

Abstract
Sepsis‐induced metabolic dysfunction contributes to organ failure and death. L‐carnitine has shown promise for septic shock, but a recent phase II study of patients with vasopressor‐dependent septic shock demonstrated a non‐significant reduction in mortality. We undertook a pharmacometabolomics study of these patients (n = 250) to identify metabolic profiles predictive of a 90‐day mortality benefit from L‐carnitine. The independent predictive value of each pretreatment metabolite concentration, adjusted for L‐carnitine dose, on 90‐day mortality was determined by logistic regression. A grid‐search analysis maximizing the Z‐statistic from a binomial proportion test identified specific metabolite threshold levels that discriminated L‐carnitine responsive patients. Threshold concentrations were further assessed by hazard ratio and Kaplan‐Meier estimate. Accounting for L‐carnitine treatment and dose, 11 1H‐NMR metabolites and 12 acylcarnitines were independent predictors of 90‐day mortality. Based on the grid‐search analysis numerous acylcarnitines and valine were identified as candidate metabolites of drug response. Acetylcarnitine emerged as highly viable for the prediction of an L‐carnitine mortality benefit due to its abundance and biological relevance. Using its most statistically significant threshold concentration, patients with pretreatment acetylcarnitine greater than or equal to 35 µM were less likely to die at 90 days if treated with L‐carnitine (18 g) versus placebo (p = 0.01 by log rank test). Metabolomics also identified independent predictors of 90‐day sepsis mortality. Our proof‐of‐concept approach shows how pharmacometabolomics could be useful for tackling the heterogeneity of sepsis and informing clinical trial design. In addition, metabolomics can help understand mechanisms of sepsis heterogeneity and variable drug response, because sepsis induces alterations in numerous metabolite concentrations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Sepsis remains a significant hazard to human health, causes a large and highly variable metabolic response, and targeted pharmacotherapy remains elusive. L‐carnitine represents a candidate therapeutic, but a recent clinical trial of L‐carnitine versus placebo in patients with septic shock demonstrated a nonsignificant reduction in mortality.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study addressed the question: are there serum metabolites that differentiate patients with septic shock who disproportionately derive a mortality benefit from L‐carnitine treatment?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Pharmacometabolomics can aid in identifying patients with sepsis that are more likely to respond to specific therapies. In this study, we identified blood concentrations of two metabolites, the acylcarnitine, acetylcarnitine (≥35 µM) and an amino acid, valine (≥88 µM) that could be used to identify these patients and for the design of a clinical trial that would test the efficacy of L‐carnitine in a specific subgroup of patients with septic shock.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
With few therapeutic options, better understanding of metabolic mechanisms that contribute to disease heterogeneity as well as patients who may respond favorably to specific treatments will move sepsis care closer to precision medicine and inform the design of clinical trials.
INTRODUCTION
Sepsis represents the leading cause of death in the intensive care unit and the single most expensive inpatient diagnosis, representing more than $17 billion in healthcare costs annually in the United States. 1 , 2 , 3 Septic shock carries a particularly poor prognosis, with short‐term mortality rates of ~ 40%. Among the many physiologic disturbances associated with sepsis is a profound shift in metabolism. 4 Hyperlactatemia represents one of the hallmarks of sepsis and is now considered a criterion for the diagnosis of septic shock. 5 However, hyperglycemia, lipolysis, and protein catabolism are also common and similarly associated with increased mortality. 4 , 6 Manipulation of these processes represents an underdeveloped but promising target for novel pharmacotherapies.
Despite the concerning sepsis mortality statistics and an increasingly focused research effort on the condition, clinical trials of novel sepsis pharmacotherapies have traditionally yielded disappointing results. Although the causes of the failure of clinical trials to further novel treatments are multifactorial, the highly heterogeneous nature of sepsis certainly contributes to these results. 7 , 8 This highlights the need to forge a better understanding of the heterogeneity and complexity of the clinical illness by identifying sepsis endotypes. 9 In doing so, strategies for enriched patient selection could be used to improve the precision of clinical trials. Importantly, predictive and prognostic enrichment strategies for clinical trials have been advocated by many and have been issued as guidance by regulatory agencies like the US Food and Drug Administration. 10 , 11 , 12
We recently completed a phase II, Bayesian adaptive dose‐finding randomized control trial comparing L‐carnitine (6, 12, or 18 g) treatment to saline (placebo) for the early treatment of septic shock. None of the tested doses of L‐carnitine resulted in a significant reduction in sequential organ failure assessment (SOFA) score at 48 h, although the highest and best performing dose (18 g) demonstrated a nonsignificant 3% and 6% absolute mortality reduction at 28 days in the intention to treat and per protocol analyses compared to saline placebo, respectively.
In parallel with the planning of the original trial, we designed an ancillary metabolomics study, the L‐Carnitine Pharmacometabolomics in Sepsis (CaPS) study, to identify candidate metabolites of drug response that could serve to endotype a heterogeneous septic shock cohort and direct the design of a clinical enrichment strategy for a phase III trial. A number of studies have demonstrated the importance of energy‐related metabolites for the differentiation of sepsis survivors and the identification of sepsis endotypes, 4 , 6 , 13 , 14 , 15 , 16 most of which are readily detected by nuclear magnetic resonance (NMR) spectroscopy 6 , 14 , 15 and targeted liquid chromatography ‐ mass spectroscopy (LC‐MS) assays. 16 Furthermore, we have previously demonstrated the utility of metabolomics in predicting drug response (pharmacometabolomics) in sepsis 15 using relatively quantified NMR metabolites and acylcarnitines generated by an LC‐MS assay. With this background in mind, we hypothesized that serum concentrations of acylcarnitines and/or other metabolites could differentiate patients that disproportionately benefit from L‐carnitine treatment as measured by mortality.
METHODS
Study design
This study utilized pretreatment serum samples collected from 236 of the 250 patients enrolled in the Rapid Administration of Carnitine (RACE) in Sepsis clinical trial. 17 The parent trial was approved by each site’s institutional review board, all patients or their surrogate gave written informed consent, and it was registered at clinicaltrials.gov prior to initiation (NCT 01665092). Details of the blood samples included in the study are provided in the Supplementary Material, Figure 1. Serum samples were assayed for acylcarnitines by LC‐MS 16 and by quantitative proton (1H) NMR as previously described. 18 , 19 More details about the methods for these measurements can be found in the Supplementary Material.
FIGURE 1.
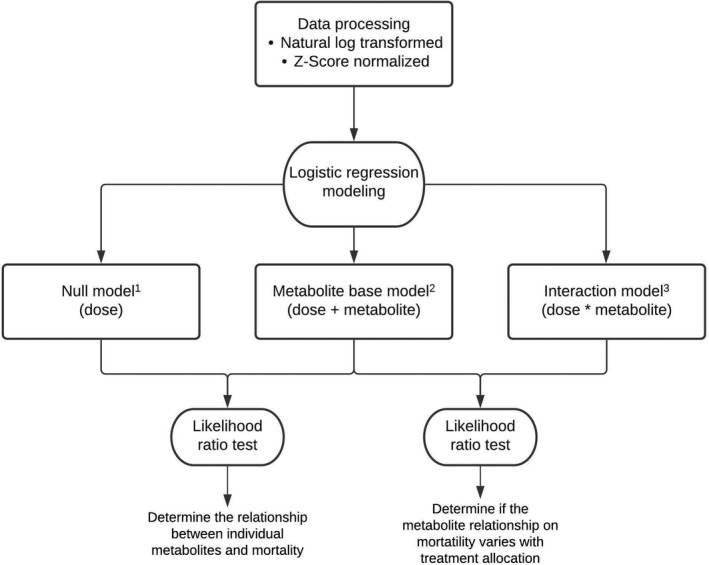
Statistical and logistic regression modeling workflow. We first natural log transformed and normalized each metabolite to have a mean of 0 and SD of 1. For each metabolite, we then considered a series of logistic regression models with an outcome of 90‐day mortality (p). The full model descriptions are provided below. In the metabolite base model, the p value corresponds to the likelihood ratio test for inclusion of the metabolite coefficient, BM, compared to the nested null model with only L‐carnitine dose (BD) as a predictor. For the interaction model, the p value corresponds to the likelihood ratio test for inclusion of the interaction coefficient, BMD, compared to a nested model with dose (BD) and metabolite concentration (BM) as predictors. 1Null model: logit(p) = B0 + BD * Dose. 2Metabolite base model: logit(p) = B0 + BD * Dose + BM * Metabolitei. 3Interaction model: logit(p) = B0 + BD * Dose + BM * Metabolitei + BMD * Metabolitei * Dose
Outcomes
We elected to use mortality as the outcome of our analysis because the primary end point of the RACE trial (reduction in SOFA score at 48 h) was not met, but the 18 g dose of L‐carnitine resulted in a trend toward a reduction in mortality. Based on data suggesting a substantial continued decline in mortality among sepsis patients beyond 28 days and preliminary data from our phase I study suggesting continued benefit from L‐carnitine treatment on longer term mortality rates, 20 we elected to assess the cumulative distribution mortality function to find the optimal time frame for assessment of mortality (28, 90, 180, or 365 days). Based on this analysis, by 90‐days, ~ 90% of the deaths had occurred (Figure S2) so we chose 90‐day mortality as the primary clinical outcome.
Statistical analyses
Descriptive data are reported as means and SDs, medians with interquartile ranges, or proportions as appropriate. Differences in categorical outcomes were compared using χ2 tests, whereas Student t‐tests and Wilcoxon rank sum tests were used to compare continuous variables. The aims of our primary analyses were to: (1) determine the relationship between individual metabolites and 90‐day mortality; (2) determine if the relationship between a predictive metabolite and mortality depends on treatment allocation; and (3) using metabolites most associated with mortality, determine the optimal (threshold) metabolite level that could be used to identify patients with septic shock most likely to respond favorably to L‐carnitine treatment. Collectively, and similar to other secondary analyses or ancillary studies of clinical and observational trials, 21 , 22 , 23 , 24 , 25 achievement of these goals would provide clinical proof of concept of a metabolically informed strategy to tackle the heterogeneity of sepsis that could also be used for a predictive enrichment design of a phase III study. 10 , 11
Because metabolomic data are on different scales due to varying abundance, in preparation for statistical analyses, data were natural‐log transformed and Z‐score normalized to have a mean of 0 and a SD of 1. 26 , 27 We began our analysis using partial least squares‐discriminant analysis (PLS‐DA) 28 to visualize the overall metabolic heterogeneity of the study participants and determine whether there were metabolic differences between sexes and the treatment groups.
We followed PLS‐DA by an assessment of the predictive value of individual metabolites on 90‐day mortality. To accomplish this, we constructed a series of logistic regression models and adjusted for treatment assignment (Figure 1). 29 We then further tested if the relationship between each metabolite’s baseline concentration and mortality varied across treatment groups using a logistic regression interaction model. The likelihood ratio test was used to determine the impact of baseline concentration and the interaction between concentration and dose for each metabolite (Figure 1). 30 Age 31 and SOFA score 32 were considered as covariates in further multivariable modeling because they are known to be associated with sepsis mortality and severity and are clinically available at the time of therapeutic decision making.
To test the potential clinical application of our pharmacometabolomics approach, after identifying metabolites strongly related to 90‐day mortality that also had a significant interaction with treatment allocation, we aimed to identify the specific concentration or levels of these candidate metabolites that could be used to predict which patients would be most likely to derive a mortality benefit from L‐carnitine (Figure 2). To achieve this, we used a grid‐search methodology to compute the Z‐statistic from the binomial proportion test at every possible threshold metabolite concentration or level. 33 For this example, because the 18 g dose of L‐carnitine was the most efficacious in the RACE trial and would be the one most likely to be tested in a phase III trial, we used the Z‐statistic to quantify the standardized difference in the proportion of deaths between those patients who received L‐carnitine (18 g) and those who received placebo. For this analysis, the metabolite level at each threshold was used as the criterion for inclusion into the proportion test. We then computed a two‐sample (binomial) proportion test, 34 which compared the proportion of patients treated with L‐carnitine who died by 90 days to those that were treated with placebo. At each threshold, we estimated the precision in the point estimate by performing jackknife resampling—systematically leaving out one observation and calculating the Z‐statistic on the remaining observations. 35 This permitted the identification of metabolite levels associated with a range of Z‐statistics, including the maximum Z‐statistic and the corresponding 95% confidence interval. The Z‐statistic simultaneously accounts for the difference in the proportion of patients who died in the treatment versus placebo groups and the sizes of each group, thereby suggesting the most optimal metabolite threshold level. Metabolites were then ranked by descending maximum Z‐statistic. Similar approaches have been used by other studies that have sought to identify the responder population in clinical and observational trials. 21 , 22 , 23 , 24 , 25 To further illustrate the implications of the use of different metabolite concentrations as predictors of mortality, hazard ratios were calculated using the Mantel‐Haenszel method, and Kaplan‐Meier curves were constructed (log rank [Mantel‐Cox] test). Metabolite concentration cut points were selected according to different trial scenarios and the grid‐search analysis described above. All statistical tests except for hazard ratios (Mantel‐Haenszel) and log rank (Mantel‐Cox) tests, which were done using PRISM, were performed in R studio (R version 3.6.2 [2019–12–12] Copyright 2019 The R Foundation for Statistical Computing) and figures were constructed in R and PRISM (version 8.4.3, June 10, 2020).
FIGURE 2.
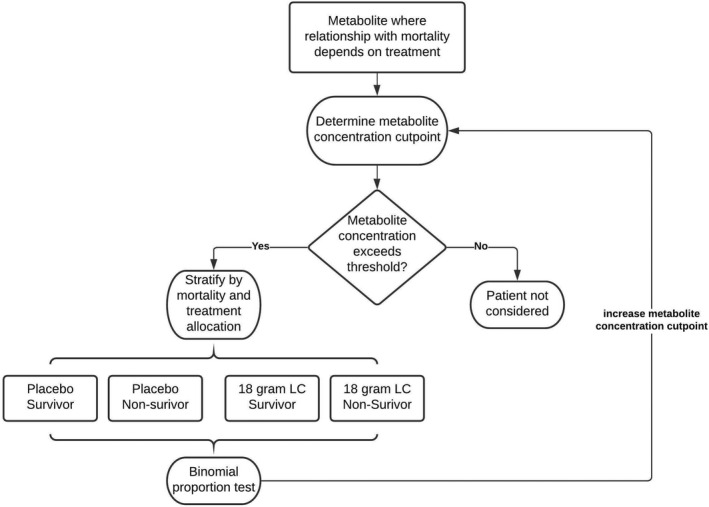
Grid‐search methodology workflow. After identifying metabolites with the strongest interaction in the logistic regression modeling, the metabolite concentration threshold or cut point that maximized the interaction was determined. For every possible threshold concentration, patients randomized to receive either placebo or 18 g L‐carnitine were considered. For patients whose values exceeded the concentration threshold, we stratified patients by treatment allocation and 90‐day mortality status and calculated the Z‐statistic from the two‐sample binomial proportion test. This was done iteratively for each metabolite, and the maximum Z‐statistic was identified from the grid‐search (see Table 4). LC, L‐carnitine
RESULTS
Of the 250 participants randomized in the parent trial, 1H‐NMR metabolomics and acylcarnitine data were available from 228 and 236 patient serum samples, respectively (Figure S1). We identified and quantified 27 serum metabolites by 1H‐NMR and 24 acylcarnitines by LC‐MS (Table S1). Representative 1H‐NMR and LC spectra are shown in Figures S3 and S4. All‐cause 90‐day mortality was 124 of 236 (52.5%), whereas 28‐day and 1‐year mortality were 104 of 236 (44.1%) and 136 of 236 (57.6%), respectively. Clinical and demographic variables of the cohort stratified by the primary outcome are summarized in Table 1. As expected, patients who died were older and had a higher SOFA score. The PLS‐DA plots of the acylcarnitine data and the NMR metabolites by treatment category (Figure S5a,b) and sex (Figure S6a,b) illustrate the metabolic heterogeneity of the study cohort and do not demonstrate any metabolic distinction between these groups.
TABLE 1.
Demographics and clinical characteristics of the cohort, stratified by 90‐day mortality
| Variable | Survived (n = 111) | Died (n = 125) | p value |
|---|---|---|---|
| Demographics | |||
| Age, years (IQR) | 61 (49, 69) | 66 (57, 76) | 0.002 |
| Male, n (%) | 60 (54) | 74 (41) | 0.43 |
| Female, n (%) | 51 (46) | 51 (59) | |
| Race | |||
| Black, n (%) | 33 (30) | 39 (31) | 0.88 |
| Asian, n (%) | 3 (3) | 2 (2) | |
| White, n (%) | 68 (61) | 74 (59) | |
| Other, n (%) | 7 (6) | 10 (8) | |
| Ethnicity | |||
| Hispanic, n (%) | 5 (4) | 7 (6) | 0.70 |
| Medical history | |||
| Diabetes, n (%) | 34 (31) | 46 (37) | 0.32 |
| Liver disease, n (%) | 11 (10) | 25 (20) | 0.03 |
| Renal disease, n (%) | 10 (10) | 24 (20) | 0.03 |
| Physiologic variables | |||
| Heart rate, beats per minute (IQR) | 100 (84, 113) | 100 (87, 114) | 0.70 |
| Respiratory rate, breaths per minute (IQR) | 20 (16, 24) | 21 (18, 26) | 0.09 |
| Cumulative vasopressor index (IQR) | 4 (3, 8) | 6 (4, 8) | <0.001 |
| Body mass index (IQR) | 28 (25, 36) | 27 (22, 35) | 0.10 |
| Laboratory values | |||
| White blood count, cells/mm3 (IQR) | 22.0 (12.3, 28.7) | 16.1 (11.4, 23.7) | 0.24 |
| Platelet count, cells/mm3 (IQR) | 161 (99, 232) | 129 (65, 210) | 0.02 |
| Creatinine, mg/dl (IQR) | 1.6 (1.1, 2.4) | 2.1 (1.4, 3.0) | 0.003 |
| Total bilirubin, mg/dl (IQR) | 0.9 (0.5, 1.7) | 1.6 (0.7, 3.7) | <0.001 |
| Clinical lactate, mmol/L (IQR) | 3.1 (2.3, 4.8) | 4.9 (2.7, 8.4) | <0.001 |
| Severity of illness | |||
| SOFA score | 10 (8, 12) | 12 (9, 15) | <0.001 |
Abbreviations: IQR, interquartile range; SOFA, sequential organ failure assessment.
We then conducted multivariable logistic regression using L‐carnitine dose and metabolites as covariates (base model) and applied a conservative Bonferroni correction for multiple comparisons. The base model identified in 11 of 27 1H‐NMR metabolites and 12 of 24 acylcarnitines that significantly discriminated 90‐day mortality (Table 2; the complete list can be found in Table S2). We then tested whether the relationship between predictive metabolites and mortality depends on treatment allocation. This was done with the addition of an interaction term between L‐carnitine dose and metabolite level (interaction model), which reduced the number of significant metabolites from 23 to 14, of which all but three metabolites were acylcarnitines (Table 3; a comprehensive list can be found in Table S3); these were not in range to withstand a conservative adjustment (e.g., Bonferroni) for multiple comparisons. In this analysis, a statistically significant and negative interaction term indicates that the predicted probability of 90‐day mortality for a given metabolic feature is lower at higher doses of L‐carnitine. To determine whether the signals found in the base and interaction models was merely due to factors associated with the risk of death, we controlled for both age 31 and SOFA score. 32 Several acylcarnitines and choline tolerated this adjustment (see Table S4 for the full list of metabolites); notably, lactate was not significant in either model (p = 0.96 and p = 0.22, respectively).
TABLE 2.
Logistic regression model for the prediction of 90‐day mortality adjusted for treatment (L‐carnitine dose or placebo)
| Metabolite predictor b | Base model a | ||
|---|---|---|---|
|
Metabolite Coefficient (βM) |
βM Standard Error |
βM p value (Bonferroni) |
|
| Acetylcarnitine (C2) c | 0.85 | 0.16 | <0.0001 |
| C18:1 c | 0.84 | 0.17 | <0.0001 |
| Acetylcarnitine (C2) d | 0.76 | 0.16 | <0.0001 |
| C20:1 c | 0.74 | 0.16 | <0.0001 |
| Tyrosine d | 0.68 | 0.16 | 0.0002 |
| Betaine d | 0.68 | 0.16 | 0.0002 |
| Propionylcarnitine (C3) c | 0.64 | 0.15 | 0.0002 |
| Propylene glycol d | 0.66 | 0.16 | 0.0003 |
| C16:1 c | 0.60 | 0.15 | 0.001 |
| Lysine d | 0.58 | 0.15 | 0.002 |
| Glycine d | 0.56 | 0.15 | 0.003 |
| C20‐carnitine c | 0.56 | 0.15 | 0.004 |
| Glutamine d | 0.55 | 0.15 | 0.01 |
| C14‐carnitine c | 0.53 | 0.15 | 0.01 |
| C16‐carnitine c | 0.52 | 0.15 | 0.01 |
| Methionine d | 0.51 | 0.15 | 0.01 |
| Lactate d | 0.51 | 0.15 | 0.02 |
| C12:1‐carnitine c | 0.51 | 0.15 | 0.02 |
| C4‐carnitine c | 0.48 | 0.14 | 0.02 |
| C20:2‐carnitine c | 0.49 | 0.15 | 0.03 |
| Proline d | 0.47 | 0.14 | 0.03 |
| C8‐carnitine c | 0.46 | 0.14 | 0.04 |
| Alanine d | 0.46 | 0.14 | 0.05 |
The base model is described as logit(p) = B0 + BD * Dose + BM * Metabolitei, where p is the probability of mortality in 90 days.
Compounds with Bonferroni adjusted p values less than or equal to 0.05 ranked in ascending order; for the complete list see Table S2 in the supplementary file.
As measured by liquid chromatography ‐ mass spectroscopy.
As measured by 1H‐nuclear magnetic resonance spectroscopy.
TABLE 3.
Logistic regression interaction model testing the relationship between metabolite predictors and mortality by treatment (L‐carnitine dose or placebo) for the prediction of 90‐day mortality ranked by ascending p value up to 0.05
| Metabolite predictor e | Interaction model a | ||
|---|---|---|---|
| Interaction coefficient (βM*D) |
βM*D Standard error |
βM*D p value d (Raw) |
|
| C10:1‐carnitine b | −1.22 | 0.37 | <0.0001 |
| C8:1‐carnitine b | −1.07 | 0.35 | 0.001 |
| C8‐carnitine b | −0.97 | 0.36 | 0.01 |
| C10‐carnitine b | −0.97 | 0.36 | 0.01 |
| C18:2‐carnitine b | −0.96 | 0.35 | 0.01 |
| C14:1‐carnitine b | −0.90 | 0.34 | 0.01 |
| C12‐carnitine b | −0.77 | 0.33 | 0.02 |
| C16:1‐carnitine b | −0.84 | 0.38 | 0.02 |
| Choline c | −0.74 | 0.33 | 0.02 |
| C16‐carnitine b | −0.82 | 0.38 | 0.02 |
| Oxoisocaproate c | −0.74 | 0.34 | 0.03 |
| C5‐carnitine b | −0.70 | 0.36 | 0.04 |
| Valine c | −0.69 | 0.35 | 0.05 |
| Acetylcarnitine (C2) b | −0.81 | 0.42 | 0.05 |
The interaction model is described as logit(p) = B0 + BD * Dose + BM * Metabolitei + BMD * Metabolitei * Dose.
As measured by liquid chromatography ‐ mass spectroscopy.
As measured by 1H‐nuclear magnetic resonance spectroscopy.
Raw p values are not adjusted for multiple comparisons.
For the complete list see supplementary Table 3 in the supplementary file.
As these findings were not evident in the parent clinical trial and they suggest that there may be a sepsis endotype that may derive a therapeutic benefit from supplement L‐carnitine, we hypothesized that a pharmacometabolomics approach may aid in defining this subgroup of patients. To identify the candidate metabolites, we took a hypothesis‐generating approach and considered all metabolites with significant (≤0.05) unadjusted p values (n = 14 in the logistic regression interaction model; Table 3) and assessed the Z‐statistic of each. Based on this analysis, the metabolites with the highest maximum Z‐statistics included a number of acylcarnitines as well as the branched chain amino acid, valine (Table 4; also see Table S5). In addition to the Z‐statistic values, to identify candidate metabolites, we also considered the prevalence of the acylcarnitine signal, the known potential of acetylcarnitine (C2) to predict drug responsiveness 15 and its close metabolic relationship with L‐carnitine. Furthermore, the maximum Z‐statistic of C12 and C8:1 represented a lower percentage of the clinical cohort than either C5 or acetylcarnitine (C2). As such, we selected acetylcarnitine (C2) as the most viable metabolite candidate to demonstrate the utility of our pharmacometabolomics approach. As examples, we assessed several concentrations of both acetylcarnitine (C2) and valine, including the ones at the maximum Z‐statistic, 35 µM (p = 0.002; as measured by LC‐MS; Figure 3) and 88 µM (p = 0.009), respectively (also see Figure S7 and Table S5). These analyses illustrate how pharmacometabolomics may aid in the design of a precision trial of L‐carnitine for the treatment of septic shock using the scheme as illustrated in Figure 4.
TABLE 4.
Significant metabolites a from the logistic regression interaction model ranked by descending maximum Z‐statistic
| Metabolite predictor | Maximum Z–statistic | 95% CI |
|---|---|---|
| C10:1‐carnitine b | 3.67 | 2.05–5.29 |
| C8:1‐carnitine b | 3.44 | 2.01–4.87 |
| C10‐carnitine b | 3.06 | 1.44–4.67 |
| Acetylcarnitine (C2) b | 3.01 | 1.93–4.09 |
| C8‐carnitine b | 2.98 | 1.24–4.72 |
| C5‐carnitine b | 2.74 | 1.76–3.73 |
| Valine c | 2.61 | 0.79–4.43 |
| C12‐carnitine b | 2.52 | 0.74–4.30 |
| C18:2‐carnitine b | 2.41 | 1.18–3.64 |
| C14:1‐carnitine b | 2.40 | 1.2–3.60 |
| C16‐carnitine b | 2.39 | 0.55–4.23 |
| C16:1‐carnitine b | 2.38 | 0.52–4.23 |
| Choline c | 1.71 | −0.24–3.67 |
| 2‐Oxoisocaproate c | 1.68 | −0.28–3.63 |
FIGURE 3.
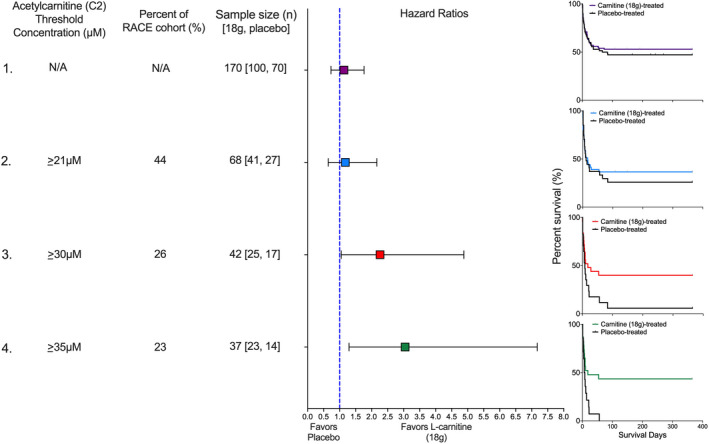
Pretreatment acetylcarnitine (C2) concentration as a predictive clinical trial enrichment strategy. Four scenarios illustrate how different threshold concentrations of acetylcarnitine (C2), a high abundant acylcarnitine would have impacted the outcome of the Rapid Administration of Carnitine (RACE) in Sepsis clinical trial in patients treated with either L‐carnitine (18 g) or placebo. In scenario one, no threshold concentration is used so the entire RACE cohort (n = 236) is eligible. The sample size of 170 patients represents those that received either L‐carnitine (18 g; n = 100) or placebo (n = 70). The hazard ratio is not significant, and consistent with the parent trial, the Kaplan‐Meier curve shows no mortality benefit of L‐carnitine (p = 0.57). In scenario two, an acetylcarnitine (C2) threshold concentration of greater than 21 µM is used. Forty‐four percent (n = 104) of the RACE cohort met this criterion and of these, 68 patients received either L‐carnitine (18 g) or placebo. The hazard ratio is not improved, and the Kaplan‐Meier curve shows no mortality benefit of L‐carnitine (p = 0.59). In scenario three, an acetylcarnitine (C2) threshold concentration of greater than 30 µM is used. Twenty‐seven percent (n = 64) of the RACE cohort met this criterion and of these, 42 patients received either L‐carnitine (18 g) or placebo. The hazard ratio is significant and favors L‐carnitine (18 g); the Kaplan‐Meier curve shows a mortality benefit of L‐carnitine (p = 0.04). Finally, scenario four uses the acetylcarnitine (C2) concentration associated with the maximum Z‐statistic (Table S4), greater than 35 µM. Twenty‐three percent (n = 54) of the RACE cohort met this criterion and of these, 37 patients received either L‐carnitine (18 g) or placebo. The hazard ratio is significant, and the Kaplan‐Meier curve shows a mortality benefit of L‐carnitine (p = 0.01). The number of patients at risk at each time point and the number of censored subjects, which was due to the end of the study (1 year), can be found here: https://doi.org/10.7302/vvqp‐ma61. N/A, not applicable
FIGURE 4.
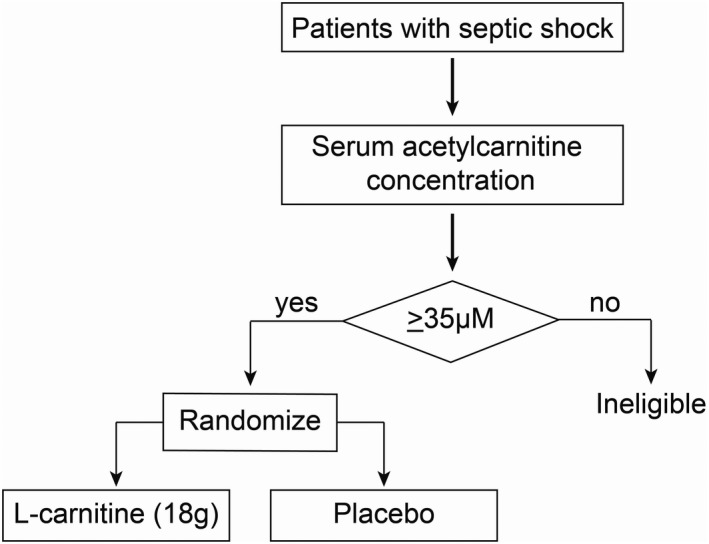
A clinical trial enrichment strategy could optimize clinical trial design for heterogeneous critical illnesses like sepsis. An example of a scheme for a hypothetical phase III clinical trial of supplement L‐carnitine for the treatment of septic shock that uses an a priori determined acetylcarnitine (C2) threshold concentration to determine whether a patient is enrolled and randomized to receive either L‐carnitine (18 g) or placebo
DISCUSSION
Our pharmacometabolomics study, CaPS, aimed to identify pretreatment, sepsis‐induced metabolic derangements in survivors and nonsurvivors treated with L‐carnitine. We found that there are likely metabolically distinct groups (endotypes) of patients that do proportionally better when they receive an 18 g dose of supplemental L‐carnitine. These findings imply that a precision, clinical trial enrichment strategy using pharmacometabolomics could help combat the heterogeneity of sepsis and drug response, which is known to have contributed to numerous negative clinical studies. 7
Here, we show that a pharmacometabolomics approach identified clinically indistinguishable sepsis endotypes that are more likely to derive a mortality benefit from treatment with L‐carnitine (18 g), a finding not evident in the metabolically naive parent trial. To accomplish this, we used a metabolomics analysis to capture high abundant polar compounds (quantitative 1H‐NMR) and acylcarnitines (LC‐MS) in serum samples collected from patients enrolled in a phase II clinical trial of L‐carnitine therapy. 17 Using this approach, similar to our prior study, 16 we found a prevalent acylcarnitine signal. From this profile, we selected acetylcarnitine (C2) and valine to illustrate how different threshold concentrations could influence mortality in patients randomized to either placebo of L‐carnitine (18 g). Specifically, patients with higher (e.g., ≥30 µM) acetylcarnitine (C2) levels at enrollment may be more likely to derive a treatment benefit as defined by decreased intermediate term (90‐day) mortality; this benefit is maximized at acetylcarnitine (C2) concentrations greater than or equal to 35 µM. Although severity of illness could contribute to this finding, clinical variables alone do not seem to account for the identification of the drug‐responsive endotype because the finding is retained when accounting for factors associated with the risk of death (age and SOFA score; see Table S4). Notably, we also found that serum concentrations of the branched chain amino acid, valine, could also be used to identify a mortality benefit of L‐carnitine but not to as great an extent as acetylcarnitine (C2). Collectively, these data suggest that there are patients that are in clinically occult subgroups. Should these data be validated, metabolically informed clinical trial design 36 and, ultimately, precision treatment strategies could represent a new paradigm of sepsis care. These data provide the groundwork and rationale for a pharmacometabolomics directed clinical trial to test L‐carnitine therapy efficacy for septic shock using a specific concentration of a key metabolite (e.g., acetylcarnitine [C2]) to guide inclusion criteria (Figure 4).
Importantly, the current study also shows that numerous metabolites may have predictive value for sepsis mortality, even after controlling for factors associated with the risk of death (see Table S4). These data provide further evidence that sepsis induces broad metabolic disruption that is linked to patient outcomes, corroborating prior studies. 37 , 38 Of note, numerous acylcarnitines, including unsaturated acylcarnitines, predicted mortality, suggesting significant disruption in fatty acid metabolic pathways. 38 Overall, the broad range in disruption of acylcarnitines may reflect differential and variable mobilization of fatty acids, 39 rather than disruption of a specific enzyme or pathway. We have previously demonstrated this in a smaller cohort of patients with septic shock. 16 Despite this variance, acetylcarnitine (C2) was the most robust predictor of overall sepsis mortality. This corroborates a previous study that identified acetylcarnitine (C2) as being associated with the severity of sepsis‐induced organ dysfunction, inflammation, and infection. 37 Acetylcarnitine (C2) also happens to be one of only two compounds (with L‐carnitine) detected by both the LC‐MS and NMR analytical platforms; regardless of the detection method, it performed similarly in the regression models.
Interestingly, acetylcarnitine (C2) outperformed the more clinically ubiquitous lactate level in predicting sepsis mortality. After correcting for age and SOFA score, lactate was not a significant independent predictor (Table S4) whereas acetylcarnitine (C2) retained its predictive value following this correction, which suggests the potential for its use as an adjunctive clinical test for risk prognosis. However, as our cohort was highly selected and involved only participants receiving vasopressors (which affect glycolysis and lactate production) 40 , 41 who were already resuscitated, it would be inappropriate to interpret these data to imply that lactate does not serve an important role in the early identification and prognosis of patients with suspected infection. In particular, serial lactate levels and its clearance rate have been used to assess the adequacy of resuscitation and lactate is included in the sepsis definition. 5 , 42 , 43 , 44 Nevertheless, limitations of lactate have been recognized 42 and, notably, others have demonstrated that acylcarnitines outperform lactate in predicting sepsis mortality. 38 Our data suggest that acetylcarnitine (C2) may represent a superior risk stratification tool in a selected cohort of fully resuscitated patients undergoing treatment with vasopressor infusions.
We also learned from the CaPS study that pretreatment serum L‐carnitine concentrations did not predict a L‐carnitine treatment mortality benefit, suggesting against the hypothesis that serum L‐carnitine deficiency drives the response to supplemental L‐carnitine in patients with sepsis. Rather, in aggregate, these data provide evidence to support the hypothesis that sepsis induces an impairment in the mobilization of acetyl groups. Although there may be a number of biologically plausible hypotheses, our findings could be due to sepsis‐induced increased intracellular accumulation of acetyl‐CoA secondary to its decreased metabolism via the tricarboxylic acid cycle (TCA) or enhanced acetyl‐CoA production via fatty acid (beta‐oxidation) metabolism (Figure S9). Consequently, increases in acetyl‐CoA are managed by several mechanisms, one of which is via the mitochondrial enzyme, carnitine acetyltransferase (EC 2.3.1.7). Carnitine acetyltransferase transfers acetyl groups to carnitine, displacing the hydrogen atom in its hydroxyl group 45 converting it to the membrane‐permeable, acetylcarnitine (C2) (Figure S9). Acetylcarnitine (C2), the shortest of the acylcarnitines, is important because it plays a controlling role over acetyl‐CoA on metabolic substrate switching and therefore, enables metabolic flexibility. 45 As the need for adenosine triphosphate (ATP) increases, acetyl‐CoA is diverted to the TCA cycle. However, in sepsis, the TCA cycle may fail to metabolize these groups resulting in excess acetyl‐CoA and subsequent elevation in measured serum acetylcarnitine (C2) concentrations. The elevation in acetylcarnitine (C2) may reflect the ability of L‐carnitine to serve as route for the disposal of excess acetyl groups, which has been demonstrated in the myocardium 46 and during exercise. 47 However, unlike acetylcarnitine (C2), the metabolic link between L‐carnitine therapeutic response and branched chain amino acid (BCAA) concentrations is less clear. We and others have shown that levels of BCAAs influence sepsis outcome 15 , 38 and shock resolution. 48 It is possible that patients with elevated BCAA blood concentrations represent those with a metabolic reserve that enables them to more efficiently utilize supplemental L‐carnitine 49 but, in general, the mechanisms of BCAA signaling and metabolic mechanisms of action are poorly understood. 50 In aggregate, our findings suggest that the magnitude of sepsis‐induced disruptions in energy metabolism may be associated with a therapeutic benefit of L‐carnitine. This relationship and the mechanisms that underlie it warrant further interrogation.
Despite the encouraging results of our study, we acknowledge that there are several important weaknesses. We recognize that “real‐time” metabolomics is not feasible in clinical practice and that routine measurement of these compounds, including acetylcarnitine (C2), for routine clinical use is not currently available. We also used a limited, focused metabolomic approach, measuring high abundant polar compounds (1H‐NMR) and acylcarnitines. We acknowledge that a broad, untargeted approach may have yielded additional compounds predictive of outcomes or treatment response. With our targeted approach, we still made multiple comparisons testing involving over 50 metabolites in this study, which opens the door to false positive findings. Our findings persisted after application of a conservative Bonferroni correction, but we acknowledge that the predictive capacity of acetylcarnitine (C2) and valine, when accounting for interactions between baseline metabolite and treatment assignment (interaction model), was not amenable to correction for multiple comparisons. As such, and given that this was an ancillary study, we acknowledge that any conclusions regarding the accurate prediction of clinical drug responsiveness are only hypothesis generating and will require rigorous prospective testing. We did, however, highlight how the use of a number of different acetylcarnitine (C2) and valine concentrations would influence the mortality outcome of the RACE trial (Figure 3 and Figure S7). These were merely used as examples to illustrate the utility of a pharmacometabolomics approach and despite including almost 250 patients, we acknowledge that our results may overestimate the true effect size and will require validation in an external cohort. Nevertheless, even though these subgroups represent less than or equal to 50% of the total RACE trial cohort, they highlight the value of a predictive enrichment strategy that could be used to design a phase III clinical trial of L‐carnitine supplementation for septic shock. Importantly, the pharmacometabolomics approach was developed concurrent with the design of the parent trial, and the conceptual model was based on and is consistent with our preliminary work in a unique, although smaller cohort, 15 strengthening the validity of the findings.
In summary, an ancillary pharmacometabolomics study, CaPS, of the parent clinical trial, RACE, found numerous predictors, independent of intervention, age, and SOFA score, for 90‐day mortality in septic shock, including many acylcarnitines and other metabolites, such as tyrosine, betaine, lysine, and glycine. We also demonstrate the translational value of the work by showing how the application of a pharmacometabolomics‐based clinical trial enrichment strategy, using pretreatment acetylcarnitine (C2) concentrations as an example, could be used to identify the responder population, a sepsis endotype, that may derive a mortality benefit from L‐carnitine supplementation. This represents a unique clinical trial enrichment strategy that could be used to improve the efficiency of a phase III L‐carnitine efficacy study in patients with septic shock 9 and other emerging therapeutics in heterogeneous critical illnesses. These findings also support the notion that distinct metabolic endotypes contribute to sepsis heterogeneity.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
M.A.P., C.E.G., T.S.J., C.R.E., A.K., C.E.M., T.L.F., A.E.J., and K.A.S. wrote the manuscript. M.A.P., A.E.J., and K.A.S. designed the research. M.A.P., A.E.J., K.A.S., C.R.E., C.E.M., and T.L.F. performed the research. M.A.P., C.E.G., and T.S.J. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge the contributions of the RACE Trial Investigators group; Nathan I. Shapiro, MD, MPH (Department of Emergency Medicine, Beth Israel Deaconess Medical Center, Boston, MA); Faheem W. Guirgis, MD (Department of Emergency Medicine, University of Florida College of Medicine–Jacksonville, FL); Michael Runyon, MD, MPH (Department of Emergency Medicine, Carolinas Medical Center, Charlotte, NC); Jason Y. Adams, MD (Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Internal Medicine, University of California, Davis, CA); Robert Sherwin, MD (Department of Emergency Medicine, Wayne State University, Detroit, MI); Ryan Arnold, MD (Department of Emergency Medicine, Christiana Care Health System, Wilmington, DE); Brian W. Roberts, MD, MSc (Department of Emergency Medicine, Cooper University Hospital, Cooper Medical School of Rowan University, Camden, NJ); Michael C. Kurz, MD, MS (Department of Emergency Medicine, The University of Alabama School of Medicine at Birmingham, Birmingham, AL); Henry E. Wang, MD, MS (Department of Emergency Medicine, The University of Texas Health Science Center at Houston, Houston, TX); Jeffrey A. Kline, MD (Department of Emergency Medicine, Indiana University School of Medicine, Indianapolis, IN); D. Mark Courtney, MD (Department of Emergency Medicine, Northwestern University, Chicago, IL); Stephen Trzeciak, MD, MPH (Department of Medicine, Cooper University Hospital, Cooper Medical School of Rowan University, Camden, NJ); Sarah A. Sterling, MD (Department of Emergency Medicine, The University of Mississippi Medical Center, Jackson, MS); Utsav Nandi, MD (Department of Emergency Medicine, The University of Mississippi Medical Center, Jackson, MS); Deepti Patki, MS (Department of Emergency Medicine, The University of Mississippi Medical Center, Jackson, MS); and Kert Viele, PhD (Berry Consultants, Austin, TX).
Puskarich MA, Jennaro TS, Gillies CE, et al; the RACE Trial Investigators . Pharmacometabolomics identifies candidate predictor metabolites of an L‐carnitine treatment mortality benefit in septic shock. Clin Transl Sci. 2021;14:2288–2299. 10.1111/cts.13088
This work was presented, in part, at the American Thoracic Society International Conference, July 2020 https://doi.org/10.1164/ajrccm‐conference.2020.201.1_Meeting Abstracts.A2604.
Funding information
This study was supported by the National Institute of General Medical Sciences (NIGMS) via R01GM103799 (A.E.J.), K23GM113041 (M.A.P.) and R01GM111400 (K.A.S.). C.E.G.’s and T.S.J.’s contributions were supported, in part, by the Michigan Institute for Data Science “Propelling Original Data Science” grant from the University of Michigan; T.S.J. also received support from the American Foundation of Pharmaceutical Education. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS or the NIH.
Contributor Information
Kathleen A. Stringer, Email: stringek@umich.edu.
for the RACE Investigators:
Nathan I Shapiro, Faheem W Guirgis, Michael Runyon, Jason Y Adams, Robert Sherwin, Ryan Arnold, Brian W Roberts, Michael C Kurz, Henry E Wang, Jeffrey A Kline, D Mark Courtney, Stephen Trzeciak, Sarah A Sterling, Utsav Nandi, Deepti Patki, and Kert Viele
DATA AVAILABILITY STATEMENT
The metabolomics data sets, subject demographics and R code used in the manuscript’s data analyses can be found at: https://github.com/UMichNMR‐Metabolomics. All analytical protocols will be made available upon request.
REFERENCES
- 1. Fleischmann C, Scherag A, Adhikari NKJ, et al. Assessment of global incidence and mortality of hospital‐treated sepsis. current estimates and limitations. Am J Respir Crit Care Med. 2016;193:259‐272. [DOI] [PubMed] [Google Scholar]
- 2. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167‐1174. [DOI] [PubMed] [Google Scholar]
- 3. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395:200‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Langley RJ, Tipper JL, Bruse S, et al. Integrative "omic" analysis of experimental bacteremia identifies a metabolic signature that distinguishes human sepsis from systemic inflammatory response syndromes. Am J Respir Crit Care Med. 2014;190:445‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA. 2016;315:801‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Z, Triba MN, Amathieu R, et al. Nuclear magnetic resonance‐based serum metabolomic analysis reveals different disease evolution profiles between septic shock survivors and non‐survivors. Crit Care. 2019;23:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seymour CW, Kennedy JN, Wang S, et al. Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA. 2019;321:2003‐2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leligdowicz A, Matthay MA. Heterogeneity in sepsis: new biological evidence with clinical applications. Crit Care. 2019;23:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. NAGMSC Working Group on Sepsis . NAGMSC Working Group on Sepsis: Final Report. 2019.
- 10.. Enrichment strategies for clinical trials to support determination of effectiveness of human drugs and biological products: guidance for industry. August 26, 2019.
- 11. Prescott HC, Calfee CS, Thompson BT, Angus DC, Liu VX. Toward smarter lumping and smarter splitting: rethinking strategies for sepsis and acute respiratory distress syndrome clinical trial design. Am J Respir Crit Care Med. 2016;194:147‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vincent JL. The coming era of precision medicine for intensive care. Crit Care. 2017;21:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johansson PI, Kiichi N, Angelo JR, et al. Plasma mitochondrial DNA and metabolomic alterations in severe critical illness. Crit Care. 2018;22:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mickiewicz B, Duggan GE, Winston BW, et al. Metabolic profiling of serum samples by 1H nuclear magnetic resonance spectroscopy as a potential diagnostic approach for septic shock. Crit Care Med. 2014;42:1140‐1149. [DOI] [PubMed] [Google Scholar]
- 15. Puskarich MA, Finkel MA, Karnovsky A, et al. Pharmacometabolomics of l‐carnitine treatment response phenotypes in patients with septic shock. Ann Am Thorac Soc. 2015;12:46‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Puskarich MA, Evans CR, Karnovsky A, et al. Septic shock nonsurvivors have persistently elevated acylcarnitines following carnitine supplementation. Shock. 2018;49:412‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones AE, Puskarich MA, Shapiro NI, et al. Effect of levocarnitine vs placebo as an adjunctive treatment for septic shock: the Rapid Administration of Carnitine in Sepsis (RACE) randomized clinical trial. JAMA Netw Open. 2018;1:e186076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Labaki WW, Tian GU, Murray S, et al. Serum amino acid concentrations and clinical outcomes in smokers: SPIROMICS metabolomics study. Sci Rep. 2019;9:11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McHugh C, Flott T, Schooff C, et al. Rapid, reproducible, quantifiable NMR metabolomics: methanol and methanol: chloroform precipitation for removal of macromolecules in serum and whole blood. Metabolites. 2018;8:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Puskarich MA, Kline JA, Krabill V, Claremont H, Jones AE. Preliminary safety and efficacy of L‐carnitine infusion for the treatment of vasopressor‐dependent septic shock: a randomized control trial. JPEN J Parenter Enteral Nutr. 2014;38:736‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anderson BJ, Calfee CS, Liu KD, et al. Plasma sTNFR1 and IL8 for prognostic enrichment in sepsis trials: a prospective cohort study. Crit Care. 2019;23:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Man M, Close SL, Shaw AD, et al. Beyond single‐marker analyses: mining whole genome scans for insights into treatment responses in severe sepsis. Pharmacogenomics J. 2013;13:218‐226. [DOI] [PubMed] [Google Scholar]
- 23. Martucci G, McNally D, Parekh D, et al. Trying to identify who may benefit most from future vitamin D intervention trials: a post hoc analysis from the VITDAL‐ICU study excluding the early deaths. Crit Care. 2019;23:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stanski NL, Stenson EK, Cvijanovich NZ, et al. PERSEVERE biomarkers predict severe acute kidney injury and renal recovery in pediatric septic shock. Am J Respir Crit Care Med. 2020;201:848‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Q, Parikh H, Butterworth MD, et al. Longitudinal metabolome‐wide signals prior to the appearance of a first islet autoantibody in children participating in the TEDDY study. Diabetes. 2020;69:465‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Everitt B, Hothorn T. An Introduction to Applied Multivariate Analysis with R. SpringerLink; 2011. [Google Scholar]
- 27. van den Berg RA , Hoefsloot HC, Westerhuis JA, Smilde AK, van der Werf MJ . Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genom. 2006;7:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Worley B, Powers R. Multivariate analysis in metabolomics. Curr Metabolomics. 2013;1:92‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Z, Seibold H, Vettore MV, Song WJ, Francois V. Subgroup identification in clinical trials: an overview of available methods and their implementations with R. Ann Transl Med. 2018;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rajasekaran S, Eric K, Richard H, et al. Red cell transfusions as an independent risk for mortality in critically ill children. J Intensive Care. 2016;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med. 2006;34:15‐21. [DOI] [PubMed] [Google Scholar]
- 32. Vincent J‐L, de Mendonca A, Cantraine F, et al. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: results of a multicenter, prospective study. Working group on "sepsis‐related problems" of the European Society of Intensive Care Medicine. Crit Care Med. 1998;26:1793‐1800. [DOI] [PubMed] [Google Scholar]
- 33. Lipkovich I, Dmitrienko A, Denne J, Enas G. Subgroup identification based on differential effect search—A recursive partitioning method for establishing response to treatment in patient subpopulations. Stat Med. 2011;30:2601‐2621. [DOI] [PubMed] [Google Scholar]
- 34. Simon R. Clinical trials for predictive medicine. Stat Med. 2012;31:3031‐3040. [DOI] [PubMed] [Google Scholar]
- 35. Efron B, Stein C. The Jackknife estimate of variance. Ann Stat. 1981;9:586‐596. [Google Scholar]
- 36. Vincent J‐L, Sakr Y. Clinical trial design for unmet clinical needs: a spotlight on sepsis. Expert Rev Clin Pharmacol. 2019;12:893‐900. [DOI] [PubMed] [Google Scholar]
- 37. Chung K‐P, Chen G‐Y, Chuang T‐Y, et al. Increased plasma acetylcarnitine in sepsis is associated with multiple organ dysfunction and mortality: a multicenter cohort study. Crit Care Med. 2019;47:210‐218. [DOI] [PubMed] [Google Scholar]
- 38. Langley RJ, Ephraim LT, van Velkinburgh JC , et al. An integrated clinico‐metabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013;5:195ra195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Costa CC, de Almeida IT , Jakobs C, Poll‐The BT, Duran M. Dynamic changes of plasma acylcarnitine levels induced by fasting and sunflower oil challenge test in children. Pediatr Res. 1999;46:440‐444. [DOI] [PubMed] [Google Scholar]
- 40. Annane D, Vignon P, Renault A, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet. 2007;370:676‐684. [DOI] [PubMed] [Google Scholar]
- 41. Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet. 2005;365:871‐875. [DOI] [PubMed] [Google Scholar]
- 42. Puskarich MA, Shapiro NI, Massey MJ, Kline JA, Jones AE. Lactate clearance in septic shock is not a surrogate for improved microcirculatory flow. Acad Emerg Med. 2016;23:690‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arnold RC, Shapiro NI, Jones AE, et al. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock. 2009;32:35‐39. [DOI] [PubMed] [Google Scholar]
- 44. Jansen TC, van Bommel J, Schoonderbeek FJ, et al. Early lactate‐guided therapy in intensive care unit patients: a multicenter, open‐label, randomized controlled trial. Am J Respir Crit Care Med. 2010;182:752‐761. [DOI] [PubMed] [Google Scholar]
- 45. Muoio D, Noland R, Kovalik J‐P, et al. Muscle‐specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schroeder MA, Atherton HJ, Dodd MS, et al. The cycling of acetyl‐coenzyme A through acetylcarnitine buffers cardiac substrate supply: a hyperpolarized 13C magnetic resonance study. Circ Cardiovasc Imaging. 2012;5:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. 2006;86:205‐243. [DOI] [PubMed] [Google Scholar]
- 48. Puskarich MA, McHugh C, Flott TL, et al. Serum levels of branched chain amino acids predict duration of cardiovascular organ failure in septic shock [published online ahead of print November 3, 2020]. Shock. 10.1097/SHK.0000000000001687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hiraoka A, Kiguchi D, Ninomiya T, et al. Can L‐carnitine supplementation and exercise improve muscle complications in patients with liver cirrhosis who receive branched‐chain amino acid supplementation? Eur J Gastroenterol Hepatol. 2019;31:878‐884. [DOI] [PubMed] [Google Scholar]
- 50. Bonvini A, Coqueiro AY, Tirapegui J, Calder PC, Rogero MM. Immunomodulatory role of branched‐chain amino acids. Nutr Rev. 2018;76:840‐856. [DOI] [PubMed] [Google Scholar]
- 51. Storey JD. A direct approach to false discovery rates. J Roy Stat Soc B. 2002;64:479‐498. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The metabolomics data sets, subject demographics and R code used in the manuscript’s data analyses can be found at: https://github.com/UMichNMR‐Metabolomics. All analytical protocols will be made available upon request.