

Abstract
CD70 is expressed in up to 80% of nasopharyngeal carcinoma (NPC) cases. Cusatuzumab is a humanized anti‐CD70 monoclonal antibody, with dual action mechanisms: induction of cytotoxicity against CD70+ tumor cells and reduction in CD70‐CD27 signaling mediated immune evasion. The aim of this study was to assess the safety, pharmacokinetic profile, immunogenicity, pharmacodynamic profile, and preliminary activity of cusatuzumab in advanced NPC. Eleven patients were enrolled: one patient was assigned to arm A (adjuvant cusatuzumab monotherapy after curative chemoradiation), nine patients to arm B (cusatuzumab monotherapy; noncurative setting), and one patient to arm C (cusatuzumab + chemotherapy; noncurative setting); irrespective of tumoral CD70 expression. Both patients in arms A and C completed the study. All patients in arm B discontinued at an early stage. Five patients experienced grade greater than or equal to 3 nondrug related treatment‐emergent adverse events, most commonly fatigue and pneumonia (18%). An infusion‐related reaction was observed in two of 11 patients. Laboratory results showed no trend over time. Seven patients were eligible for response evaluation. No objective response to cusatuzumab was observed with stable disease being the best response. The current study indicates that the safety profile of cusatuzumab (with or without concurrent chemotherapy) is manageable in patients with advanced NPC, which is consistent with known safety profile. Limited activity of cusatuzumab in advanced NPC was observed. Combination therapies of cusatuzumab and other types of therapy should be explored for the improvement of activity in NPC and other CD70‐expressing malignancies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Currently, a multitude of immunotherapies are studied for their possible use in various solid carcinomas. Extensive research of the tumor microenvironment is done to determine possible immunological key mechanisms, which are related to tumor growth and progression. In this matter, chronic tumoral CD70 expression enhances proliferation of inhibitory regulatory T‐cells within the tumor microenvironment via activation of the CD70/CD27 axis. Upregulated CD70 expression, reported in high incidence in Epstein‐Barr virus‐related nasopharyngeal carcinoma (NPC), is considered a possible key pathophysiologic mechanism of NPC carcinogenesis and has emerged as a potential target.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study evaluated if cusatuzumab, an anti‐CD70 antibody, administered in patients with NPC at 5 mg/kg every 3 weeks intravenously, is safe and shows any clinical activity. Next, several biomarkers related to NPC (Epstein‐Barr virus DNA copy numbers) or the tumor microenvironment (soluble CD27) were assessed for any possible indication for clinical activity of cusatuzumab.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
A manageable safety profile was observed, comparable to previously reported grade greater than or equal to three treatment‐emerging adverse events for cusatuzumab. No response was observed with stable disease as best overall response, and median progression‐free survival was 11.6 weeks. No changes in biomarkers were observed, which may indicate the beneficial effect of cusatuzumab given 5 mg/kg every 3 weeks intravenously.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
In light of these results, activity of cusatuzumab should be evaluated in combination with other therapies, such as chemotherapy or an anti‐PD‐(L)1 agent. This should be done especially in Epstein‐Barr virus‐induced malignancies with confirmed high tumoral CD70 expression as this might increase the chance of clinical activity of cusatuzumab. Moreover, as a manageable safety profile was observed for the administration of cusatuzumab at 5 mg/kg every 3 weeks intravenously, it should be considered to evaluate activity of cusatuzumab doses higher than 5 mg/kg every 3 weeks. This as recent data in other malignancies shows an acceptable safety profile at a dose as high as 20 mg/kg every 2 weeks.
INTRODUCTION
Nasopharyngeal carcinoma (NPC) arises from the epithelial lining of the nasopharynx, which is the narrow passage behind the nasal cavity. It represents up to 95% of malignancies originating from the nasopharynx. The treatment of choice for early stage NPC is single‐modality radiotherapy. However, most patients are diagnosed with locally advanced stage disease and thus, require a more aggressive approach, based on concurrent chemoradiotherapy, combined with neoadjuvant or adjuvant (platinum‐based) chemotherapy. In recurrent or metastatic disease, chemotherapy‐based systemic therapy is the most common strategy, although with limited success. 1 Advances in the treatment of NPC have been hampered by its relatively low prevalence. Comparative studies indicate that such advances are unlikely to arise from variations in radiochemotherapy regimens. 2 , 3 On the other hand, immune checkpoint inhibition with anti‐programmed cell death protein‐1 (PD‐1) agents for metastatic NPC is being tested in phase I and II trials, with acceptable safety profiles and promising response rates up to 34%. 4 , 5 , 6 Currently, the molecular underpinnings of Epstein‐Barr virus (EBV)‐induced malignancies, which are correlated to poor prognosis, 7 are being unraveled and upregulated CD70 expression was reported as a potential target in NPC. 8
CD70 is a member of the tumor necrosis factor ligand family. In physiological conditions, CD70 is upregulated on activated T‐cells, B‐cells, and dendritic cells, serving as a unique ligand of CD27, which is expressed on early thymocytes, naive T‐cells, and activated B‐cells. The CD70/CD27 costimulatory pathway plays a crucial part in the development of T‐ and B‐cells. 9 , 10 , 11 Interaction between CD70 and CD27 acts as a transient, rapid‐response component of the normal immune response. Conversely, the chronic expression of CD70 on tumor cells has been shown to enhance proliferation of regulatory T‐cells (Tregs), known for hosting an immunosuppressive environment and, subsequently, promoting tumor survival and progression. 12 , 13 , 14 CD70 is expressed at very low levels in normal tissues, including all vital organs, but increased CD70 expression has been documented in a broad range of malignancies. 15 , 16 , 17 Anti‐CD70 immunotherapy may therefore induce direct tumor cytotoxicity and stimulate an antitumor immune response. 18 , 19
Cusatuzumab (ARGX‐110) is a humanized monoclonal antibody (mAb) of camelid origin, that binds to human CD70. It has been glyco‐engineered (through de‐fucosylation) using Potelligent technology to induce enhanced Ab‐dependent cell‐mediated cytotoxicity (ADCC). 20 , 21 It was demonstrated that cusatuzumab has a dual mechanism of action: induction of cytotoxicity against CD70+ tumor cells via various effector functions (enhanced ADCC, complement‐dependent cytotoxicity and Ab‐dependent cellular phagocytosis), and improving the antitumor immune response by interrupting the CD70‐CD27 signaling with Tregs. 22 The tolerability of cusatuzumab, pharmacokinetics (PKs), and preliminary antitumor activity in heavily pretreated patients with advanced CD70+ malignancies have already been demonstrated. 23 The relative contribution of these two mechanisms of action regarding antineoplastic activity is yet to be established.
Inhibition of CD70 signaling, reducing immunosuppression by Tregs and eradication of NPC cells expressing CD70, aims at a possible key pathophysiologic mechanism of NPC carcinogenesis and may be associated with clinical benefit. The primary objective of this study was to determine the safety profile of cusatuzumab in a cohort of patients with advanced NPC. Secondary objectives were determining the PK profile, immunogenicity, pharmacodynamic profile, and preliminary activity.
METHODS
Study design
This single‐site open‐label, nonrandomized, phase Ib feasibility trial was conducted in Belgium. The inclusion and exclusion criteria for the study are provided as Supplementary Information. The study was approved by an independent ethics committee, the Belgian health authorities, and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from every patient. Inclusion of 15 patients with high‐risk or advanced NPC was planned. After conducting the trial for over 2 years, enrollment was discontinued as it was concluded based on the interim results that the outcome of the study would not change the assessment of the risk‐benefit profile for cusatuzumab observed to date.
Patients were assigned to one of the following three arms:
Study arm A: Patients with locally advanced NPC, who recently completed radiochemotherapy with curative intent. Cusatuzumab was given as adjuvant therapy within 12 weeks of the primary therapy.
Study arm B: Patients with recurrent or metastatic NPC. Cusatuzumab was given in second‐ or later‐line setting as monotherapy.
Study arm C: Patients with recurrent or metastatic NPC. Cusatuzumab was given in second‐ or later‐line setting as add‐on to chemotherapy chosen by the treating physician following standard of care (capecitabine 1500 mg daily every 2 of 3 weeks).
Cusatuzumab, provided by argenx B.V., was given 5 mg/kg intravenously every 3 weeks on day 1 of each cycle (Q3W dosing regimen). Given the absence of toxicity associated with peak concentrations, a 3‐weekly dosing interval was chosen to minimize the clinical burden of frequent infusions and to support an eventual pharmacoeconomic benefit. The PK data confirm that drug concentrations are active in experimental models with a 5 mg/kg Q3W dosing regimen, as suggested by the results of the phase I dose‐escalation study of cusatuzumab. 23 All treatment cycles were administered in the outpatient setting following premedication with an antihistamine (diphenhydramine 50 mg equivalent orally, 12 h and 30 min prior to study mediation infusion) and a glucocorticoid (hydrocortisone 100 mg equivalent intravenously 30 min prior to study mediation infusion) to minimize the risk of infusion‐related reactions (IRRs). In addition to minimize the risk for IRRs, the infusion ratio of cusatuzumab during cycle 1 was set at 10 ml/h and doubled every 30 min for a maximum infusion ratio of 160 ml/h. Total infusion time during cycle one was approximately 3 h. From cycle two onward, the infusion rate was set at 160 ml/h for a total infusion time of ~ 100 min.
For every arm, administration of cusatuzumab was continued for a maximum of 18 cycles. Premature discontinuation was allowed in case of progressive disease (PD) according to immune‐related response criteria (irRC), 24 intolerable drug‐related toxicity (as per physician assessment), or withdrew consent to receive further treatment.
Study objectives
The primary study objective was to determine the safety and feasibility of administering cusatuzumab as a monotherapy or in combination with chemotherapy. Treatment‐emergent adverse events (TEAEs) were characterized using Common Terminology Criteria for Adverse Events (CTCAE), version 4.03. Clinical laboratory parameters, electrocardiograms, and vital signs were followed up. Patients were included in the safety analysis if administered at least one dose of cusatuzumab.
Secondary objectives included characterization of the cusatuzumab PK profile, immunogenicity to cusatuzumab, and characterization of pharmacodynamic biomarkers of cusatuzumab activity through EBV DNA copy number as well as soluble CD27 (sCD27). Quantification of cusatuzumab in serum for PK analysis was done via a validated enzyme‐linked immunosorbent assay (ELISA) assay (M08.ARGX‐110.huse.1; ICON Laboratory Services, USA) with a lower limit of quantification of 0.5 μg/ml. Serum for PK analysis was collected at cycle one predose, cycle one postdose (within 60 min of end of cusatuzumab infusion for determination of the maximum concentration [Cmax]), cycle one 168 h postdose, cycle three predose, and onward every odd cycle predose. Immunogenicity via the measurement of antidrug antibodies (ADAs) to cusatuzumab was evaluated in serum samples using an electrochemiluminescent assay method that can detect any class of ADA. Reactive samples were analyzed in a confirmatory assay for verification of specificity (ICON Laboratory Services, USA). An extensive method description for PK and ADA analysis is provided as Supplementary Information. Serum for ADA analysis was collected predose at every odd cycle. EBV DNA was measured as this is a causative agent for NPC and can upregulate CD70 expression. Changes in EBV viral load may therefore be related to an improved clinical activity. EBV was measured in EDTA blood samples through an in‐house DNA amplification test. Samples were considered positive if higher than the lower limit of quantification (LLOQ; =300 copies/µg DNA). The sCD27 was measured using the human CD27/TNFRSF7 DuoSet ELISA kit (cat no. DY382‐05, R&D systems) as this would give an insight into the inhibition of the CD70/CD27 axis by cusatuzumab. Serum samples were measured in replicate at a 1/10 dilution, next to control serum samples with an sCD27 concentration in serum of 2045 ± 520 pg/ml. Measurement runs with a control sample value outside of the range 1525 pg/ml (min) to 2561 pg/ml (max) were be repeated. Urine samples were measured in replicate at a 1/100 dilution, next to control urine samples with an sCD27 concentration in urine of 67,689 ± 2248 pg/ml. Measurement runs with a control sample value outside the range of 46,705 pg/ml (min) to 102,210 pg/ml (max) were be repeated. All samples for the pharmacodynamic analysis were collected predose during every odd cycle. Preliminary evidence of cusatuzumab‐mediated antitumor activity in different subsets of patients with NPC was documented through assessment of tumor response via computed tomography (CT) and/or magnetic resonance imaging (MRI) at the start of the study and every 12 weeks until disease progression or treatment termination. Antitumor activity was reported by objective response rate (ORR) according to irRC, 24 progression‐free survival (PFS), and disease‐free survival (defined as time from start of therapy to PD or death due to any cause, whichever occurred first).
Statistical Methods
Formal statistical calculation of sample size was not applied to this pilot feasibility study, due to the limited incidence of NPC in the Belgian population. PK analyses were performed using R 3.3.1 (R Foundation for Statistical Computing). All statistical analyses were performed using SAS 9.3 or higher. All patients who received at least one dose, complete or incomplete, of cusatuzumab were included in the analysis. The majority of statistical analyses were descriptive and exploratory. Relative dose intensity (RDI) was calculated as the delivered dose intensity divided by the standard dose intensity. A comparison of sCD27 concentrations in serum and urine between baseline and cycle three was made using a Wilcoxon test. The correlation between EBV DNA copy number, sCD27 in serum, and sCD27 in urine was evaluated using Pearson’s correlation coefficient, r. The PFS estimate was calculated using Kaplan‐Meier analysis.
RESULTS
Patient characteristics
A total of 11 patients were enrolled in the study between March 2015 and December 2017. The Consolidated Standards of Reporting Trials diagram is shown in Figure 1.
FIGURE 1.
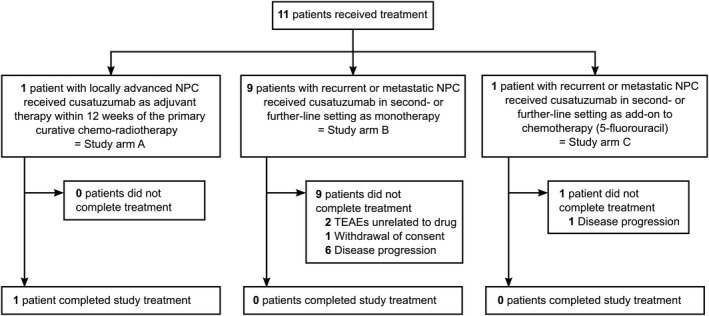
Consolidated Standards of Reporting Trials (CONSORT) Diagram. Patients were not randomized between different study arms. Patients completed study treatment if all 18 cycles were completed. Monotherapy (arm B) and combination therapy (arm C) in palliative noncurative setting were not comparator arms. TEAEs, treatment‐emerging adverse events; NPC, nasopharyngeal carcinoma
Detailed patient demographics per study arm are presented in Table 1. All patients were White, and the majority were men (82%). Median weight was 75.0 kg (range 49.4–94.5 kg) and median body mass index was 24.5 kg/m2 (range 16.3–31.3 kg/m2).
TABLE 1.
Patient characteristics
| Characteristic | Study arm A | Study arm B | Study arm C |
|---|---|---|---|
| Patients included, N (%) | 1 (100) | 9 (100) | 1 (100) |
| Completed treatment, N (%) | 1 (100) | 0 (0) | 1 (100) |
| Gender, N (%) | |||
| Male | 1 (100) | 7 (78) | 1 (100) |
| Female | 0 (0) | 2 (22) | 0 (0) |
| Age (years) | |||
| Median (range) | 39 (–) | 47 (23, 76) | 60 (–) |
| Weight (kg) | |||
| Median (range) | 80.8 (–) | 74.4 (49.4, 94.5) | 93.7 (–) |
| Body mass index (kg/m2) | |||
| Median (range) | 26.4 (–) | 24.2 (16.3, 31.3) | 30.2 (–) |
| Time since diagnosis (days) | |||
| Median (range) | 130 (–) | 623 (185, 6066) | 1156 (–) |
| Eastern Cooperative Oncology Group performance status, N (%) | |||
| 0 | 0 (0) | 2 (22) | 0 (0) |
| 1 | 0 (0) | 4 (44) | 1 (100) |
| 2 | 1 (100) | 3 (33) | 0 (0) |
| Laterality, N (%) | |||
| Right | 1 (100) | 2 (22) | 1 (100) |
| Left | 0 (0) | 4 (44) | 0 (0) |
| Bilateral | 0 (0) | 1 (11) | 0 (0) |
| Unknown | 0 (0) | 2 (22) | 0 (0) |
| Differentiation, N (%) | |||
| Good | 0 (0) | 1 (11) | 0 (0) |
| Moderate | 0 (0) | 0 (0) | 0 (0) |
| Poor | 0 (0) | 3 (33) | 1 (100) |
| Undifferentiated | 1 (100) | 2 (22) | 0 (0) |
| Not specified | 0 (0) | 3 (33) | 0 (0) |
| Recurrent disease, N (%) | |||
| No | 0 (0) | 3 (33) | 0 (0) |
| Yes | 1 (100) | 6 (67) | 1 (100) |
| Clinical T stage, N (%) | |||
| 1 | 0 (0) | 3 (33) | 0 (0) |
| 2 | 1 (100) | 1 (11) | 0 (0) |
| 3 | 0 (0) | 1 (11) | 0 (0) |
| 4 | 0 (0) | 3 (33) | 1 (100) |
| Unknown | 0 (0) | 1 (11) | 0 (0) |
| Clinical N stage, N (%) | |||
| 0 | 0 (0) | 1 (11) | 0 (0) |
| 1 | 0 (0) | 1 (11) | 0 (0) |
| 2 | 1 (100) | 4 (44) | 1 (100) |
| 3 | 0 (0) | 2 (22) | 0 (0) |
| Unknown | 0 (0) | 1 (11) | 0 (0) |
| Clinical M stage, N (%) | |||
| 0 | 1 (100) | 6 (67) | 1 (100) |
| 1 | 0 (0) | 2 (22) | 0 (0) |
| Unknown | 0 (0) | 1 (11) | 0 (0) |
| Prior cancer surgery, N (%) | |||
| No | 1 (100) | 7 (78) | 0 (0) |
| Yes | 0 (0) | 2 (22) | 1 (100) |
| Prior lines of chemotherapy, N (%) | |||
| 0 | 0 (0) | 1 (11) | 0 (0) |
| 1 – 2 | 1 (100) | 4 (44) | 1 (100) |
| 3 – 4 | 0 (0) | 4 (44) | 0 (0) |
| Prior radiotherapy, N (%) | |||
| 1 | 1 (100) | 4 (44) | 1 (100) |
| 2 | 0 (0) | 3 (33) | 0 (0) |
| 3+ | 0 (0) | 2 (22) | 0 (0) |
All data are N = number of patients (% = percentage of patients), except for age, weight, body mass index, and time since diagnosis: median (range).
Treatment exposure
Patients received a median number of four cycles (range 1–18). The dose of cusatuzumab administered per visit ranged between 243.5 and 473.6 mg, and median dose was 377 mg (median doses of 387.4, 369.6, and 473.6 mg for arms A, B, and C, respectively). The median treatment duration was 9.1 weeks (range 0.1 to 66.1 weeks; median duration of 66.1, 6.9, and 15.6 weeks for arms A, B, and C, respectively). The RDI could be calculated for 10 of 11 patients and median RDI was equal to 98.7%, ranging between 70.7% and 100% (median RDI of 70.7%, 99.9%, and 97.5% for arms A, B, and C, respectively).
Safety
A total of 96 TEAEs were reported in all patients, of which four patients (36%) experienced serious TEAEs (1 patient in arm A and 3 patients in arm B). A total of nine serious TEAEs were reported in these patients of which none were drug related. Eight of nine serious TEAEs were considered severe (grade ≥3) of which two resulted in subsequent non‐drug‐related deaths of two patients (both arm B, due to pneumonia and embolism, respectively). In addition, one patient also reported a nonserious grade greater than or equal to 3 TEAEs.
In total, five patients (45%) encountered grade greater than or equal to 3 TEAEs, of which all received cusatuzumab in monotherapy, with a total of 14 non‐drug‐related grade greater than or equal to 3 TEAEs (fatigue and pneumonia being the most frequent; both 18%). An IRR was observed in two of 11 patients (18%, both grade 2). Back pain (36%), diarrhea (27%), and productive cough (27%) were the most frequent grade 1–2 TEAEs. All observed TEAEs are shown in Table 2.
TABLE 2.
Overview of TEAEs
| TEAE | Monotherapy (Arm A + B; N = 10) | Combination therapy (Arm C; N = 1) | ||||
|---|---|---|---|---|---|---|
| Grade description |
Any n, N (%) |
1–2 n, N (%) |
3–5 n, N (%) |
Any n, N (%) |
1–2 n, N (%) |
3–5 n, N (%) |
| Number of TEAEs | 88, 10 (100) | 74, 10 (100) | 14, 5 (50) | 8, 1 (100) | 8, 1 (100) | 0, 0 (0) |
| Number of serious TEAEs | 9, 4 (40) | 1, 1 (10) | 8, 4 (40) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Blood and lymphatic system disorders | ||||||
| Anemia | 5, 1 (10) | 3, 1 (10) | 2, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Cardiac disorders | ||||||
| Palpitations | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Ear and labyrinth disorders | ||||||
| Deafness | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Ear disorder | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 2, 1 (100) | 2, 1 (100) | 0, 0 (0) |
| Vertigo | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Eye disorders | ||||||
| Dry eye | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Gastrointestinal disorders | ||||||
| Abdominal pain | 1, 1 (10) | 0, 0 (0) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Constipation | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Diarrhea | 3, 2 (20) | 3, 2 (20) | 0, 0 (0) | 1, 1 (100) | 1, 1 (100) | 0, 0 (0) |
| Dyspepsia | 2, 2 (20) | 2, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Gastritis | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Nausea | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Toothache | 3, 2 (20) | 3, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Vomiting | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| General disorders and administration site conditions | ||||||
| Asthenia | 1, 1 (10) | 0, 0 (0) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Chills | 1, 1 (10) a | 1, 1 (10) a | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Fatigue | 4, 2 (20) | 2, 1 (10) | 2, 2 (20) | 1, 1 (100) | 1, 1 (100) | 0, 0 (0) |
| Influenza‐like illness | 4, 2 (20) | 4, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Malaise | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Mucosal inflammation | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Infections and infestations | ||||||
| Abdominal infection | 1, 1 (10) | 0, 0 (0) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Bronchitis | 3, 2 (20) | 3, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Erysipelas | 1, 1 (10) | 0, 0 (0) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Mucosal infection | 1, 1 (10) | 0, 0 (0) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Nasopharyngitis | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Pharnyngitis | 1, 1 (10) | 0, 0 (0) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Pneumonia | 2, 2 (20) | 0, 0 (0) | 2, 2 (20) b | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Rhinitis | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Skin infection | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Viral infection | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Injury, poisoning and procedural complications | ||||||
| Infusion‐related reaction | 1, 1 (10) a | 1, 1 (10) a | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Investigations | ||||||
| Blood bilirubin increased | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 1, 1 (100) | 1, 1 (100) | 0, 0 (0) |
| Neutrophil count increased | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Metabolism and nutrition disorders | ||||||
| Decreased appetite | 2, 2 (20) | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 3, 2 (20) | 3, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Back pain | 5, 4 (40) | 5, 4 (40) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Bone pain | 2, 1 (10) | 2, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Musculoskeletal chest pain | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Pain in extremity | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Nervous system disorders | ||||||
| Anesthesia | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 1, 1 (100) | 1, 1 (100) | 0, 0 (0) |
| Dysesthesia | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Facial neuralgia | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Headache | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Hypoesthesia | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Third nerve disorder | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 1, 1 (100) | 1, 1 (100) | 0, 0 (0) |
| Neuropathy peripheral | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Peripheral sensory neuropathy | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Tremor | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Psychiatric disorders | ||||||
| Insomnia | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Renal and urinary disorders | ||||||
| Urinary retention | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Cough | 2, 2 (20) | 2, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Dysphonia | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Dyspnea | 2, 1 (10) | 2, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Epistaxis | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Laryngeal inflammation | 3, 2 (20) | 3, 2 (20) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Nasal congestion | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Productive cough | 2, 2 (20) | 2, 2 (20) | 0, 0 (0) | 1, 1 (100) | 1, 1 (100) | 0, 0 (0) |
| Skin and subcutaneous tissue disorders | ||||||
| Pruritus | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Skin ulcer | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Surgical and medical procedures | ||||||
| Removal of inert matter from skin or subcutaneous tissue | 1, 1 (10) | 1, 1 (10) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
| Vascular disorders | ||||||
| Embolism | 2, 2 (20) | 1, 1 (10) | 1, 1 (10) b | 0, 0 (0) | 0, 0 (0) | 0, 0 (0) |
All data are n = number of events, N = number of patients, (% = percentage of patients per therapy cohort).
Abbreviation: TEAEs, treatment‐emerging adverse events.
Considered treatment‐related adverse events.
Two not drug‐related deaths occurred (grade 5 TEAEs), one due to pneumonia and one due to embolism.
No clinically significant changes in hematology or biochemistry were noticed, nor was any trend toward abnormal values observed. There was only a clinically significant aberration increase in total bilirubin value from 0.7 to 1.3 mg/dl in one patient (arm C, cycle 6, day 1). All other liver enzymes were within normal ranges.
PK and immunogenicity
The cusatuzumab concentration‐time profiles are shown in Figure 2. Following the first administration, the median Cmax of cusatuzumab was 123.0 µg/ml (range 72.2–152.0 µg/ml), with a median volume of distribution equal to 3.28 L (range 2.26–5.15 L). The median total drug exposure (area under the curve [AUC]) during the first 336 h following study drug administration was 16.8 µg × h/ml (range 6.8–24.0 µg × h/ml). Median trough serum concentration (Ctrough) was 12.6 µg/ml (range 0.7–58.7 µg/ml) from cycle three onward, and 48.6 µg/ml (range 27.8–92.8 µg/ml) for patients who reached cycle five. The patient in arm C showed no increase in cusatuzumab serum concentration at cycle five due to an unevaluable postdose sample. The observed PK profile appeared similar between cusatuzumab monotherapy and when cusatuzumab was administered in combination with chemotherapy. An overview of the noncompartmental PK analysis is provided as Supplementary Information Table S1.
FIGURE 2.
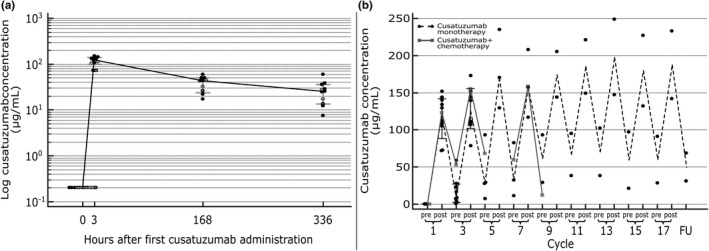
Concentration‐time PK profile of cusatuzumab 5 mg/kg. (a) PK profile after the first administration of cusatuzumab. Cusatuzumab serum concentrations are represented for all ADA‐negative patients (n = 8, full black dots), ADA‐positive patients at start of cycle one (n = 8, open black dots) and ADA‐positive patients at start of cycle three (n = 2, open grey dots). The black line represents median cusatuzumab serum concentration with 95% confidence interval (in grey). (b) PK profile throughout study treatment. Round black and reactangle grey dots represent patient cusatuzumab serum concentrations for monotherapy (arm A plus B), and combination therapy (arm C), respectively. Dashed black and full grey lines represent median cusatuzumab serum concentrations for monotherapy (arm A plus B), and combination therapy (arm C), respectively. Per cycle, the predose and postdose concentrations are depicted. For the patient in the combination therapy arm (arm C), the postdose PK sample during cycle five was not evaluable. ADAs, anti‐drug antibodies; PK, pharmacokinetic
Some immunogenicity against cusatuzumab was shown in four patients (36%; all in arm B). One patient was ADA positive at baseline and showed second lowest Cmax among patients (72.4 µg/ml). The three remaining patients were ADA negative at baseline and ADA positivity was observed at the start of cycle three (n = 2) and cycle seven (n = 1). All patients that were ADA positive at the beginning of cycle three (n = 3), showed Ctrough (range 0.7–8.7 µg/ml) lower than the median Ctrough observed for the study (12.6 µg/ml) as well as lower postdose concentration at cycle three (range 78.6–111.0 µg/ml) than the median concentrations (139.0 µg/ml). No difference for these patients was observed in the maximum exposure between cycle one and cycle three. Aforementioned patients were all ADA positive at the conclusion of the study.
Pharmacodynamics
All pharmacodynamics profiles are shown in Figure 3.
FIGURE 3.
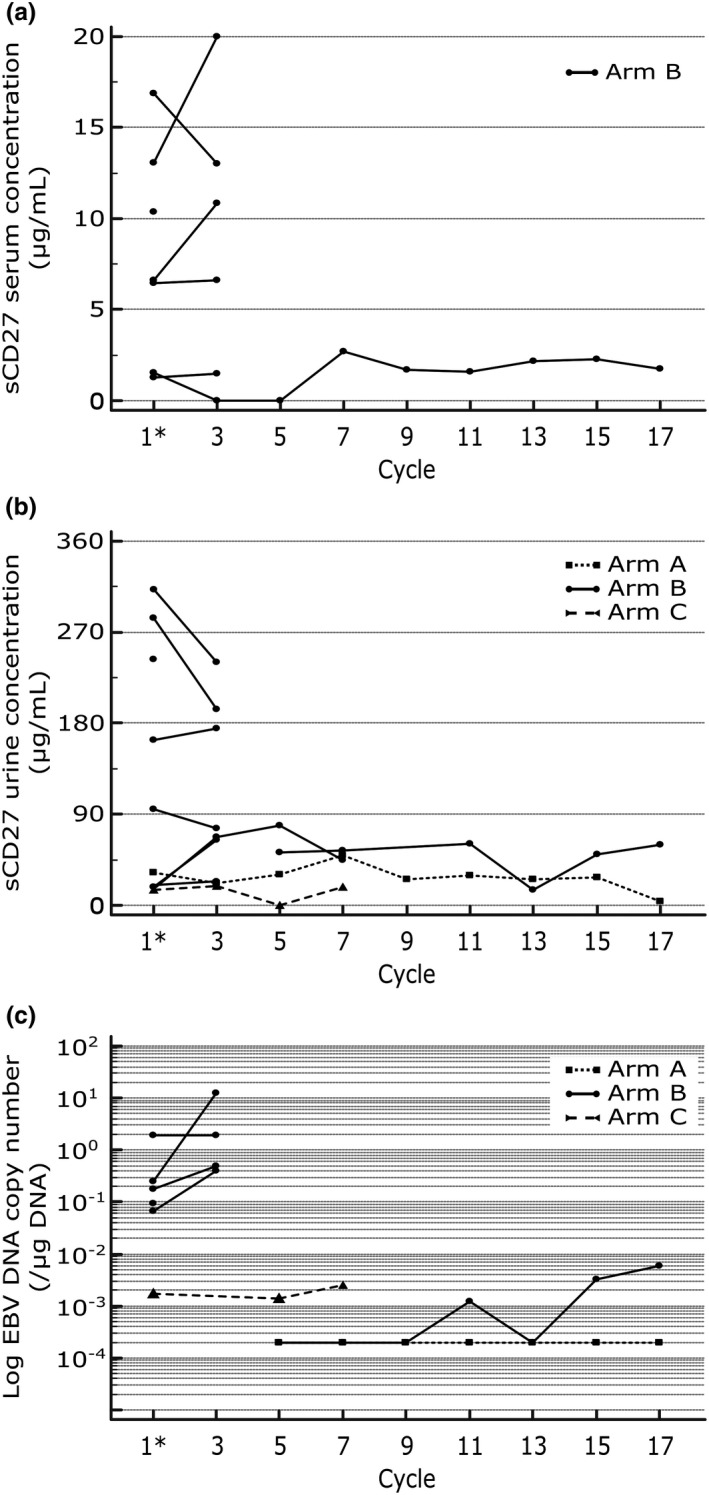
Concentration‐time biomarker profile throughout study treatment. Rectangle, round, and triangle dots represent patient concentrations in arms A, B, and C, respectively. Dotted, full, and dashed lines represent intrapatient concentration changes in arms A, B, and C, respectively. All samples were taken predose. Asterisk indicates baseline measurement for comparison. (a) Pharmacodynamic profile for sCD27 in serum. Serum concentrations were undetectable in arms A and C, as well as for two patients in arm B (not depicted). (b) Pharmacodynamic profile for sCD27 in urine. No sCD27 urine concentrations at baseline and at cycle three are depicted for one patient in arm B due to missing values. (c) Pharmacodynamic profile for EBV DNA. EBV DNA copy numbers were below the limit of detection in arm A, as well as for three patients in arm B, and are depicted at a concentration of 102 copies/µg DNA (below limit of detection). EBV, Epstein‐Barr virus; sCD27, soluble CD27
Six patients in arm B had measurable sCD27 serum concentrations at baseline and in cycle three. These patients had a baseline median sCD27 serum concentration of 6.52 µg/ml (range 1.29–16.86 µg/ml), which was comparable to the median sCD27 serum concentration at the start of cycle three (8.74 µg/ml; range 1.25–19.98 µg/ml; p = 0.5625). One patient had a baseline sCD27 serum concentration due to onset of a serious TEAE and subsequent death. The sCD27 serum concentration was below the lower detection limit for all other patients (in arm A, in arm C, and in 3 patients in arm B) throughout the study.
All patients had measurable sCD27 urine concentrations, except for one patient in arm B with missing sCD27 urine concentration at baseline and in cycle three. Baseline median sCD27 urine concentration was 63.49 ng/ml (range 14.83–312.33 ng/ml). Comparable to sCD27 in serum, no change in sCD27 urine concentrations were found at the beginning of cycle three (median sCD27 urine concentration = 67.74 ng/ml; range 19.14–240.11 ng/ml; p = 0.8203).
Limit of detection for EBV DNA copy number was not reached for the patient in arm A and for four patients in arm B at baseline. The baseline number of EBV DNA copies in all other patients ranged between 1.7 × 103 and 1.883 × 106 copies/µg DNA. For the case of the five patients with no measurable baseline EBV DNA copy number, the number of EBV DNA copies remained unmeasurable during the study for all but one patient. The sudden and extreme increase in EBV DNA copy numbers for one patient at cycle three was attributed to progressive disease, rather than to an EBV reactivation.
At baseline, EBV DNA copy number is borderline significantly correlated to the sCD27 serum concentration (r = 0.8367, p = 0.0773; n = 6). No correlation exists between EBV DNA copy number and sCD27 urine concentration (r = 0.5021, p = 0.3101), nor between sCD27 serum concentration and sCD27 urine concentration (r = 0.4817, p = 0.3334). After one administration of cusatuzumab, no correlation could be found between EBV DNA copy number and sCD27 serum concentration (r = 0.4718, p = 0.5282; n = 5), EBV DNA copy number and sCD27 urine concentration (r = 0.6988, p = 0.3012), and sCD27 serum concentration and sCD27 urine concentration (r = 0.0509, p = 0.9352).
Preliminary clinical activity
Seven of 11 patients were eligible for evaluation of preliminary activity (6 in arm B and 1 in arm C). No objective response according to irRC was observed in all cusatuzumab‐treated patients. Two subjects in arm B experienced stable disease, which was considered the best overall response (BOR). All other evaluable patients showed PD. Four patients were not evaluable. One patient in arm A received additional local therapy prior to start of the trial and was categorized as “disease‐free”; in three other patients from arm B, treatment was discontinued early (due to TEAEs [n = 2] and withdrawal of consent [n = 1]). BOR rate is given in Table 3 and a waterfall plot for BOR and swimmer plot are depicted in Figure 4.
TABLE 3.
Preliminary efficacy results for cusatuzumab in patients with advanced nasopharyngeal carcinoma
| Outcome |
Study arm A N (%) |
Study arm B N (%) |
Study arm C N (%) |
|---|---|---|---|
| Complete response | 0 (0) | 0 (0) | 0 (0) |
| Partial response | 0 (0) | 0 (0) | 0 (0) |
| Stable disease | 0 (0) | 2 (22) | 0 (0) |
| Progressive disease | 0 (0) | 4 (44) | 1 (100) |
| Not evaluable | 0 (0) | 3 (33) | 0 (0) |
| No disease | 1 (100) | 0 (0) | 0 (0) |
Best overall response was determined using the immune‐related response criteria (24). The patient in study arm A had no measurable disease at baseline and was therefore categorized as “no disease.” Three patients in study arm B were not evaluable due to early treatment discontinuation. All data are N = number of patients (% = percentage of patients).
FIGURE 4.
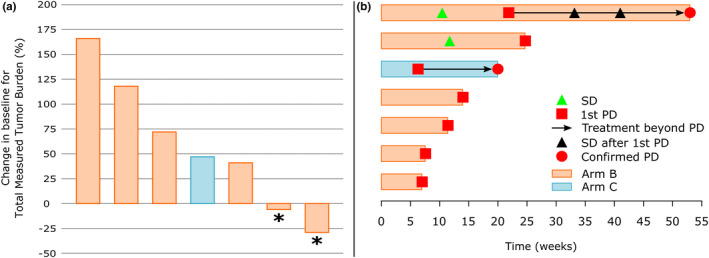
Preliminary activity results. (a) Waterfall plot for BOR. Waterfall plot shows the change in baseline for total measured tumor burden in evaluable patients. Only seven patients (arm B [red bars] and C [blue bars]) were eligible for efficacy evaluation. No response (complete or partial) was observed. Patients reaching stable disease as BOR according to immune‐related response criteria (24) are highlighted with an asterisk. (b) Swimmer plot. Each bar represents one (eligible) study patient. X‐axis depicts treatment time in weeks. Items are explained in the figure legend. BOR, best overall response; PD, progressive disease; SD, stable disease
Of all patients with recurrent/metastatic disease (arm B and C), seven of 10 showed PD during the course of their treatment. Median PFS was equal to 11.6 weeks (95% confidence interval = 6.4–24.9 weeks). Median PFS was prolonged in patients who received three or four prior lines of chemotherapy compared to those who received a maximum of two prior lines of chemotherapy, although this observation lacked statistical significance (7.7 vs. 11.6 weeks, p = 0.6193). Similar results were found for the number of prior radiotherapies received (7.7 vs. 14.1 weeks for one versus two or more radiotherapies received, p = 0.1328) and for total prior radiotherapeutic dose received (7.7 vs. 24.9 weeks for 70 or less Gy vs. more than 70 Gy, p = 0.1351).
DISCUSSION
The objective of this study was to determine the safety profile of cusatuzumab in patients with NPC when administered at a 5 mg/kg Q3W dosing regimen. Secondary objectives included PK, pharmacodynamics, immunogenicity, and preliminary activity.
As NPC remains a rare disease in Europe, the distribution among the different arms depended on the number and disease state of the patients who were referred to the trial. The rarity of NPC explains why the number of patients in this phase I study was relatively low.
Cusatuzumab was generally well‐tolerated. All patients had TEAEs that were mostly mild or moderate and manageable. Two non‐drug‐related deaths were observed, which was as expected in this heavily pretreated and advanced cancer population. The seriousness, frequency, and severity of TEAEs reported in this study were consistent with the safety profile of cusatuzumab in a previous trial in solid carcinoma conducted by Aftimos et al. 23 In our study, 36% of patients had a serious TEAE and 15% of TEAEs were grade greater than or equal to 3. These numbers are similar to the number observed by Aftimos et al. (38% and 18%, respectively). 23 Fatigue was the most frequent grade greater than 3 TEAE (in 18% of patients) observed in current trial, which is comparable to the findings from the phase I dose‐escalation study of cusatuzumab in advanced malignancies, reporting grade 3–4 fatigue in 12% of patients. IRRs were the only drug‐related TEAE and were observed in two of 11 patients (18%). This was lower than the number of IRRs (38%) reported by Aftimos et al. 23 but equivalent to the data (17%) reported by Riether et al. 25 The difference in these results could be attributed to the use of premedication prior to cusatuzumab administration, which effectively lowered the occurrence of IRRs.
PK analysis of cusatuzumab demonstrated median serum Cmax of 123.0 µg/ml and Ctrough of 12.6 µg/ml. This is similar to previous data reported for cusatuzumab administration in a variety of advanced malignancies. 23 Although the PK profile appeared to be similar with and without chemotherapy, no conlusion can be drawn with regard to drug‐drug interaction. In contrast to this previous study, some immunogenicity was observed in 36% of patients (n = 4), two of which showed positive postdosing ADA titers after the first two dose administrations of cusatuzumab (18%). To our knowledge, ADA titers were not previously reported in the context of cusatuzumab administration. Therefore, ADA should be evaluated in future administrations of cusatuzumab with regard to PK profile and antitumoral activity.
Pharmacodynamic evaluation consisted of the measurement of sCD27 in serum, sCD27 in urine, and EBV DNA copy number. Measurement of sCD27 in serum and urine showed no trends over time. Note that sCD27 concentrations in urine are highly dependent on the corresponding volume of urine obtained from each patient, and may therefore be biased as no data were available about the collected urine volume. The sCD27 in serum was lower than the detection limit in four of 11 patients. It can be hypothesized that the low serum levels are likely due to the fact that these patients demonstrated no to very low EBV DNA copy numbers. This was a somewhat peculiar finding as NPC is considered to be mainly an EBV‐induced malignancy, 8 especially in other (endemic) regions of the world. In addition, EBV DNA copy numbers were positively correlated to sCD27 in serum at baseline in patients with measurable EBV DNA copy number (borderline significant), which might be attributed to the fact that an EBV infection triggers CD70 expression which, in turn, increases activity of CD27‐CD70 axis. 8 Interestingly, this correlation was absent following the first two administrations of cusatuzumab. This could indicate that cusatuzumab possibly exerted some suboptimal effect in the tumor. Nevertheless, our sample size is to small to allow strong statistical inference and to validate this hypothesis in the current study.
Unfortunately, no objective response to treatment with cusatuzumab according to irRC was observed, with stable disease as BOR in two of seven evaluable patients. This might be due to the upfront use of a glucocorticoid prior to study mediation, as has been hypothesized that glucocorticoid medication may limit the activity of immune‐oncology agents, 26 and low EBV positivity as only half of patients (n = 6) showed measurable EBV DNA copy numbers. As it has been proven that EBV presence can upregulate CD70 expression, one can hypothesize that there was insufficient tumoral CD70 expression to target with an anti‐CD70 immunotherapy. 27 The link between low EBV copy number and tumoral CD70 expression should be further validated by determination of CD70 expression via immunohistochemistry or reverse transcription quantitative polymerase chain reaction (RT‐qPCR). Moreover, as we evaluated a heavily pretreated and advanced patient population, this can account for the fact that some patients showed rapid progression on therapy (within 9 weeks), prior to any possible effect of treatment with cusatuzumab. In addition, median PFS was 11.6 weeks, which correlated to almost four cycles of cusatuzumab administration. This corresponds to the median PFS of 12.0 weeks that has been reported for anti‐PD‐1 therapy in NPC, but is lower compared to the median PFS of 22.3–23.1 weeks reported for various chemotherapeutic regimens studied in similar patient populations. 5 , 28 , 29
When subdividing patients among the number of prior chemotherapies, the number of prior radiotherapies, and total prior therapeutic dose received, patients who received higher number of treatments and higher doses had numerically longer PFS, although not statistically significant. It could be hypothesized that having received numerous prior chemo‐ and radiotherapies, which are known to have immunomodulating effects, can trigger the CD70‐CD27 axis. 18 CD70 blockade in these patients might be more effective, although no statements can be made due to the limited patient sample size. Nevertheless, this would indicate that cusatuzumab might function as a treatment regimen in heavily pretreated patients. Moreover, including cusatuzumab in a combination regimen could further boost therapeutic activity with regard to ORR and survival rate, as combinations of Ab‐based therapies have been shown superior over Ab‐based monotherapies in various solid and hematological malignancies. Combinations of mAbs, such as cetuximab, trastuzumab, or rituximab, were already successfully applied in the treatment of patients with colorectal cancer, breast cancer, or non‐Hodgkin’s lymphoma; respectively. 30 , 31 To this end, several NPC‐related studies investigated combinations of radiotherapy or chemotherapy with targeted therapies or immune checkpoint inhibitors. 5 , 6 , 32 Even more, it has been reported that EBV tumors can also upregulate PD‐L1 expression in tumors. 33 Therefore, it would be worthwhile to evaluate the activity of cusatuzumab in combination with chemotherapy or an anti‐PD‐(L)1 agent in patients with NPC or other EBV‐induced malignancies with confirmed high tumoral CD70 expression. In addition, it should be considered to evaluate activity of doses of cusatuzumab higher than the dose of 5 mg/kg every 3 weeks studied in our trial, as cusatuzumab even showed an acceptable safety profile at a dose as high as 20 mg/kg every 2 weeks. 25 Increasing the dose frequency to, for example, once weekly might also be an opportunity as this would align the treatment exposure to the previously determined half‐life of the compound. To our knowledge, this can be done safely as all grade greater than or equal to 3 TEAEs were not considered to be treatment related.
Major factors affecting this study were the low number of patients in the study, although this is attributable to the low incidence of NPC in Belgium (0.5–2 cases per 100,000). 1 Next, the majority of patients were heavily pretreated and no control arm was established, which may also affect the safety profile and activity assessment. Last, no patient selection based on tumoral CD70 expression was done before administration of the anti‐CD70 therapy. As EBV titers were unmeasurable in five patients, it is possible that these patients also would have low or absent tumoral CD70 expression and might therefore have resulted in bias in the activity assessment of this trial. Similarly, patients were administered an immunotherapeutic agent irrespective of lymphocyte count, which is currently more and more a criterion in clinical trials. This might also reflect why no response was observed in this setting.
In summary, the current study indicates that cusatuzumab, when administered at 5 mg/kg every 3 weeks with or without concurrent chemotherapy, is safe and tolerable in patients with advanced NPC. The safety data collected affirm the risk‐benefit profile for cusatuzumab observed to date. Unfortunately, only a limited activity of cusatuzumab in advanced NPC was observed. Nonetheless, the results on outcome seem to be in line with those documented in patients with NPC who have been treated with other targeted therapies. As chemotherapy and radiotherapy can have immunomodulatory properties, combination therapies of cusatuzumab and chemotherapy/targeted therapy/immunotherapy should be explored for the improvement of activity in NPC as well as other CD70‐positive malignancies.
CONFLICT OF INTEREST
T.D. is an employee of argenx B.V. S.F. reports personal fees from argenx B.V., personal fees from Rheacell GmbH & Co KG, personal fees from ISA Therapeutics B.V, personal fees from Kiadis Pharma Netherlands B.V., personal fees from Molecular Partners AG, personal fees from Novateur Ventures, personal fees from Polyphor, personal fees from Synteract GmbH, other from Immutep, personal fees from Pharmalog GmbH, outside the submitted work. All other authors declared no competing interest for this work.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. P.D., F.D., L.F., and S.R. designed the research. A.D.M., T.V., L.F., and S.R. performed the research. T.V., T.D., and S.F. analyzed the data. D.C., T.D., P.P., and L.F. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge all the participating patients, their families, and their referring physicians. In addition, we would like to thank the argenx medical writing and scientific communication teams for their review of the article.
De Meulenaere A, Vermassen T, Creytens D, et al. An open‐label, nonrandomized, phase Ib feasibility study of cusatuzumab in patients with nasopharyngeal carcinoma. Clin Transl Sci. 2021;14:2300–2313. 10.1111/cts.13089
Astrid De Meulenaere and Tijl Vermassen contributed equally to this paper.
Funding information
This investigator‐driven research was supported by the TGO fund, granted by the Flemish Agency for Innovation by Science and Technology (IWT). argenx provided additional financial support for analytical and statical analysis.
REFERENCES
- 1. Rottey S, Madani I, Deron P, Van Belle S. Modern treatment for nasopharyngeal carcinoma: current status and prospects. Curr Opin Oncol. 2011;23:254‐258. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Sarraf M, LeBlanc M, Giri PG, et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: phase III randomized Intergroup study 0099. J Clin Oncol. 1998;16:1310‐1317. [DOI] [PubMed] [Google Scholar]
- 3. Lin JC, Jan JS, Hsu CY, et al. Phase III study of concurrent chemoradiotherapy versus radiotherapy alone for advanced nasopharyngeal carcinoma: positive effect on overall and progression‐free survival. J Clin Oncol. 2003;21:631‐637. [DOI] [PubMed] [Google Scholar]
- 4. Lv JW, Li JY, Luo LN, Wang ZX, Chen YP. Comparative safety and efficacy of anti‐PD‐1 monotherapy, chemotherapy alone, and their combination therapy in advanced nasopharyngeal carcinoma: findings from recent advances in landmark trials. J Immunother Cancer. 2019;7:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma BBY, Lim WT, Goh BC, et al. Antitumor activity of nivolumab in recurrent and metastatic nasopharyngeal carcinoma: an international, multicenter study of the Mayo Clinic phase 2 consortium (NCI‐9742). J Clin Oncol. 2018;36:1412‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yeo ELL, Li YQ, Soo KC, Wee JTS, Chua MLK. Combinatorial strategies of radiotherapy and immunotherapy in nasopharyngeal carcinoma. Chin Clin Oncol. 2018;7:15. [DOI] [PubMed] [Google Scholar]
- 7. Lin JC, Wang WY, Chen KY, et al. Quantification of plasma Epstein‐Barr virus DNA in patients with advanced nasopharyngeal carcinoma. N Engl J Med. 2004;350:2461‐2470. [DOI] [PubMed] [Google Scholar]
- 8. Agathanggelou A, Niedobitek G, Chen R, et al. Expression of immune regulatory molecules in Epstein‐Barr virus‐associated nasopharyngeal carcinomas with prominent lymphoid stroma. Evidence for a functional interaction between epithelial tumor cells and infiltrating lymphoid cells. Am J Pathol. 1995;147:1152‐1160. [PMC free article] [PubMed] [Google Scholar]
- 9. Bowman MR, Crimmins MA, Yetz‐Aldape J, et al. The cloning of CD70 and its identification as the ligand for CD27. J Immunol. 1994;152:1756‐1761. [PubMed] [Google Scholar]
- 10. Hintzen RQ, Lens SM, Beckmann MP, et al. Characterization of the human CD27 ligand, a novel member of the TNF gene family. J Immunol. 1994;152:1762‐1773. [PubMed] [Google Scholar]
- 11. Grewal IS. CD70 as a therapeutic target in human malignancies. Expert Opin Ther Targets. 2008;12:341‐351. [DOI] [PubMed] [Google Scholar]
- 12. Claus C, Riether C, Schürch C, Matter MS, Hilmenyuk T, Ochsenbein AF. CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res. 2012;72:3664‐3676. [DOI] [PubMed] [Google Scholar]
- 13. Jak M, Mous R, Remmerswaal EB, et al. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T‐cells in chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:788‐801. [DOI] [PubMed] [Google Scholar]
- 14. Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. CD70+ non‐Hodgkin lymphoma B cells induce Foxp3 expression and regulatory function in intratumoral CD4+CD25 T cells. Blood. 2007;110:2537‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Law CL, Gordon KA, Toki BE, et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti‐CD70 antibody‐drug conjugates. Cancer Res. 2006;66:2328‐2337. [DOI] [PubMed] [Google Scholar]
- 16. Ryan MC, Kostner H, Gordon KA, et al. Targeting pancreatic and ovarian carcinomas using the auristatin‐based anti‐CD70 antibody‐drug conjugate SGN‐75. Br J Cancer. 2010;103:676‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flieswasser T, Camara‐Clayette V, Danu A, et al. Screening a broad range of solid and haematological tumour types for CD70 expression using a uniform IHC methodology as potential patient stratification method. Cancers. 2019;11:1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobs J, Deschoolmeester V, Zwaenepoel K, et al. CD70: An emerging target in cancer immunotherapy. Pharmacol Ther. 2015;155:1‐10. [DOI] [PubMed] [Google Scholar]
- 19. Jacobs J, Zwaenepoel K, Rolfo C, et al. Unlocking the potential of CD70 as a novel immunotherapeutic target for non‐small cell lung cancer. Oncotarget. 2015;6:13462‐13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masuda K, Kubota T, Kaneko E, et al. Enhanced binding affinity for FcgammaRIIIa of fucose‐negative antibody is sufficient to induce maximal antibody‐dependent cellular cytotoxicity. Mol Immunol. 2007;44:3122‐3131. [DOI] [PubMed] [Google Scholar]
- 21. Yamane‐Ohnuki N, Kinoshita S, Inoue‐Urakubo M, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody‐dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614‐622. [DOI] [PubMed] [Google Scholar]
- 22. Silence K, Dreier T, Moshir M, et al. ARGX‐110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs. 2014;6:523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aftimos P, Rolfo C, Rottey S, et al. Phase I dose‐escalation study of the anti‐CD70 antibody ARGX‐110 in advanced malignancies. Clin Cancer Res. 2017;23:6411‐6420. [DOI] [PubMed] [Google Scholar]
- 24. Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune‐related response criteria. Clin Cancer Res. 2009;15:7412‐7420. [DOI] [PubMed] [Google Scholar]
- 25. Riether C, Pabst T, Höpner S, et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26:1459‐1467. [DOI] [PubMed] [Google Scholar]
- 26. Ma Y, Yang H, Kroemer G. Endogenous and exogenous glucocorticoids abolish the efficacy of immune‐dependent cancer therapies. Oncoimmunology. 2020;9:1673635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Izawa K, Martin E, Soudais C, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70‐CD27 pathway in immunity to Epstein‐Barr virus infection. J Exp Med. 2017;214:73‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prawira A, Oosting SF, Chen TW, et al. Systemic therapies for recurrent or metastatic nasopharyngeal carcinoma: a systematic review. Br J Cancer. 2017;117:1743‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato H, Fushimi C, Okada T, et al. Investigation of the efficacy and safety of nivolumab in recurrent and metastatic nasopharyngeal carcinoma. Vivo. 2020;34:2967‐2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corraliza‐Gorjón I, Somovilla‐Crespo B, Santamaria S, Garcia‐Sanz JA, Kremer L. New strategies using antibody combinations to increase cancer treatment effectiveness. Front Immunol. 2017;8:1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jazirehi AR, Bonavida B. Cellular and molecular signal transduction pathways modulated by rituximab (rituxan, anti‐CD20 mAb) in non‐Hodgkin's lymphoma: implications in chemosensitization and therapeutic intervention. Oncogene. 2005;24:2121‐2143. [DOI] [PubMed] [Google Scholar]
- 32. Chen C, Zhou Y, Zhang X, et al. Anti‐epidermal growth factor receptor monoclonal antibody plus palliative chemotherapy as a first‐line treatment for recurrent or metastatic nasopharyngeal carcinoma. Cancer Med. 2020;9:1721‐1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fang W, Zhang J, Hong S, et al. EBV‐driven LMP1 and IFN‐gamma up‐regulate PD‐L1 in nasopharyngeal carcinoma: implications for oncotargeted therapy. Oncotarget. 2014;5:12189‐12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material