

Abstract
The highly selective, covalent Bruton’s tyrosine kinase inhibitor evobrutinib is under investigation for treatment of patients with multiple sclerosis (MS). Early clinical studies in healthy participants and patients with relapsing MS indicated that evobrutinib is well‐tolerated and effective. We undertook a mass balance study in six men who received a single 75‐mg oral dose of evobrutinib containing ~ 3.6 MBq (100 μCi) 14C‐evobrutinib, to determine the absorption, metabolic pathways, and routes of excretion of evobrutinib. The primary objectives of this phase I study (NCT03725072) were to (1) determine the rates and routes of total radioactivity excretion, including the mass balance of total drug‐related radioactivity in urine and feces, (2) assess the pharmacokinetics (PKs) of total radioactivity in blood and plasma, and (3) characterize the plasma PKs of evobrutinib. Exploratory end points included identifying and quantifying evobrutinib and its metabolites in plasma and excreta (urine and feces) and exploring key biotransformation pathways and clearance mechanisms. Evobrutinib was primarily eliminated in feces (arithmetic mean percentage, SD, 71.0, 2.1) and, to a lesser extent, in urine (20.6, 2.0), with most of the total radioactivity (85.3%) excreted in the first 72 h after administration. No unchanged evobrutinib was detected in excreta. Evobrutinib was rapidly absorbed and substantially metabolized upon absorption. Only one major metabolite M463‐2 (MSC2430422) was identified in plasma above the 10% of total drug exposure threshold, which classifies M463‐2 (MSC2430422) as a major metabolite according to the US Food and Drug Administration (FDA; metabolites in safety testing [MIST]) and the European Medicines Agency (EMA; International Conference on Harmonization [ICH] M3). These results support further development of evobrutinib and may help inform subsequent investigations.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Multiple sclerosis (MS) is associated with a high frequency of relapse and a need for effective therapies. The pathogenesis of MS is driven by the proinflammatory action of B cells and myeloid cells. The highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor evobrutinib targets both B cells and myeloid cells and is therefore under investigation as a treatment for autoimmune diseases, including MS.
WHAT QUESTION DID THIS STUDY ADDRESS?
This phase I human mass balance study was conducted to better understand the absorption, metabolic pathways, and routes of excretion of a single oral dose of 14C‐radiolabeled evobrutinib in healthy male participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study provides a better understanding of the drug disposition and pharmacokinetic (PK) characteristics of evobrutinib and a comprehensive characterization of its metabolites in human participants.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The PK characteristics and metabolic pathways of evobrutinib elucidated in this study support the further investigation of a promising BTK inhibitor for the treatment of patients with MS.
INTRODUCTION
Multiple sclerosis (MS) is commonly diagnosed during early adulthood, at an average age of 30 years. 1 Early onset and the chronic, progressively incapacitating nature of the disease mean high costs to patients, families, and society. 2 Disability progression 3 , 4 , 5 and relapse frequency 6 erode patients’ quality of life. Finding an effective treatment can also be challenging due to the highly heterogeneous presentation of MS and the variable evolution in the clinical course of the disease, which requires treatment adjustments. 7 Treatment‐switching is frequent in MS, typically because of a lack of sustained efficacy, resulting relapse, 8 , 9 , 10 intolerance to therapy, or adverse events. 11
At the cellular level, relapsing MS is characterized by blood–brain barrier disruption and inflammation caused by T cells, B cells, and myeloid cells infiltrating the central nervous system (CNS), where autoreactive T cells are reactivated through local antigen presentation with activated CNS‐resident microglia and astrocytes. 12 , 13 Bruton’s tyrosine kinase (BTK) has an important role in the development and function of immune cells, including B cells and myeloid cells, 14 and is a key mediator of B cell and myeloid cell signaling. 15 , 16
Evobrutinib is an orally administered, highly selective, covalent BTK inhibitor 17 currently under investigation for treatment of MS. In clinical studies with healthy participants and patients with relapsing MS, evobrutinib was well‐tolerated and effective. 18 , 19
In a first‐in‐human, phase I, double‐blind, dose‐escalation study, healthy participants were randomized to receive evobrutinib (25, 50, 100, 200, 350, or 500 mg) or placebo as a single dose, or evobrutinib 25, 75, or 200 mg or placebo once daily (q.d.) for 14 days. 18 Treatment‐emergent adverse events (TEAEs) were reported in nine participants (25%) after single‐dosing and in 13 (48.1%) after multiple‐dosing. Most TEAEs were mild, with no dose‐dependency with respect to frequency or type of TEAE. Absorption was rapid, with a time to reach maximum plasma concentration (T max) of ~ 0.5 h, a short half‐life of ~ 2 h, and pharmacokinetics (PKs) were dose‐proportional, with no accumulation or time‐dependency with repeat dosing. 18 BTK occupancy was dose‐dependent, with maximum occupancy (>90%) reached within ~ 4 h after single evobrutinib doses of greater than or equal to 200 mg; an occupancy of greater than 50% was observed at 96 h with doses of greater than or equal to 100 mg. Full BTK occupancy in healthy participants was achieved with 25 mg evobrutinib after multiple daily dosing. 18
In a phase II, randomized, double‐blind study, 267 patients with relapsing MS received evobrutinib 25 or 75 mg q.d., 75 mg twice daily (b.i.d.), or placebo, or open‐label dimethyl fumarate as reference treatment. 19 Evobrutinib 75 mg q.d. and b.i.d. significantly reduced the total number of gadolinium‐enhancing lesions on T1‐weighted magnetic resonance imaging over 24 weeks versus placebo. At week 24, evobrutinib 75 mg q.d. and b.i.d. also showed evidence of reduced annualized relapse rate, an effect that was sustained at week 48 and maintained further, at 108 weeks in the open‐label study extension. 20 The majority of TEAEs in this study were mild or moderate. 19
An in vitro study investigating the metabolism of evobrutinib in rat and human hepatocytes determined that evobrutinib was metabolized into 23 different metabolites. 21 There were species‐related metabolic differences with the main metabolite in human hepatocytes identified as evobrutinib‐diol, whereas in rat hepatocytes the most abundant metabolite was hydroxyl‐evobrutinib. 21
To better understand these clinical properties, drug disposition, and PK characteristics of evobrutinib, we conducted a mass balance study to evaluate the absorption, metabolic pathways, and routes of excretion of evobrutinib in healthy male participants after administration of a single 75‐mg oral dose of 14C‐radiolabeled evobrutinib.
METHODS
Study participants
Eligible participants were men, 18–55 years, with body weight 50–120 kg, and body mass index (BMI) of 19.0–30.0 kg/m2, who were healthy based on comprehensive clinical assessments. Study inclusion/exclusion criteria are in the Supplementary Material. All participants provided written informed consent before initiation of study. The study protocol and consent forms were approved by the Independent Ethics Committee of Evaluation of Ethics in Biomedical Research, Assen, The Netherlands. The study was conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Council for Harmonization E6 Guideline for Good Clinical Practice.
Study design
This was a phase I, open‐label, single‐dose study (NCT03725072) to determine the absorption, metabolism, and routes of excretion of 14C‐radiolabeled evobrutinib. Following eligibility screening, participants were admitted to the study center on day −1. The study phase was from day 1 up to a maximum of day 35, with final assessments up to day 37 depending on participant status (Figure 1).
FIGURE 1.
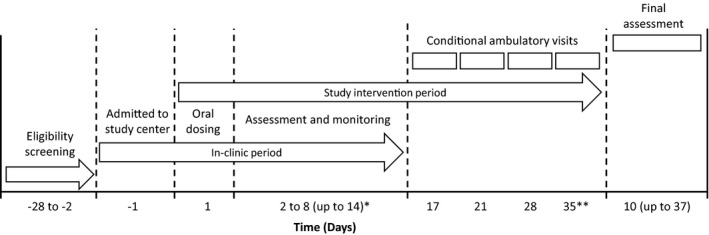
Study design. *If, on the morning of day 8, recovery of radioactivity in excreta was greater than or equal to 90% of the dose, the participant was to be released from the study at the discretion of the Principal Investigator or delegate (in agreement with the Sponsor). If radioactivity recovery was less than 90% on the morning of day 8, the participant was required to remain in the study center until radioactivity recovery in excreta was greater than or equal to 90%, which could require an additional in‐house period of up to 6 days. During the study, no patient was required to remain in the study center past day 10. **On day 14 (13 days after dosing), any remaining participants could be released irrespective of radioactivity recovery but could be asked to return to the study center for one or more conditional follow‐up visits planned for days 17, 21, 28, and 35
On the morning of day 1 (evobrutinib dosing day), participants underwent predose safety evaluations and provided blood, urine, and feces for PK assessments. Serial blood samples for determination of clinical laboratory variables and PK assessments were collected throughout the study. TEAEs, clinical laboratory variables, electrocardiograms, and vital signs were also assessed throughout the study. A final assessment of each participant took place 1–2 days after discharge from the study center.
Recovery of radioactivity in excreta was monitored daily throughout the study. Participants with a recovery of radioactivity greater than or equal to 90% on day 8 were released from the study, those with a recovery rate less than 90% remained in the study center for up to 6 additional days, until a recovery rate of greater than or equal to 90% was obtained.
Study medication
Participants received a single 75‐mg oral dose of evobrutinib as a 30 ml solution from a glass drinking bottle. The bottle was rinsed twice (50 ml each) and the additional fluid was also taken by the participant. This single 75‐mg oral dose contained in total ~ 3.6 MBq (~100 μCi) of 14C‐evobrutinib. The structural formula of 14C‐evobrutinib is shown in Figure 2.
FIGURE 2.
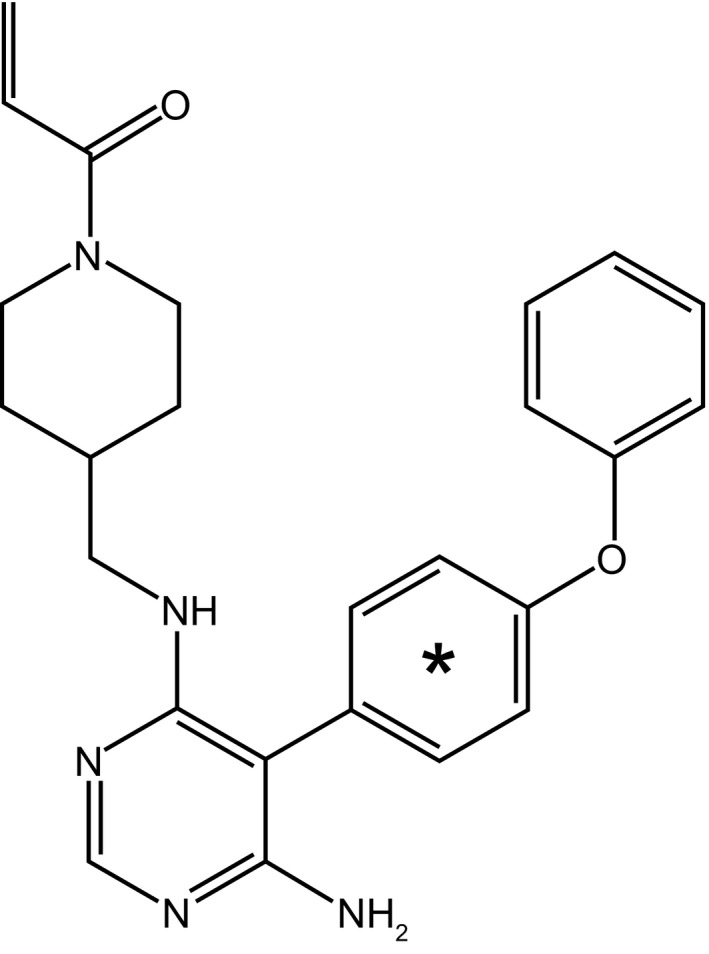
Structure of 14C‐evobrutinib. *Denotes the position of the radiolabel. The material is ring labeled, with an average of one carbon‐14 per molecule
Dosimetry assessment to calculate radiation burden
The effective radiation burden after a single oral dose of 3.6 MBq (~ 100 μCi) of 14C‐evobrutinib was calculated by dosimetry assessment. Calculations were based on previous excretion and PK data for evobrutinib and quantitative tissue distribution studies performed on mice, and on data from additional preclinical and clinical studies, assuming a worst‐case absorbed fraction of 1.0. According to these calculations, a single oral dose of 3.6 MBq of 14C‐evobrutinib carries an estimated effective radiation burden of ~ 0.02 mSv, which is below the effective dose considered trivial and well acceptable (0.1 mSv) for investigations in human participants. 22
Study objectives
The primary objectives were to (1) determine the rates and routes of excretion of total radioactivity, including mass balance of total evobrutinib‐related radioactivity in urine and feces, (2) assess the PK of total radioactivity in blood and plasma, and (3) characterize the plasma PKs of evobrutinib. An exploratory endpoint was to identify and quantify evobrutinib and its metabolites in excreta (urine and feces) and plasma to investigate key biotransformation pathways and clearance mechanisms in humans.
Sample collection
The collection schedule for whole blood, plasma, urine, and feces is shown in Tables S1 and S2.
Liquid scintillation counting of total radioactivity
Total radioactivity in whole blood, plasma, urine, and feces samples were determined by liquid scintillation counting (LSC). Scintillation cocktail was directly spiked into plasma and urine samples. Whole blood samples were solubilized and decolorized before addition of the scintillation cocktail. Feces samples were homogenized with weight equivalents of water, dried, and combusted with an absorber agent for trapping carbon dioxide. At the end of the combustion cycle, the absorber was mixed with scintillant cocktail. All scintillation counting using either low level or normal count modes was carried out in duplicate using a Tri‐Carb 3100 TR liquid scintillation analyzer (Perkin Elmer, Waltham, MA, USA) in compliance with analytical procedures validated by PRA Health Sciences.
Bioanalysis of evobrutinib
Bioanalysis of total plasma concentrations of unlabeled evobrutinib was performed by Nuvisan (Grafing, Germany) by validated liquid chromatography–mass spectrometry (LC‐MS/MS). In brief, the isotopically labeled internal standard [2H]‐MSC2364447C was added to 100 µL K2EDTA plasma. Evobrutinib was extracted with 500 µL ethyl acetate, then gently mixed. Samples were centrifuged, 200 µL aliquots of the upper organic phase were transferred, evaporated to dryness (15 min, 40℃), and reconstituted in 300 µL acetonitrile/water (3+7, V/V). An aliquot of 10 µl was injected onto an Acquity BEH C18 column (2.1 × 50 mm; 1.7 µm; Waters, Milford, MA, USA). Separation was carried out using a mobile phase gradient (eluent A: 10 mM ammonium formate buffer; eluent B: acetonitrile) with a flow rate of 0.8 ml/min. Detection was performed in positive ionization mode on an AB Sciex 5500 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) with selected reaction monitoring (m/z 430.2 to 279.0 for evobrutinib and 433.2 to 279.0 for its deuterated internal standard). The lower limit of quantification (LLOQ) was 0.600 ng/ml and the calibration standard responses were linear over the range between 0.600 and 600 ng/ml using a weighted (1/x2) linear regression.
Metabolite profiling and identification
Metabolite profiling and identification was performed by ultra‐performance liquid chromatography‐high‐resolution mass spectrometry (UPLC‐HR‐MS; Acquity UPLC and Xevo G2‐XS QTof (quadrupole time‐of‐flight) mass spectrometer; Waters) and simultaneously the eluent was fractionated (CTC HTS PAL Fraction Collector; Axel Semrau, Sprockhövel, Germany) with a collection interval of 7 s for each well. Radioactivity in the fractions was determined off‐line by microplate scintillation counting using the Topcount NXT counter (Perkin Elmer, Waltham, MA, USA) and the radio‐chromatograms were recompiled from those data using Laura 4.1 software (Lablogic, Sheffield, UK).
For urine and feces, metabolites were identified in a single pooled sample per matrix and participant that covered at least 90% of the total radioactivity of the respective matrix excreted. Intervals with low radioactivity were not used for generation of the pooled samples, to avoid dilution and to increase the signal‐to‐noise ratio of the respective chromatograms. For plasma, metabolites were identified in individual samples (at 1, 2, 4, 8, 12, and 24 h). Plasma samples were analyzed after an exhaustive extraction process using ethanol and acetonitrile in multiple steps, processed by gentle evaporation under nitrogen flow (37°C), and redissolved in ACN/MeOH/water (1+1+8, V/V/V) for injection. Feces sample pools were extracted as followed: first with an n‐hexane wash step to degrease the samples, followed by stepwise extraction with EtOH (2×), ACN/water (1+1, V/V), and ACN/water/formic acid (FA) (49+49+2, V/V/V; 2×). Urine sample pools were directly injected without extraction after evaporation under nitrogen (37°C) and re‐dissolution in ACN/MeOH/water (1+1+8, V/V/V). Extraction process and efficiency was checked in all samples by LSC counting of the final precipitate after solubilization (by Solvable), homogenization (using Ultra‐Turrax) and de‐colorization (by hydrogen peroxide). These values (as percentage of total radioactivity in the sample) are reported as “nonextractable radioactivity” in plasma and feces.
Pharmacokinetic assessments
The following PK parameters were assessed: (1) total radioactivity recovery, percentage excretion in urine and feces, clearance of parent, and total radioactivity in urine and feces; (2) whole blood and plasma total radioactivity, maximum concentration (Cmax), T max, terminal half‐life (t 1/2), and area under the curve (AUC); and (3) evobrutinib plasma Cmax, T max, AUC, t 1/2, apparent volume of distribution during the terminal phase (V z/F), and apparent plasma clearance of evobrutinib (CL/F).
Calculations and statistical analyses
All PK parameters for total radioactivity, evobrutinib, and its metabolites were calculated using noncompartmental methods with Phoenix WinNonlin 8.1 (Certara, Sheffield, UK), based on the actual sampling times. Results for all parameters were reported using descriptive statistics. Individual plasma and whole‐blood concentrations for total radioactivity (by LSC) and individual plasma concentrations for evobrutinib (by validated LC‐MS/MS), and 14C‐evobrutinib and its metabolites (by metabolite profiling) were reported with descriptive statistics using the nominal sampling times for all time points. Similarly, individual urine and feces concentrations for total radioactivity, 14C‐evobrutinib, and its metabolites (by metabolite profiling) were reported with descriptive statistics using the nominal sampling times for all time intervals. Excretion of total radioactivity was calculated based on the administered radioactive dose and the relative amount of radioactivity recovered in urine and feces. All participants who received a dose of evobrutinib were included in the PK analysis set.
RESULTS
Study participants
In total, 19 healthy men were screened. Six were eligible for inclusion, dosed with evobrutinib, and completed per protocol. They were of mean (SD) age of 31 (8) years, with a body weight of 85.0 (14.45) kg, and BMI 24.3 (3.39) kg/m2. There were no clinically relevant findings in medical history or previous medications.
Mass balance of 14C‐evobrutinib
Recovery criteria for the collection of radioactivity were met for all participants on day 7 (168 h after dosing); for one participant, collection of urine and feces was prolonged until day 8 (192 h). Mass balance was achieved within 168 h after a single oral 75‐mg dose of 14C‐evobrutinib with a mean total radioactivity recovery of 91.6% (range 90.1–92.8%). The majority of radioactivity was excreted in feces (71.0%) and less in urine (20.6%). Individual total recovery ranged from 69.2 to 74.1% in feces and from 18.6 to 23.1% in urine. No unchanged evobrutinib was recovered from urine or feces (Figure 3). The majority of radioactivity (85.3%) was excreted within 72 h after evobrutinib administration.
FIGURE 3.
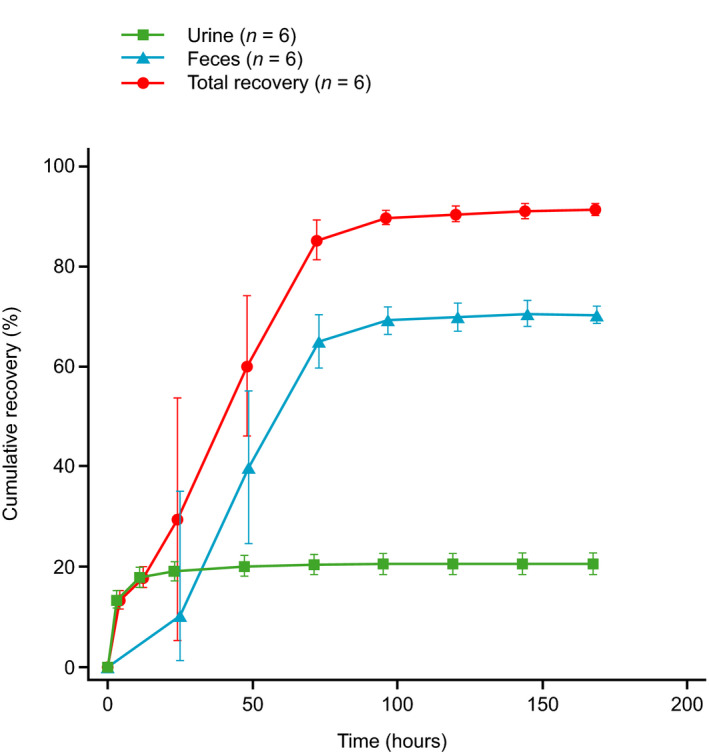
Cumulative total radioactivity excreted in urine and feces, and total radioactivity recovery. Values are arithmetic mean with SD
Total radioactivity and evobrutinib concentrations in systemic circulation
Total radioactivity in plasma was detected at the first sampling time, 0.25 h postdose, in all participants and was close to or below the LLOQ (9.91 ng eq/ml [30 dpm/ml]) of the plasma LSC method in samples from 48 to 192 h. The individual maximum total radioactivity concentration in plasma ranged from 740 to 1445 ng eq/ml and was observed 0.25–1‐h postdose.
Evobrutinib in plasma was detected at the first sampling time point after evobrutinib administration (0.25 h postdose) for all participants and was quantifiable up to 8 h in five participants and up to 12 h in one participant. The individual maximum evobrutinib plasma concentration ranged from 181 to 465 ng/ml and was observed 0.25–0.50 h postdose.
The mean concentration–time profile for evobrutinib in plasma rapidly declined with little differentiation between distribution and elimination phases (Figure 4a and b). In contrast, the mean concentration–time profiles for total radioactivity in plasma and whole blood displayed a slower elimination phase. Although total radioactivity concentrations in blood and plasma decreased in a similar manner, the mean blood to plasma ratios of total radioactivity increased from 0.6 at 0.25 h to 1.6 at 72 and 96 h postdose.
FIGURE 4.
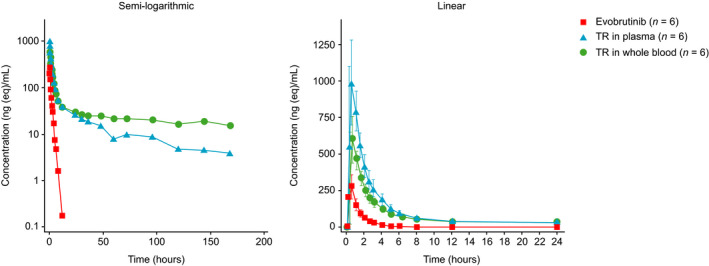
Plasma and whole‐blood concentration–time profiles for evobrutinib and total radioactivity. Values are arithmetic mean and, on linear scale, standard deviation. TR, total radioactivity. Time course on linear scale is limited to first 24 h after dosing
Pharmacokinetic parameters
The Cmax of total radioactivity in plasma was approximately threefold higher than that for evobrutinib in plasma (Table 1). The whole blood to plasma ratio for mean Cmax of total radioactivity was ~ 0.6. Plasma AUC0–24 and AUC0–t for evobrutinib represented 13% and 10% of the corresponding AUC total radioactivity in plasma at those time points, respectively, indicating extensive metabolism. Absorption was rapid with a median T max of 0.5 h for total radioactivity in whole blood and plasma, as well as evobrutinib in plasma (Table 1). The geometric mean t1/2 of evobrutinib, at 1.33 h, was short relative to the t 1/2 for total radioactivity, as shown in the concentration–time profiles. The t1/2 of the prolonged elimination phase for total radioactivity (~ 102 h in plasma and 290 h in whole blood) was too long relative to the PK sampling duration to be reliably estimated.
TABLE 1.
Summary of PK parameters in whole blood and plasma following a single 75‐mg oral dose of 14C‐radiolabeled evobrutinib
| Parameter | Evobrutinib | Total radioactivity | |
|---|---|---|---|
| Plasma | Plasma | Whole blood | |
| Cmax, ng (eq)/ml, GM (GM CV, %) a | 321 (33.5) | 1047 (22.8) | 623 (25.5) |
| AUC0–24h, ng (eq) h/ml, GM (GM CV, %) a | 378 (19.4) | 2805 (14.8) | 2001 (9.9) |
| AUC0‐t, ng (eq) h/ml, GM (GM CV, %) a | 375 (19.4) | 3915 (32.7) | 4944 (13.6) |
| T max, h, median (min, max) | 0.50 (0.25, 0.55) | 0.50 (0.25, 1.00) | 0.50 (0.25, 1.00) |
| T 1/2, h, GM (GM CV, %) | 1.33 (27.9) | NC b | NC b |
| CL/F, L/h, GM (GM CV, %) | 199 (19.4) | – | – |
| V z/F, L, GM (GM CV, %) | 381 (33.9) | – | – |
Abbreviations: AUC0‐24, area under the plasma/whole blood concentration–time curve from time zero (dosing) to 24 h after dosing of unchanged drug or radioactivity; AUC0‐t, area under the plasma/whole blood concentration–time curve from time zero (dosing) to the last sampling time at which the concentration is at or above the lower limit of quantification of unchanged drug or radioactivity; CL/F, apparent total body clearance of drug from plasma following oral administration; Cmax, maximum observed concentration of unchanged drug or radioactivity; GM, geometric mean; GM CV, geometric coefficient of variation; NC, not calculated; PK, pharmacokinetic; T max, time to reach the maximum observed concentration of unchanged drug or radioactivity; T 1/2, terminal half‐life of unchanged drug or radioactivity; V z/F, apparent volume of distribution of drug during the terminal phase following oral administration.
ng eq/ml for total radioactivity (by liquid scintillation counting) and ng/mL for evobrutinib (by validated LC‐MS/MS method).
Did not meet acceptable diagnostic criteria.
Metabolite profiling and identification
Chemical structures of 40 phase I and 18 phase II metabolites of evobrutinib were fully or partially elucidated. The phase I reactions that occurred were single and multiple oxygenations, hydroxylation, and further oxidation to the ketone, dehydrogenation, reduction, cleavage of either the acrylamide, or the piperidine together with the acrylamide moiety, and oxidative piperidine ring opening. Phase II metabolites were formed by glucuronidation, sulfation, N‐acetylation, and cysteine‐, cysteinylglycine‐, or N‐acetyl cysteine‐conjugation of the acrylamide moiety. All metabolites elucidated were more polar than evobrutinib. Biotransformation of evobrutinib to form the major metabolite, M463‐2 (MSC2430422) is shown in Figure 5. A comprehensive investigation of the metabolism of evobrutinib is ongoing.
FIGURE 5.
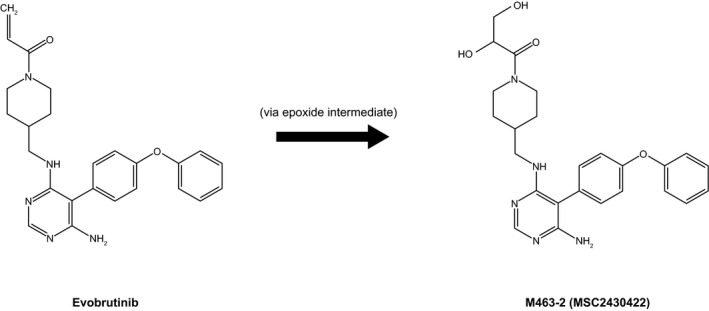
Proposed metabolism of evobrutinib to its major metabolite in healthy humans
Metabolites in plasma
A total of 24 metabolites were detected and elucidated in plasma (Figure 6). Fourteen of these were formed by oxidations and 10 by conjugation. The most abundant plasma metabolite, M463‐2 (MSC2430422), which was presumably formed by epoxidation and hydrolysis of the acrylamide moiety, was observed up to 8 h after evobrutinib administration in all six participants and attained its Cmax after 1 h. Concentrations of M463‐2 (MSC2430422) ranged from 92.8 to 210 ng eq/ml at Cmax. The geometric mean AUC0–24 of the metabolite relative to total radioactivity and evobrutinib was 14.9% and 202.5%, respectively. Each of the other circulating metabolites was below 10% of the total radioactivity AUC0–24. No single metabolite other than M463‐2 (MSC2430422) exceeded 100% of the evobrutinib (parent) AUC0–24. The geometric mean AUC0–24 of nonextractable radioactivity relative to total radioactivity AUC0–24, at 1085 h٠ng eq/ml, was 40.9%. Nonextractable radioactivity was determined after exhaustive extraction procedures, as detailed in the Methods section. Plasma samples after 48 h were not analyzed further as total radioactivity was too low (<23.1 ng eq/ml [70 dpm/ml]) to facilitate investigation of radioactive drug‐related material.
FIGURE 6.
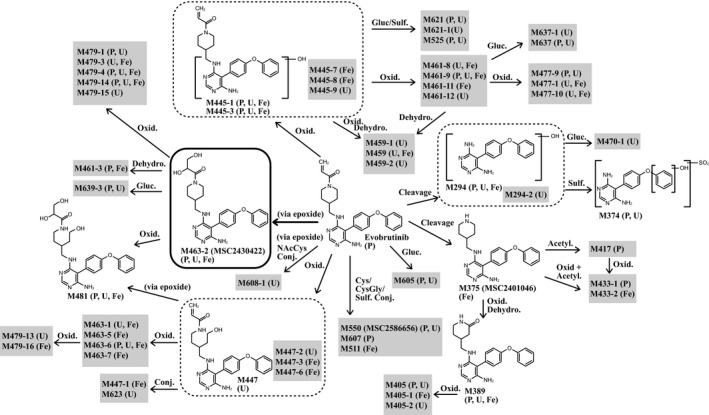
Proposed metabolic pathway of evobrutinib in humans. Identified metabolites and their occurrence in the corresponding matrices: plasma, urine, and feces. The thickness of lines is indicative of the relative importance of the metabolic pathways, as deduced from concentrations of circulating metabolites at their maximum plasma concentration (Cmax). Note that due to the high number of metabolites found, not all metabolites identified in the present study are displayed. Acetyl, acetylation; Conj, conjugation; Cys, cysteine; CysGly, cysteinyl glycine; Dehydro, dehydrogenation; F, feces; Gluc, glucuronidation; NAcCys, N‐acetyl cysteine; Oxid, oxidation; P, plasma; Sulf, sulfation; U, urine
Metabolites in urine
In urine, between 17.2% and 21.8% of the radioactive dose was excreted within 24 h after evobrutinib administration. Unchanged evobrutinib was not detected in any of the urine metabolite profiles. In total, 41 metabolites were detected and elucidated in urine (Figure 6). Twenty‐nine of these were formed by oxidations and 12 by conjugation. The most abundant metabolites excreted that did not arise from a cluster of coeluting metabolites were M445‐3 (formed by hydroxylation; 1.22–2.32% of the administered dose) and M374 (cleavage of the piperidine and acrylamide moiety, hydroxylation, and sulfation; 1.53–1.94% of the administered dose).
The most prominent coeluting metabolites were M525 (oxygenation and sulfation) together with M447‐2 (reduction and oxygenation) comprising 1.42–2.34% of the administered dose, and an additional alternating metabolite cluster comprising M463‐2 (MSC2430422), M445‐1 (hydroxylation), M463‐6 (oxidative ring opening of piperidine moiety and oxygenation), M608‐1 (epoxidation and N‐acetyl cysteine conjugation of acrylamide moiety), M459 (double oxygenation and further oxidation to the ketone on piperidine moiety), and M389 (cleavage of acrylamide moiety with oxygenation and further oxidation to ketone on piperidine moiety) comprising 2.03–3.34% of the administered dose.
Metabolites in feces
In feces, 66.4–73.5% of the radioactive dose was excreted within 96 h after administration. Unchanged evobrutinib was not detected. In total, 30 metabolites were detected and elucidated in feces, with 26 formed by oxidations, three by conjugation, and one by cleavage of the acrylamide moiety (Figure 6). M463‐2 (MSC2430422; 6.18–9.28% of the administered dose) was identified as the main fecal metabolite, followed (in rank order of the amount excreted) by M445‐1 (3.67–8.14% of the administered dose), M479‐3 (epoxidation and hydrolysis of acrylamide moiety and hydroxylation; 1.27–5.92% of the administered dose), M445‐3 (2.74–5.05% of the administered dose), M481 (double hydroxylation of acrylamide moiety and oxidative piperidine ring opening; 1.31–2.96% of the administered dose), M375 (MSC2401046; 1.58–2.40% of the administered dose), and M463‐6 (1.45–2.17% of the administered dose). Moreover, an alternating cluster of M461‐11 (double oxygenation), M479‐16 (multiple oxygenation), M447‐3 (reduction of acrylamide moiety and oxygenation), M461‐3 (epoxidation and hydrolysis of acrylamide and dehydrogenation of piperidine moiety), and M445‐7 (oxygenation) was observed in the fecal samples accounting for 1.53–5.01% of the administered dose. The coeluting metabolites M461‐8 (double oxygenation), M477‐1 (double hydroxylation of piperidine moiety and hydroxylation), and M463‐1 (oxidative ring opening of piperidine moiety and hydroxylation) represented 1.58–3.01% of the administered dose.
Safety
In total, four (67%) participants experienced an adverse event (AE) following the single dose of 14C‐evobrutinib. There were no serious AEs, deaths, or withdrawals. AEs reported were oropharyngeal pain (2 participants), nausea, headache, epistaxis, and nasopharyngitis (1 participant each). All AEs were mild and not considered study treatment related.
DISCUSSION
This mass balance study investigated the absorption, metabolism, and excretion routes of a single 75‐mg oral dose of 14C‐radiolabeled evobrutinib in six healthy male participants. Evobrutinib metabolites in plasma and eliminated in urine and feces were also evaluated and characterized for comprehensive understanding of the metabolic pathways of evobrutinib in humans.
Most of the administered radioactivity (mean of 85.3%) was excreted within 72 h after dosing. Greater than 90% was recovered from two participants within 96 h (mean of 89.7%), and a mean of 91.6% was recovered from all participants within 168 h. Total radioactivity was eliminated primarily in feces (71.0%) and to a lesser extent in urine (20.6%). No unchanged evobrutinib was detected in urine or feces. Similar results were reported for ibrutinib, which, in 2013, was designated as a BTK inhibitor breakthrough therapy for treatment of refractory mantle cell lymphoma that received accelerated approval for treatment of chronic leukemia. 23 In the mass balance study with 14C‐radiolabeled ibrutinib, a structurally related BTK inhibitor, 88.5% of total radioactivity was excreted within the first 168 h following dosing. 23 As in our study, most radioactivity was excreted in feces (80.6%), the majority within 48 h. Much less (7.8%) radioactivity was excreted in urine, mostly within 24 h. 23
The Cmax for total radioactivity was threefold higher than the Cmax for evobrutinib, which demonstrated that evobrutinib was rapidly and substantially metabolized upon absorption. Evobrutinib also was extensively metabolized, with the parent drug comprising only 10–13% of the total radioactivity plasma exposure (AUC) following a single dose. The absorption of evobrutinib was rapid, with a median T max for both total radioactivity in whole blood and plasma, and for evobrutinib in plasma, of 0.5 h, as expected following administration of evobrutinib as a single oral dose in solution. A similar rapid absorption of evobrutinib was also seen in a placebo‐controlled study of 48 healthy participants who received single oral doses (25–500 mg) in solution, in which the Cmax of evobrutinib was found within a median of 0.5–1 h after administration for all dose cohorts. 18 The short evobrutinib half‐life of 1.33 h in our study is also consistent with the half‐life of 1.8–2.6 h for the 25–200 mg doses reported in this previous study. The main metabolite, M463‐2 (MSC2430422), shows similar PK characteristics (T max: 1 h; mean half‐life: 2.4 h) and is not active on BTK (Merck data on file). Therefore, it is not relevant for the pharmacodynamic effect of evobrutinib, as reported previously. 18 , 24 , 25
Evobrutinib metabolites and nonextractable radioactivity were eliminated more slowly than evobrutinib from plasma. However, because the majority of the administered dose was recovered in urine and feces within 72 h postdose, a relatively small percentage of the dose accounted for the prolonged detection of metabolites and nonextractable radioactivity in plasma. The nonextractable radioactivity in plasma, which was measured after an exhaustive extraction process, was not unexpected due to the acrylamide moiety. Similar results have been reported for other covalent BTK inhibitors (ibrutinib 23 ) and targeted covalent inhibitors (pyrotinib, 26 osimertinib, 27 and neratinib 28 ) where binding to albumin was described as a common feature for the Michael acceptor moiety. On this basis, we would therefore presume that the evobrutinib nonextractable radioactivity was similarly bound to protein.
Comprehensive metabolite profiling in this study allowed us to propose a metabolic pathway for evobrutinib. A total of 58 metabolites, mainly formed by oxidations, were identified in urine, feces, and plasma. Of these, 24 were detected and elucidated in plasma, indicating that evobrutinib is extensively metabolized. Only one metabolite, M463‐2 (MSC2430422), was considered major. This was the main fecal metabolite, eliminated at less than 10% of the administered dose. All other metabolites were excreted individually below 6.4% of the administered dose. No unchanged evobrutinib was detected in urine or feces. Further investigation of the metabolic pathway of evobrutinib that will provide more detailed information on these metabolites is currently being undertaken and will be reported separately.
As this study was conducted in healthy male participants, the results may not extrapolate to women or patients with MS. However, there is no evidence to suggest PK differences between these groups in the ongoing clinical studies of evobrutinib.
In summary, the results of this study advance our understanding of the PK and metabolic pathways of the highly selective, covalent BTK inhibitor evobrutinib and can help inform its further investigation. Studies to date suggest that evobrutinib has the potential to become a valuable treatment option for patients with a highly heterogeneous and variable disease, such as MS. Two phase III randomized controlled trials (NCT04338022 and NCT04338061) evaluating the efficacy and safety of evobrutinib in relapsing MS commenced in 2020.
CONFLICTS OF INTEREST
H.S. is employed by the healthcare business of Merck KGaA, Darmstadt, Germany. M.D. is employed by EMD Serono, Billerica, MA, USA. A.S.‐K. is employed by the healthcare business of Merck KGaA, Darmstadt, Germany. E.H.‐M. is employed by the healthcare business of Merck KGaA, Darmstadt, Germany. N.M. is employed by Cytel Inc., Paris, France. A.P. is employed by the healthcare business of Merck KGaA, Darmstadt, Germany. A.B. is employed by the healthcare business of Merck KGaA, Darmstadt, Germany. J.D. is employed by EMD Serono, Billerica, MA, USA. J.J.v.L. is employed by PRA Health Sciences, Groningen, The Netherlands. W.T. is employed by Nuventra Inc., Exton, PA, USA. D.M. is employed by Nuventra Inc., Denver, CO, USA.
AUTHOR CONTRIBUTIONS
H.S. wrote the manuscript. H.S., M.D., A.P., A.B., W.T., and D.M. designed the research. H.S. and J.J.v.L. performed the research. H.S., M.D., A.S.‐K., E.H.‐M., N.M., J.D., W.T., and D.M. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Dr. Lisa Steinhauser, Study Director for the bioanalysis of evobrutinib at Nuvisan GmbH (Am Feld 32, 85567, Grafing, Germany), Maximilian Kranz, Study Director for metabolite profiling and quantification at Nuvisan GmbH, L.P. van Tilburg, Study Director for radioactivity measurement at PRA Health Sciences (Early Development Services, Bioanalytical Laboratory, Amerikaweg 18, 9407 TK Assen, The Netherlands), and Dr Katrin Georgi (the healthcare business of Merck KGaA, Darmstadt, Germany) for contributing to the conception of the study, review of the study report, and discussion and interpretation of results. The authors would also like to thank the participants and their families, investigators, co‐investigators, and the study teams at the participating center. Bioscript Stirling Ltd provided medical writing support, funded by the healthcare business of Merck KGaA, Darmstadt, Germany.
Scheible H, Dyroff M, Seithel‐Keuth A, et al. Evobrutinib, a covalent Bruton’s tyrosine kinase inhibitor: Mass balance, elimination route, and metabolism in healthy participants. Clin Transl Sci. 2021;14:2420–2430. 10.1111/cts.13108
Funding information
This study was sponsored by Merck KGaA, Darmstadt, Germany. Bioscript Stirling Ltd. provided medical writing support, funded by Merck KGaA, Darmstadt, Germany.
Contributor Information
Martin Dyroff, Email: holger.scheible@merckgroup.com.
Jennifer Dong, Email: holger.scheible@merckgroup.com.
REFERENCES
- 1. Multiple Sclerosis International Federation . Atlas of MS 2013. 2013.
- 2. Fattore G, Lang M, Pugliatti M. The treatment experience, burden, and unmet needs (TRIBUNE) study – measuring the socioeconomic consequences of multiple sclerosis. Mult Scler. 2012;18:5‐6. [DOI] [PubMed] [Google Scholar]
- 3. Naci H, Fleurence R, Birt J, Duhig A. The impact of increasing neurological disability of multiple sclerosis on health utilities: a systematic review of the literature. J Med Econ. 2010;13:78‐89. [DOI] [PubMed] [Google Scholar]
- 4. Kobelt G, Thompson A, Berg J, et al. New insights into the burden and costs of multiple sclerosis in Europe. Mult Scler. 2017;23:1123‐1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parkin D. Treatment of multiple sclerosis with interferon beta: an appraisal of cost‐effectiveness and quality of life. J Neurol Neurosurg Psychiatry. 2000;68:144‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baumstarck K, Pelletier J, Boucekine M, Auquier P, & MusiQoL Study Group. Predictors of quality of life in patients with relapsing‐remitting multiple sclerosis: a 2‐year longitudinal study. Rev Neurol (Paris). 2015;171:173‐180. [DOI] [PubMed] [Google Scholar]
- 7. Disanto G, Berlanga AJ, Handel A, Para AE. Heterogeneity in multiple sclerosis: scratching the surface of a complex disease. Autoimmune Dis. 2010;2011:932351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coppola G, Lanzillo R, Florio C, et al. Long‐term clinical experience with weekly interferon beta‐1a in relapsing multiple sclerosis. Eur J Neurol. 2006;13:1014‐1021. [DOI] [PubMed] [Google Scholar]
- 9. Rotstein DL, Healy BC, Malik MT, Chitnis T, Weiner HL. Evaluation of no evidence of disease activity in a 7‐year longitudinal multiple sclerosis cohort. JAMA Neurol. 2015;72:152‐158. [DOI] [PubMed] [Google Scholar]
- 10. Parks NE, Pittock SJ, Mandrekar J, et al. Population‐based study of "no evident disease activity" in MS. Neurol Neuroimmunol Neuroinflamm. 2018;5:e495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Visaria J, Thomas N, Gu T, Singer J, Tan H. Understanding the patient’s journey in the diagnosis and treatment of multiple sclerosis in clinical practice. Clin Ther. 2018;40:926‐939. [DOI] [PubMed] [Google Scholar]
- 12. Filippi M, Bar‐Or A, Piehl F, et al. Multiple sclerosis. Nat Rev Dis Primers. 2018;4:43. [DOI] [PubMed] [Google Scholar]
- 13. Mishra MK, Yong VW. Myeloid cells – targets of medication in multiple sclerosis. Nat Rev Neurol. 2016;12:539‐551. [DOI] [PubMed] [Google Scholar]
- 14. López‐Herrera G, Vargas‐Hernández A, González‐Serrano ME, et al. Bruton’s tyrosine kinase – an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. 2014;95:243‐250. [DOI] [PubMed] [Google Scholar]
- 15. Hendriks RW. Drug discovery: new BTK inhibitor holds promise. Nat Chem Biol. 2011;7:4‐5. [DOI] [PubMed] [Google Scholar]
- 16. Alankus YB, Grenningloh R, Haselmayer P, Bender A, Bruttger J. BTK inhibition prevents inflammatory macrophage differentiation: a potential role in MS. Poster presented at the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), October 10‐12, Berlin, Germany. P557 (2018)
- 17. Caldwell RD, Qiu H, Askew BC, et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 2019;62:7643‐7655. [DOI] [PubMed] [Google Scholar]
- 18. Becker A, Martin EC, Mitchell DY, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration‐QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin Transl Sci. 2020;13:325‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380:2406‐2417. [DOI] [PubMed] [Google Scholar]
- 20. Montalban X, Arnold DL, Weber MS, et al. Clinical relapse rates in relapsing MS patients treated with the BTK inhibitor evobrutinib: results of an open‐label extension to a Phase II study. Poster presented at MS Virtual 2020: 8th Joint ACTRIMS–ECTRIMS Meeting, October 9‐12. P0197 (2020).
- 21. Li Z, Zhang L, Yuan Y, Yang Z. Identification of metabolites of evobrutinib in rat and human hepatocytes by using ultra‐high performance liquid chromatography coupled with diode array detector and Q Exactive Orbitrap tandem mass spectrometry. Drug Test Anal. 2019;11:129‐139. [DOI] [PubMed] [Google Scholar]
- 22. International Commission on Radiological Protection . The 2007 recommendations of the International Commission on Radiological Protection. Annals ICRP. 2007;103:1‐332. [DOI] [PubMed] [Google Scholar]
- 23. Scheers E, Leclercq L, de Jong J et al. Absorption, metabolism, and excretion of oral (1)(4)C radiolabeled ibrutinib: an open‐label, phase I, single‐dose study in healthy men. Drug Metab Dispos. 2015;43:289‐297. [DOI] [PubMed] [Google Scholar]
- 24. Haselmayer P, Camps M, Liu‐Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202:2888‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boschert U, Crandall T, Pereira A, et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by evobrutinib (M2951) a novel Bruton’s tyrosine kinase inhibitor. Mult Scler J. 2017;23:327 (Abstract P678). [Google Scholar]
- 26. Meng J, Liu X‐Y, Ma S, et al. Metabolism and disposition of pyrotinib in healthy male volunteers: covalent binding with human plasma protein. Acta Pharmacol Sin. 2019;40:980‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickinson PA, Cantarini MV, Collier J, et al. Metabolic disposition of osimertinib in rats, dogs, and humans: insights into a drug designed to bind covalently to a cysteine residue of epidermal growth factor receptor. Drug Metab Dispos. 2016;44:1201‐1212. [DOI] [PubMed] [Google Scholar]
- 28. Chandrasekaran A, Shen L, Lockhead S, Oganesian A, Wang J, Scatina J. Reversible covalent binding of neratinib to human serum albumin in vitro. Drug Metab Lett. 2010;4:220‐227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material