that develop during chronic or latent infections have a less activated/dysfunctional phenotype, and contribute to the pool of TCF-1+ CD8+ T cells in a chronic LCMV infection model (He et al., 2016). Thus, it was possible that RTEs were an important reservoir of new CD8+ T cells that maintained tumor-specific CD8+ T cells in the dLN and tumor. In order to investigate the contribution of RTEs, we used an non-immunosuppresive bone marrow transplantation technique using anti-CD117 conjugated to saporin toxin (Czechowicz et al., 2019). This strategy selectively eliminates HSCs, and, with a lower dose of anti-CD117/toxin, does not cause quantitative or qualitative changes in the circulating myeloid or lymphoid compartments (Czechowicz et al., 2019) (Figure S7K). Donor-derived (CD45.1+) CD8+ T cells became detectable in the peripheral blood around four weeks after bone marrow transplant and increased over time, consistent with production of new donor T cells in the thymus (Figure S7L). Sixteen weeks after initiation, donor-derived CD8+ T cells comprised ~20% of total CD8+ T cells within the dLN (Figure S7M). However, donor-derived CD8+ T cells were only ~0.5% and ~0.1% of SIINFEKL-specific CD8 T cells in the dLN and tumor-bearing lungs, respectively (Figure S7M). Thus, recent thymic emigrants do not appear to contribute to the response in the KP model of lung adenocarcinoma, suggesting that continued division of tumor-specific CD8+ T cells in the dLN is important for maintaining the anti-tumor immune response.
The quantity and state of migratory cDC1s change over tumor progression and can be boosted to drive improved tumor-specific CD8+ T cell responses
Because tumor-specific CD8+ T cell division decreased in the dLN despite preserved numbers and functionality, we hypothesized that these cells were insufficiently activated by migratory dendritic cells (DCs) as tumors progressed. After activation within tissues, immature DCs undergo a maturation process, allowing for egress and migration to the dLN (Worbs et al., 2017). Type 1 conventional DCs (cDC1) are critical regulators of the CD8+ T cell response, and cDC1 are critical for driving anti-tumor CD8+ T cell responses after checkpoint blockade therapy (Mayoux et al., 2020; Oh SA, 2020; Salmon et al., 2016). However, it is unknown how the quantity or function of cDC1s changed within dLNs over the course of tumor progression.
In the KP LucOS model, we found that migratory cDC1 numbers in the dLN increased at 5 weeks post-initiation compared to naïve KP mice (Figure 7A, S7N–P), but decreased ~5- and 10-fold (vs. week 5) at weeks 8 and 12 post-initiation, respectively (Figure 7A). Moreover, expression of the IR ligand programmed cell death-ligand 1 (PD-L1) on migratory cDC1 cells increased at week 8 compared to week 5, which was sustained at week 12 (Figure 7B). By contrast, expression of the co-stimulatory ligand CD86 was decreased at 12 weeks post-initiation compared to 5 weeks (Figure 7C). Collectively, these data demonstrate that the number of migratory cDC1s in the dLN decrease and the cells became less stimulatory as tumors progress.
Figure 7. Migratory cDC1 quantity and quality in the dLN are important for maintaining the SlamF6+ TCF-1+ tumor-specific CD8+ T cell response.
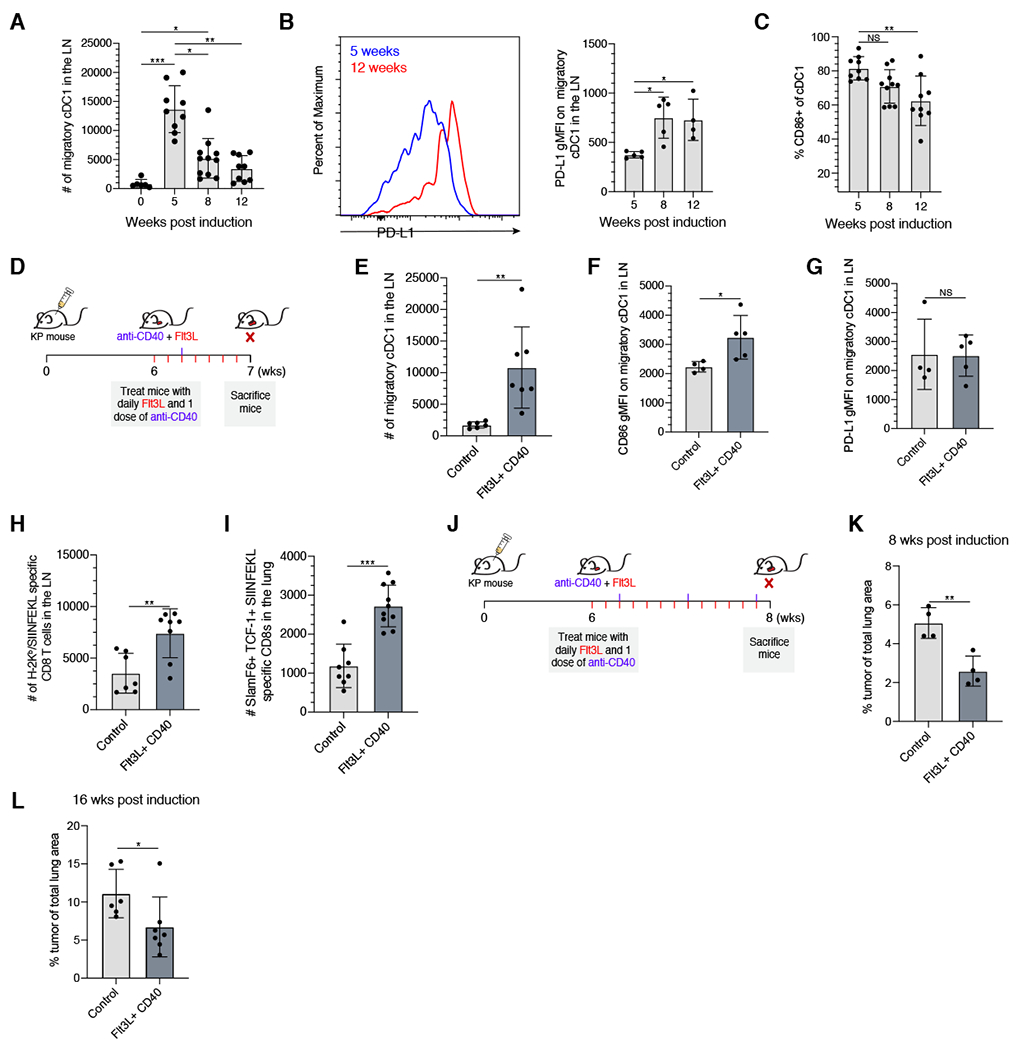
(A) Number of migratory cDC1 isolated from the dLN at the designated timepoint post initiation. (B) Representative histogram showing PD-L1 expression on migratory cDC1 in the dLN node at 5 weeks (blue) and 12 weeks (red), and PD-L1 geometric MFI on migratory cDC1 in the dLN at the indicated timepoints. (C) Percent of cDC1 expressing CD86 by flow cytometry. (D) Boosting cDC1 numbers with Flt3L and agonistic anti-CD40; experimental schematic. (E) Number of migratory cDC1 in the dLN in control or Flt3L/agonistic CD40 treated animal after 1 week of treatment. (F & G) CD86 or PD-L1 geometric MFI on migratory cDC1 in the dLN in control or Flt3L/agonistic anti-CD40 treated KP mice after 1 week of treatment. (H) Total numbers of H-2Kb/SIINFEKL tetramer+ CD8+ T cells in dLN of control or Flt3L/agonistic anti-CD40 treated KP mice after 1 week of treatment. (I) Total numbers of SlamF6+ TCF-1+ H-2Kb/SIINFEKL tetramer+ CD8 T cells in tumor bearing lungs of control or Flt3L/agonistic anti-CD40 treated KP mice after 1 week of treatment. (J) Extended treatment with Flt3L and anti-CD40 antibody to assess tumor burden; experimental schematic. (K) Percent of total lung occupied by tumor at 8 weeks post induction in mice treated with Flt3L/anti-CD40 treated starting at 6 weeks post-induction or control mice. (L) Percent of total lung occupied by tumor at 16 weeks post induction in mice treated with Flt3L/anti-CD40 treated starting at 14 weeks post-induction or control mice. Tukey’s multiple comparisons test for A and B. Student t-test for D-H. Data from at least 2 experiments or 1 experiment representative of 2 experiments (geometric mean fluorescence intensity analysis) (dot: one mouse), Error bars: SD. *p<0.05, **p<0.001, ***p<0.001, NS: non-significant (p>0.05). See also Supplemental Figure 7.
Because the number of migratory cDC1s decreased over time, we hypothesized that increasing their numbers and improving their stimulatory potential could drive improved CD8+ T cell responses within the dLN and ultimately in tumor-bearing lungs. To test this hypothesis, we treated mice with a combination of Flt3L, which stimulates increased production of pre-DCs from the bone marrow, and agonistic anti-CD40 antibody, which can activate DCs (Figure 7D) (Karsunky et al., 2003; Li and Ravetch, 2011). Supporting our hypothesis, treatment of KP LucOS mice 6 weeks after initiation with Flt3L and agonistic CD40 antibody increased the number of migratory cDC1 in the dLN ~10 fold compared to control KP mice (Figure 7E). Moreover, CD86, but not PD-L1, was increased on cDC1s in treated KP mice compared to control mice, suggesting that this regimen could restore quantity and boost the quality of migratory cDC1s in the mediastinal lymph node (Figure 7F,G).
Finally, we asked whether therapeutically augmenting migratory cDC1s improved the anti-tumor CD8+ T cell response. After one week of treatment with Flt3L/anti-CD40, we observed a ~2.5 fold increase in the number of tumor-specific CD8+ T cells in the dLN (Figure 7H) and a ~3 fold increase in the number of SlamF6+ TCF-1+ tumor-specific CD8+ T cells within the tumor-bearing lungs (Figure 7I). Next, we determined if a more prolonged treatment course could drive protective anti-tumor CD8+ T cell responses (Figure 7J). Two weeks after treatment, tumor area had decreased ~50% in Flt3L/anti-CD40 treated mice compared to control KP mice (Figure 7K). Moreover, starting treatment at 14 weeks post induction and treating for two weeks resulted in a similar percent burden reduction (Figure 7L). Collectively, these data demonstrate that restoring cDC1 quantity and stimulatory capacity is sufficient to increase SlamF6+ TCF-1+ CD8+ T cells, and can drive improved anti-tumor immunity and decreased tumor burden.
DISCUSSION
T cell dysfunction is a major obstacle for harnessing the immune response against cancer (Ribas and Wolchok, 2018; Wei et al., 2018). Studies examining CD8+ T cell responses in chronic viral models have been critical in defining a framework for T cell dysfunction, especially the temporal changes in CD8+ T cell states (Angelosanto et al., 2012; Pauken et al., 2016; Penaloza-MacMaster et al., 2015; Schietinger et al., 2016). These changes have consequences for how well CD8+ T cells can be therapeutically harnessed, particularly with respect to checkpoint blockade (Pauken et al., 2016). However, because human tumors typically grow over the course of years, extending the experimental time course in preclinical models to study CD8+ T cell dysfunction has tremendous value for basic and translational tumor immunology.
Using the KP-LucOS mouse model of lung adenocarcinoma, we longitudinally monitored tumor-specific CD8+ T cells over the course of several months. Paralleling some of the observations in chronic viral models (Pauken et al., 2016; Penaloza-MacMaster et al., 2015), there is an early time window spanning several weeks after tumorigenesis where the CD8+ T cell response undergoes rapid functional and transcriptional changes that sets CD8+ T cells on different dysfunctional trajectories. Following these dysfunctional trajectories over time in the KP-LucOS model revealed diversity within dysfunctional CD8+ T cell populations, most notably for the TCF-1+ population, which was not previously described in other animal models, but corresponded to cell states observed in advanced human tumors.
We focused on a “progenitor-like” TCF-1+ subset, previously shown to maintain a cytotoxic Tim-3+ population and drive the response to checkpoint blockade (He et al., 2016; Im et al., 2016; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016). It was unknown if and how the TCF-1+ population changed over longer time scales. Our work showed heterogeneity in TCF-1+ tumor-specific CD8+ T cells which changes with tumor progression. Importantly, our data suggests that rather than simply being a source of proliferative, cytokine-secreting cells, a substantial subset of TCF-1+ cells (e.g. the SlamF6− population) does not actively divide and has altered functional attributes. This SlamF6− TCF-1+ population increased proportionately during tumor progression. This SlamF6− subset expressed genes encoding the inflammatory chemokines CCL3, CCL4 and CCL5, which are broadly recognized by a diverse array of both innate and adaptive leukocytes (Griffith et al., 2014), and genes encoding several immunoregulatory proteins, such as Galectin-1, Galectin-3 and Fgl-2, all of which can dampen the immune response to tumors (Henderson and Sethi, 2009; Sundblad et al., 2017).
While it is appreciated that the priming of effector CD8+ T cell responses occurs within dLNs and that effector CD8+ T cells traffic through peripheral blood to tumors, how dLN and blood populations change over the course of tumor progression and how they relate to CD8+ T cells within the tumor microenvironment has not been well established. Our work demonstrates that the dLN and blood harbor a substantial fraction of functional anti-tumor CD8+ T cells. Blocking the egress of CD8+ T cells from dLNs caused a significant loss of the proliferative SlamF6+ TCF-1+ tumor-specific CD8+ T cell population within the tumor, implicating the dLNs as an important contributor to anti-tumor immunity. These findings are in line with recent reports of the importance of the peripheral immune system in driving productive immune responses after checkpoint blockade in humans (Huang et al., 2017; Kamphorst et al., 2017; Valpione et al., 2020; Wu et al., 2020; Yost et al., 2019), and that activation of peripheral CD8+ T cells outside the tumor has positive prognostic value for response to checkpoint blockade (Huang et al., 2017; Kamphorst et al., 2017; Valpione et al., 2020; Wu et al., 2020; Yost et al., 2019).
Dendritic cells are potent regulators of T cell responses, and can be broadly defined as type 1 and type 2 conventional dendritic cells (cDC1 and cDC2, respectively). Previous work has demonstrated the importance of cDC1s in priming CD8+ T cells and for driving CD8+ T cell responses to checkpoint blockade immunotherapy (Mayoux et al., 2020; Oh SA, 2020; Salmon et al., 2016). In our model, we found that migratory cDC1 number and stimulatory phenotype in the dLN decreased with tumor progression. Moreover, therapeutically restoring the migratory cDC1 axis drove expansion of SlamF6+ TCF-1+ tumor-specific CD8+ T cells in the dLN and tumor-bearing lung, highlighting the importance of sustaining activation by cDC1 within the dLN in order to continue to propagate the tumor-specific CD8+ T cell response in tumors.
Our findings also help to explain the diminished response to checkpoint blockade therapy during tumor progression recently reported in the KP model of lung adenocarcinoma (Burger et al, in press at Cell). This work demonstrated that while tumor-specific CD8+ T cells initially numerically increased after checkpoint blockade, this boost did not occur 12 weeks after initiation (Burger et al, in press at Cell). Given the role cDC1s play in regulating the response to checkpoint blockade, our data demonstrating a substantial reduction in migratory cDC1 in the dLN is consistent with the diminished checkpoint response seen at later timepoints in the KP model.
Overall, our study provides new insights into the dynamic nature of TCF-1+ tumor-specific CD8+ T cells along tumor progression. Our data support a model where T cell populations within the dLN are an important functional reservoir for the anti-tumor immune response. These findings are conistent with studies in humans, where patients who respond to checkpoint blockade have demonstrated the emergence of new T cell clones, which likely represents activation events within the dLNs (Valpione et al., 2020; Wu et al., 2020; Yost et al., 2019). Collectively, these findings advance a paradigm where anti-tumor CD8+ T cells in the dLN represent important immune effectors that could be harnessed for therapeutic applications in cancer, and provide further rationale for targeting the migratory cDC1 axis to improve anti-tumor immunity.
LIMITATIONS OF STUDY
We are currently unable to parse apart the lineage relationship between the SlamF6+ and SlamF6− TCF-1+ CD8+ T cell populations as well as their functional contributions in anti-tumor immunity in the KP tumor model. Additionally, while we are able to show boosting migratory cDC1 numbers in the dLN is sufficient to drive increase numbers of SlamF6+ TCF-1+ CD8+ T cells in tumor bearing lungs, we did not have the tools to ablate migratory cDC1 and examine the resultant effect on the CD8+ T cell response. Future studies attempting to further parse both of these areas are warranted.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tyler Jacks (tjacks@mit.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Original single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper also analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All original code has been deposited on Github and is publicly available as of the date of this publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CD45, APC-eFlour780 | ThermoFisher | Cat #:47-0451-80; clone: 30-F11; RRID: AB_1548790; dilution: 1 to 40 |
| Rat anti mouse/human CD44, Brilliant Violet 785 | Biolegend | Cat #: 103059; clone: IM7; RRID: AB_2571953; dilution: 1 to 400 |
| Rat anti mouse/human CD44, Alexa Flour 700 | Biolegend | Cat #: 103026; clone: IM7; RRID: AB_493713; dilution 1 to 200 |
| Mouse anti human Ki-67, Alexa Flour 700 | BD Biosciences | Cat #: 561277; clone: B56; RRID: AB_10611571; dilution: 1 to 200 |
| Mouse anti Ki-67, BV786 | BD Biosciences | Cat #: 563756; clone: B56; RRID: AB_2732007; dilution: 1 to 200 |
| Rat anti mouse CD8a, BUV395 | BD Biosciences | Cat# 563786; Clone: 53-6.7; RRID: AB_2732919; dilution: 1 to 400 |
| Rat anti mouse CD8a, BUV737 | BD Biosciences | Cat #: 612759; Clone: 53-6.7; RRID: AB_2722580; dilution 1 to 400 |
| Rat anti mouse CD8a, APC | Biolegend | Cat #: 100712; Clone: 53-6.7; RRID: AB_312751; dilution 1 to 200 |
| Rat anti mouse CD366 (Tim-3), Brilliant Violet 605 | Biolegend | Cat #: 119721; Clone: RMT3-23; RRID: AB_2616907; dilution: 1 to 200 |
| Rat anti mouse CD366 (Tim-3), Brilliant Violet 785 | Biolegend | Cat #: 119725; Clone: RMT3-23; RRID: AB_2716066; dilution 1 to 100 |
| Rat monoclonal anti TIGIT, FITC | Thermo Fisher | Cat #: 11-9501-82; Clone: GIGD7; RRID: AB_2637373; dilution: 1 to 200 |
| Rat anti mouse CD279 (PD-1), Brilliant Violet 510 | Biolegend | Cat #: 135241; Clone: 29F.1A12; RRID: AB_2715761; dilution: 1 to 400 |
| Rat anti mouse CD223 (Lag-3), BUV737 | BD Biosciences | Cat #: 741820; Clone: C9B7W; dilution: 1 to 200 |
| Rat anti mouse CD244.2 (2B4), PE-Cyanine7 | Thermo Fisher | Cat #: 25-2441-82; Clone: eBio244F4; RRID: AB_2573432; dilution: 1 to 200 |
| Rat anti mouse TNF-alpha, PE | Biolegend | Cat #: 506306; Clone: MP6-XT22; RRID: AB_315427; dilution: 1 to 200 |
| Rat anti mouse IFN-gamma, FITC | Biolegend | Cat #: 505806; Clone: XMG1.2; RRID: AB_315400; dilution 1 to 100 |
| Rat anti mouse IFN-gamma, PerCP/Cyanine5.5 | Biolegend | Cat #: 505822; Clone: XMG1.2; RRID: AB_961359; dilution: 1 to 100 |
| Rat anti mouse CD8b (Ly-3), Alexa Flour 647 | Biolegend | Cat #: 126612; Clone: YTS156.7.7; RRID: AB_2075777; dilution 1 to 200 |
| Rat anti mouse CD326 (Ep-CAM) | Biolegend | Cat #: 118208; Clone: G8.8; RRID: AB_1134107; dilution 1 to 200 |
| Streptavidin, R-Phycoerythrin Conjugate (SAPE) | Invitrogen | Cat #: S21388 |
| Rabbit monoclonal anti TCF1/TCF7, Alexa Flour 647 | Cell Signaling Technology | Cat #: 6709S; Clone: C63D9; RRID: AB_2797631; dilution: 1 to 250 |
| Rabbit monoclonal anti TCF1/TCF7, Alexa Flour 488 | Cell Signaling Technology | Cat #: 6444S; Clone: C63D9; RRID: AB_2797627; dilution 1 to 400 |
| Armenian hamster anti mouse CD183 (CXCR3), BV421 | BD Biosciences | Cat #: 562937; Clone: CXCR3-173; RRID: AB_2687551; dilution: 1 to 200 |
| Armenian hamster anti mouse CD183 (CXCR3), BUV395 | BD Biosciences | Cat #: 745689; Clone: CXCR3-173; RRID: AB_2743174; dilution: 1 to 100 |
| Mouse anti mouse Ly-108 (SlamF6), BB700 | BD Biosciences | Cat #: 742272; Clone: 13G3; dilution: 1 to 400 |
| Armenian hamster anti CD196 (CCR6), PE/Dazzle 594 | Biolegend | Cat #: 129822; Clone: 29-2L17; RRID: AB_2687019; dilution: 1 to 200 |
| Chemicals, peptides, and recombinant proteins | ||
| SIINFEKL-Kb monomer | NIH Tetramer Core | |
| SIINFEKL peptide | Genscript | |
| Critical commercial assays | ||
| Thru-Plex-FD Library Prep Kit | Rubicon Genomics | |
| Nextera XT Library Prep Kit | Illumina | FC-131-1096 |
| Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit v1.1 | 10X Genomics | 1000167 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10X Genomics | 1000071 |
| Deposited data | ||
| 10X and SmartSeq2 Single Cell RNA-seq | This Paper | GSE182028 |
| Sade-Feldman et al SmartSeq2 Single Cell RNA-seq | Sade-Feldman et al., 2018 | GSE120575 |
| Tirosh et al SmartSeq2 Single Cell RNA-seq | Tirosh et al., 2016 | GSE89567 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J | Jackson Laboratory | RRID: IMSR_JAX:008179 |
| Mouse: B6.129P2-Trp53tm1Brn/J | Jackson Laboratory | RRID: IMSR_JAX:008462 |
| Recombinant DNA | ||
| Plasmid: Lenti-LucOS | Addgene | Addgene_22777 |
| Software and algorithms | ||
| RSEM v1.2.8 | Li and Dewey, 2011 | http://bmcbioinformatbmc.biomedcentral.coc/articles/10.1186/1147-2105-12-323 |
| Prism | GraphPad | |
| Original Code | This Paper | https://github.com/RebeccaHerbst/TCF7_Tumor_Specific_CD8_T_cells |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
KP mice were used for all studies and have previously been described (Jackson et al., 2005). Both male and female mice were used for all experiments, and mice were gender and age-matched within experiments. Adult mice at 10-16 weeks of age were used to initiate all studies. Experimental and control mice were co-housed whenever appropriate under SPF conditions. All studies were performed under an animal protocol approved by the Massachusetts Institute of Technology (MIT) Committee on Animal Care. Mice were assessed for morbidity according to MIT Division of Comparative Medicine guidelines and humanely sacrificed prior to natural expiration.
METHOD DETAILS
In vivo treatments
For in vivo labeling of circulating immune cells, anti-CD4-PE (eBioscience, RM4-4, 1:400) and anti-CD8β-PE (eBioscience, 1:400) were diluted in PBS and administered by IV injection 5 minutes before harvest(Anderson et al., 2014) Alternatively, anti-CD45-PE-CF594 (30-F11, BD Biosciences, 1:200) or anti-CD45-APC-ef780 (30-F11, eBioscience 1:40) were also used for intravascular labeling and were administered 2 or 5 minutes, respectively, before sacrifice. Mice were exposed ad libitum to drinking water containing dissolved FTY720 at a concentration of 2 μg/mL for 2 weeks starting at 6 weeks post-initiation.
Lentiviral production and tumor induction
The lentiviral backbone Lenti-LucOS has been described previously(DuPage et al., 2009). Lentiviral plasmids and packaging vectors were prepared using endo-free maxiprep kits (Qiagen). Lentiviruses were produced by co-transfection of 293FS* or 293T cells with Lenti-LucOS, psPAX2 (gag/pol), and VSV-G vectors at a 4:3:1 ratio, respectively, with Mirus TransIT LT1 (Mirus Bio, LLC). Virus-containing supernatant was collected 48 and 72h after transfection and filtered through 0.45mm filters before concentration by ultracentrifugation (25,000 RPM for 2 hours with low decel). Virus was then resuspended in 1:1 Opti-MEM (Gibco) HBSS. Aliquots of virus were stored at −80°C and titered using the GreenGo 3TZ cell line.
For tumor induction, mice between 10-16 weeks of age received 2.5 x104 PFU of Lenti-LucOS intratracheally as described previously (DuPage et al., 2009).
Isolation/preparation of cell suspensions
After sacrifice, lungs were placed in 5 mL collagenase/DNAse buffer in gentleMACS C tubes (Miltenyi) and processed using program m_impTumor_01.01. Lungs were then incubated at 37°C for 30 minutes with gentle agitation. The tissue suspension was filtered through a 70 μm cell strainer and centrifuged at 2,000 RPM for 5 minutes. Red blood cell lysis was performed by incubation with ACK Lysis Buffer (Life Technologies) for 1 minute. Samples were centrifuged again, followed by resuspension in RPMI 1640 (VWR) supplemented with 5% heat-inactivated FBS.
Spleens and LNs were dissociated using the blunt ends of syringe backs into RPMI 1640 supplemented with 5% heat-inactivated FBS. Spleen cell suspensions were spun down at 2,000 RPM for 5 minutes, and red blood cell lysis with ACK Lysis Buffer was performed for 1 minute. Cells were filtered through 70 μm nylon mesh and, after centrifugation, resuspended in supplemented RPMI 1640. LN suspensions were spun down at 2,000 RPM for 5 minutes, and resuspended in supplemented RPMI 1640.
Staining for flow cytometric analysis
Approximately 0.5-1 x 106 cells were stained for 1 hour at 4°C in 96-well U-bottom plates (BD Biosciences) with directly conjugated antibodies (below) diluted in DPBS, 1x without calcium and magnesium (VWR) supplemented with 2% heat inactivated FBS. SIINFEKL-Kb tetramer was prepared using streptavidin-PE (Invitrogen) and SIINFEKL-Kb monomer from the NIH Tetramer Core.
After staining, cells were fixed with a Fixation/Permeabilization kit (eBioscience). Intracellular cytokine and transcription factor staining was performed overnight at 4°C in Permeabilization Buffer (eBioscience). Analysis of tissue tumor specific CD8+ T cells (i.v.negCD8a+CD44hiSIINFEKL+) was performed on a LSR Fortessa (BD) with 355, 405, 488, 561, and 640 lasers. Data analysis was performed using FlowJo software.
Plate-based scRNA-seq
Tetramer+ (CD8a+ CD45 IV− DAPI− SIINFEKL Tetramer+) and Tetramer− (CD8a+ CD45 IV− DAPI− SIINFEKL Tetramer-) cells were isolated from ~4 mice per timepoint and single-cell sorted into TCL buffer (Qiagen) plus 1% β-mercaptoethanol in 96-well plates using a MoFlo Astrios cell sorter. Each plate had a 30-100 cell population well and an empty well as controls. Following sorting, plates were spun down for 1 minute at 2,000 RPM and frozen immediately at −80°C.
Plates were thawed and RNA was purified using 2.2X RNAclean SPRI beads (Beckman Coulter) without final elution SMART-seq2 and Nextera library preparation was performed as previously described with some modifications as described in a previous study(Singer et al., 2017). Plates were pooled into 384 single-cell libraries, and sequenced 50 x 25 paired end reads using a single kit on the NextSeq500 5 instrument.
Droplet-based scRNA-seq
After sacrifice, lungs were placed in 5 mL collagenase/DNAse buffer and cut into small pieces. Lungs were then incubated at 37°C for 30 minutes with gentle agitation. The tissue suspension was filtered through a 70 μm cell strainer and centrifuged at 2,000 RPM for 5 minutes. Red blood cell lysis was performed by incubation with ACK Lysis Buffer (Life Technologies) for 1 minute. Samples were centrifuged again, followed by resuspension in DPBS, 1x without calcium and magnesium (VWR) supplemented with 2% heat inactivated FBS and 1mM calcium chloride. Dead cells were then depleted using EasySep Dead Cell Removal (Annexin V) Kit (Stemcell) per the manufacturer’s instructions.
tumor specific CD8+ T cells (live, i.v.negCD8a+CD44hiSIINFEKL+) cells were isolated from 3 mice per timepoint and sorted into Eppendorf LoBind Microcentrifuge Tubes (Eppendorf) with RPMI 1640 (VWR) supplemented with 5% heat-inactivated FBS using an Aria Cell Sorter (BD) with 405, 488, 561, and 640 lasers before proceeding to droplet based scRNA-Seq.
Single cells were processed through the 10X Genomics Single Cell 5′ platform using the Chromium Single Cell 5′ Library & Gel Bead Kit V2 kit (10X Genomics), per manufacturer’s protocol. Briefly, 6,000 cells were loaded onto each channel and partitioned into Gel Beads in Emulsion in the Chromium instrument. Cell lysis and barcoding occur, followed by amplification, fragmentation, adaptor ligation and index library PCR. Libraries were sequenced on an Illumina HiSeqX at a read length of 98 base pairs.
In situ peptide-MHC Class I tetramer staining
Lung lobes allocated for in situ tetramer staining were cut into 300-700 μm sections using a surgical blade. Tissue sections were then stained overnight at 4°C in DPBS, 1x without calcium and magnesium (VWR) supplemented with 2% heat inactivated FBS with PE-SIINFEKL tetramer diluted to 2 g/mL. Tissues were then fixed in 2% PFA in DPBS, 1x without calcium and magnesium (VWR) for 2 hours at 4°C. Samples were then moved to 30% sucrose in DPBS, 1x without calcium and magnesium (VWR) and incubated overnight at 4°C. The tissue was then snap frozen in Tissue Tek OCT (VWR) using a 2− methylbutane (Sigma-Aldrich) solution cooled by dry ice. Frozen tissue was then wrapped in parafilm and stored at −80°C until ready to section.
5μm sections were cut using a CryoStar NX70 cryostat (Thermo), and air-dried for 60-90 minutes at room temperature. Sections were incubated in ice-cold acetone (Sigma) for 10 minutes at −20°C and then washed 3 x 5 minutes with PBS. Samples were blocked with 5% BSA in PBS. Primary antibodies were incubated for 1 hour at room temperature. Primary antibodies used were Rat anti mouse CD8b (Ly-3) in Alexa Flour 647 (Biolegend, 126612, 1:200) and Rat anti mouse CD326 (Ep-CAM) in FITC (Biolegend, 118208, 1:2000). Sections were then mounted using Prolong Diamond Antifade Mountant (ThermoFisher).
In situ tetramer stained tissue section images were acquired using a Nikon 80 Eclipse 80i fluorescence microscope using 10x and 20x objectives and an attached Andor camera.
In vitro stimulation/cytokine measurements
For ex vivo T cell stimulation experiments to detect intracellular cytokines, lung cells were incubated for 20 minutes at 4°C with monoclonal Ly-6G (1A8, Biolegend, 1:100), EpCAM (G8.8, Biolegend, 1:100, and F4/80 (BM8, Biolegend, 1:100). Samples then underwent negative selection using Dynabeads Sheep Anti-Rat IgG (Invitrogen) as per the manufacturer’s suggestions. 20% of remaining cells were plated in a 96-well flat-bottom plate (BD Biosciences) in RPMI 1640 (VWR) supplemented with 10% heat-inactivated FBS, 1X penicillin-streptomycin (Gibco), 1X HEPES (Gibco), 1X GlutaMAX (Gibco), 1mM sodium pyruvate (ThermoFisher), 1X MEM non-essential amino acids (Sigma), 50μM β-mercaptoethanol (Gibco), 1X monensin (BioLegend), and 1X Protein Transport Inhibitor (Containing Brefeldin A) (BD Biosciences). The remaining 80% of cells were plated in a 96-well flat-bottom plate (BD Biosciences), with the same media previously described and 10 millimoles of SIINFEKL peptide. Cells were incubated in a tissue culture incubator at 37°C with 5% CO2 for 4-5 hours.
QUANTIFICATION AND STATISTICAL ANALYSIS
Pre-processing of SMART-Seq2 scRNA-seq data
BAM files were converted to de-multiplexed FASTQs using Illumina’s Bcl2Fastq software package v2.17.1.14. Paired-end reads were mapped to the UCSC mm10 mouse transcriptome using Bowtie with parameters ‘-n 0 -m 10’, which allows alignment of sequences with zero mismatches and allows at most 10 multi-mapping instances per read.
Gene expression levels were quantified using TPM values calculated by RSEM v1.2.8 in paired-end mode that were corrected for lower library complexity to compute normalized expression log2((TPM/100) + 1) denoted as log2(TP10K+1). For each cell, the number of detected genes (TP10K > 0) was calculated and cells with less than 500 or more than 5,000 genes detected were excluded, as well as cells that had a transcriptome mapping < 15%. To further remove potential doublets or ambient-RNA contaminated cells (mostly of B cells and epithelial cells), we calculated the sum log2(TP10K+1) over Cd79a, Cd19, Lyz1, Lyz2 and Sftpc, and excluded any cell that scored higher than 3. We retained only genes expressed in at least five cells in the entire dataset. Selection of variable genes was performed using the function FindVariableFeatures() of the Seurat package in R with default parameters.
Pre-processing of droplet-based scRNA-seq
De-multiplexing, alignment to the mm10 transcriptome (mm10-3.0.0 version) and unique molecular identifier (UMI)-collapsing were performed using the CellRanger toolkit from 10X Genomics version 3.1.0. For each cell, we quantified the number of genes for which at least one read was mapped, and excluded all cells with fewer than 300 detected genes. Next, genes that were detected in less than 3 cells were excluded. Expression values Ei,j for gene i in cell j were calculated by dividing UMI counts for gene i by the sum of the UMI counts in cell j, to normalize for differences in coverage, and then multiplying by 10,000 to create TPM-like values (TP10K), and finally computing log2(TP10K + 1). Cells with B cell-related contamination (through doublets or ambient RNA) were removed by summing log2(TP10K+1) over Cd79a, H2-Aa, H2-Eb1, H2-Ab1, Ly86, Ly6d, H2-Ob, H2-DMb2 and excluding cells with a total expression of at least 5.
Selection of variable genes was performed by fitting a logistic regression to the cellular detection fraction (often referred to as α), using the total number of UMIs per gene as a predictor. Outliers from this curve are genes that are expressed in a lower fraction of cells than expected given the total number of UMIs mapping to that gene, and thus might be cell-type or state-specific genes. We used a threshold of deviance of <−0.15 and a minimum of 100 total UMIs. Furthermore, because of the 5’ Chromium chemistry, we detected the V-region transcripts in the mRNA fraction. In order to remove the variance due to T cell receptor sequences, V-region genes were removed from the list of variable genes for downstream analysis.
Reduction and visualization using UMAP
We restricted the expression matrix to the subsets of variable genes and high-quality cells noted above, and then centered and scaled values before performing principal component analysis (PCA), implemented using ‘RunPCA’ in Seurat which runs the irlba function. The cell embeddings were either the singular vectors themselves or the singular vectors multiplied with the singular values depending on the cells. After PCA, significant principal components (PCs) were identified using the elbow-method when looking at the distribution of singular values. Scores from only those significant PCs were used as input to further analysis. For visualization, the dimensionality of the datasets was further reduced to 2D embeddings using the RunUMAP() function of the Seuratpackage in R on the significant PCs.
Identifying programs and their time dependence
LDA was separately performed on all cells and on only Tcf7+ T cells from droplet-based scRNA-seq, as well as on all cells from plate-based scRNA-seq. We used the FitGoM() function from the CountClust R package (Dey et al., 2017) to fit LDA topic models to the UMI counts for the droplet-based data or on the rounded log2(TP10K + 1) values for the plate-based scRNA-seq data (Bielecki et al., 2018). To improve topic signals, genes expressed in less than 2% of cells and TCR-V region genes were removed from the count matrix prior to fitting a topic model (analogous to the removal of highly abundant words or extremely rare words in document analysis). The number of topics to fit (K) and the tolerance value are required to run FitGoM() function. We fit a range of K and tolerance values and picked values that gave us robust topics and where we mostly saturated the number of informative topics found. This was achieved with a tolerance of 0.01, and a K of 16 for Tcf-7+ cells, K of 18 for all other droplet-based datasets, and K of 14 for plate-based cells. The top 50 genes to highlight for each topic were selected using the ExtractTopFeatures() function and allowed for genes to get highly scored in more than one topic. These top feature genes can be found in Table S1 for all datasets analyzed in this manuscript. To compare inferred topics between the plate-based and droplet-based data, we centered the gene weights across topics within each dataset to normalize for differences between the methods and then computed the Spearman correlation of the centered gene weights across topics. The naming of the different topics was derived from examining the top 25 genes and looking at which gene weights were dominant within an individual topic, and then manually curating the known functions associated with each of the genes that were identified in these lists and categorized the general predominant themes associated with each topic.
To test if a program’s expression changed during tumor progression, we estimated a linear mixed-effects model using the lmer function of the lme R package. For each topic, we fitted the model: omega (weights over cells) ~ detected genes + time point + (1|mouse). For the time point covariate, the earliest time point of the dataset was taken as reference.
Comparing Topics across datasets
Topic were computed for Tcf7+ CD8+ T cells of human melanoma samples(Sade-Feldman et al., 2018; Tirosh et al., 2016) with the following parameters; Sade-Feldman k=16, tol=0.01, Tirosh et al. k=14, tol=0.01. In order to compare the topics identified in the human datasets to ours, we scored the human cells using the top 50 features of each topic identified in our Tcf7+ tumor-specific CD8+ T cells, using the signature scoring method as described below. We could then compute a spearman correlation between the human topic weights, and the signature z-score across the two human datasets. Similarly, when comparing topic to signatures taken from literature, the same procedure was followed.
Subsetting Tcf7+ tumor-specific CD8+ T cells
For most of the analysis in the manuscript, we subsetted the Tcf7+ tumor-specific CD8+ T cells by detection of Tcf7 gene expression. However, due to drop-out, there is a concern that we did not include cells that should have been included and therefore biased our downstream analysis. In order to test if we lost a specific flavor of Tcf7+ CD8+ tumor-specific T cells by filtering this way, we also subsetted tumor-specific CD8+ T cells that scored > 0.1 for Topic 10 (Quiescent) and 12 (Th-17 like) in Figure S3B. We then recomputed topic models for these cells, and correlated these topic to the topics of Tcf7+ tumor-specific CD8+ T cells by correlating the gene weights (theta) across topics between the two datasets.
Testing for differential Slamf6 expression
We wanted to test if topics are differentially expressed as a function of Slamf6 expression. As Slamf6 expression is associated with cell-cycle, and therefore cellular complexity, we had to normalize for that. To this end, we ran a linear regression on topic weights normalizing for UMI. Specifically, we ran the lm() function with the following model: topic weight ~ Slamf6 expression + UMI.
Computing gene signatures
Mean program expression was calculated by averaging over the genes in each program of the centered and scaled gene expression table and transforming to a z-score over 1,000 randomly selected gene sets with matched mean-variance patterns. First, genes were grouped into 10 bins based on their mean expression, and into 10 (separate) bins based on their variance of expression across all cells. Given a list of genes (e.g. genes in a program), a cell-specific signature score was computed for each cell as follows: First, 1,000 random gene lists were generated, where each instance of a random gene-list was generated by sampling (with replacement) for each gene in the gene-list a gene from the equivalent mean and variance bin it was placed in. Then, the sum of centered and scaled gene expression in the given cell was computed for all 1000 random gene-lists generated and the z-score of the original gene-list for the generated 1,000 sample distribution is returned, as in(Singer et al., 2017).
Additional statistical analyses
Unpaired, two-tailed Student’s t tests, Mann-Whitney tests, Tukey’s multiple comparisons tests, and Sidak’s multiple comparisons tests were used for all statistical comparisons using GraphPad Prism software.
Supplementary Material
Supplementary Table 1: A Summary of Data Related to All Single-Cells Analysis, Related to Figures 2–4 and S4
ST1A – Reference signatures derived from previously published datasets (Related to Figures 2–4 and S4)
ST1B – 10x Tumor infiltrating Tcf7+ CD8 T cells topic modeling top features (Related to Figure 4)
STIC – 10x Tumor infiltrating Tcf7+ CD8 T cells expression top features (Related to Figure 4)
ST1D – Tirosh et al topic modeling top features of tumor infiltrating TCF-1+ CD8 T cells (Related to Figure S4)
ST1E – Sade-Feldman et al topic modeling top features of tumor infiltrating TCF-1+ CD8 T cells (Related to Figure S4)
ST1F – SmartSeq2 topic modeling top features of tumor specific CD8 T cells over time (Related to Figure 2)ST1G – 10X topic modeling top features of tumor specific CD8 T cells over time (Related to Figure 3)
Highlights:
Longitudinal scRNA-seq of tumor specific TCF-1+ CD8+ T cells in KP lung adenocarcinoma
Identified a proliferative Slamf6+ TCF-1+ T cell subset and a non-cycling SlamF6− subset
The lymph node contains a recruitable reservoir of functional TCF-1+ CD8+ T cells
Flt3L+CD40 boosts cDC1, increases TCF-1+CD8+ T cell frequencies, decreases tumor burden
ACKNOWLEDGEMENTS
We thank M. Singer and S. Riesenfeld for thoughtful discussion and technical advice; S. Levine at the MIT BioMicro Center for sequencing support; C. Otis and S. Saldi in the Broad Flow Cytometry Core and G. Paradis in the Koch Institute Flow Cytometry Facility for flow cytometry assistance; K. Cormier and C. Condon from the Hope Babette Tang (1983) Histology Facility for histology assistance; L. Gaffney and A. Hupalowska for artwork and advice on figures; K. Anderson, J. Teixeira, M. Magendantz, and K. Yee for administrative and logistical support. This work was supported by the Howard Hughes Medical Institute (T.J. and A.R.), the Klarman Cell Observatory at the Broad Institute (A.R.), and the Ludwig Center at MIT (A.R.). This work was supported by grant PO1-CA42063 from the National Institutes of Health and supported in part by Cancer Center Support (Gordon et al.) grant P30-CA14051 from the National Cancer Institute, NIH Pre-Doctoral Training Grant T32GM007287 (A.L.), Oncopathology Training Grant T32CA251062 (J.M.S), NCI K08CA256044 (J.M.S.), a National Science Foundation Fellowship (D.C.). T.J. is a a Daniel K. Ludwig Scholar. AV was supported by a fellowship from the American Italian Cancer Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific. He is also a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. He is the President of Break Through Cancer. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. T.J. laboratory currently also receives funding from the Johnson & Johnson Lung Cancer Initiative and The Lustgarten Foundation for Pancreatic Cancer Research, but this funding did not support the research described in this manuscript. A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov until July 31, 2020. From August 1, 2020, A.R. is an employee of Genentech.
REFERENCES
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, and Masopust D (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelosanto JM, Blackburn SD, Crawford A, and Wherry EJ (2012). Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. Journal of virology 86, 8161–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019). Defining ‘T cell exhaustion’. Nature reviews. Immunology 19, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhankar S, Chen Y, Lapato A, Dotson AL, Wang J, Vandenbark AA, Saugstad JA, and Offner H (2015). PD-L1 Monoclonal Antibody Treats Ischemic Stroke by Controlling Central Nervous System Inflammation. Stroke 46, 2926–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czechowicz A, Palchaudhuri R, Scheck A, Hu Y, Hoggatt J, Saez B, Pang WW, Mansour MK, Tate TA, Chan YY, et al. (2019). Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun 10, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey KK, Hsiao CJ, and Stephens M (2017). Visualizing the structure of RNA-seq expression data using grade of membership models. PLoS Genet 13, e1006599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer cell 19, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, and Jacks T (2012). Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, van Hall T, and Ossendorp F (2018). Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annual review of immunology 32, 659–702. [DOI] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–428. [DOI] [PubMed] [Google Scholar]
- Henderson NC, and Sethi T (2009). The regulation of inflammation by galectin-3. Immunological reviews 230, 160–171. [DOI] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, and Jacks T (2005). The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer research 65, 10280–10288. [DOI] [PubMed] [Google Scholar]
- Kallies A, Zehn D, and Utzschneider DT (2020). Precursor exhausted T cells: key to successful immunotherapy? Nature reviews. Immunology 20, 128–136. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. (2017). Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proceedings of the National Academy of Sciences of the United States of America 114, 4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsunky H, Merad M, Cozzio A, Weissman IL, and Manz MG (2003). Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med 198, 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50, 181–194 e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Herbst RH, Canner D, Schenkel JM, Smith OC, Kim JY, Hillman M, Bhutkar A, Cuoco MS, Rappazzo CG, et al. (2019). IL-33 Signaling Alters Regulatory T Cell Diversity in Support of Tumor Development. Cell Rep 29, 2998–3008 e2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, and Ravetch JV (2011). Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 333, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D, and Schenkel JM (2013). The integration of T cell migration, differentiation and function. Nature reviews. Immunology 13, 309–320. [DOI] [PubMed] [Google Scholar]
- Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, Jost C, Fransen MF, Buser RB, Kowanetz M, et al. (2020). Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Science translational medicine 12. [DOI] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nature immunology 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Gebhardt T, Carbone FR, and Heath WR (2013). Memory T cell subsets, migration patterns, and tissue residence. Annual review of immunology 31, 137–161. [DOI] [PubMed] [Google Scholar]
- Oh SA WD, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, Leader AM, Merad M, Roose-Germa M, Warming S, Yan M, Kim JM, Rutz S, Mellman I (2020). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer 1, 681–691. [DOI] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, and Wherry EJ (2012). Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, and Wherry EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaloza-MacMaster P, Provine NM, Blass E, and Barouch DH (2015). CD4 T Cell Depletion Substantially Augments the Rescue Potential of PD-L1 Blockade for Deeply Exhausted CD8 T Cells. J Immunol 195, 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, and Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hammerling GJ, et al. (2016). Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 45, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab SR, and Cyster JG (2007). Finding a way out: lymphocyte egress from lymphoid organs. Nature immunology 8, 1295–1301. [DOI] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AH, and Pauken KE (2017). The diverse functions of the PD1 inhibitory pathway. Nature reviews. Immunology. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110. [DOI] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2017). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 171, 1221–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundblad V, Morosi LG, Geffner JR, and Rabinovich GA (2017). Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J Immunol 199, 3721–3730. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Valpione S, Galvani E, Tweedy J, Mundra PA, Banyard A, Middlehurst P, Barry J, Mills S, Salih Z, Weightman J, et al. (2020). Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer 1, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Duffy CR, and Allison JP (2018). Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nature reviews. Immunology 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worbs T, Hammerschmidt SI, and Forster R (2017). Dendritic cell migration in health and disease. Nature reviews. Immunology 17, 30–48. [DOI] [PubMed] [Google Scholar]
- Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O’Gorman WE, Au-Yeung A, et al. (2020). Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. [DOI] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nature medicine 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: A Summary of Data Related to All Single-Cells Analysis, Related to Figures 2–4 and S4
ST1A – Reference signatures derived from previously published datasets (Related to Figures 2–4 and S4)
ST1B – 10x Tumor infiltrating Tcf7+ CD8 T cells topic modeling top features (Related to Figure 4)
STIC – 10x Tumor infiltrating Tcf7+ CD8 T cells expression top features (Related to Figure 4)
ST1D – Tirosh et al topic modeling top features of tumor infiltrating TCF-1+ CD8 T cells (Related to Figure S4)
ST1E – Sade-Feldman et al topic modeling top features of tumor infiltrating TCF-1+ CD8 T cells (Related to Figure S4)
ST1F – SmartSeq2 topic modeling top features of tumor specific CD8 T cells over time (Related to Figure 2)ST1G – 10X topic modeling top features of tumor specific CD8 T cells over time (Related to Figure 3)
Data Availability Statement
Original single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper also analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All original code has been deposited on Github and is publicly available as of the date of this publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-CD45, APC-eFlour780 | ThermoFisher | Cat #:47-0451-80; clone: 30-F11; RRID: AB_1548790; dilution: 1 to 40 |
| Rat anti mouse/human CD44, Brilliant Violet 785 | Biolegend | Cat #: 103059; clone: IM7; RRID: AB_2571953; dilution: 1 to 400 |
| Rat anti mouse/human CD44, Alexa Flour 700 | Biolegend | Cat #: 103026; clone: IM7; RRID: AB_493713; dilution 1 to 200 |
| Mouse anti human Ki-67, Alexa Flour 700 | BD Biosciences | Cat #: 561277; clone: B56; RRID: AB_10611571; dilution: 1 to 200 |
| Mouse anti Ki-67, BV786 | BD Biosciences | Cat #: 563756; clone: B56; RRID: AB_2732007; dilution: 1 to 200 |
| Rat anti mouse CD8a, BUV395 | BD Biosciences | Cat# 563786; Clone: 53-6.7; RRID: AB_2732919; dilution: 1 to 400 |
| Rat anti mouse CD8a, BUV737 | BD Biosciences | Cat #: 612759; Clone: 53-6.7; RRID: AB_2722580; dilution 1 to 400 |
| Rat anti mouse CD8a, APC | Biolegend | Cat #: 100712; Clone: 53-6.7; RRID: AB_312751; dilution 1 to 200 |
| Rat anti mouse CD366 (Tim-3), Brilliant Violet 605 | Biolegend | Cat #: 119721; Clone: RMT3-23; RRID: AB_2616907; dilution: 1 to 200 |
| Rat anti mouse CD366 (Tim-3), Brilliant Violet 785 | Biolegend | Cat #: 119725; Clone: RMT3-23; RRID: AB_2716066; dilution 1 to 100 |
| Rat monoclonal anti TIGIT, FITC | Thermo Fisher | Cat #: 11-9501-82; Clone: GIGD7; RRID: AB_2637373; dilution: 1 to 200 |
| Rat anti mouse CD279 (PD-1), Brilliant Violet 510 | Biolegend | Cat #: 135241; Clone: 29F.1A12; RRID: AB_2715761; dilution: 1 to 400 |
| Rat anti mouse CD223 (Lag-3), BUV737 | BD Biosciences | Cat #: 741820; Clone: C9B7W; dilution: 1 to 200 |
| Rat anti mouse CD244.2 (2B4), PE-Cyanine7 | Thermo Fisher | Cat #: 25-2441-82; Clone: eBio244F4; RRID: AB_2573432; dilution: 1 to 200 |
| Rat anti mouse TNF-alpha, PE | Biolegend | Cat #: 506306; Clone: MP6-XT22; RRID: AB_315427; dilution: 1 to 200 |
| Rat anti mouse IFN-gamma, FITC | Biolegend | Cat #: 505806; Clone: XMG1.2; RRID: AB_315400; dilution 1 to 100 |
| Rat anti mouse IFN-gamma, PerCP/Cyanine5.5 | Biolegend | Cat #: 505822; Clone: XMG1.2; RRID: AB_961359; dilution: 1 to 100 |
| Rat anti mouse CD8b (Ly-3), Alexa Flour 647 | Biolegend | Cat #: 126612; Clone: YTS156.7.7; RRID: AB_2075777; dilution 1 to 200 |
| Rat anti mouse CD326 (Ep-CAM) | Biolegend | Cat #: 118208; Clone: G8.8; RRID: AB_1134107; dilution 1 to 200 |
| Streptavidin, R-Phycoerythrin Conjugate (SAPE) | Invitrogen | Cat #: S21388 |
| Rabbit monoclonal anti TCF1/TCF7, Alexa Flour 647 | Cell Signaling Technology | Cat #: 6709S; Clone: C63D9; RRID: AB_2797631; dilution: 1 to 250 |
| Rabbit monoclonal anti TCF1/TCF7, Alexa Flour 488 | Cell Signaling Technology | Cat #: 6444S; Clone: C63D9; RRID: AB_2797627; dilution 1 to 400 |
| Armenian hamster anti mouse CD183 (CXCR3), BV421 | BD Biosciences | Cat #: 562937; Clone: CXCR3-173; RRID: AB_2687551; dilution: 1 to 200 |
| Armenian hamster anti mouse CD183 (CXCR3), BUV395 | BD Biosciences | Cat #: 745689; Clone: CXCR3-173; RRID: AB_2743174; dilution: 1 to 100 |
| Mouse anti mouse Ly-108 (SlamF6), BB700 | BD Biosciences | Cat #: 742272; Clone: 13G3; dilution: 1 to 400 |
| Armenian hamster anti CD196 (CCR6), PE/Dazzle 594 | Biolegend | Cat #: 129822; Clone: 29-2L17; RRID: AB_2687019; dilution: 1 to 200 |
| Chemicals, peptides, and recombinant proteins | ||
| SIINFEKL-Kb monomer | NIH Tetramer Core | |
| SIINFEKL peptide | Genscript | |
| Critical commercial assays | ||
| Thru-Plex-FD Library Prep Kit | Rubicon Genomics | |
| Nextera XT Library Prep Kit | Illumina | FC-131-1096 |
| Chromium Next GEM Single Cell 5′ Library and Gel Bead Kit v1.1 | 10X Genomics | 1000167 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10X Genomics | 1000071 |
| Deposited data | ||
| 10X and SmartSeq2 Single Cell RNA-seq | This Paper | GSE182028 |
| Sade-Feldman et al SmartSeq2 Single Cell RNA-seq | Sade-Feldman et al., 2018 | GSE120575 |
| Tirosh et al SmartSeq2 Single Cell RNA-seq | Tirosh et al., 2016 | GSE89567 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.129S4-Krastm4Tyj/J | Jackson Laboratory | RRID: IMSR_JAX:008179 |
| Mouse: B6.129P2-Trp53tm1Brn/J | Jackson Laboratory | RRID: IMSR_JAX:008462 |
| Recombinant DNA | ||
| Plasmid: Lenti-LucOS | Addgene | Addgene_22777 |
| Software and algorithms | ||
| RSEM v1.2.8 | Li and Dewey, 2011 | http://bmcbioinformatbmc.biomedcentral.coc/articles/10.1186/1147-2105-12-323 |
| Prism | GraphPad | |
| Original Code | This Paper | https://github.com/RebeccaHerbst/TCF7_Tumor_Specific_CD8_T_cells |