

Abstract
Following a decision to require label warnings for concurrent use of opioids and benzodiazepines and increased risk of respiratory depression and death, the US Food and Drug Administratioin (FDA) recognized that other sedative psychotropic drugs may be substituted for benzodiazepines and be used concurrently with opioids. In some cases, data on the ability of these alternatives to depress respiration alone or in conjunction with an opioid are lacking. A nonclinical in vivo model was developed that could detect worsening respiratory depression when a benzodiazepine (diazepam) was used in combination with an opioid (oxycodone) compared to the opioid alone based on an increased arterial partial pressure of carbon dioxide (pCO2). The current study used that model to assess the impact on respiration of non‐benzodiazepine sedative psychotropic drugs representative of different drug classes (clozapine, quetiapine, risperidone, zolpidem, trazodone, carisoprodol, cyclobenzaprine, mirtazapine, topiramate, paroxetine, duloxetine, ramelteon, and suvorexant) administered alone and with oxycodone. At clinically relevant exposures, paroxetine, trazodone, and quetiapine given with oxycodone significantly increased pCO2 above the oxycodone effect. Analyses indicated that most pCO2 interaction effects were due to pharmacokinetic interactions resulting in increased oxycodone exposure. Increased pCO2 recorded with oxycodone‐paroxetine co‐administration exceeded expected effects from only drug exposure suggesting another mechanism for the increased pharmacodynamic response. This study identified drug‐drug interaction effects depressing respiration in an animal model when quetiapine or paroxetine were co‐administered with oxycodone. Clinical pharmacodynamic drug interaction studies are being conducted with these drugs to assess translatability of these findings.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Benzodiazepines exacerbate opioid‐induced respiratory depression. The impact of other sedative psychotropic drugs on respiration alone or co‐administered with an opioid is often unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study addressed whether a rat model could detect increased partial pressure of carbon dioxide, indicative of respiratory depression, for sedative psychotropic drugs with previously unknown influence on respiration, alone or in combination with an opioid.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study suggests that drug‐drug interactions between designated sedative psychotropic drugs and opioids could exacerbate opioid‐induced respiratory depression and that additional clinical studies would be informative.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Dependent upon outcomes in clinical studies, this work could define a model for early screening of drug‐drug interactions and respiratory depression risk as well as inform the need for additional clinical interaction studies with respiration as the end point.
INTRODUCTION
More than 10 million people age 12 years and older misused opioids in the United States in 2018. 1 In 2018, 46,802 overdose deaths in the United States involved opioids, and opioids were involved in 69.5% of all drug overdose deaths during that year. 2 Co‐use of sedative‐hypnotics, including benzodiazepines, was found in about 30% of long‐term opioid users. 3 Of the 46,802 opioid overdose deaths in 2018, 23% or 10,724 deaths were associated with concomitant use of a benzodiazepine. 4 In response to a citizen petition, 5 the US Food and Drug Administration (FDA) conducted an extensive review of the scientific evidence about the risk and in August 2016 required the addition of boxed warnings to labeling of opioids and benzodiazepines—including generic versions, ~ 400 drugs—with information about risks associated with their concurrent use, including respiratory depression and death. 6
Although the boxed warnings focused on the risks of concurrent benzodiazepine and opioid use, there were concerns that the addition of these boxed warnings could lead to other drugs being used concurrently with opioids in place of benzodiazepines. The risk of respiratory depression with these potential combinations is unknown for many of these drugs. These drugs were termed sedative psychotropic drugs (SPDs) for this investigation, and a multiple‐step process was developed to address this question for drugs that could be used in place of benzodiazepines (e.g., for the treatment of insomnia, anxiety disorders, or other neuropsychiatric conditions) and that frequently cause sedation in patients. First, a review of SPDs was performed to identify drugs that have inadequate information on their effects on respiratory depression when combined with an opioid. Second, an animal model was developed to study respiratory depression induced by orally administered drugs or combinations of drugs. 7 Third, a clinical study 8 was designed to study the drugs identified as having a positive signal from the animal studies reported here.
A rat model to detect respiratory depression induced by oral administration of oxycodone and diazepam individually or in combination has been described. 7 At clinically relevant exposures, diazepam alone was shown to have no effect on respiration but to exacerbate opioid‐induced respiratory depression when co‐administered with oxycodone. A pharmacokinetic (PK) interaction was observed, with diazepam increasing oxycodone plasma concentration. However, the observed increase in partial pressure of carbon dioxide (pCO2) could not be solely explained by this increase in plasma concentration, suggesting a potential direct pharmacodynamic interaction. Using this rat model, the present study proposed to assess the respiratory effects of 13 additional SPDs, administered alone and in combination with oxycodone. The tested drugs included atypical antipsychotics, a selective serotonin reuptake inhibitor (SSRI), skeletal muscle relaxants, a melatonin receptor agonist, an orexin receptor antagonist, an anticonvulsant, a serotonin receptor antagonist and reuptake inhibitor, a tetracyclic antidepressant, a non‐benzodiazepine sedative/hypnotic, and a serotonin and norepinephrine reuptake inhibitor.
METHODS
Selection of drugs for testing
A wide net was cast for drugs that might be used in a psychiatric or neurological treatment regimen that have a primary effect in the central nervous system, that might be used in place of a benzodiazepine (on or off‐label) and that may have respiratory effects. All drugs were being marketed in the United States as of February 2017. Table 1 shows the selected drugs for investigation. Details of the selection process are provided in Supplementary File pages 2–25.
TABLE 1.
Drug groups and index drugs selected for studies in a rat model
| Class | Drug(s) |
|---|---|
| Atypical antipsychotic | Clozapine, Quetiapine, Risperidone |
| Benzodiazepine | Diazepam (positive control) |
| Imidazopyridine | Zolpidem |
| SARI | Trazodone |
| Skeletal muscle relaxant | Carisoprodol, Cyclobenzaprine |
| Tetracyclic antidepressant | Mirtazapine |
| Anticonvulsant | Topiramate |
| SSRI | Paroxetine |
| SNRI | Duloxetine |
| Melatonin receptor agonist | Ramelteon |
| Orexin receptor agonist | Suvorexant |
Abbreviations: SARI, serotonin receptor antagonist and reuptake inhibitor; SNRI, serotonin and norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor.
Animals
Adult male Sprague‐Dawley rats with a surgically pre‐implanted intra‐femoral‐artery cannula (Taconic Biosciences) were used in this study. Animal procedures were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited facility in accordance with the Guide for the Care and Use of Laboratory Animals 9 under an animal study protocol approved by the FDA’s White Oak Animal Care and Use Committee. Cannula care, husbandry, and other animal care details were previously described in detail. 7
Single drug studies
Three doses of each of the SPDs were tested to determine blood drug concentrations and drug effects on pCO2 and pO2. The three single drug doses were selected based on: (1) human dose equivalent according to the FDA conversion guidance, 10 (2) concentrations cited in previous studies in rats, 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 and (3) recorded lethal dose (mg/kg) in 50% of a test population (LD50) in rats where available. In most cases, the human dose equivalent was the approximate low dose, commonly cited nontoxic literature concentrations were used as the middle dose, and the high dose was chosen to stay below the LD50 to avoid severe toxicity. Selected oral dosing solution formulations and analytical methods for each drug are described in Supplementary File page 26. For single drug studies, arterial blood samples were collected prior to and at 15, 30, 60, 120, 180, 240, 360, and 480 min after oral dosing. Six rats were dosed at each concentration and serial arterial blood samples were taken at the times mentioned above. At each time point, SPD plasma concentrations were measured as well as pharmacodynamic (PD) effects, arterial pCO2 and pO2. Analysis of samples allowed subsequent identification of PK parameters to help determine dose and dose scheduling for combined drug experiments.
Combination studies
For each SPD, oxycodone 150 mg/kg was given with the SPD, at a dose based on results of the single drug studies; the oxycodone dose selection was previously described. 7 The relative timing for dosing of oxycodone and the SPD was also based on single drug studies and was established with the goal of having both drugs approach maximal plasma concentration (C max) at approximately the same time. In the combination studies, arterial blood samples were collected prior to and at 15, 30, 60, 120, and 180 min after dosing of the second drug in the combination treatment. In the oxycodone alone control arm, arterial blood samples were collected prior to and at 15, 30, 60, 120, and 180 min after oxycodone dosing. Time points beyond 180 min of combined drug exposure were not considered necessary to see the maximum respiratory effect in combined studies. At each time point, SPD and/or oxycodone serum concentrations were measured. For combination studies of SPD drug with oxycodone and oxycodone alone, there were 6 or 12 rats for each group; some experiments with equivocal data were repeated and data combined for analysis. Blood sampling and blood gas analysis procedures were previously described in detail. 7
Plasma drug concentration measurements
The plasma concentrations of all drugs were measured by validated liquid chromatographic tandem mass spectrometric methods. A few of these methods have previously been published. 29 , 30 The remaining method details are summarized in Supplementary File 0001 pages 27–32.
End points and statistical methods
For single drug and combination drug studies, plasma drug concentration was measured for each drug at every timepoint. These measures from single drug studies were used to estimate C max and the time at which C max is reached (T max). Single drug study C max and T max determined the timing of the combined study dosing. These PK parameters were calculated by noncompartmental analysis using Phoenix software.
Both pCO2 and pO2 were measured at each timepoint, since respiratory depression can cause increased pCO2 and decreased pO2. The normal range of arterial pCO2 in humans and rats is between 35 and 45 mm of mercury (mmHg). The normal range for pO2 is between 75 and 100 mmHg. Although both parameters reflect changes in respiration, pCO2 is a more sensitive measure of decreased respiration because it is inversely proportional to the alveolar ventilation rate. 31 Therefore, pCO2 was selected as the primary PD end point for this study. Because statistical significance could be established for changes that might be of irrelevant magnitude, a threshold of at least 10% change from baseline or control was set as relevant change. This value was based on the narrow normal range of pCO2 (35–45), with 10% change equal to an approximate 40% shift within the normal range or possibly excursion outside of the normal range.
In single drug studies, one‐way analysis of variance (ANOVA) was used to assess whether pCO2 and drug concentrations were significantly different among the three dose groups (data not shown). A two‐way ANOVA (treatment and time) was used to determine statistically significant differences in pCO2 and change from baseline pCO2 between oxycodone alone and oxycodone‐SPD combinations. A t‐test was used to compare the maximum pCO2 observed in combination experiments with the maximum pCO2 measurement from the corresponding oxycodone control group (regardless of timepoint). Significance level for all analyses was p less than 0.05. Area under the curve (AUC) calculations, AUC0‐tlast, were used to compare percent change in systemic exposures in single drug versus combination studies. For five scenarios (diazepam, paroxetine, quetiapine, ramelteon, and trazodone) in which the SPD with oxycodone showed an increase in pCO2 compared to oxycodone alone, a linear‐mixed effect analysis was performed with all available data from the SPD alone, oxycodone alone (including oxycodone alone arms from different SPD experiments), and the two drugs together. Fixed effects were included for oxycodone concentration, SPD concentration, an interaction between oxycodone and SPD concentration, SPD on the intercept, and baseline pCO2. Random effects by subject were included on all intercept and concentration fixed effects (individual and interaction slopes). The purpose of this assessment was to approximately determine the contribution of oxycodone to the observed changes in pCO2 compared to the SPD alone, as co‐administration altered exposures of oxycodone and the SPD in some cases. The contribution of oxycodone to the overall effect in these cases was calculated as the model‐predicted effect on pCO2 at mean oxycodone C max divided by the total effect on pCO2 at mean C max for oxycodone and the SPD. Data, data dictionary, and code are provided in [Link], [Link], [Link].
RESULTS
Single drug studies
The different doses administered for each SPD yielded distinguishable dose‐dependent concentration curves. Calculated PK parameters for all SPDs and single drug study pO2 and pCO2 measures are presented on Supplementary File 0001 pages 37–64. These measures show that some SPDs had a statistically significant increase of 10% or greater in pCO2 from baseline value at all doses and, for others, no significant increases were observed at any dose. A significant increase from baseline in resting arterial pCO2 at all three doses was observed with carisoprodol, duloxetine, and paroxetine. Zolpidem had significant increases in pCO2 observed at the middle and high doses and, for trazodone and clozapine, significant increases were noted only at the high dose. The middle dose of suvorexant showed a significant increase in arterial pCO2. Despite a large increase in pCO2 from baseline with the high dose of suvorexant, the findings were of high variability and nonsignificant. Although the middle dose of zolpidem demonstrated a significant increase in pCO2, this increase did not reach the predetermined 10% change to be considered relevant. A significant increase in arterial pCO2 was seen at the low dose of cyclobenzaprine, but not at the middle or high doses.
Based on these single drug study PD results and review of doses previously reported in the literature, the middle dose of each SPD, except for topiramate, was selected for initial use in combination studies with oxycodone. The low dose was selected for topiramate because the low dose had no PD effect, whereas the middle and high doses of topiramate decreased pCO2. Oxycodone, for combination and control experiments, was dosed at 150 mg/kg as described in a previous study with the benzodiazepine, diazepam. 7 The T max in single drug studies varied between 15 and 360 min. The T max for oxycodone and each SPD, and the combination dosing sequences selected using these data, are presented in Supplementary File page 33.
Combination studies
Plasma concentrations of oxycodone and SPDs when administered alone and in combination were compared. This information is presented for all study drugs in Supplementary File pages 33–36. Table 2 summarizes percentage changes in exposure and response between SPD and oxycodone administered alone and in combination. Most drug combinations resulted in a larger oxycodone AUC value and a lower SPD AUC value with the exceptions of zolpidem, clozapine, and cyclobenzaprine. When cyclobenzaprine and oxycodone were co‐administered, both drugs had larger C max and AUC values. For zolpidem and clozapine, co‐administration with oxycodone resulted in lower values for both the SPD and oxycodone.
TABLE 2.
Differences in mean PK and PD measures in combined studies in relation to single study measures
| Drug given with oxycodone 150 mg/kg | Percentage difference in AUC | Percentage difference in C max | Percentage difference in pCO2 | Difference in pCO2 change from baseline | ||
|---|---|---|---|---|---|---|
| Oxycodone | SPD | Oxycodone | SPD | Combined versus oxycodone alone | Combined versus oxycodone alone (mmHg) | |
| Paroxetine 50 mg/kg | 357 | −53 | 520 | −49 | 49* | 13.2# |
| Paroxetine 5 mg/kg | 58 | −75 | 38 | −75 | 16* | 5.1# |
| Risperidone 10 mg/kg | 23 | −23 | −12 | −15 | −4 | −2.9 |
| Cyclobenzaprine 30 mg/kg | 99 | 30 | 70 | 45 | 7 | 2.2# |
| Mirtazapine 50 mg/kg | 20 | −14 | 0 | −20 | 2 | −3.4# |
| Zolpidem 50 mg/kg | −12 | −52 | −26 | −56 | 7 | 3.2 |
| Duloxetine 50 mg/kg | 210 | −42 | 136 | −30 | 4 | 0.2 |
| Clozapine 25 mg/kg | −10 | −84 | −20 | −85 | −13§ | −0.6 |
| Quetiapine 250 mg/kg | 680 | −62 | 1004 | −55 | 53* | 12.3# |
| Quetiapine 25 mg/kg | 81 | −92 | 92 | −85 | −1 | 0.1 |
| Trazodone 100 mg/kg | 43 | −78 | 1 | −82 | 13* | 3.8# |
| Topiramate 20 mg/kg | 68 | −73 | 64 | −72 | 4 | 4.0# |
| Carisoprodol 50 mg/kg | 75 | −14 | 173 | 77 | 3 | 0.3 |
| Ramelteon 30 mg/kg | 88 | −57 | 45 | −43 | 11* | 9.8# |
| Suvorexant 60 mg/kg | 24 | −66 | −4 | −60 | 0 | 2.4 |
Abbreviations: AUC, area under the curve; Cmax, maximum plasma concentration; PD, pharmacodynamic; PK, pharmacokinetic; pCO2, partial pressure of carbon dioxide; SPD, sedative psychotropic drug.
Significant differences between single drug and combination drug values were determined by 2‐way ANOVA. *Significant increase (p < 0.05) in pCO2 of > 10%; §significant decrease (p < 0.05) in pCO2 of > 10%; changes of less than 10% were not considered relevant regardless of p‐value; #significant difference (p < 0.05) in pCO2 change from baseline.
Figure 1 is a depiction of the pCO2 profile of the combination studies for paroxetine (a) and quetiapine (b), demonstrating an increase in arterial pCO2 with the combination of each drug with oxycodone compared to oxycodone alone. Each combined dosing experiment was performed with an oxycodone only control group. Similar graphs were created for each combination experiment and are available in Supplementary File 0001 pages 37−64. Clozapine was the only SPD that, when co‐administered with oxycodone, resulted in significantly lower pCO2 across the experimental timeline (ANOVA) compared to oxycodone given alone. Significantly higher pCO2 between the combination versus oxycodone alone (two‐way ANOVA) and at least a 10% maximum difference was noted with trazodone 100 mg/kg, ramelteon 30 mg/kg, higher dose quetiapine (250 mg/kg), and both the higher (50 mg/kg) and lower (5 mg/kg) dose paroxetine compared to oxycodone given alone.
FIGURE 1.
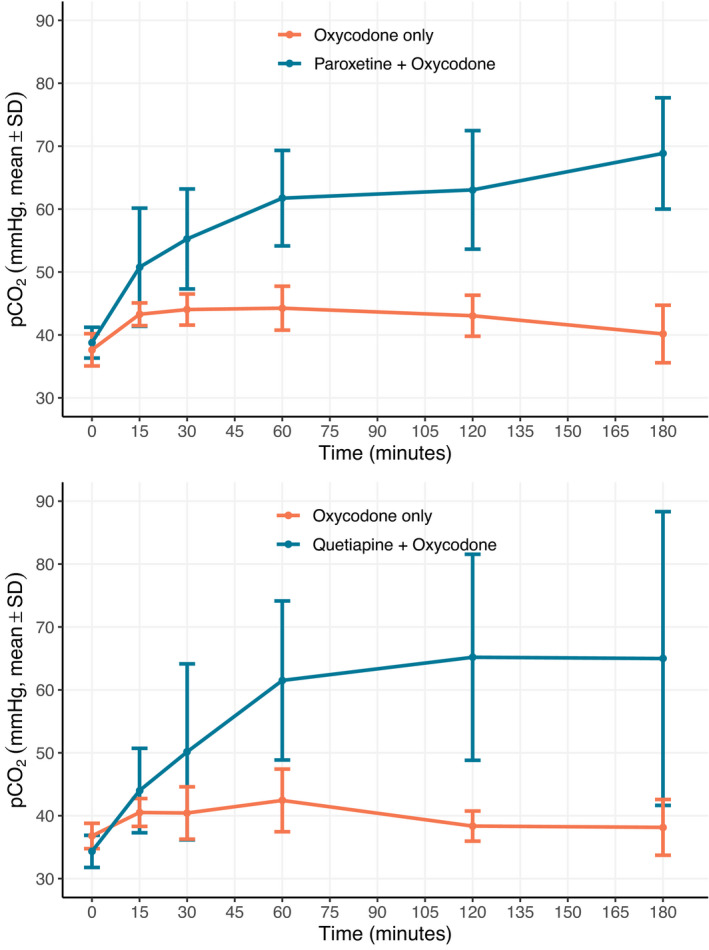
Comparison of arterial partial pressure of carbon dioxide (pCO2). Changes between oxycodone alone and paroxetine (a) or Quetiapine (b) co‐administered with oxycodone. Plots of mean pCO2 at each timepoint following administration shows a significant increase in pCO2 with co‐administration of oxycodone (150 mg/kg) with (a) paroxetine (50 mg/kg) and (b) quetiapine (250 mg/kg) compared to oxycodone alone (150 mg/kg). Values are mean ± SD; significant difference (p < 0.05) was determined by analysis of variance across the entire experimental times series for both paroxetine and quetiapine; n = 6 per experimental group. Paroxetine was given 3 h prior to time zero when oxycodone was administered. Quetiapine and oxycodone were given concurrently
Exposure‐response analyses
To further explore these drug‐drug interactions, linear‐mixed effect analyses (Figure 2) were evaluated for oxycodone and paroxetine, oxycodone and quetiapine, oxycodone and trazodone, and oxycodone and ramelteon, as well as oxycodone alone (oxycodone effect by itself) and oxycodone with diazepam (previously demonstrated combination effect). These results indicated significant slopes for oxycodone alone and in the combination analyses. The oxycodone slope was consistent across all cases and across all SPD studies, with an estimated slope ranging between 0.075 ando 0.088 mmHg*ml/ng oxycodone. Diazepam, paroxetine, quetiapine, and trazadone had significant positive slope parameters. The interaction slope parameters were positive for oxycodone with diazepam, with paroxetine, and with trazodone, whereas they were negative for oxycodone with quetiapine and with ramelteon.
FIGURE 2.
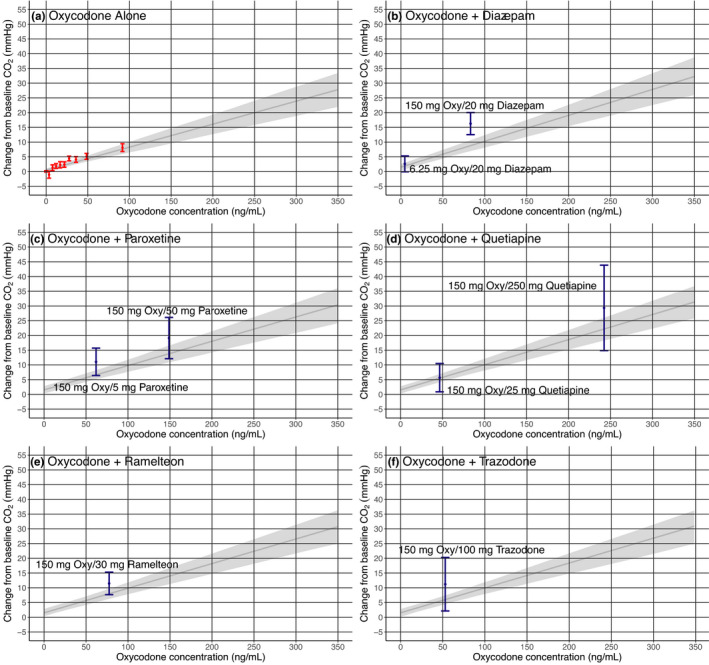
Effects of oxycodone (Oxy) alone (a) versus combined effects of oxycodone with diazepam (b), paroxetine (c), quetiapine (d), ramelteon (e), and trazodone (f) on change from baseline arterial partial pressure of carbon dioxide (pCO2). Shown are the univariate (a) and multivariate (b–f) linear regression results using data from the animal experiments with oxycodone alone, the sedative psychotropic drug (SPD) alone, and the SPD combined with oxycodone. The univariate linear regression for oxycodone (dark gray with 95% confidence interval [light gray]) is shown to display model‐predicted effects on change from baseline arterial pCO2 from oxycodone alone. Multivariate linear regression models were developed for each combination of oxycodone and SPD. The resulting oxycodone relationship from the multivariate linear regression is shown on each plot for comparison with the effects from oxycodone combined with the SPD. Shown in blue are the mean change from baseline arterial pCO2 with 95% confidence for the SPD and oxycodone combination arms (amount administered is labeled on each figure). Each point is represented on the x‐axis based on the geometric maximum concentration of all rats at that dose and/or combination. If the combination treatment is less than or overlaps with the mean effect of oxycodone alone (dark gray), this would suggest oxycodone alone could explain the observed effects on change from baseline arterial pCO2. Likewise, if the combination effect is greater, this would suggest the drug combination is having an additional effect, which could be due to the SPD alone or synergy
With co‐administration of 20 mg/kg diazepam and 150 mg/kg oxycodone, oxycodone C max increased 2.0‐fold over oxycodone alone. Diazepam C max at 20 mg/kg was 53% lower when administered with oxycodone. An approximate contribution of oxycodone to the overall change in pCO2 during co‐administration was calculated based on the estimated linear mixed effect model. For 20 mg/kg diazepam, 50–55% of the observed increase in pCO2 would be predicted based on the observed oxycodone concentration. This analysis supports that, at this dose and resulting exposures, diazepam is having an additive or synergistic effect on pCO2 when combined with oxycodone.
This approach was repeated for the paroxetine, quetiapine, ramelteon, and trazodone results. With co‐administration of 50 mg/kg and 5 mg/kg paroxetine, oxycodone C max increased ~ 6.2‐fold and 1.4‐fold, respectively, over oxycodone alone. Paroxetine C max results at 50 mg/kg and 5 mg/kg were 49% and 75% lower, respectively, when administered with oxycodone compared to when administered alone. For 5 mg/kg paroxetine, almost the entire observed effect on pCO2 was associated with changes in oxycodone concentration. With 50 mg/kg paroxetine, close to 50% of the observed increase in pCO2 was associated with an increased oxycodone concentration (similar to diazepam). Like diazepam, this analysis supports that paroxetine may be having an additive or synergistic effect on pCO2 when combined with oxycodone.
For the quetiapine experiments, co‐administration with 250 mg/kg and 25 mg/kg quetiapine resulted in ~ 11.0‐fold and 1.9‐fold higher oxycodone C max, respectively, compared to oxycodone alone. Quetiapine C max results were 55% and 85% lower when 250 mg/kg and 25 mg/kg quetiapine, respectively, were administered with oxycodone compared to when they were administered alone. Using the same approach as outlined above, the entire observed effect on pCO2 at both concentrations could be explained by the observed oxycodone exposure. Results for quetiapine suggest that the observed combination effect is entirely the result of a PK interaction with oxycodone. Further exploration of the data would be necessary to fully describe the exposure‐pCO2 time course profiles from these experiments.
Co‐administration of trazodone 100 mg/kg with oxycodone had no effect (1% increase) on oxycodone C max and lowered trazodone C max by 82% but resulted in a significant 13% increase in pCO2 compared to oxycodone alone. In ramelteon experiments, co‐administration resulted in a 1.4‐fold increase in oxycodone C max and a 43% decrease in ramelteon C max with a significant 11% increase in pCO2. Using the same estimated linear mixed effect model approach, the oxycodone concentration could explain at least 95% of the observed combined effect with trazodone and at least 65% of the observed combined effect with ramelteon. Notably, ramelteon was an SPD where the individual drug decreased pCO2 (i.e., negative concentration‐slope relationship) so the combined effect may be underestimated.
Assessment of potential clinical relevance
With these studies completed, a more detailed evaluation of the potential clinical relevance of the findings was undertaken. The results of that examination are summarized in Table 3. Human plasma concentration data from literature reports and regulatory submissions to the FDA were evaluated for human exposure levels associated with labeled dosing recommendations. These values were then compared to the maximal plasma concentrations measured in the rat single drug studies, and parallels were drawn to approximate human equivalent exposures.
TABLE 3.
Comparison between experimental rat C max and human C max to assess clinical relevance of findings in rats
| Drug/dose administered to rats | Mean rat C max (ng/ml) | Drug and dosage administered to human | Mean human C max (ng/ml) | Potential clinical relevance category |
|---|---|---|---|---|
| Oxycodone 150 mg/kg | 51.5 | Oxycodone 40 mg extended‐release tablet (single dose) | 48 47 | A |
| Clozapine 25 mg/kg | 65.3 | Clozapine 100 mg tablet nightly (steady‐state) | 275 48 | D |
| Cyclobenzaprine 30 mg/kg | 6.4 | Cyclobenzaprine 15 mg extended‐release capsule (single dose) | 8.3 49 | A |
| Duloxetine 50 mg/kg | 465.1 | Duloxetine 60 mg capsule Q12H (steady‐state) | 128.5 50 | C |
| Mirtazapine 50 mg/kg | 87.5 | Mirtazapine 30 mg tablet nightly (steady‐state) | 69.7 51 | B |
| Paroxetine 5 mg/kg | 54.9 | Paroxetine HCl 30 mg tablet daily (steady‐state) | 61.7 52 | B |
| Paroxetine 50 mg/kg | 1090.0 | Paroxetine HCl 30 mg tablet daily (steady‐state) | 61.7 53 | C |
| Quetiapine 25 mg/kg | 46.3 | Quetiapine 25 mg tablet (single dose) | 79 54 | A |
| Quetiapine 250 mg/kg | 512.3 | Quetiapine 150 mg tablet twice daily (steady‐state) | 445.7 55 | B |
| Ramelteon 30 mg/kg | 768.2 | Ramelteon 8 mg tablet (single dose) | 5.73 56 | C |
| Risperidone 10 mg/kg | 108.5 | Risperidone 8 mg tablet daily (steady‐state) | 155 57 | B |
| Suvorexant 60 mg/kg | 1668.0 | Suvorexant 20 mg tablet nightly (steady‐state) | 258.8 58 | C |
| Topiramate 20 mg/kg | 8830.3 | Topiramate 100 mg tablet Q12H (steady‐state) | 8400 59 | B |
| Trazodone 100 mg/kg | 991.1 | Trazodone 50 mg tablet (single dose) | 755 60 | A |
| Trazodone 100 mg/kg | 991.1 | Trazodone 300 mg extended‐release tablet daily (steady‐state) | 1812 61 | D |
| Zolpidem 50 mg/kg | 1243.4 | Zolpidem 10 mg tablet (single dose) | 111.6 62 | C |
| Carisoprodol 50 mg/kg | 115.3 | Carisoprodol 250 mg tablet (single dose) | 1200 63 | D |
Abbreviation: Cmax, maximum plasma concentration.
Four test drugs were given at doses approximating human single dose exposure (oxycodone, cyclobenzaprine, quetiapine 25 mg/kg, and trazodone). Five test drugs were given at doses approximating human steady‐state exposure (mirtazapine, paroxetine 5 mg/kg, quetiapine 250 mg/kg, risperidone, and topiramate). Experimental doses of duloxetine, paroxetine (50 mg/kg), ramelteon, suvorexant, and zolpidem resulted in rat exposures substantially higher than reported in humans receiving treatment at labeled dosages. Experimental doses of clozapine and carisoprodol resulted in rat exposures substantially lower than reported in humans receiving treatment at labeled dosages.
For the four drugs, which were observed in rats to cause a significant increase in arterial pCO2 in combination with oxycodone as compared to oxycodone alone, ramelteon was assessed to have exposures much larger than relevant to human use, whereas three had exposures potentially relevant to human use:
Paroxetine 5 mg/kg was associated with exposure in rats similar to that humans may experience when taking paroxetine 30 mg daily as a treatment for major depressive disorder, obsessive‐compulsive disorder, panic disorder, post‐traumatic stress disorder, or social anxiety disorder.
Quetiapine 250 mg/kg was associated with exposure in rats similar to that humans may experience when taking quetiapine 150 mg twice daily as a treatment for schizophrenia (labeled recommended dose: 150–750 mg/day).
Trazodone 100 mg/kg was associated with exposure in rats similar to that humans may experience when taking trazodone 50 mg (i.e., as a nonapproved off‐label treatment for insomnia 32 ) but substantially lower than steady‐state achieved by 300 mg of an extended‐release formulation given daily for major depressive disorder.
DISCUSSION
Clinically relevant exposures of paroxetine, quetiapine, and trazodone in rats with oxycodone significantly increased resting arterial pCO2 compared to oxycodone alone. At exposures less than expected clinically, clozapine co‐administered with oxycodone decreased resting arterial pCO2 compared to oxycodone alone. However, this finding is believed to be an experimental artifact. There was a difference in baseline (lower pCO2 for clozapine combination) that was maintained throughout the experiment. When compared as change from baseline there was no difference between the combination and oxycodone alone. Paroxetine and quetiapine pCO2 findings corresponded with increased oxycodone exposure (C max and AUC) when paroxetine and quetiapine were co‐administered. When these SPDs were administered on their own, paroxetine increased pCO2 at all tested doses and quetiapine did not alter pCO2 at any of the tested concentrations. Exposure‐response modeling supported that the respiratory depression signal (i.e., increased pCO2) seen with either quetiapine or trazodone in combination with oxycodone could be explained by the increased oxycodone plasma concentration, whereas the signal seen with paroxetine in combination with oxycodone was largely but not completely due to increased oxycodone plasma concentration. As PK and PD drug‐drug interactions can differ significantly between species, clinical studies will assess the translatability of these findings.
The assessments of potential clinically relevant exposures are a means to guide dosing regimens in future rat studies as well as to generate hypotheses for investigation in clinical studies. In addition to paroxetine, quetiapine, and trazodone, ramelteon was observed to significantly increase arterial pCO2 in combination with oxycodone. Although paroxetine 5 mg/kg, quetiapine 250 mg/kg, and trazodone resulted in potentially relevant clinical exposures, ramelteon experiments were at exposures significantly higher than humans would be expected to experience. Although no changes in pCO2 were observed with co‐administration of clozapine or carisoprodol with oxycodone, clozapine and carisoprodol exposures were well below clinically relevant exposures. Future studies with ramelteon, clozapine, and carisoprodol at more clinically relevant concentrations would better assess their potential for respiratory depression in combination with oxycodone. With all other SPDs evaluated in this study, human relevant or substantially higher than human relevant exposures were obtained without a significant increase in pCO2 of a 10% or greater magnitude.
No clinical or nonclinical literature relevant to respiratory depression was found specifically for the atypical antipsychotic, quetiapine (Supplementary File 0001 pages 5–6). Limited evidence for respiratory depression with typical antipsychotics has been identified in overdose patients. 33 There was no nonclinical data on the ability of trazodone to cause respiratory depression and a single clinical study reporting an increase in the rate of oxygen consumption following trazodone administration (Supplementary File 0001 pages 11–12). Paroxetine also had no clinical or nonclinical literature addressing respiratory depression (Supplementary File 0001 pages 13–14). Another drug in the same class as paroxetine (SSRI), fluoxetine, had nonclinical evidence of respiratory depression when used alone 34 but negative findings in a single clinical study in use with morphine. 35 In the present study, dose‐dependent increase in pCO2 was observed with paroxetine in rats. The study did not identify a mechanism for this PD observation. Whether a similar exacerbation of opioid‐induced respiratory depression would be observed clinically is unknown.
Literature was reviewed on mechanisms that may have accounted for the largest observed interactions (paroxetine, quetiapine, and trazodone). Metabolism of oxycodone is via CYP3A4 and CYP2D6, 36 , 37 and in vitro inhibition of CYP3A4 resulted in increased oxycodone exposure primarily through delayed elimination. 38 Inhibition of CYP2D6 did not influence oxycodone exposures. 39 In humans and rats, paroxetine is also metabolized by these two as well as several other CYP enzymes. 40 Paroxetine is a prototypical CYP2D6 inhibitor 41 used for inhibition studies. Clinically, paroxetine co‐administration has been shown to increase oxycodone exposure but, in that study, paroxetine did not impact the PD end point, analgesia. 42 In another study reporting increased oxycodone exposures with paroxetine co‐administration, CYP2D6 inhibition was deemed of only minor importance in increasing oxycodone exposure. 43 In a study with CYP2D6 extensive metabolizers, paroxetine pretreatment did not increase oxycodone C max or AUC, although it did reduce measured oxycodone pharmacodynamic effects (pupillometry, cold pressor test). 44 Quetiapine and trazodone are also metabolized by CYP3A4 and CYP2D6, but a clear mechanism for interaction of quetiapine or trazodone and oxycodone was not identified in the existing literature.
The results of this study must be viewed in the light of its limitations. Intersubject variability was not consistent across individual experiments. Experiments were not adequately powered in all cases. The oral route, typical of prescribed routes for these drugs, introduces absorption and distribution variabilities avoided by intravenous administration. For the combination experiments, aside from the pretreatment timepoint, the experimental time course was initiated with administration of the second drug whether that was an SPD or oxycodone. In combinations in which oxycodone was given prior to SPD, the timepoints are not truly aligned in terms of duration of oxycodone exposure. This misalignment was endured, attempting to achieve maximum concentrations at near the same time and to have adequate time to monitor for PD effects. Humans differ from animals with regard to isoform composition, expression, and catalytic activities of drug‐metabolizing enzymes, 45 and translatability of PK and PD interactions must be considered in that context. These studies were designed to negatively impact respiratory exchange but not necessarily simulate severe respiratory distress. Drug‐associated increased resting arterial pCO2 suggests that the normal ventilatory response to compensate for increased CO2 is blunted, but we presently do not know how this relates to respiratory distress. Different experimental conditions, such as an environment of higher inhaled CO2, 46 may have been more revealing of the impact these combination drugs have on respiration.
CONCLUSION
This study shows that sedative psychotropic drugs, both alone and when combined with oxycodone, have differing ability to increase resting arterial pCO2 and lower pO2 in a rat model. At clinically relevant exposures, paroxetine, quetiapine, and trazodone administered with oxycodone increased the resting arterial pCO2, which was associated with large increases in oxycodone plasma concentration. Ramelteon co‐administered with oxycodone increased pCO2 at exposures higher than relevant clinical exposures. This study identifies drug‐drug interactions between oxycodone and specific SPDs that could result in exacerbated opioid‐induced respiratory depression. Clinical studies are being conducted to assess the translatability of data from this rodent model.
DISCLAIMER
The conclusions in this article are the opinions of the authors, have not been formally disseminated by the FDA and should not be construed to represent any Agency determination or policy. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the Department of Health and Human Services.
Access to the ToxIC and NPDS datasets were obtained via contracts between the FDA and the respective organizations. The American Association of Poison Control Centers (AAPCC) maintains NPDS, and case records in this database are from self‐reported calls. Exposures do not necessarily represent a poisoning or overdose, and AAPCC is not able to verify the accuracy of every report made to member centers. All data produced from NPDS during the year in which exposures occur is considered preliminary.
FDA DISCLAIMER
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
L.X., R.L. Racz., J.W., D.A.V., J.F., M.K.M., M.S., M.C.D., D.G.S., and R.L. Rouse wrote the manuscript. L.X., J.W., V.P., H.Z., M.S., M.C.D., D.G.S., and R.L. Rouse designed the research. L.X., A.K., S.S., K.S., R.L. Racz., J.W., D.A.V., N.R.P., S.N., J.F., V.P., M.K.M., H.Z., M.C.D., D.G.S., and R.L. Rouse performed the research. L.X., R.L. Racz., J.W., N.R.P., S.N., J.F., M.K.M., and R.L. Rouse analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Naomi Kruhlak and Marlene Kim for performing structural similarity analysis to assist in drug selection; and Dan Mellon for reviewing the manuscript.
Xu L, Krishna A, Stewart S, et al. Effects of sedative psychotropic drugs combined with oxycodone on respiratory depression in the rat. Clin Transl Sci. 2021;14:2208–2219. 10.1111/cts.13080
Funding information
The study was funded by US Food and Drug Administration.
REFERENCES
- 1. SAMSA: key substance use and mental health indicators in the United States: results from the 2018 National Survey on Drug Use and Health. 2020. https://www.samhsa.gov/data/sites/default/files/cbhsq‐reports/NSDUHNationalFindingsReport2018/NSDUHNationalFindingsReport2018.pdf. Accessed March 17, 2020.
- 2. Hedegaard H, Miniño AM, Warner M. Drug overdose deaths in the United States, 1999–2018. NCHS Data Brief, no 356. Hyattsville, MD: National Center for Health Statistics; 2020.
- 3. Boudreau D, Von Korff M, Rutter CM, et al. Trends in long‐term opioid therapy for chronic non‐cancer pain. Pharmacoepidemiol Drug Saf. 2009;18:1166‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. NIDA: overdose death rates (attached supplemental document: drug overdoses data document), revised March 2020. National Institute of Drug Abuse. 2020. https://www.drugabuse.gov/related‐topics/trends‐statistics/overdose‐death‐rates. Accessed March 17, 2020.
- 5. Baltimore Commissioner of Health , Citizens petition. 2016. http://health.baltimorecity.gov/sites/default/files/Final%20Draft%20FDA%20petition‐Full%20Co‐Signers‐2.19.16%20(2)%20(1).pdf. Accessed March 12, 2020.
- 6. FDA 2016, New safety measures announced for opioid analgesics, prescription opioid cough products, and benzodiazepines. https://www.fda.gov/drugs/information‐drug‐class/new‐safety‐measures‐announced‐opioid‐analgesics‐prescription‐opioid‐cough‐products‐and. Accessed March 18, 2020.
- 7. Xu L, Chockalingam A, Stewart S, et al. Developing an animal model to detect drug‐drug interactions impacting drug‐induced respiratory depression. Toxicol Rep. 2020;7:188‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. National Institutes of Health (NIH), US National Library of Medicine . ClinicalTrials.gov. Clinical study to investigate the effect of the combination of psychotropic drugs and an opioid on ventilation. https://clinicaltrials.gov/ct2/show/NCT04310579. Accessed May 15, 2020.
- 9. Guide for the Care and Use of Laboratory Animals, 8th ed. 2011. https://grants.nih.gov/grants/olaw/guide‐for‐the‐care‐and‐use‐of‐laboratory‐animals.pdf. Accessed March 12, 2020. [Google Scholar]
- 10. FDA, Guidance for Industry . Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. 2005. https://www.fda.gov/media/72309/download. Accessed March 17, 2020.
- 11. Batra VR, Schrott LM. Acute oxycodone induces the pro‐emetic pica response in rats. J Pharmacol Exp Ther. 2011;339(3):738‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merchant KM, Dobner PR, Dorsa DM. Differential effects of haloperidol and clozapine on neurotensin gene transcription in rat neostriatum. J Neurosci. 1992;12(2):652‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao XM, Hashimoto T, Cooper TB, Tamminga CA. The dose‐response characteristics of rat oral dyskinesias with chronic haloperidol or clozapine administration. J Neural Transm. 1997;104:97‐104. [DOI] [PubMed] [Google Scholar]
- 14. Sun L, Lau CE. Intravenous and oral clozapine pharmacokinetics, pharmacodynamics, and concentration‐effect relations: acute tolerance. Eur J Pharmacol. 2000;398(2):225‐238. [DOI] [PubMed] [Google Scholar]
- 15. Invernizzi R, Morali F, Pozzi L, Samanin R. Effects of acute and chronic clozapine on dopamine release and metabolism in the striatum and nucleus accumbens of conscious rats. Br J Pharmacol. 1990;100(4):774‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ezzeldin E, Asiri YA, Iqbal M. Effects of green tea extracts on the pharmacokinetics of quetiapine in rats. Evid Based Complement Alternat Med. 2015;2015:615285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aravagiri M, Marder SR. Brain, plasma and tissue pharmacokinetics of risperidone and 9‐hydroxyrisperidone after separate oral administration to rats. Psychopharmacology. 2002;159(4):424‐431. [DOI] [PubMed] [Google Scholar]
- 18. Diaz‐Garcia JM, Oliver‐Botana J, Galve DF. Pharmacokinetics of diazepam in the rat: influence of an experimentally induced hepatic injury. Eur J Drug Metab Pharmacokinet. 1991;3:94‐101. [PubMed] [Google Scholar]
- 19. Brisbare‐Roch C, Dingemanse J, Koberstein R, et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13:150‐155. [DOI] [PubMed] [Google Scholar]
- 20. Me Abdel Salam O, Sleem AA, Shafee N. Effect of trazodone and nefazodone on hepatic injury induced by carbon tetrachloride. Drug Discov Ther. 2010;4(4):285‐297. [PubMed] [Google Scholar]
- 21. Gonzalez LA, Gatch MB, Taylor CM, Bell‐Horner CL, Forster MJ, Dillon GH. Carisoprodol‐mediated modulation of GABAA receptors. In vitro and in vivo studies. J Pharmacol Exp Ther. 2009;329(2):827‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flexeril® (Cyclobenzaprine HCl) Tablets – FDA NDA 17‐821/S‐045. https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/017821s045lbl.pdf. Accessed May 15, 2020.
- 23. Lightowler S, Kennett GA, Williamson IJ, Blackburn TP, Tulloch IF. Anxiolytic‐like effect of paroxetine in a rat social interaction test. Pharmacol Biochem Behav. 1994;49(2):281‐285. [DOI] [PubMed] [Google Scholar]
- 24. Rouini MR, Lavasani H, Sheikholeslami B, Owen H, Giorgi M. Pharmacokinetics of mirtazapine and its main metabolites after single intravenous and oral administrations in rats at two dose rates. DARU J Pharmaceut Sci. 2014;22(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wauquier A, Zhou S. Topiramate: a potent anticonvulsant in the amygdala‐kindled rat. Epilepsy Res. 1996;24(2):73‐77. [DOI] [PubMed] [Google Scholar]
- 26. Iyengar S, Webster AA, Hemrick‐Luecke SK, Xu JY, Simmons RMA. Efficacy of duloxetine, a potent and balanced serotonin‐norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther. 2004;311(2):576‐584. [DOI] [PubMed] [Google Scholar]
- 27. Hirai K, Kita M, Ohta H, et al. Ramelteon (TAK‐375) accelerates reentrainment of circadian rhythm after a phase advance of the light‐dark cycle in rats. J Biol Rhythms. 2005;20(1):27‐37. [DOI] [PubMed] [Google Scholar]
- 28. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant‐a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1–2):52‐61. [DOI] [PubMed] [Google Scholar]
- 29. Pilli NR, Narayanasamy S, Lin XU, et al. A high‐throughput bioanalytical assay to support pharmacokinetic interaction study of oxycodone and diazepam in Sprague Dawley rats. RSC Adv. 2020;10:886‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayanasamy S, Pilli NR, Lin XU, et al. An alternating polarity switching assay for quantification of oxycodone and topiramate: An application of LC‐MS/MS method in support to PK/PD study in rodents. J Chromatogr B. 2019;1119:93‐100. [DOI] [PubMed] [Google Scholar]
- 31. Pandya NK, Sharma S. Capnography and pulse oximetry. NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health. Treasure Island, FL: StatPearls Publishing; 2020. [Google Scholar]
- 32. Jaffer KY, Chang T, Vanle B, et al. Tradozone for insomnia: a systematic review. Innov Clin Neurosci. 2017;14(7–8):24‐34. [PMC free article] [PubMed] [Google Scholar]
- 33. Ciranni MA, Kearney TE, Olson KR. Comparing acute toxicity of first‐ and second‐generation antipsychotic drugs: a 10‐year, retrospective study. J Clin Psychiat. 2009;70:122‐129. [DOI] [PubMed] [Google Scholar]
- 34. Henderson DR, Konkle DM, Mitchell GS. Effects of serotonin re‐uptake inhibition on ventilator control in goats. Resp Physiol. 1999;115:1‐10. [DOI] [PubMed] [Google Scholar]
- 35. Erjavec MK, Coda BA, Nguyen Q, Donaldson G, Risler L, Shen DD. Morphine‐fluoxetine interactions in healthy volunteers: analgesia and side effects. J Clin Pharmacol. 2000;40:1286‐1295. [PubMed] [Google Scholar]
- 36. Lalovic B, Phillips B, Risler LL, Howald W, Shen DD. Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos. 2004;32(4):447‐454. [DOI] [PubMed] [Google Scholar]
- 37. Andreassen TN, Klepstad P, Davies A, et al. Influences on the pharmacokinetics of oxycodone: a multicentre cross‐sectional study in 439 adult cancer patients. Eur J Clin Pharmacol. 2011;67(5):493‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saari TI, Grönlund J, Hagelberg NM, et al. Effects of itraconazole on the pharmacokinetics and pharmacodynamics of intravenously and orally administered oxycodone. Eur J Clin Pharmacol. 2010;66(4):387‐397. [DOI] [PubMed] [Google Scholar]
- 39. Heiskanen T, Olkkola KT, Kalso E. Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Clin Pharmacol Ther. 1998;64(6):603‐611. [DOI] [PubMed] [Google Scholar]
- 40. Jornil J, Jensen KG, Larsen K, Linnet K. Identification of cytochrome P450 isoforms involved in the metabolism of paroxetine and estimation of their importance for human paroxetine metabolism using a population‐based simulator. Drug Metab Dispos. 2010;38(3):376‐385. [DOI] [PubMed] [Google Scholar]
- 41. VandenBrink BM, Foti RS, Rock DA, Wienkers LC, Wahlstrom JL. Prediction of CYP2D6 Drug interactions from in vitro data: evidence for substrate‐dependent inhibition. Drug Metab Dispos. 2012;40(1):47‐53. [DOI] [PubMed] [Google Scholar]
- 42. Lemberg KK, Heiskanen TE, Neuvonen M, et al. Does co‐administration of paroxetine change oxycodone analgesia: an interaction study in chronic pain patients. Scand J Pain. 2010;1(1):24‐33. [DOI] [PubMed] [Google Scholar]
- 43. Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Olkkola KT, Laine K. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70(1):78‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kummer O, Hammann F, Moser C, Schaller O, Drewe J, Krähenbühl S. Effect of the inhibition of CYP3A4 or CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Eur J Clin Pharmacol. 2011;67:63‐71. [DOI] [PubMed] [Google Scholar]
- 45. Martignoni M, Groothuis GMM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP‐mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875‐894. [DOI] [PubMed] [Google Scholar]
- 46. Read DJ. A clinical method for assessing the ventilatory response to carbon dioxide. Australas Ann Med. 1967;16(1):20‐32. [DOI] [PubMed] [Google Scholar]
- 47. NDA 22272 Clinical Pharmacology/Biopharmaceutics Review, Table 2.3 (page 28). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022272s000ClinPharmR.pdf. Accessed May 15, 2020.
- 48. Glue P, Gale C, Menkes DB, Hung N. Evaluation of bioequivalence between clozapine suspension and tablet formulations. Clin Drug Invest. 2012;32(11):723‐727. [DOI] [PubMed] [Google Scholar]
- 49. NDA 21777 clinical pharmacology/biopharmaceutics review, Table 1 (page 8). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/021777s000_ClinPharmR.pdf. Accessed May 15, 2020.
- 50. NDA 21427 clinical pharmacology/biopharmaceutics review, Table 39 (page 2). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021427_s000_Cymbalta_BioPharmr_P2.pdf. Accessed May 15, 2020.
- 51. Spaans E, van den Heuvel MW, Schnabel PG, et al. Concomitant use of mirtazapine and phenytoin: a drug–drug interaction study in healthy male subjects. Eur J Clin Pharmacol. 2002;58(6):423‐429. [DOI] [PubMed] [Google Scholar]
- 52. NDA 21299 clinical pharmacology/biopharmaceutics review, Table 5 (page 22). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21‐299.pdf_Paroxetine%20Mesylate_BioPharmr.pdf. Accessed May 15, 2020.
- 53. NDA 21299 clinical pharmacology/biopharmaceutics review, Table 5 (page 22). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21‐299.pdf_Paroxetine%20Mesylate_BioPharmr.pdf. Accessed May 15, 2020.
- 54. Thyrum PT, Wong YW, Yeh C. Single‐dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521‐533. [DOI] [PubMed] [Google Scholar]
- 55. NDA 20639 S016 and S017 clinical pharmacology/biopharmaceutics review, Table 1 (page 313). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/020639_S016&S017_SEROQUEL%20TABLETS.pdf. Accessed May 15, 2020.
- 56. Karim A, Tolbert D, Cao C. Disposition kinetics and tolerance of escalating single doses of ramelteon, a high‐affinity MT1 and MT2 melatonin receptor agonist indicated for treatment of insomnia. J Clin Pharmacol. 2006;46(2):140‐148. [DOI] [PubMed] [Google Scholar]
- 57. NDA 20272 Clinical Pharmacology/Biopharmaceutics Review, Table 9 (page 11). https://www.accessdata.fda.gov/drugsatfda_docs/nda/97/20‐272S007_Biopharm%20Review%20Rwperdal_BioPharmr_P1.pdf. Accessed May 15, 2020.
- 58. NDA 204569 Clinical Pharmacology/Biopharmaceutics Review, page 58. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204569Orig1s000ClinPharmR.pdf. Accessed May 15, 2020.
- 59. Bialer M, Shekh‐Ahmad T, Braun TL, Halvorsen MB. Comparative steady‐state pharmacokinetic evaluation of immediate‐release topiramate and USL 255, a once‐daily extended‐release topiramate formulation. Epilepsia. 2013;54(8):1444‐1452. [DOI] [PubMed] [Google Scholar]
- 60. Gammans RE, Mackenthun AV, Russell JW. Comparative bioavailability of trazodone formulations using stable isotope methodology. Br J Clin Pharmacol. 1984;18(3):431‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oleptro (trazodone hydrochloride) extended‐release tablets. Full Prescribing Information, Section 12.3 (Pharmacokinetics). https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022411s008lbl.pdf. Accessed May 15, 2020.
- 62. NDA 19908 clinical pharmacology/biopharmaceutics review, page 11. https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/019908_S000_BIOR.pdf. Accessed May 15, 2020.
- 63. NDA 11792 Clinical Pharmacology Review, Table 1 (page 3; page 337 of PDF), https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/011792Orig1s041.pdf. Accessed May 15, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material