

Abstract
The evolution of evidence and availability of Clinical Pharmacogenetic Implementation Consortium (CPIC) guidelines have enabled assessment of pharmacogenetic (PGx) actionability and clinical implementation. However, population‐level actionability is not well‐characterized. We leveraged the Alabama Genomic Health Initiative (AGHI) to evaluate population‐level PGx actionability. Participants (>18 years), representing all 67 Alabama counties, were genotyped using the Illumina Global Screening array. Using CPIC guidelines, actionability was evaluated using (1) genotype data and genetic ancestry, (2) prescribing data, and (3) combined genotype and medication data. Of 6,331 participants, 4230 had genotype data and 3386 had genotype and prescription data (76% women; 76% White/18% Black [self‐reported]). Genetic ancestry was concordant with self‐reported race. For CPIC level A genes, 98.6% had an actionable genotype (99.4% Blacks/African; 98.5% White/European). With the exception of DPYD and CYP2C19, the prevalence of actionable genotypes by gene differed significantly by race. Based on prescribing, actionability was highest for CYP2D6 (70.9%), G6PD (54.1%), CYP2C19 (53.5%), and CYP2C9 (47.5%). Among participants prescribed atenolol, carvedilol, or metoprolol, ~ 50% had an actionable ADRB1 genotype, associated with decreased therapeutic response, with higher actionability among Blacks compared to Whites (62.5% vs. 47.4%; p < 0.0001). Based on both genotype and prescribing frequencies, no significant differences in actionability were observed between men and women. This statewide effort highlights PGx population‐level impact to help optimize pharmacotherapy. Almost all Alabamians harbor an actionable genotype, and a significant proportion are prescribed affected medications. Statewide efforts, such as AGHI, lay the foundation for translational research and evaluate “real‐world” outcomes of PGx.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Genotype‐guided prescribing can improve medication‐related outcomes. Although medications with pharmacogenetic (PGx) recommendations are commonly prescribed in acute settings, population‐level actionability is not well‐characterized.
WHAT QUESTION DID THIS STUDY ADDRESS?
Population‐level PGx actionability in Alabama Genomic Health Initiative participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This state‐wide effort enrolled participants across Alabama, many in clinics and/or the community as opposed to acute care settings. By evaluating real‐world PGx actionability, we have identified (1) 98.6% of participants have an actionable genotype, (2) medications affected by PGx recommendations are commonly prescribed, and (3) combining genetic and medication data allows for the identification of associations that may be more actionable based on race and/or gender.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Understanding population‐level PGx actionability can enable optimization of pharmacotherapy to improve outcomes. Evaluating actionability based on true genotype and prescribing frequencies by race and gender allows for the identification of areas, which can be prioritized for implementation efforts. Such efforts lay the foundation for translational research and evaluate “real‐world” outcomes of PGx.
INTRODUCTION
Medications, the most common modality for treatment of disease, account for over $330 billion of annual healthcare costs. 1 , 2 , 3 However, despite the use of patient‐specific factors (e.g., age, renal/hepatic function, etc.) to tailor therapy, drug response remains highly variable. The spectrum of drug response ranges from lack of efficacy to toxicity, both of which are common occurences. 4 , 5 , 6 Although adverse drug reactions (ADRs) contribute significantly to the burden and costs of medication therapy, 7 , 8 lack of therapeutic benefit also increases healthcare costs and resource utilization, and is common across disease states and drug classes. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16
Differences in efficacy and susceptibility to ADRs by race and gender are recognized. For example, Blacks are more susceptible to certain ADRs, such as angiotensin converting enzyme (ACE) inhibitor‐induced angioedema and cough, thrombolytic‐associated intracranial hemorrhage, 17 drug‐induced liver injury, 18 and antipsychotic‐associated adverse effects. 19 Furthermore, differences in first‐line recommended antihypertensive therapy, increased occurrence of treatment‐resistant hypertension, 20 and reduced efficacy of antihypertensive therapy 21 in Blacks, have also been noted. Similarly, women are approximately two times more likely to experience, and be hospitalized for, an ADR compared to men. 22 , 23 Specifically, premenopausal women report more ADRs than men, although men typically experience more severe and fatal ADRs. 24 Similar to Blacks, women have also been reported to have a higher risk of ACE‐inhibitor associated ADRs. 25 Recent assessments of gender‐related differences in ADRs found that 88% (76/86) of the drugs evaluated displayed higher pharmacokinetic (PK) parameters in women, independent of weight. 26 Furthermore, reduced efficacy due to higher clearance, higher plasma concentrations due to reduced clearance, and more ADRs despite comparable plasma concentrations have been noted for women. 27
Incorporating pharmacogenetic (PGx) information into clinical decisions has been shown to improve outcomes in a cost effective manner. 28 , 29 Whereas clinical trials leading to the approval of many medications have lacked racial diversity. 30 , 31 PGX researchers, recognizing differences in underlying genetic architecture, have worked to understand genetic influences that lead to differential medication effects by race/ancestry. 32 , 33 , 34 , 35 Although further work is still needed, these efforts have identified variants unique to individuals of African ancestry (e.g., DPYD), 36 genes with differential effects by race (e.g., CYP2C/CYP4F2 and warfarin), 37 and genes that may be more important by gender due to being located on the X chromosome (e.g., G6PD). 38 The Clinical Pharmacogenetics Implementation Consortium (CPIC) leads efforts to enable translation of PGx associations into clinical practice through the development and dissemination of gene/drug clinical practice guidelines. 39 Gene/drug pairs classified as CPIC level A or B have a recommended prescribing action and are likely to be actionable. Although much work has been done evaluating CPIC actionability, most efforts have focused on academic health systems. 40 , 41 Population‐level PGx actionability combining genetic and medication data, and differences in actionability by race and gender have not been evaluated. Herein, we evaluate exposure to medications with high‐level evidence of PGx actionability, identify genes with differential medication exposure by race and/or gender, and comprehensively evaluate PGx actionability combining medication and genetic data in a general population cohort of participants in the Alabama Genomic Health Initiative (AGHI).
METHODS
Alabama Genomic Health Initiative
Supported by the State of Alabama, AGHI is aimed at preventing and treating conditions with a genetic basis. This joint University of Alabama at Birmingham (UAB) ‐ HudsonAlpha Institute for Biotechnology effort provides genetic testing, interpretation, and counseling free of charge to residents in each of Alabama’s 67 counties. Under the approval of the UAB’s Institutional Review Board, Alabama residents over the age of 18 years were enrolled in AGHI at sites throughout Alabama, including UAB Hospital (Birmingham), UAB Medicine clinics (Birmingham, Huntsville, Selma, and Montgomery), and additional recruitment events held at various locations. During enrollment, demographic information (e.g., age, race, gender, etc.), medical history, family health history, and a blood sample were collected. Samples were genotyped through the HudsonAlpha laboratory using the Illumina Global Screening Array (GSA; version 1 or 2, depending on time of enrollment).
The pharmacogenetic actionability assessment
The PGx actionability assessment was conducted (1) using genotype data and genetic ancestry; (2) using medication data from participants with an ambulatory care encounter at UAB with or without genotype data; and (3) combining genotype and medication data in participants with both available. For the medication and combined assessments, self‐reported race was used. Because the assessments utilized both genetic ancestry and self‐reported race, we evaluated the genetic ancestry composition of participants among self‐reported race groups to determine concordance prior to conducting the actionability assessment. Genetic admixture for each participant was determined using the ADMIXTURE package in R and reference data obtained from 1000 Genomes Project, Simons Genetic Diversity Project, and the Ashkenazi Genetics Consortium. Overall composition of European, African, Asian, and Native American ancestry was evaluated for each participant, and any other components of ancestry were classified as other for the analysis. Within each self‐reported race group, the overall average components of genetic ancestry were calculated.
Gene/drug pairs were selected for evaluation if (1) they had a CPIC evidence level of A or B, or (2) they were CPIC level B/C and also had either an informative or actionable PGx information on the US Food and Drug Administration (FDA) package label or a reported level of evidence in the Pharmacogenomics Knowledgebase (PharmGKB). CPIC guidelines were reviewed to determine actionable genotypes and/or variants. In instances where a CPIC guideline was not available, PharmGKB was utilized to select variants, with variants with a 1A/B–2A/B PharmGKB level of evidence prioritized for evaluation. 42 For gene/drug pairs with no CPIC guideline where only lower level PharmGKB associations were reported (example: POLG [CPIC level A/B] and rs3087374 [PharmGKB level 3]) the highest level variant was used for the evaluation. Associations with no guideline and no PharmGKB clinical annotation were excluded. CFTR was excluded because recommendations are unique to patients with cystic fibrosis. Additionally, we excluded genes located in the HLA region, as the specific risk alleles (e.g., HLA‐B *57:01) were not directly genotyped. Since G6PD is located on the X‐chromosome, we also evaluated expected phenotypes, because phenotype assignment differs among men and women. The prevalence of actionable genotypes/variants was evaluated overall, by genetic ancestry, and by gender collectively by gene. Finally, we evaluated overall PGx actionability among AGHI participants based on genes/variants designated CPIC level A. Variants selected for evaluation and available on the GSA are listed in Table S1.
Medication data were ascertained through Informatics for Integrating Biology and the Bedside (i2b2) for AGHI participants with a recorded ambulatory care encounter with the UAB Health System. Visits were limited to ambulatory care visits to exclude occurrences of acute medication exposure (e.g., single doses received during emergency/inpatient encounter). Because RYR1, CACNA1S, and BCHE influence inhaled anesthetics and neuromuscular blockers, we excluded these gene/drug pairs because they would likely be administered in clinical settings. Furthermore, RYR1 and CACNA1S are considered medically actionable by the American College of Medical Genetics and Genomics (ACMG). Variation in these genes was rare in the cohort and has previously been reported. 43 For the medication assessment, medication exposure was evaluated overall, and by self‐reported race and gender collectively by gene. Genes that influenced the same set of medications were combined for the assessment (e.g., NUDT15 and TPMT for thiopurines). For each gene, participants were considered to have actionable medication exposure if they had ever been prescribed at least one affected medication prior to November 2020. RxNorm terminology was used to compile a list of ingredients (single and combination) for each medication influenced by a PGx recommendation. Genes and corresponding drugs included in the medication assessment are presented in Table S2.
The final analysis combined medication and genotype data to determine the percent of participants who were both (1) exposed to affected medications, and (2) also had an actionable genotype. For the PGx actionability assessment, we included medications utilized by at least 1% of participants overall or in any of the evaluated groups. Additionally, because warfarin has a race‐specific CPIC guideline, we evaluated warfarin actionability overall, stratified by race, and stratified by race and gender in AGHI participants prescribed warfarin.
Statistical analysis
Differences in PGx genotype frequencies and medication exposure by race and gender were evaluated using the χ2 test and Fisher’s exact test as appropriate. Odds ratios (ORs) and 95% confidence intervals (CIs) are reported with respect to Blacks and women for the race and gender stratified assessments, respectively.
RESULTS
Patient population
From June 2017 to January 2020, AGHI enrolled 6331 participants, representing all 67 Alabama counties. At the time of this analysis, genotype data was available for 4230 participants (74.0% women; self‐reported White [70%]; Black [19.8%]). Among participants, 3567 (76.0% women) had a UAB encounter and 3386 had both genotype and drug exposure data (75.9% female; Figure 1a). When self‐reported race and genetic ancestry were compared, the highest average percent of genetic ancestry was concordant with self‐reported race (Figure 1b). Given that ~ 94% of participants with both genotype and medication data self‐reported as White or Black, the assessment was limited to these groups.
FIGURE 1.
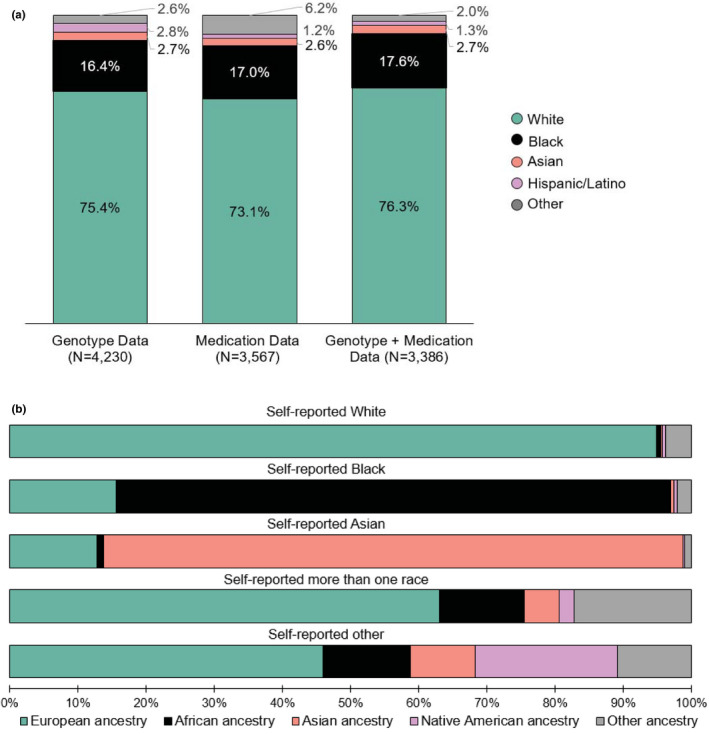
Self‐reported race (a) and genetic ancestry (b) among Alabama Genomic Health Initiative participants. Other races include American Indian/Alaskan Native, Hawaiian/Pacific Islander, more than one race, and other. Participants with unknown or those who declined/refused to provide race were not included in the assessment. For the genetic ancestry evaluation, other ancestry includes any composition of genetic ancestry apart from European, African, Asian, or Native American
Pharmacogenetic actionability based on genotype frequencies
Among genes designated CPIC level A, 98.6% (n = 4171/4230) of participants had an actionable genotype. The prevalence of level A actionable genotypes was similar between participants of African (99.4%; n = 637/641) and European ancestry (98.5%; n = 2853/2895). Among CPIC level A or B genes (Figure 2), ABCG2, influencing rosuvastatin efficacy, had the highest actionability, based on homozygosity for the major allele. Other genes with a high prevalence of actionable genotypes included CYP2C19 (61.1%), VKORC1 (55.2%), IFNL3 (53.7%), and NAT2 (52.6%). Although the prevalence of actionable genotypes for CYP2D6 was only 7.8%, data were only available for a limited number of CYP2D6 alleles (*6, *7, *9, *17, and *41).
FIGURE 2.
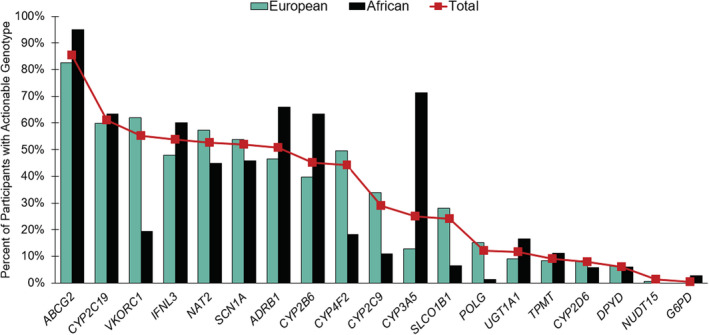
Actionability across pharmacogenes meeting Clinical Pharmacogenetics Implementation Consortium (CPIC) level A or B evidence threshold. Red squares indicate the overall prevalence of actionable genotypes in the cohort. ABCG2 actionability is based on homozygosity for the major allele. For G6PD, actionability was defined as men with one variant allele and women with two variant alleles. No participants had variation in MT‐RNR1, and no participants of African ancestry had an actionable NUDT15 genotype. CYP2D6 was not adequately interrogated on the array, and only a limited number of actionable genotypes could be determined. The actionable variant for SLC6A4 was not available
With the exception of CYP2C19 and DPYD, actionability by gene significantly differed among participants of African and European ancestry (Table S3). Notably, DPYD actionability did not differ by ancestry, primarily due to rs115232898, an African‐specific variant. Among participants of African ancestry with an actionable DPYD genotype, 89.5% (n = 34/38) had the rs115232898 variant. Actionable CYP2C9 genotypes were more common in European participants, however, CYP2C9*8, the most common variant in individuals of African ancestry, 44 was not assessed by the GSA. Additionally, 1.35% of participants had an actionable NUDT15 genotype, however, actionable NUDT15 genotypes were rare in European participants (0.6%) and absent in African participants.
PGx actionability was similar by gender, with 99.2% (n = 3106/3131) of women, and 99.2% (n = 1090/1099) of men having an actionable genotype (p = 0.95). When assessed by gene, differences in actionable genotype frequencies were observed for ABCG2, CYP2B6, CYP4F2, G6PD, and IFNL3 (Table S4). Compared to men, women were less likely to have an actionable CYP4F2 genotype (43.1% vs. 47.3%; p = 0.02), but more likely to have actionable ABCG2 (86.1% vs. 83.3%; p = 0.02), CYP2B6 (45.7% vs. 43.0%; p < 0.0001), and IFNL3 (55.1% vs. 49.9%; p = 0.003) genotypes. Some of these findings may also be related to ancestry differences, as ~ 83% (n = 535/641) of participants of African ancestry were women.
Variation in G6PD is associated with G6PD deficiency, and can lead to drug‐induced hemolytic anemia upon exposure to numerous medications. Variants are defined based on expected decrease in G6PD enzyme activity. Among evaluated variants, G6PD A‐ (class III) is associated with 10 to 60% of normal enzyme activity, and the Mediterranean variant (class II) is associated with less than 10% of normal enzyme activity, without chronic nonspherocytic hemolytic anemia (CNSHA). CNSHA is a clinical condition that can result in hemolytic anemia absent exposure to an offending agent. Men with one of these alleles and women with two alleles likely have a G6PD deficient phenotype, whereas women who with only one of these alleles have a variable phenotype. 38 Overall, ~ 5% of participants harbored either the A‐ or Mediterranean G6PD variants. However, only 0.5% were expected to have a deficient G6PD phenotype. When evaluated by ancestry, 28.5% of participants of African ancestry, and 0.1% of participants of European ancestry had a G6PD variant. Although no participants of European ancestry were expected to have a deficient phenotype, 2.8% of African ancestry participants were expected to be G6PD deficient. Additionally, as expected, women were more likely to have a G6PD variant, but less likely to have the actionable G6PD deficient phenotype (OR 0.35, 95% CI 0.12–1.00; p = 0.04).
Pharmacogenetic actionability‐based medication prescribing
Assessment of actionability‐based on prescribing demonstrated the importance of CYP2D6, CYP2C19, G6PD, and CYP2C9 overall, by race, and by gender (Figure 3 and Table S5). Over 40% of participants had been prescribed a medication influenced by one of these four genes. Among these, CYP2C9 showed differential prescribing by race and gender, with Black participants and women more commonly prescribed CYP2C9 affected medications. Additionally, women were more likely to be prescribed medications affected by CYP2C19 (OR 1.19, 95% CI 1.02–1.40; p = 0.03). Among other genes, Black participants were ~ 3‐fold more likely to be prescribed the CYP3A5 affected medication, tacrolimus (OR 3.72, 95% CI 1.87–7.32; p = 0.0003), and fivefold more commonly prescribed medications influenced by UGT1A1 (OR 5.28, 95% CI 2.24–12.66; p = 0.0002). Black participants and women were 37% (OR 0.63, 95% CI 0.42–0.94; p = 0.02) and 52% (OR 0.48, 95% CI 0.36–0.63; p < 0.0001) less likely to be prescribed rosuvastatin (ABCG2), respectively, with women also less 35% (OR 0.65, 95% CI 0.49–0.87; p = 0.004) less commonly prescribed simvastatin (SLCO1B1). Differences in rosuvastatin prescribing by race were primarily due to lower prescribing in women. Additionally, the analysis did not take into account the indication for statin therapy, such as clinical atherosclerotic cardiovascular disease, which may have been more common in certain groups of participants, and led to more frequent statin prescribing.
FIGURE 3.
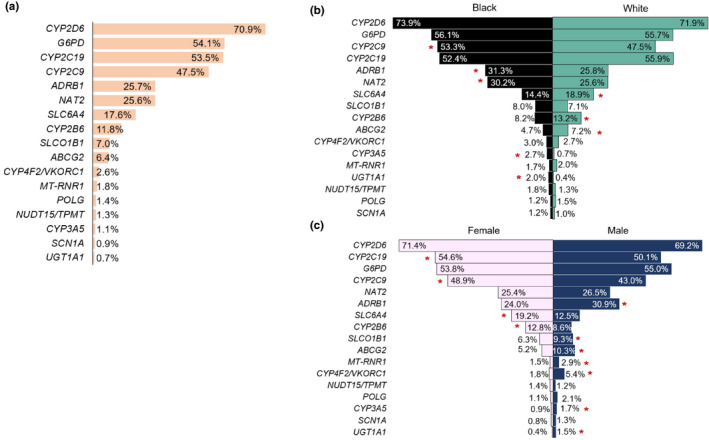
Potential for pharmacogenomic actionability among Alabama Genomic Health Initiative participants assessed by medication prescribing (a) overall, (b) by race, and (c) by gender. Red asterisks indicate a significant difference in prescribing frequency and the group with higher exposure. Genes with less than 1% exposure in all groups and not included: DPYD (fluoropyrimidines) and IFNL3/IFNL4 (peginterferon alfa 2a/2b)
Black participants and men were more frequently prescribed medications influenced by ADRB1, which included the beta blockers, atenolol, carvedilol, and metoprolol. Conversely, White participants and women were more commonly prescribed the antidepressants, citalopram and escitalopram, affected by SLC6A4. Gene/drug pairs with less than 1% exposure in all groups and not further evaluated included DPYD and IFNL3/IFNL4.
Pharmacogenetic actionability based on combined medication exposure and genetic variation
When medication and genotype data were combined (Figure 4), ABCG2 had the highest actionability due to the major allele being the actionable genotype. Among participants prescribed atenolol, carvedilol, or metoprolol, ~ 50% had an actionable ADRB1 rs1801253 genotype, associated with decreased therapeutic response. Furthermore, Black participants were more commonly prescribed these medications (31.3% vs. 25.8%; p = 0.02), had a higher frequency of actionable genotypes (66.1% vs. 46.5%; p < 0.0001), and also had higher combined actionability (62.5% vs. 47.4%; p < 0.0001). Similar results were observed for CYP3A5 and the immunosuppressant, tacrolimus, also more frequently prescribed in Black participants. Among participants prescribed tacrolimus (n = 35), 62.5% (n = 10/16) of Black participants had an actionable CYP3A5 genotype, compared to only 5.3% (n = 1/19) of White participants (p = 0.0008). Based on combined actionability, no significant differences were observed between men and women.
FIGURE 4.
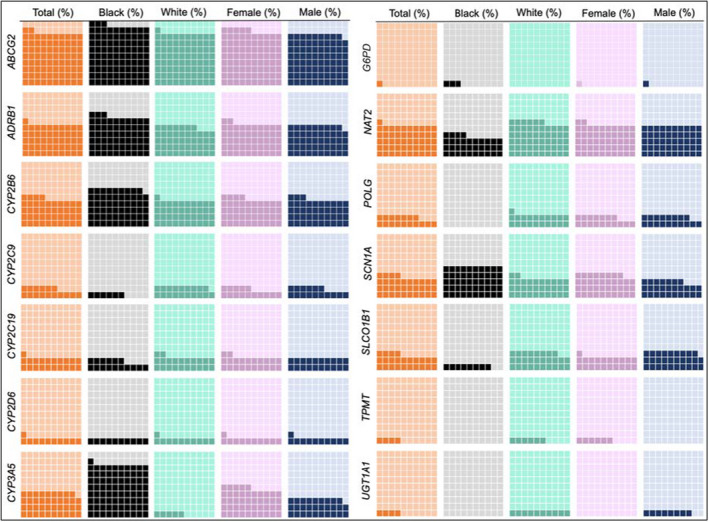
Pharmacogenetic actionability among Alabama Genomic Health Initiative participants prescribed an affected medication. Each large square represents 100% of participants. Dark shaded boxes represent the percent of participants on affected medications who also harbored an actionable genotype. Due to race specific guidelines, warfarin was not included in the CYP2C9 medication assessment, and CYP4F2 and VKORC1 are also not included. CYP2D6 was not adequately interrogated on the array, and therefore reported actionability is likely much lower that true actionability. For UGT1A1 and TPMT, there were less than five participants with an actionable genotype prescribed an affected medication. G6PD includes only participants expected to have a deficient phenotype. For women, G6PD actionability was 0.4%
Warfarin and corresponding genes were evaluated separately due to race‐specific differences in actionability (Table 1). The assessment was also stratified by race and gender to account for these differences in the gender‐specific evaluation. Among AGHI participants prescribed warfarin (n = 89), 59.6% had an actionable genotype for at least one of the genes that influences warfarin dosing. However, 72.2% (n = 13/18) of Black participants had an actionable genotype, compared to 45.8% (n = 33/72) of White participants (p = 0.05). Similar results were observed when stratified by both race and gender with 66.7% (n = 10/15) of Black and 43.3% (n = 13/30) of White women having an actionable genotype. Similarly, although all Black men prescribed warfarin also had an actionable genotype, this was due to only a small number (n = 3) being prescribed warfarin. Given that data were not available on the GSA for CYP2C9*8, common in Blacks, true warfarin actionability for Black participants may be higher than observed.
TABLE 1.
Pharmacogenetic actionability among AGHI participants prescribed warfarin overall, stratified by race, and stratified by race and gender
| Gene and variant | Total (n = 89) | Black (n = 18) | White (n = 72) | Black women (n = 15) | White women (n = 30) | Black men (n = 3) | White men (n = 42) |
|---|---|---|---|---|---|---|---|
| N (%) | N (%) | N (%) | N (%) | N (%) | N (%) | N (%) | |
| CYP2C9 (*2, *3) and VKORC1 rs9923231 | 30 (33.7%) | 2 (11.1%) | 28 (38.9%) | 2 (13.3%) | 9 (30.0%) | 0 (0.0%) | 19 (45.2%) |
| CYP2C9 a *5, *6, *11 | 2 (2.2%) | 1 (5.6%) | 1 (1.4%) | 1 (6.7%) | 1 (3.3%) | 0 (0.0%) | 0 (0.0%) |
| CYP4F2 rs2108622 | 8 (9.0%) | Not actionable | 8 (11.1%) | Not actionable | 6 (20.0%) | Not actionable | 2 (4.8%) |
| CYP2C rs12777823 | 27 (30.3%) | 12 (66.7%) | Not actionable | 9 (60.0%) | Not actionable | 3 (100%) | Not actionable |
| Overall actionability | 53 (59.6%) | 13 (72.2%) | 33 (45.8%) | 10 (66.7%) | 13 (43.3%) | 3 (100%) | 20 (47.6%) |
Abbreviation: AGHI, Alabama Genomic Health Initiative.
Data for CYP2C9*8, the most common CYP2C9 variant in Blacks, was not included on the global screening array.
Modification of pharmacogenetic actionability based on race versus ancestry
Given that 1.3% of participants had an actionable NUDT15 genotype, but only 0.6% of European participants and no African participants had an actionable genotype, we further evaluated NUDT15. Among participants with an actionable NUDT15 genotype, 42.9% (n = 18) reported White, 31% (n = 13) reported Asian, 23.8% (n = 10) reported other race, and 2.4% (n = 1) reported more than one race. Additionally, 35.7% of participants with an actionable genotype reported Hispanic ethnicity, and all participants who reported other race (n = 10) also reported Hispanic ethnicity. This was similar to results observed in the larger cohort, where participants who reported other race had the highest fraction of Native American genetic ancestry (Figure 1b). Genetic variation in NUDT15 and TPMT is associated with thiopurine‐related myelosuppression. It is recognized that NUDT15 variation is commonly responsible for these effects in Hispanics and Asians, whereas TPMT variation is commonly implicated in individuals of European and African ancestry. 45 , 46 If a race‐based approach to genotyping was employed, for example, only testing NUDT15 in patients who self‐reported Asian or Hispanic, ~ 31% (n = 13) of participants with an actionable NUDT15 genotype would have been missed due to reporting White/non‐Hispanic. Similarly, if both race and ethnicity were not considered, this would have failed to identify an additional 23.8% (n = 10) of participants with an actionable NUDT15 genotype who reported other race/Hispanic ethnicity. Given these results, the genetic ancestry of participants with a NUDT15 variant was evaluated (Figure 5). As expected, the majority of participants with the variant had primarily Asian and Native American ancestry; however, participants with predominately European ancestry were also found to have a variant. Among these participants, Native American, African, Middle Eastern, Ashkenazi, and other ancestral admixture was observed.
FIGURE 5.
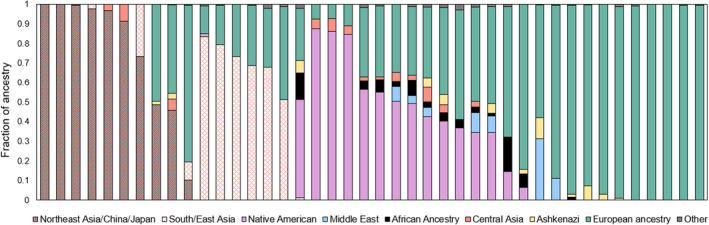
Genetic ancestry composition of Alabama Genomic Health Initiative participants with an actionable NUDT15 genotype. European and African ancestry include the total of all combined fractions of European and African ancestry, respectively
Additionally, we evaluated self‐reported race and genetic ancestry among 26 participants with the African‐specific DPYD rs115232898 variant and genetic ancestry data. Notably, one participant with the variant self‐reported White. When genetic ancestry was evaluated, the participant had ~ 60% European, 32% Middle Eastern, and 8% Ashkenazi ancestry. Furthermore, six additional participants of predominately African ancestry harboring the variant, also had components of Middle Eastern and/or Ashkenazi ancestry (data not shown).
DISCUSSION
This state‐wide population‐based evaluation of PGx actionability was done to create a defendable value proposition for pharmacogenomics in tailoring medication therapy at a population level. We do so by demonstrating that PGx potentially impacts (almost) every person; 98.6% of Alabamians have an actionable genotype influencing response to one or more medications. By assessing PGx actionability in the context of medication use, we demonstrate its current impact in optimizing medication selection and dosing.
In addition to differences in PGx actionability resulting from allele frequency differences across ancestral groups, prescribing frequencies of affected medications also differed. Black participants were more likely to harbor the ADRB1 variant (rs1801253), associated with decreased metoprolol response (for hypertension), and decreased response to carvedilol, atenolol, and metoprolol (for heart failure). 47 Although we did not evaluate the indication for beta blocker therapy among AGHI participants, over 30% of Black participants were prescribed these medications, of which, 63% also had an actionable genotype associated with decreased response. Given the disparities between Blacks and Whites in cardiovascular‐related outcomes, identifying PGx associations that may be more actionable in Black patients may help tailor therapy. Future investigations should evaluate associations with lower level evidence (CPIC level B) that may lack and/or have conflicting evidence due to under‐representation or ancestral differences in allele frequencies. This may offer insight into observed differences in medication efficacy and/or adverse effects.
Although it has been shown that self‐identified race ethnicity and genetic ancestry are highly concordant for PGx risk stratification, 48 we evaluated the ancestry composition of participants among self‐reported race groups to confirm these findings in our cohort prior to conducting the PGx assessment. Whereas the predominant ancestry fraction among each self‐reported race group was concordant, we present examples where the use of self‐reported race in lieu of PGx testing for variants that are thought to be unique to a specific ancestry group, may fail to identify participants who harbor the variant due to genetic admixture. Misclassifying such actionable PGx associations could undermine the promise of PGx in preventing severe/fatal adverse effects, such as thiopurine‐related myelosuppression and fluoropyrimidine toxicity. With increasing admixture in the United States, self‐reported race as a proxy for ancestry will become less reliable. This underscores the importance of testing all actionable variants regardless of self‐reported race.
By assessing PGx actionability in the context of prevalent chronic conditions we highlight its potential future impact on medical management by optimizing medication (selection and dosing) regimens. Figure 6 presents acute and chronic conditions among AGHI participants. Approximately 15% of participants in this nonmedical cohort have previously experienced adverse drug effects resulting in a medical diagnosis. In addition to medications with current PGx actionability, this provides a glimpse into future actionability as PGx research and evidence evolves.
FIGURE 6.
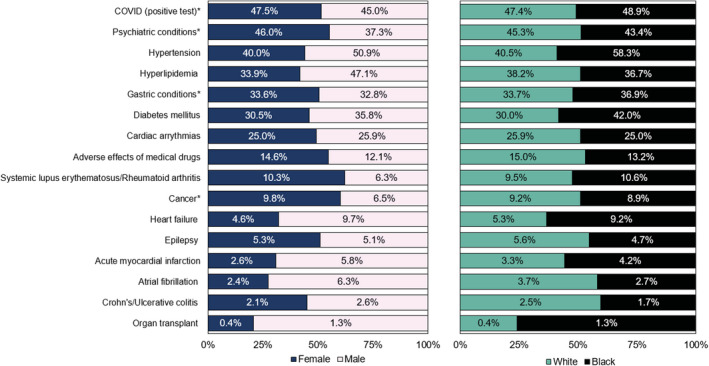
Prevalence of selected acute and chronic conditions among Alabama Genomic Health Initiative participants. Conditions in which the treatment had a high‐level pharmacogenetic association were prioritized for inclusion, in addition to adverse effects of medical drugs. Conditions of interest that may be more actionable as further evidence emerges (e.g., diabetes), and those that may provide insight into the potential utility of pre‐emptive pharmacogenetic data for research and/or treatment for unseen circumstances (e.g., coronavirus disease 2019 [COVID‐19]) are presented. *For COVID, prevalence represents the number of participants who tested positive. Among participants who tested positive, 3.8% (6.8% of Black COVID (+) participants; 3.2% of White COVID (+) participants; 2.9% of women with COVID (+) participants; 6.9% of men with COVID (+) participants) had severe disease requiring intervention (e.g., mechanical ventilation). Psychiatric conditions include adjustment disorders, anxiety disorders, attention deficit disorders, delirium dementia/other cognitive disorders, mood disorders, personality disorders, schizophrenia, and other psychotic disorders. Cancer includes breast, colorectal, gastrointestinal cancers, leukemia, and lung cancer. Gastric conditions: gastroesophageal reflux disease and gastroparesis
Finally, population‐level efforts, such as AGHI, can provide insight into understanding, and, where applicable, deploying PGx during emerging public health crises, such as the pandemic of coronavirus disease 2019 (COVID‐19). Genetic variation influences the PKs of several drugs investigated as potential therapies, including hydroxychloroquine/chloroquine (CYP2C8, CYP2D6, SLCO1A2, and SLCO1B1), azithromycin (ABCB1), ribavirin (SLC29A1, SLC28A2, and SLC28A3), and lopinavir/ritonavir (SLCO1B1, ABCC2, and CYP3A). In addition, variation influences the propensity for adverse effects, most notably for hydroxychloroquine/chloroquine (G6PD; hemolysis), ribavirin (ITPA; hemolysis), and interferon β −1b (IRF6; liver toxicity). 49 Having PGx data available prior to prescribing, especially in the case of COVID‐19 where many therapies had limited/no evidence of efficacy for the indication, has the potential to help mitigate certain adverse effects, such as G6PD‐associated hemolytic anemia.
A major limitation of this study was the lack of coverage for CYP2D6 on the genotyping array. CYP2D6 is the most actionable gene based on prescribing frequencies, but true actionability based on combined genetic variation and medications could not be adequately evaluated. This highlights the importance of comprehensive CYP2D6 evaluation and for programs using arrays to interrogate the genome should the plan include a separate assessment of CYP2D6 variation. We were also unable to evaluate UGT1A1 rs8175347, which determines the *28, *36, and *37 alleles, depending on the number of TA repeats present. However, UGT1A1 rs887829 is in strong linkage disequilibrium (LD) with rs8175347, with the rs887829 T allele in LD with the decreased function TA7 (*28) and TA8 (*38) alleles, and the rs887829 C allele in LD with the increased function and reference TA5 (*36) and TA6 (*1) alleles, respectively. For atazanavir, the only CPIC level A medication influenced by UGT1A1 with a guideline available, therapeutic recommendations are the same for *28/*38, and *1/*36. Due to this, an expected poor metabolizer phenotype can be inferred based on homozygosity for the rs887829 T allele. 50 However, other medications, including irinotecan, belinostat, dolutegravir, nilotinib, pazopanib, and raltegravir, are also influenced by UGT1A1 variation. 39 Because CPIC guidelines are medication specific, expected UGT1A1 phenotypes for these medications may differ from atazanavir, making it necessary to interrogate UGT1A1 rs8175347. This is important to consider as PGx evidence emerges for UGT1A1 affected medications.
State‐level population‐based initiatives, such as AGHI, have the potential to improve health‐related outcomes through pre‐emptive genotyping. With greater than 6,000 Alabama residents enrolled in AGHI since its inception in 2017, AGHI will now transition from a population‐based initiative to enrolling patients in UAB primary care clinics, with the aim of genotyping and returning actionable PGx results to patients and clinicians. Results will be stored in the electronic medical record so data will be available to help inform current and/or future prescribing decisions.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
B.H.D. and N.A.L. wrote the manuscript. B.H.D., D.A., B.K., and N.A.L. designed the research. B.H.D., D.A., and K.W. performed the research. B.H.D. and N.A.L. analyzed the data.
Supporting information
Table S1
Table S2
Table S3
Table S4
Table S5
Davis BH, Williams K, Absher D, Korf B, Limdi NA. Evaluation of population‐level pharmacogenetic actionability in Alabama. Clin Transl Sci. 2021;14:2327–2338. 10.1111/cts.13097
Funding information
This study was supported by funding from the State of Alabama, in part by NIH grants (T32HG008961 [B.H.D.] and K24HL133373 [N.A.L]), and the UAB Center for Clinical and Translational Science, funded by a National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) grant (UL1TR003096), the UAB‐HudonAlpha Center for Genomic Medicine, and the Hugh Kaul Precision Medicine Institute.
REFERENCES
- 1. Buttorff C, Ruder T, Bauman M. Multiple Chronic Conditions in the United States. Santa Monica, CA: RAND. Corporation, 2017. https://www.rand.org/pubs/tools/TL221.html. Accessed July 8, 2019. Web site. Published Updated. [Google Scholar]
- 2. Centers for Disease Control and Prevention: Data and Statistics . https://www.cdc.gov/datastatistics/index.html. Accessed July 8, 2019. Web site. Published Updated.
- 3. Hartman M, Martin AB, Espinosa N, Catlin A. National health care spending in 2016: spending and enrollment growth slow after initial coverage expansions. Health Aff. 2018;37(1):150‐160. [DOI] [PubMed] [Google Scholar]
- 4. Spear BB, Heath‐Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med. 2001;7(5):201‐204. [DOI] [PubMed] [Google Scholar]
- 5. Weiss A, Freeman W, Heslin K, Barrett M. Adverse Drug Events in U.S. Hospitals, 2010 Versus 2014‐2018. https://psnet.ahrq.gov/issue/adverse‐drug‐events‐us‐hospitals‐2010‐versus‐2014.
- 6. Weiss AJ, Freeman WJ, Heslin KC, Barrett ML. Adverse Drug Events in U.S. Hospitals, 2010 Versus 2014. HCUP Statistical Brief #234. January 2018. Agency for Healthcare Research and Quality, Rockville, MD. www.hcup‐us.ahrq.gov/reports/statbriefs/sb234‐Adverse‐Drug‐Events.pdf.
- 7. Budnitz DS, Shehab N, Kegler SR, Richards CL. Medication use leading to emergency department visits for adverse drug events in older adults. Ann Intern Med. 2007;147(11):755‐765. [DOI] [PubMed] [Google Scholar]
- 8. Budnitz D, Lovegrove M, Shehab N, Richards C. Emergency hospitalizations for adverse drug events in older Americans. N Engl J Med. 2011;365:2002‐2012. [DOI] [PubMed] [Google Scholar]
- 9. Zhdanava M, Kuvadia H, Joshi K, et al. Economic burden of treatment‐resistant depression in privately insured U.S. patients with physical conditions. J Manag Care Spec Pharm. 2020;26(8):996‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pilon D, Joshi K, Sheehan JJ, et al. Burden of treatment‐resistant depression in Medicare: A retrospective claims database analysis. PLoS One. 2019;14(10):e0223255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amos TB, Tandon N, Lefebvre P, et al. Direct and indirect cost burden and change of employment status in treatment‐resistant depression: a matched‐cohort study using a US commercial claims database. J Clin Psychiatry. 2018;79(2):24‐32. [DOI] [PubMed] [Google Scholar]
- 12. Olfson M, Amos TB, Benson C, McRae J, Marcus SC. Prospective service use and health care costs of Medicaid beneficiaries with treatment‐resistant depression. J Manag Care Spec Pharm. 2018;24(3):226‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pilon D, Sheehan JJ, Szukis H, et al. Medicaid spending burden among beneficiaries with treatment‐resistant depression. J Comp Eff Res. 2019;8(6):381‐392. [DOI] [PubMed] [Google Scholar]
- 14. Pilon D, Szukis H, Joshi K, et al. US Integrated delivery networks perspective on economic burden of patients with treatment‐resistant depression: A retrospective matched‐cohort study. Pharmacoecon Open. 2020;4(1):119‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pan IW, Lam S, Clarke DF, Shih YT. Insurance transitions and healthcare utilization for children with refractory epilepsy. Epilepsy Behav. 2018;89:48‐54. [DOI] [PubMed] [Google Scholar]
- 16. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905‐1917. [DOI] [PubMed] [Google Scholar]
- 17. McDowell SE, Coleman JJ, Ferner RE. Systematic review and meta‐analysis of ethnic differences in risks of adverse reactions to drugs used in cardiovascular medicine. BMJ. 2006;332(7551):1177‐1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chalasani N, Reddy KRK, Fontana RJ, et al. Idiosyncratic drug induced liver injury in African‐Americans is associated with greater morbidity and mortality compared to Caucasians. Am J Gastroenterol. 2017;112(9):1382‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jerome RN, Pulley JM, Sathe NA, et al. Exploring biologic pred response disparities to atypical antipsychotics among blacks: A quasi‐systematic review. Ethn Dis. 2020;30(Suppl 1):229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brewster LM, van Montfrans GA , Oehlers GP, Seedat YK. Systematic review: antihypertensive drug therapy in patients of African and South Asian ethnicity. Intern Emerg Med. 2016;11(3):355‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogedegbe G, Shah NR, Phillips C, et al. Comparative effectiveness of angiotensin‐converting enzyme inhibitor‐based treatment on cardiovascular outcomes in hypertensive blacks versus whites. J Am Coll Cardiol. 2015;66(11):1224‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tharpe N. Adverse drug reactions in women's health care. J Midwifery Womens Health. 2011;56(3):205‐213. [DOI] [PubMed] [Google Scholar]
- 23. Nakagawa K, Kajiwara A. Female sex as a risk factor for adverse drug reactions. Nihon Rinsho. 2015;73(4):581‐585. [PubMed] [Google Scholar]
- 24. Watson S, Caster O, Rochon PA, den Ruijter H . Reported adverse drug reactions in women and men: Aggregated evidence from globally collected individual case reports during half a century. EClinicalMedicine. 2019;17:100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bots SH, Groepenhoff F, Eikendal ALM, et al. Adverse drug reactions to guideline‐recommended heart failure drugs in women: A systematic review of the literature. JACC Heart Fail. 2019;7(3):258‐266. [DOI] [PubMed] [Google Scholar]
- 26. Zucker I, Prendergast BJ. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol Sex Differ. 2020;11(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tannenbaum C, Day D, Alliance M. Age and sex in drug development and testing for adults. Pharmacol Res. 2017;121:83‐93. [DOI] [PubMed] [Google Scholar]
- 28. Cavallari LH, Lee CR, Beitelshees AL, et al. Multisite investigation of outcomes with implementation of CYP2C19 genotype‐guided antiplatelet therapy after percutaneous coronary intervention. JACC Cardiovasc Interv. 2018;11(2):181‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Limdi NA, Cavallari LH, Lee CR, et al. Cost‐effectiveness of CYP2C19‐guided antiplatelet therapy in patients with acute coronary syndrome and percutaneous coronary intervention informed by real‐world data. Pharmacogenomics J. 2020;20(5):724‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khan MS, Shahid I, Siddiqi TJ, et al. Ten‐year trends in enrollment of women and minorities in pivotal trials supporting recent US Food and Drug Administration approval of novel cardiometabolic drugs. J Am Heart Assoc. 2020;9(11):e015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geller SE, Koch A, Pellettieri B, Carnes M. Inclusion, analysis, and reporting of sex and race/ethnicity in clinical trials: have we made progress? J Womens Health (Larchmt). 2011;20(3):315‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asiimwe IG, Zhang EJ, Osanlou R, et al. Genetic factors influencing warfarin dose in Black‐African patients: a systematic review and meta‐analysis. Clin Pharmacol Ther. 2020;107(6):1420‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu N, Irvin MR, Zhi D, et al. Influence of common and rare genetic variation on warfarin dose among African‐Americans and European‐Americans using the exome array. Pharmacogenomics. 2017;18(11):1059‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shendre A, Dillon C, Limdi NA. Pharmacogenetics of warfarin dosing in patients of African and European ancestry. Pharmacogenomics. 2018;19(17):1357‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oetting WS, Schladt DP, Guan W, et al. Genomewide association study of tacrolimus concentrations in African American kidney transplant recipients identifies multiple cyp3a5 alleles. Am J Transplant. 2016;16(2):574‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elraiyah T, Jerde CR, Shrestha S, et al. Novel deleterious dihydropyrimidine dehydrogenase variants may contribute to 5‐fluorouracil sensitivity in an East African population. Clin Pharmacol Ther. 2017;101(3):382‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson JA, Caudle KE, Gong L, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for pharmacogenetics‐guided warfarin dosing: 2017 update. Clin Pharmacol Ther. 2017;102(3):397‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Relling MV, McDonagh EM, Chang T, et al. Clinical pharmacogenetics implementation consortium (CPIC) guidelines for rasburicase therapy in the context of G6PD deficiency genotype. Clin Pharmacol Ther. 2014;96(2):169‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. CPIC . https://cpicpgx.org. Accessed March 25, 2021.
- 40. Ramsey LB, Ong HH, Schildcrout JS, et al. Prescribing prevalence of medications with potential genotype‐guided dosing in pediatric patients. JAMA Netw Open. 2020;3(12):e2029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hicks JK, El Rouby N, Ong HH, et al. Opportunity for genotype‐guided prescribing among adult patients in 11 US health systems. Clin Pharmacol Ther. 2021. 10.1002/cpt.2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics knowledge base. Methods Mol Biol. 2013;1015:311‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. East KM, Kelley WV, Cannon A, et al. A state‐based approach to genomics for rare disease and population screening. Genet Med. 2021;23:777‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu Y, Jeong H, Takahashi H, et al. Decreased warfarin clearance associated with the CYP2C9 R150H (*8) polymorphism. Clin Pharmacol Ther. 2012;91(4):660‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Relling MV, Schwab M, Whirl‐Carrillo M, et al. Clinical pharmacogenetics implementation consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther. 2019;105(5):1095‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang JJ, Landier W, Yang W, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33(11):1235‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Very Important Pharmacogene: ADRB1 . PharmGKB website. https://www.pharmgkb.org/vip/PA166170369. Accessed March 30, 2021.
- 48. Nagar SD, Conley AB, Jordan IK. Population structure and pharmacogenomic risk stratification in the United States. BMC Biol. 2020;18(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takahashi T, Luzum JA, Nicol MR, Jacobson PA. Pharmacogenomics of COVID‐19 therapies. NPJ Genomic Medicine. 2020;5(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gammal RS, Court MH, Haidar CE, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for UGT1A1 and atazanavir prescribing. Clin Pharmacol Ther. 2016;99(4):363‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Table S5