

Abstract
Milademetan is a small‐molecule inhibitor of murine double minute 2 (MDM2) that is in clinical development for advanced solid tumors and hematological cancers, including liposarcoma and acute myeloid leukemia. Milademetan is a CYP3A and P‐glycoprotein substrate and moderate CYP3A inhibitor. The current study aims to understand the drug‐drug interaction (DDI) risk of milademetan as a CYP3A substrate during its early clinical development. A clinical DDI study of milademetan (NCT03614455) showed that concomitant administration of single‐dose milademetan with the strong CYP3A inhibitor itraconazole or posaconazole increased milademetan mean area under the curve from zero to infinity (AUCinf) by 2.15‐fold (90% confidence interval [CI], 1.98–2.34) and 2.49‐fold (90% CI, 2.26–2.74), respectively, supporting that the milademetan dose should be reduced by 50% when concomitantly administered with strong CYP3A inhibitors. A physiologically‐based pharmacokinetic (PBPK) model of milademetan was subsequently developed to predict the magnitude of CYP3A‐mediated DDI potential of milademetan with moderate CYP3A inhibitors. The PBPK model predicted an increase in milademetan exposure of 1.72‐fold (90% CI, 1.69–1.76) with fluconazole, 1.91‐fold (90% CI, 1.83–1.99) with erythromycin, and 2.02‐fold (90% CI, 1.93–2.11) with verapamil. In addition, it estimated that milademetan’s original dose (160 mg once daily) could be resumed from its half‐reduced dose 3 days after discontinuation of concomitant strong CYP3A inhibitors. The established PBPK model of milademetan was qualified and considered to be robust enough to support continued development of milademetan.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Milademetan is a CYP3A and P‐gp substrate and moderate CYP3A inhibitor. Evaluation of drug‐drug interaction (DDI) risk of milademetan by combining clinical studies and physiologically‐based pharmacokinetic (PBPK) modeling has not previously been described.
WHAT QUESTION DID THIS STUDY ADDRESS?
Will milademetan PK be affected by the concomitant administration of strong or moderate CYP3A inhibitors? When can the original dose of milademetan be resumed after the discontinuation of strong CYP3A inhibitors?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study illustrates the use of a clinical DDI study and PBPK modeling in the early clinical development of milademetan to assess DDI risks in scenarios that have not yet been tested clinically at the time.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
PBPK modeling integrates in vitro and clinical data to facilitate the mechanistic understanding of PKs. Recommendations from PBPK modeling can support the design of clinical studies for the investigation of DDIs.
INTRODUCTION
Milademetan (DS‐3032) is an orally bioavailable small‐molecule inhibitor of murine double minute 2 (MDM2) that disrupts the interactions between MDM2 and the tumor suppressor protein p53 in tumor cells. Multiple clinical trials are currently ongoing to evaluate milademetan, either as a monotherapy or in combination with other anticancer agents, as a potential treatment for liposarcoma and acute myeloid leukemia. In the phase I, multiple ascending–dose study in subjects with advanced solid tumors or lymphomas (NCT01877382), milademetan’s maximum tolerated dose was 160 mg in the once‐daily (q.d.) 21/28 schedule and 260 mg in the q.d. 3/14 × 2 schedule (1 cycle was 28 days). Following oral administration of repeated doses of 120 mg q.d. 21/28, a 1.8‐fold accumulation at steady‐state was observed after 18–21 days of continuous dosing. In general, milademetan showed a dose‐proportional increase in systemic drug exposure over a dose range of 15–340 mg.
In vitro data (unpublished in‐house data) indicated that milademetan is a substrate of cytochrome P450 3A (CYP3A) and P‐glycoprotein (P‐gp). The clearance mechanism of milademetan in humans mainly involves hepatic metabolism. Incubation of [14C]milademetan (1 µM) with recombinant human CYPS for 60 min revealed that the remaining parent was 61% and 84% of the total radioactivity when incubated with CYP3A4 and CYP3A5, respectively, but was not noticeably different from controls when incubated with other CYP isoenzymes; this suggests that the major metabolic pathway of milademetan is oxidation via CYP3A. Hence, concomitant use of other drugs that can modulate CYP3A activities may influence the pharmacokinetics (PKs) of milademetan.
Early understanding of clinically relevant drug‐drug interactions (DDIs) is critical in the optimal development of anticancer drugs, particularly those given orally, as a number of concomitant medications are routinely used in patients with cancer. Physiologically‐based PK (PBPK) modeling has been used successfully to predict CYP‐mediated metabolic DDIs. 1 , 2 In addition, many PBPK simulations have been submitted to regulatory agencies and accepted as the basis for dose adjustment in drug labels. 3 , 4 , 5 , 6 The objective of this study was to assess the DDI risk of milademetan as a CYP3A substrate using a combination of clinical DDI studies and PBPK modeling. This work was used to guide the dose adjustment of milademetan when co‐administered with strong or moderate CYP3A inhibitors.
MATERIALS AND METHODS
Clinical DDI study (DS3032‐A‐U107; U107)
This was an open‐label, randomized, three‐treatment, two‐period, two‐sequence crossover study (NCT03614455). The primary objective was to evaluate the single‐dose PK of milademetan when administered alone and concomitantly with the strong CYP3A inhibitor itraconazole or posaconazole. The secondary objective was to assess milademetan safety and tolerability.
The study design is shown in Figure S1. In sequence AB, subjects were given a single dose of milademetan 100 mg on days 1 and 14 under fasted conditions and 14 doses of itraconazole 200 mg (twice daily [b.i.d.] on day 8 and q.d. on days 9 through 20), also under fasted conditions. On study day 14, milademetan was administered 1 h after the morning dose of itraconazole. In sequence AC, subjects were administered a single dose of milademetan 100 mg on days 1 and 14, 2 h following a full meal, and 39 doses of posaconazole 200 mg (3 times daily [t.i.d.] on study days 8 through 20) administered with a full meal. On study day 14, milademetan was administered 2 h after the morning dose of posaconazole. The institutional review board of the investigational site reviewed and approved the clinical study protocol. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All subjects provided written informed consent before participating in any study procedures.
Plasma concentration of milademetan was measured using a validated turbo ion spray liquid chromatography/tandem mass spectrometry bioanalytical assay. The linear calibration ranged from 0.500 to 1000 ng/ml, with a correlation coefficient of greater than or equal to 0.9972. The intra‐ and interassay accuracy and precision were less than or equal to ±6.3% and less than or equal to 6.2%, respectively. The lower limit of quantification of the bioassay was 0.5 ng/ml.
PK parameters were calculated by noncompartmental methods using Phoenix WinNonlin version 8.1 (Pharsight; Certara, Princeton, NJ, USA).
PBPK model development for milademetan
The PBPK model for milademetan was developed using the Simcyp population‐based Simulator version 17.1 (Simcyp; a Certara company, Sheffield, UK). The graphical plots were generated in R (version 3.5.1) with RStudio (version 1.1.463). The modeling strategy is outlined in Figure 1.
FIGURE 1.
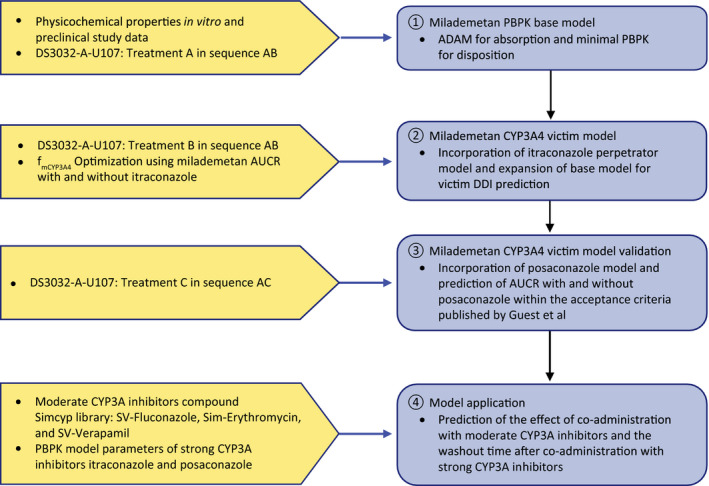
Summary of PBPK modeling strategy. Treatment A: single dose of milademetan 100 mg on study day 1 administered under fasted conditions; treatment B: itraconazole 200 mg twice daily on study day 8 and 200 mg once daily on study days 9 through 20 administered under fasted conditions and single dose of milademetan 100 mg on study day 14 administered 1 h post itraconazole dose; treatment C: posaconazole 200 mg three times daily on study days 8 through 20 administered with a full meal and single dose of milademetan 100 mg on study day 14 administered 2 h post posaconazole dose. ADME, absorption, distribution, metabolism, and excretion; AUCR, area under the concentration‐time curve ratio; CYP3A, cytochrome P3A; fmCYP3A4, fraction metabolized by CYP3A4; PBPK, physiologically based pharmacokinetics
Physicochemical parameters
The physicochemical and absorption, distribution, metabolism, and excretion (ADME) parameters used for the milademetan PBPK model development are summarized in Table 1.
TABLE 1.
Physicochemical and pharmacokinetic parameter input for PBPK model of milademetan
| Parameters (units) | Definition | Values | Data source |
|---|---|---|---|
| Physicochemical properties | |||
| MW (g/mol) | Molecular weight | 618.5 | Calculated |
| logP | Octanol‐to‐water partition coefficient | 3.7 | Predicted by PCModels CLOGP V.4.83 (MOLSIS Inc) |
| pKa | Ionization coefficient | 1.91, 2.26 | Predicted by Pallas UNIX V.4.1.1 (CompuDrug Ltd.) |
| f u,plasma | Plasma protein unbound fraction | 0.02 | Experimental data measured in vitro |
| B/P | Blood‐to‐plasma ratio | 0.784 | Predicted |
| Absorption (ADAM model) | |||
| f u,gut | Unbound fraction in gut enterocyte | 0.02 | Assumed to be equal to fu,plasma |
| Peff,man (10−4 cm/s) | Effective permeability | 1.891 | Predicted based on permeability measured in Caco−2 cell |
| Permeability assay | Caco‐2 | ||
| Apical:basolateral pH | 7.4:7.4 | ||
| Activity | Passive | ||
| PappaA:B (10−6 cm/s) | Apparent permeability | 17 | |
| Reference compound | Propranolol | ||
| Reference compound PappaA:B (10−6 cm/s) | 43 | ||
| Scalar | 1 | Assumed | |
| Transporter | ABCB1 (P‐gp/MDR1) | Exploratory data | |
| Jmax (pmol/min/cm2) | Maximal efflux rate | 13.4 | Calculated from in vitro Caco‐2 transport assay |
| K m (µM) | Michaelis‐Menten constant | 0.324 | Calculated from in vitro Caco‐2 transport assay |
| Distribution (minimal PBPK model) | |||
| V ss (L/kg) | Volume of distribution at steady‐state | 1.8 | Optimized by fitting clinical data (U107) for milademetan alone under fasted conditions |
| k in (1/h) | First‐order rate constant for distribution to the single adjusting compartment | 0.07 | Optimized by fitting clinical data (U107) for milademetan alone under fasted conditions |
| k out (1/h) | First‐order rate constant for distribution from the single adjusting compartment | 0.07 | Optimized by fitting clinical data (U107) for milademetan alone under fasted conditions |
| V sac (L/kg) | Volume of single adjusting compartment | 0.5 | Optimized by fitting clinical data (U107) for milademetan alone under fasted conditions |
| Elimination | |||
| CLint,CYP3A4 (µL/min/pmol of isoform) | CYP3A4‐specific intrinsic clearance | 0.459 | Optimized by fitting clinical data (U107) for milademetan and itraconazole sequence |
| CLint (HLM) (µL/min/mg protein) | Additional hepatic microsomal intrinsic clearance | 40.2 | Optimized by fitting clinical data (U107) for milademetan and itraconazole sequence |
| CLR | Renal clearance | 0 | Assumed |
| CLRbile | Bile clearance | 0 | Assumed |
| Interaction with CYP3A4 | |||
| K i (µM) | CYP3A4 reversible inhibition constant | 4.2 | Experimental data |
| K app (µM) | Apparent inactivation constant for CYP3A4 time‐dependent inhibition | 60.5 | Experimental data |
| K inact (h−1) | Maximal rate constant for enzyme inactivation | 3.71 | Experimental data |
Abbreviations: ADAM, advanced dissolution, absorption, and metabolism; PBPK, physiologically‐based pharmacokinetic.
Absorption
Milademetan absorption was described using an advanced dissolution, absorption, and metabolism (ADAM) model to incorporate intestinal P‐gp kinetics into milademetan absorption. The clinical food effect study (NCT03647202) estimated that the exposure of milademetan (area under the concentration‐time curve [AUC]) was reduced by 24% when administered with a high‐calorie, high‐fat meal compared with the fasted state (unpublished in‐house data). The negative food effect suggested that membrane permeability, but not drug dissolution, is the rate‐limiting step in absorption; therefore, solution was selected as the formulation in the ADAM model. P‐gp transport kinetic parameters, maximal efflux rate of milademetan (Jmax), and Michaelis Menten Constant (Km) shown in Table 1, were estimated by kinetic analysis using a three‐compartment model consisting of apical, cellular, and basal compartments along with Phoenix WinNonlin version 6.3 (Pharsight; Certara, Princeton, NJ, USA) based on the apparent permeability coefficient (Papp) of milademetan obtained by bidirectional transport studies across Caco‐2 cell monolayers. For prediction of the effective permeability (Peff,man) within Simcyp, the Caco‐2 transport assay results (Papp) measured at an apical pH of 7.4 and a basolateral pH of 7.4 were used with “Passive” option. Details of the P‐gp transport assay and parameter estimation are provided in the Supplementary Methods (Part A).
Disposition and elimination
Milademetan disposition and elimination were described by a minimal PBPK model with a single adjusting compartment (SAC). PK parameters, volume of distribution at steady‐state (V ss), first‐order rate constants into and out of the SAC (k in and k out, respectively), volume of SAC (V sac), and oral clearance (CLpo) were optimized by fitting to the milademetan concentration‐time data observed in period one of sequence AB (milademetan alone under fasted conditions) in study U107. The optimized CLpo of 8.5 L/h was used in the retrograde model to calculate intrinsic clearance (CLint) values for CYP3A4 and additional metabolic clearance. The value of fraction metabolized by CYP3A4 (f mCYP3A4) and additional metabolic clearance were optimized to recover the observed area under the concentration‐time curve from zero to infinity (AUCinf) and maximum serum concentration (Cmax) ratios (AUCR and CmaxR) of milademetan with and without co‐administration of itraconazole in study U107. The urinary excretion of milademetan in humans is not available. However, given that urinary excretion observed in rats was negligible (1.6 ± 0.7%), the renal clearance (CLR) was assumed to be zero. The bile clearance in humans was also assumed to be zero, as the biliary excretion of radiolabeled milademetan was minimal in rats (20.4 ± 1.8%) (unpublished in‐house data).
The minimal PBPK model included competitive inhibition of CYP3A4 (K i, 4.2 µM) and time‐dependent CYP3A4 inhibition (K app, 60.5 µM and k inact, 3.71 h−1). The inhibitory effect of milademetan on the activities of human hepatic CYP isoenzymes was characterized in vitro using pooled human hepatic microsomes. Details and parameter estimations of the experiment are provided in the Supplementary Methods (Part B).
CYP3A4 perpetrator model development
Itraconazole
The internally modified library model (SV‐Itraconazole_Fasted Soln, version 17) was used to describe itraconazole PK. The K i for itraconazole inhibition of intestinal and hepatic P‐gp was estimated to be 0.03 µM based on the clinical DDI study of digoxin with itraconazole. 7 The metabolite hydroxyitraconazole was also included in the model (K i, 0.0023 µM), as it has been shown to contribute to CYP3A inhibition. 8 , 9 The model parameter input for itraconazole and hydroxyitraconazole are provided in Tables S1 and S2. In a simulation scenario of DDIs with itraconazole, a single dose of milademetan 100 mg was administered under fasted conditions either alone or 1 h after itraconazole dosing on day 7. The dosing regimen of itraconazole was 200 mg b.i.d. on day 1 followed by 200 mg q.d. on days 2 to 13. The plasma concentration‐time profiles of milademetan were simulated up to 168 h for milademetan given alone or with itraconazole.
Posaconazole
The PBPK model for posaconazole as a strong CYP3A inhibitor was built according to the publication by Cleary et al. 10 that describes a minimal PBPK model with a first‐order absorption. In a simulation scenario of DDIs with posaconazole, a single dose of milademetan 100 mg was administered under fasted conditions either alone or 2 h after posaconazole dosing on day 7. The dosing regimen of posaconazole was 200 mg t.i.d. on days 1 to 13. The plasma concentration‐time profiles of milademetan were simulated up to 168 h for milademetan given alone or with posaconazole. Because posaconazole absorption is enhanced by food and it was administered under fed conditions, the fraction absorbed (f a) value of 0.85 derived from the mechanistic absorption model in GastroPlus (Simulations Plus) by Cleary et al. 10 was used for multiple doses of posaconazole under fed conditions. The final model parameters for posaconazole are summarized in Table S3.
Milademetan CYP3A4 victim DDI model validation
The performance of the milademetan CYP3A4 victim DDI model developed using data from sequence AB in study U107 was verified by comparing the simulated AUCR and CmaxR with those observed in sequence AC in study U107 (milademetan co‐administered with or without posaconazole). The model was considered to be verified when predicted AUCR and CmaxR were within the acceptance criteria calculated using the equations published by Guest et al. 11 , 12 :
where R obs is the observed ratio of AUC+inhibitor and AUCcontrol and δ is a parameter that accounts for variability, and is fixed to 1.25 in our study.
Simcyp simulations of coadministration with moderate CYP3A inhibitors
The effects of co‐administration of fluconazole (moderate CYP3A inhibitor), erythromycin (moderate CYP3A mechanism–based inhibitor), and verapamil (moderate CYP3A mechanism–based inhibitor and P‐gp inhibitor) on the PKs of milademetan were simulated. The input parameters of these CYP3A inhibitors are available in the compound library of Simcyp version 17: SV‐Fluconazole, Sim‐Erythromycin, SV‐Verapamil, and SV‐Norverapamil. The simulation design is provided in Table S4. Ten trials were simulated for each simulation scenario, with 10 subjects per trial. The default dosing regimens of moderate CYP3A inhibitors in Simcyp, which are the recommended clinical dosing regimens, were used in the simulations. All simulations were conducted with a virtual population of healthy subjects (Sim‐Healthy Volunteers in Simcyp version 17) under fasted conditions.
Simcyp simulations of washout period after discontinuation of strong CYP3A inhibitors
The appropriate time for resuming milademetan 160 mg q.d. from the reduced dose (80 mg q.d.) after discontinuing concomitant use of itraconazole or posaconazole was estimated. The simulated trial design is provided in Table S5. The simulation scenarios included milademetan 80 mg administered once daily with itraconazole or posaconazole for 8 days and returned to 160 mg q.d. on either day 9 or 11, and milademetan 160 mg q.d. without itraconazole or posaconazole on days 1 to 14 served as the reference. The dosing regimen of itraconazole was 200 mg b.i.d. on day 1 followed by 200 mg q.d. on days 2 to 8. The dosing regimen of posaconazole was 200 mg t.i.d. on days 1 to 8. In each simulation scenario, the plasma concentration‐time profiles of milademetan were simulated up to day 14, and milademetan daily AUCtau was calculated using a noncompartmental approach.
RESULTS
PK of milademetan when concomitantly administered with itraconazole or posaconazole
A total of 36 subjects were enrolled in study U107 and included in the PK analysis. The majority of subjects were men (66.7%) and White (50.0%) or Black (44.4%). The overall mean age was 40.0 years. The PK parameters of milademetan administered alone or with strong CYP3A4 inhibitor itraconazole or posaconazole are summarized in Table 2.
TABLE 2.
Summary of PK parameters of milademetan administered alone or concomitantly with itraconazole (treatment sequence AB) or posaconazole (treatment sequence AC)
| Parameter (unit) | Sequence AB | Sequence AC | ||
|---|---|---|---|---|
|
Treatment A (n = 18) |
Treatment B (n = 17) |
Treatment A (n = 18) |
Treatment C (n = 18) |
|
| AUCinf (ng/ml•h) | 14,698 (33.5) | 31,914 (35.4) | 13,595 (32.5) | 34,398 (25.5) |
| AUClast (ng/ml•h) | 14,627 (33.3) | 30,282 (34.5) | 13,529 (32.4) | 32,684 (24.5) |
| Cmax (ng/ml) | 718 (26.4) | 778 (33.8) | 675 (33.6) | 802 (30.3) |
| Tmax (h) | 3.52 (2.50–8.02) | 3.52 (2.97–6.08) | 3.03 (2.00–6.00) | 3.77 (1.02–8.03) |
| t 1/2 (h) | 20.7 (3.74) | 38.7 (6.23) | 21.3 (3.24) | 37.5 (7.09) |
| CL/F (L/h) | 6.80 (33.5) | 3.13 (35.4) | 7.36 (32.5) | 2.91 (25.5) |
| V z/F (L/h) | 200 (29.4) | 173 (32.3) | 223 (33.7) | 154 (25.6) |
PK parameters are presented as geometric mean (CV%) except median (minimum–maximum) for Tmax and arithmetic mean (SD) for t 1/2.
Treatment A: single dose of milademetan 100 mg on study day 1; treatment B: single dose of milademetan 100 mg on study day 14 administered 1 h post‐itraconazole dose; treatment C: single dose of milademetan 100 mg on study day 14 administered 2 h post‐posaconazole dose.
Abbreviations: AUCinf, area under the concentration‐time curve from zero to infinity; AUClast, area under the concentration‐time curve from time of administration up to the time of the last quantifiable concentration; CL/F, apparent clearance; Cmax, maximum concentration; PK, pharmacokinetic; t 1/2, terminal half‐life; Tmax, time to reach the maximum concentration; Vz/F, apparent volume of distribution.
Following a single, oral, 100 mg dose in healthy subjects, milademetan was absorbed, with a time to reach the maximum concentration (Tmax) of 3.52 h. Distribution of milademetan to tissues was moderate, with the geometric mean of apparent volume of distribution (Vz /F) estimated to be 200 L. The mean terminal half‐life (t 1/2) and apparent clearance (CL/F) of milademetan were 20.7 h and 6.8 L/h, respectively.
Concomitant administration of itraconazole appeared to have a minimal effect on the absorption of milademetan, manifested by the same median Tmax (3.52 h) and similar Cmax (CmaxR, 1.077 [90% confidence interval [CI], 98.8–117.4]). The AUCinf of milademetan was increased following concomitant administration of itraconazole. The AUCR was 2.152, with a 90% CI that fell outside the range of 80%–125%. This was consistent with the decrease in CL/F (3.13 vs. 6.80 L/h) and increase in t 1/2 (38.7 vs. 20.7 h) observed in the presence of itraconazole versus milademetan alone.
Concomitant administration of posaconazole slightly delayed the absorption of milademetan (3.77 vs. 3.03 h). Statistical analysis of PK parameters showed that the AUCinf and Cmax of milademetan were higher in the presence of posaconazole. The ratios of AUCinf and Cmax were 2.490 and 1.188, respectively, and their corresponding 90% CIs fell outside the range of 80%–125%. This was consistent with the decrease in CL/F (2.91 vs. 7.36 L/h) and increase in t 1/2 (37.5 vs. 21.3 h) observed in the presence of posaconazole versus milademetan alone.
Milademetan PBPK model
The milademetan base PBPK model well captured the biphasic shape of the plasma concentration‐time profiles observed in healthy subjects, manifested by the agreement between simulated PK profiles of milademetan and those observed from period one of sequence AB in the clinical DDI study (Figure 2). Following oral administration of milademetan alone, the predicted absorption parameters of the fraction of drug absorbed across the apical membrane of the enterocytes (fa ), fraction of the drug escaping gut metabolism (fg ), and first order absorption rate constant (ka ) were 0.98, 1, and 0.826 h−1, respectively. The predicted milademetan geometric mean Cmax of 734 ng/ml and AUCinf of 14,166 ng/ml•h were within 20% of the corresponding observed values (Cmax of 718 ng/ml and AUCinf of 14,698 ng/ml•h; Table 3).
FIGURE 2.
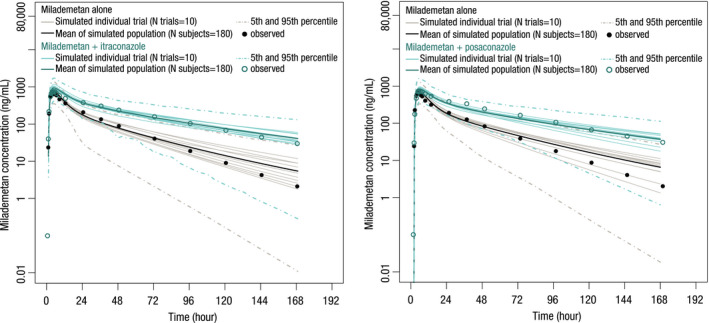
Predicted and observed mean plasma concentration‐time profiles of milademetan in the presence of multiple daily doses of itraconazole or posaconazole
TABLE 3.
Summary of predicted and observed PK parameters for interaction of milademetan with itraconazole and posaconazole
| AUCinf (ng/ml•h) | Cmax (ng/ml) | AUCR | CmaxR | |
|---|---|---|---|---|
| Inhibitory effect of itraconazole on PK of milademetan | ||||
| Control | ||||
| Observed | 14,698 (33.5) | 718 (26.4) | ||
| Predicted | 14,166 | 734 | ||
| With itraconazole | ||||
| Observed | 31,914 (35.4) | 778 (33.8) | 2.15 (1.98–2.34) | 1.08 (0.99–1.17) |
| Predicted | 32,908 | 860 | 2.32 (2.26–2.39) | 1.17 (1.16–1.18) |
| Acceptance criteria | 1.30–3.55 | 0.83–1.41 | ||
| Inhibitory effect of posaconazole on PK of milademetan | ||||
| Control | ||||
| Observed | 13,595 (32.5) | 675 (33.6) | ||
| Predicted | 13,999 | 674 | ||
| With posaconazole | ||||
| Observed | 34,398 (25.5) | 802 (30.3) | 2.49 (2.26–2.74) | 1.19 (1.10–1.28) |
| Predicted | 29,663 | 762 | 2.12 (2.06–2.18) | 1.13 (1.12–1.14) |
| Acceptance criteria | 1.47–4.23 | 0.87–1.63 | ||
AUCinf and Cmax are presented as geometric mean (CV%). AUCR and CmaxR are presented as geometric mean (90% CI).
Acceptance criteria calculated using equations published by Guest et al. 12
Abbreviations: AUCinf, area under the concentration‐time curve from zero to infinity; AUCR, area under the concentration‐time curve ratio; Cmax, maximum concentration; CmaxR, maximum serum concentration ratio; PK, pharmacokinetic.
The base PBPK model was then extended to include an in vivo estimate of the f mCYP3A4 of milademetan to allow CYP3A victim DDI simulations. The value of f mCYP3A4 was optimized to be 0.58 to recover the AUCR and CmaxR observed from sequence AB in the clinical DDI study. The predicted ratios of AUCinf and Cmax were 2.32 (90% CI, 2.26–2.39) and 1.17 (90% CI, 1.161–1.18), which are in good agreement with the observed ratios of 2.15 (90% CI, 1.98–2.34) and 1.08 (90% CI, 0.99–1.17), respectively (Table 3). Therefore, the developed PBPK model could capture the clinical observed DDIs of strong CYP3A4 inhibitor itraconazole with milademetan.
The performance of the milademetan CYP3A4 victim DDI model was verified by comparing the simulated plasma concentration‐time profiles of milademetan with those observed in sequence AC in the clinical DDI study (Figure 2). The predicted AUCR and CmaxR after co‐administration of posaconazole were 2.12 (90% CI, 2.06–2.18) and 1.13 (90% CI, 1.12–1.14) versus the observed ratios of 2.49 (90% CI, 2.26–2.74) and 1.19 (90% CI, 1.10–1.28), respectively (Table 3). Given that the predicted ratios were within the acceptance criteria calculated using the methods proposed by Guest et al., 12 the milademetan CYP3A4 victim model with the fmCYP3A4 value of 0.58 was considered to be verified and can be used to predict the effects of moderate CYP3A inhibitors on milademetan exposure that have not been studied in clinical trials.
Effect of moderate CYP3A inhibitors
The effects of co‐administration of fluconazole, erythromycin, and verapamil on the PKs of milademetan were simulated using the verified milademetan CYP3A4 victim model. The milademetan PK profiles after repeated doses of fluconazole (400 mg on day 1, 200 mg q.d. from days 2 to 13), erythromycin (500 mg t.i.d. from days 1 to 13), and verapamil (100 mg t.i.d. from days 1 to 12) are shown in Figure 3. The milademetan AUCR after concomitant administration with fluconazole, erythromycin, and verapamil was predicted to be 1.72 (90% CI, 1.69–1.76), 1.91 (90% CI, 1.83–1.99), and 2.02 (90% CI, 1.93–2.11), and the milademetan CmaxR was 1.13 (90% CI, 1.12–1.14), 1.11 (90% CI, 1.10–1.12), and 1.17 (90% CI, 1.15–1.18; Table S6).
FIGURE 3.
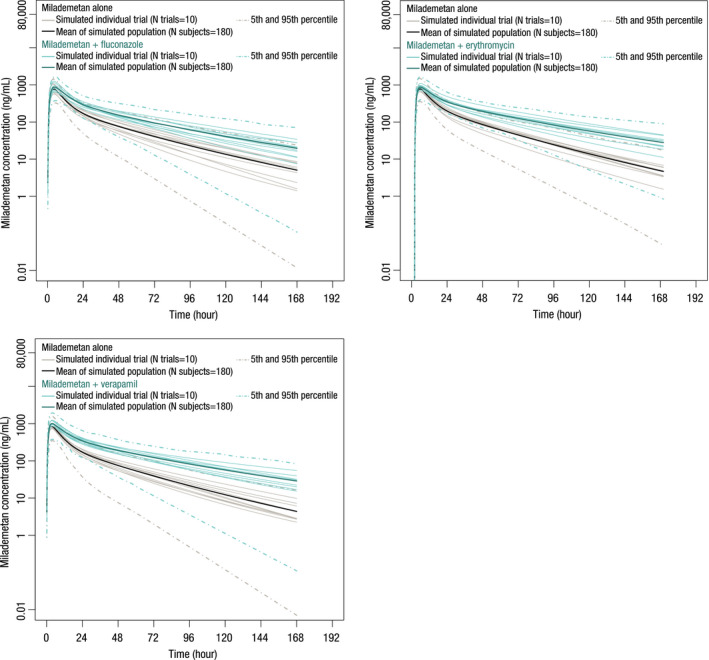
Predicted and observed mean plasma concentration‐time profiles of milademetan in the presence of multiple daily doses of fluconazole, erythromycin, and verapamil
Estimation of washout period after discontinuation of strong CYP3A inhibitors
The simulated milademetan plasma concentration‐time profiles with or without concomitant administration of itraconazole or posaconazole are shown in Figure 4. The milademetan daily AUCtau on days 8 to 14 for each simulation scenario are provided in Table S7. Half of the original dose of milademetan (80 mg q.d.) was administered with itraconazole or posaconazole. When the dose level of milademetan was returned to 160 mg q.d. on day 9 (1 day after the last dose of itraconazole), the AUCtau of milademetan on day 9 was estimated to be 1.56‐fold higher than the AUCtau of the reference (32,126 ng/ml•h vs. 20,641 ng/ml•h) and continued to increase to 34,959 ng/ml•h (1.69‐fold) on day 10 even if itraconazole was already discontinued on day 8. In contrast, when the milademetan dose was returned to 160 mg q.d. on day 11 (3 days after the last dose of itraconazole), the milademetan daily AUCtau after discontinuation of itraconazole was predicted to be comparable to the reference AUCtau (fold change ranging from 1.01 to 1.13). Similar simulation results were found for posaconazole. Resuming the milademetan dose at 160 mg 3 days after the last dose of posaconazole yielded an exposure comparable to that of the reference (fold change ranging from 0.92 to 1.22).
FIGURE 4.
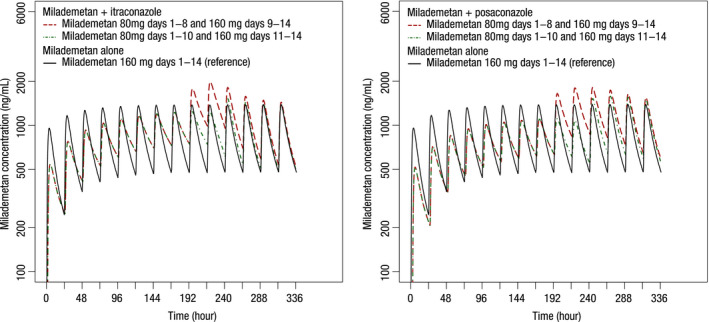
Simulated mean plasma concentration‐time profiles of milademetan with or without coadministration of itraconazole or posaconazole
DISCUSSION
This work studied the metabolic DDIs of milademetan and strong CYP3A inhibitors (itraconazole and posaconazole) in the clinical DDI study (U107). A PBPK model of milademetan was developed, and its predictive capability for DDI risk was qualified by comparing model‐predicted exposure ratios with those observed in the clinical DDI study. The verified PBPK model was used to assess the potential DDI risks of milademetan with coadministration of moderate CYP3A inhibitors (fluconazole, erythromycin, and verapamil) and predict the appropriate time when milademetan could resume its clinical dose (e.g., 160 mg q.d.) from the reduced dose (80 mg q.d.) after discontinuation of concomitant use of strong CYP3A inhibitors.
Milademetan is a substrate of both CYP3A4/5 and P‐gp in vitro. The U107 clinical DDI study evaluated the DDI of milademetan with itraconazole or posaconazole. Concomitant administration with itraconazole (a strong CYP3A and P‐gp dual inhibitor) and posaconazole (a strong CYP3A inhibitor) increased the plasma exposure (AUCinf) of milademetan by 2.15‐fold (90% CI, 1.98–2.34) and 2.49‐fold (90% CI, 2.26–2.74), respectively. This clinical study clearly demonstrated the involvement of CYP3A in the PK of milademetan in vivo and the minimal contribution of P‐gp as demonstrated by the sensitivity analysis and discussed later. In addition, on the basis of the DDI results from this study, it was recommended that the milademetan dose be reduced to half of the assigned dose when co‐administered with strong CYP3A inhibitors.
The application of PBPK modeling to predict DDI has gained recognition by health authorities, as exemplified by published guidance from the US Food and Drug Administration (FDA), European Medicines Agency (EMA), and Pharmaceuticals and Medical Devices Agency (PMDA) as well as a recent publication reporting the successful application of a PBPK model in predicting CYP‐mediated DDI. 4 , 10 To understand potential DDI risk with moderate CYP3A inhibitors, a human PBPK model for milademetan was subsequently developed using in vitro and in vivo PK parameters and verified by clinical DDI results from study U107. P‐gp–mediated efflux of the milademetan absorption process in the gut was incorporated into the PBPK model of milademetan and described by a Michaelis‐Menten type equation, where Jmax (maximum efflux rate) and K m (substrate concentration giving half‐Jmax) were determined by the model analysis of transport experiment across Caco‐2. 13 , 14 , 15 To assess the modeling hypothesis that the contribution of P‐gp efflux on milademetan PK is limited, a sensitivity analysis was performed on a single dose of milademetan 80 mg in a representative population (Sim‐Healthy Volunteers). The P‐gp efflux effect on milademetan fa , Cmax, and AUC was evaluated by changing the efflux parameters Km and Jmax from one‐tenth‐ to 10‐fold of their respective values estimated from transport assay (Km , 0.324 µM; Jmax, 13.4 pmol/min/cm2). The simulation reveals the Km and Jmax had minimal effect on the fa , Cmax, and AUC of milademetan, suggesting the limited effect of P‐gp efflux on milademetan PKs.
Milademetan oral absorption was described by ADAM model with in vitro data as model inputs (e.g., physiochemical properties of compound, intestinal permeability of human Caco‐2 cell system). The model estimates the fraction of absorption (e.g., fraction of the dose absorbed from the gastrointestinal tract [fa ]), however, these parameters were not verified due to limited milademetan ADME data. Milademetan rat ADME was studied after a single oral dose of [14C]milademetan 30 mg/kg. In noncannulated rats, drug‐derived radioactivity was primarily recovered in the feces (94.2 ± 0.7%) after 168 hours. In bile duct–cannulated rats, drug‐derived radioactivity was primarily recovered in the bile and feces, accounting for 20.4 ± 1.8% and 75.1 ± 3.3% of the dose, respectively, up to 48 h after administration. No [14C]‐ADME study conducted in humans. Of note, the PBPK oral absorption model predicted milademetan geometric means of Cmax and AUCinf within 20% of those observed from period 1 in study U107; however, it does not address the performance of this PBPK model of oral absorption. Successful application of milademetan PBPK models of oral absorption require iterative cycles of model verification. The performance of the model will be improved as more mechanistic information at late stages of milademetan development are available.
The PBPK modeling analysis in this study predicted the magnitude of DDIs with milademetan as a substrate. The model predicted less than twofold increase in milademetan exposure after co‐administration with fluconazole and erythromycin, suggesting the weak inhibitory effect of fluconazole and erythromycin on milademetan. Regarding verapamil, our model predicted an approximately twofold increase in milademetan AUC similar to those predicted for the strong CYP3A inhibitors itraconazole and posaconazole. The simulation was then performed using the DDI study of verapamil with midazolam published in the SV‐verapamil validation report but changed the verapamil dosing scheme to the one in our study (verapamil 100 mg t.i.d. for 13 days). The simulation predicted a 9.5‐fold increase in midazolam AUC, suggesting verapamil could have a strong CYP3A inhibitory effect, and yielded a DDI effect on milademetan similar to those predicted for strong CYP3A inhibitors. Furthermore, our PBPK model also estimated that the milademetan dose can be returned to its originally assigned dose 3 days after discontinuing the concomitant strong CYP3A inhibitors itraconazole and posaconazole. Recommendations based on this PBPK modeling analysis will be incorporated into future milademetan clinical studies. In the meantime, the PBPK model will be further refined using additional data collected from clinical studies.
CONFLICT OF INTEREST
All authors are paid employees of Daiichi Sankyo. Y.H., F.L., H.I., and M.A. own stock in Daiichi Sankyo.
AUTHOR CONTRIBUTIONS
Y.H., T.I., A.W., M.T., and M.L. wrote the manuscript. Y.H., A.W., M.T., and T.I. designed the research. Y.H., A.W., M.T., and T.I. performed the research. Y.H., T.I., A.W., M.T., M.L., H.I., F.L., and M.A. analyzed the data.
Supporting information
Figure S1
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Ming Zheng of Daiichi Sankyo, Inc. for his scientific review of and feedback on the manuscript. Editorial support in the preparation of this manuscript was provided by SciMentum and was funded by Daiichi Sankyo, Inc.
Hong Y, Ishizuka T, Watanabe A, et al. Model‐based assessments of CYP3A‐mediated drug‐drug interaction risk of milademetan. Clin Transl Sci. 2021;14:2220–2230. 10.1111/cts.13082
Y.H. and T.I. contributed equally to this work.
Funding information
The study was funded by Daiichi Sankyo.
REFERENCES
- 1. Yeo KR, Jamei M, Rostami‐Hodjegan A. Predicting drug‐drug interactions: application of physiologically based pharmacokinetic models under a systems biology approach. Expert Rev Clin Pharmacol. 2013;6:143‐157. [DOI] [PubMed] [Google Scholar]
- 2. Fahmi OA, Hurst S, Plowchalk D, et al. Comparison of different algorithms for predicting clinical drug‐drug interactions, based on the use of CYP3A4 in vitro data: predictions of compounds as precipitants of interaction. Drug Metab Dispos. 2009;37:1658‐1666. [DOI] [PubMed] [Google Scholar]
- 3. Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision‐making at the European Medicines Agency. Clin Pharmacol Ther. 2017;102:98‐105. [DOI] [PubMed] [Google Scholar]
- 4. Yu J, Zhou Z, Owens KH, Ritchie TK, Ragueneau‐Majlessi I. What can be learned from recent new drug applications? A systematic review of drug interaction data for drugs approved by the US FDA in 2015. Drug Metab Dispos. 2017;45:86‐108. [DOI] [PubMed] [Google Scholar]
- 5. Rowland M, Peck C, Tucker G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45‐73. [DOI] [PubMed] [Google Scholar]
- 6. Jamei M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609‐613. [DOI] [PubMed] [Google Scholar]
- 8. Templeton IE, Thummel KE, Kharasch ED, et al. Contribution of itraconazole metabolites to inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2008;83:77‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32:1121‐1131. [DOI] [PubMed] [Google Scholar]
- 10. Cleary Y, Gertz M, Morcos PN, et al. Model‐based assessments of cyp‐mediated drug–drug interaction risk of alectinib: physiologically based pharmacokinetic modeling supported clinical development. Clin Pharmacol Ther. 2018;104:505‐514. [DOI] [PubMed] [Google Scholar]
- 11. Shebley M, Sandhu P, Emami Riedmaier A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104:88‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guest EJ, Aarons L, Houston JB, Rostami‐Hodjegan A, Galetin A. Critique of the two‐fold measure of prediction success for ratios: application for the assessment of drug‐drug interactions. Drug Metab Dispos. 2011;39:170‐173. [DOI] [PubMed] [Google Scholar]
- 13. Neuhoff S, Yeo KR, Barter Z, Jamei M, Turner DB, Rostami‐Hodjegan A. Application of permeability‐limited physiologically‐based pharmacokinetic models: part I–digoxin pharmacokinetics incorporating P‐glycoprotein‐mediated efflux. J Pharm Sci. 2013;102:3145‐3160. [DOI] [PubMed] [Google Scholar]
- 14. Mikkaichi T, Yoshigae Y, Masumoto H, et al. Edoxaban transport via p‐glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42:520‐528. [DOI] [PubMed] [Google Scholar]
- 15. Tachibana T, Kitamura S, Kato M, et al. Model analysis of the concentration‐dependent permeability of P‐gp substrates. Pharm Res. 2010;27:442‐446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Supplementary Material