

Abstract
Perineural invasion (PNI), a pathologic feature defined as cancer cell invasion in, around and through nerves, is an indicator of poor prognosis and survival in prostate cancer (PC). Despite widespread recognition of the clinical significance of PNI, the molecular mechanisms are largely unknown. Here, we report that monoamine oxidase A (MAOA) is a clinically and functionally important mediator of PNI in PC. MAOA promotes PNI of PC cells in vitro and tumor innervation in an orthotopic xenograft model. Mechanistically, MAOA activates SEMA3C in a Twist1-dependent transcriptional manner, which in turn stimulates cMET to facilitate PNI via autocrine or paracrine interaction with coactivated PlexinA2 and NRP1. Further, MAOA inhibitor treatment effectively reduces PNI of PC cells in vitro and tumor-infiltrating nerve fiber density along with suppressed xenograft tumor growth and progression in mice. Collectively, these findings characterize the contribution of MAOA to the pathogenesis of PNI and provide a rationale for using MAOA inhibitors as a targeted treatment for PNI in PC.
Introduction
Prostate cancer (PC) currently is the most commonly diagnosed noncutaneous malignancy and the second leading cause of cancer-related death in American men [1]. While organ-confined PC can be effectively controlled, the metastatic disease originating from extracapsular extension is inevitably incurable [2]. Perineural invasion (PNI), a complex process of neoplastic invasion of nerves, has been recognized as a significant route for the metastatic spread of PC, independent of lymphatic or vascular involvement [3]. PNI is highly prevalent in PC, observed in up to 75% of surgical resection specimens [4]. Moreover, extracapsular extension by cancer spreading entirely or predominantly within perineural spaces was present in >50% of PC specimens [5]. PNI is an ominous clinical and pathological characteristic of PC, which has been associated with cancer pain, adverse pathological features, elevated biochemical recurrence rates, increased risk for bone metastasis and diminished overall survival [4, 6–8]. However, no targeted treatment modalities to date are aimed at PNI and its associated metastatic process in PC.
Recent studies have identified several molecular mechanisms that likely facilitate PNI evoked by either a predominant cancer-induced event or two-way cancer-nerve communication in multiple cancer types. These include cellular adhesion molecules and diverse signaling driven by neurotropic factors, chemokines and axon guidance molecules in a ligand-receptor interactive mode between cancer cells and the neural microenvironment [9, 10]. Specifically, semaphorin axon guidance signaling, including semaphorin (SEMA) 4D and SEMA4F, was recently demonstrated to have an active role promoting PNI in PC [11, 12]. The semaphorins are a large family of secreted axon guidance molecules, which regulate directional migration of axons during embryonic development through interactions with their receptors and co-receptors, plexins and neuropilins respectively [13]. Despite these mechanistic investigations, the molecular mechanisms controlling PNI remain still largely unknown, which hampers the development of novel therapeutics targeting PNI in PC.
Monoamine oxidase A (MAOA) is a key enzyme responsible for metabolism of monoamine neurotransmitters and modulation of neurotransmission, neural circuits and brain function. MAOA has been widely studied in the context of neuropsychiatric disorders such as depression. MAOA inhibitors are currently used as anti-depressants in the clinic [14, 15]. Recent studies including ours demonstrated MAOA’s essential roles in mediating PC growth and metastasis primarily through activation of the downstream central transcription factor Twist1 [16–19]. However, whether and how MAOA contributes to PNI in PC is not clear. In this study, we explored the expanding role of MAOA in regulating PNI in PC. We showed that MAOA promotes PNI and enhances cancer-nerve cell crosstalk in PC cells and xenograft tumor models. Mechanistically, MAOA facilitates PNI of PC cells via upregulation of class 3 semaphorin signaling composed of the SEMA3C/PlexinA2/NRP1 triad in a cMET-dependent manner. These findings establish the functional role of MAOA in PNI pathogenesis and also suggest that MAOA and its associated molecules are a potential therapeutic target for PNI in PC.
Results
MAOA expression levels are elevated along with PNI in clinical specimens
To seek initial evidence of MAOA’s role in PNI of PC, we performed histological analysis of a tissue panel comprised of primary human PC tumors, including 12 PNI-positive and 104 PNI-negative samples. We found that MAOA protein levels were elevated in the tumor samples where PNI was present compared to those where PNI was not observed (Figs. 1a and 1b). Intriguingly, in all PNI-positive tumor samples, MAOA expression levels were further increased in tumor cells within perineural spaces compared to those distal to nerves (Figs. 1c and 1d). These results suggest the association of tumor MAOA with PNI in PC.
Fig. 1.
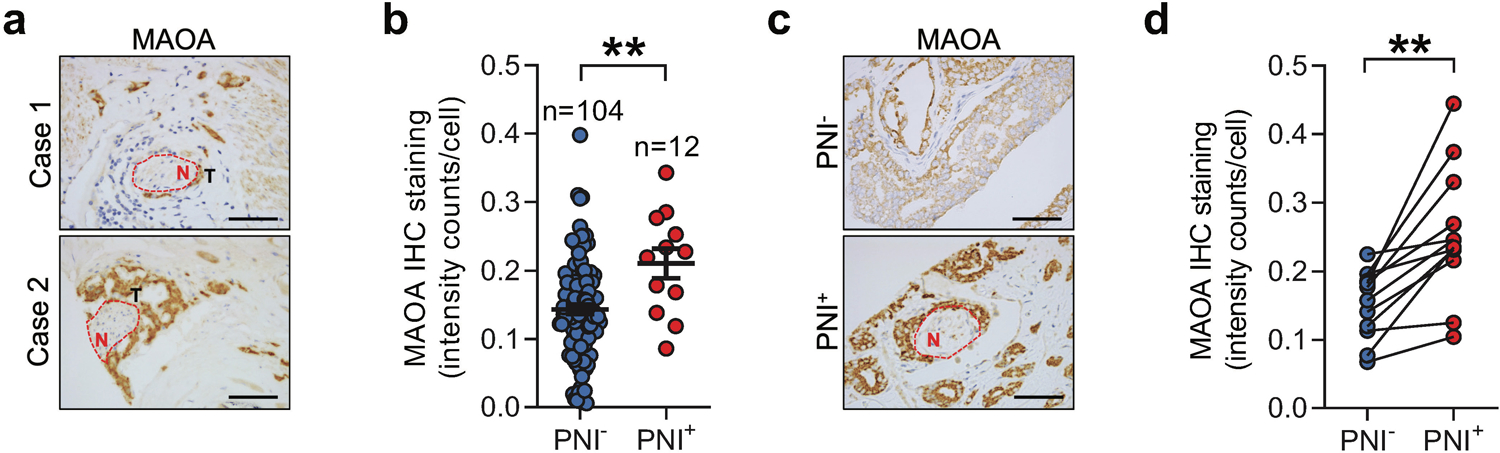
Elevated MAOA expression is associated with PNI in PC. a IHC images of MAOA expression in tumors that invade nerves (N) in 2 representative PNI-positive PC patient samples. Scale bars: 20 μm. b Quantification of MAOA expression in a cohort of clinical PC tissue samples categorized by PNI positivity (PNI−, n=104; PNI+, n=12). c IHC images of MAOA expression in cancerous areas distal from (PNI−) or adjacent to (PNI+) cancer-invading perineural space from a representative PNI-positive patient sample. Scale bars: 20 μm. d Quantification of MAOA expression in cancerous areas in the absence or presence of PNI from 12 PNI-positive patient samples of the cohort described in (b). Average cell-based IHC staining intensity was analyzed by HALO software for making comparisons in (b) and (d). Data represent the mean ± SEM. *p<0.05.
MAOA promotes PNI in cancer-nerve cell co-cultures
To test whether MAOA upregulation in nerve-invading PC cells controls PNI of PC cells, we established a 3D cancer-nerve cell co-culture model using PC-12 neuronal-like cells or 50B11 neurons. PC-12 is a classical neuronal cell model that resembles the phenotype of sympathetic ganglion neurons upon differentiation with NGF [20], while 50B11 is an immortalized dorsal root ganglion sensory neuronal line that undergoes differentiation in the presence of forskolin [21]. The acquired neuronal properties of these cells upon extrinsic induction accord with innervation of the prostate and prostate tumor by sensory or autonomic nerve fibers [22], which makes these cells suitable for use to mimic the clinical observation of PNI. We grew the differentiated PC-12 or 50B11 cells in Matrigel and then added fluorescence dye-labeled cancer cells to the media, allowing cancer cells to widely disperse for an enhanced opportunity to interact with a large number of individual neurites. Concurrently, we seeded fluorescent cancer cells surrounding a blank Matrigel for parallel assessment of autonomous cancer cell invasion independent from extrinsic interaction with nerve cells under the same experimental conditions. We determined the perineural invasive ability of cancer cells by quantification of the fluorescent cancer cells confined in the neurite-bearing Matrigel from cancer-nerve cell co-culture after subtraction of the fluorescent cancer cells invading the blank Matrigel from parallel cancer cell culture alone. We showed that enforced MAOA expression in two MAOA-low PC cell lines, PC-3 and LAPC-4, significantly promoted PNI by 2- to 3.1-fold compared to controls expressing an empty vector (Figs. 2a–2d). Conversely, we stably silenced MAOA expression using two shRNAs targeting separate non-overlapping MAOA coding regions in LNCaP and 22Rv1 cells, which both have abundant MAOA levels. After confirming successful knockdown by Western blot analysis, we observed decreases of PNI ranging from 28% to 87% in both PC cells when MAOA expression was abolished compared to controls expressing a scrambled shRNA (Figs. 2e–2h). These data in aggregate indicate the PNI-promoting role of MAOA in PC cells.
Fig. 2.
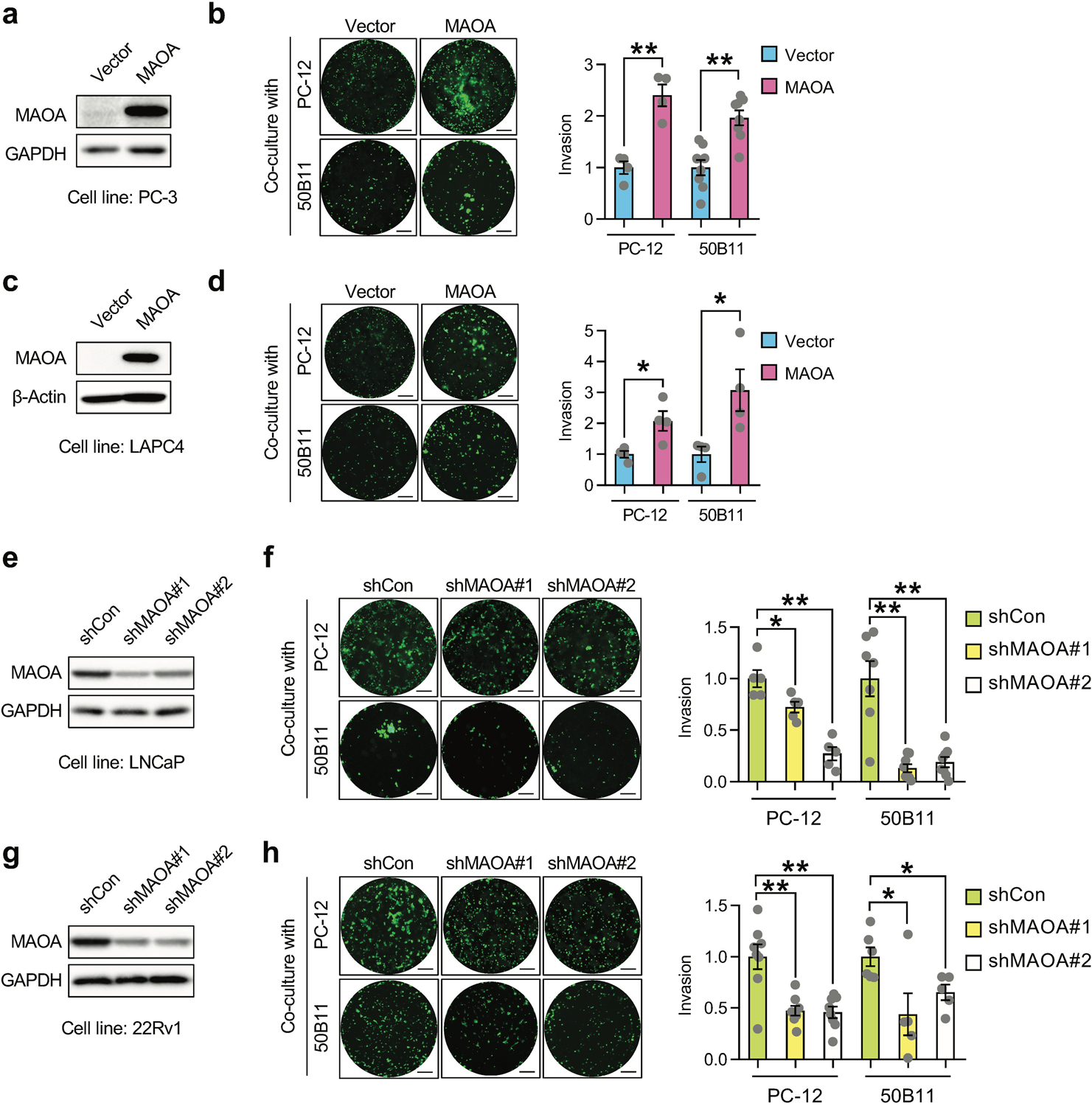
MAOA promotes PNI of PC cells in cancer-nerve cell co-cultures. a, c Western blot analysis of MAOA protein expression in control and MAOA-OE PC-3 (a) or LAPC4 (c) cells. b, d Quantification of PNI of control and MAOA-OE PC-3 (b, n=4–8) or LAPC4 (d, n=4) cells in co-cultures with PC-12 or 50B11 cells by fluorescence measurement. Cancer cell invasion in the control group was set as 1. Representative images of fluorescence dye-labeled cancer cells that invaded nerve cells from each group are shown. Scale bars: 50 μm. e, g Western blot analysis of MAOA protein expression in control (shCon) and MAOA-knockdown (shMAOA) LNCaP (e) or 22Rv1 (g) cells. f, h Quantification of PNI of control and MAOA-knockdown LNCaP (f, n=5–8) or 22Rv1 (h, n=5–8) cells in co-cultures with PC-12 or 50B11 cells by fluorescence measurement. Cancer cell invasion in the control group was set as 1. Representative images of fluorescent cancer cells that invaded nerve cells from each group are shown. Scale bars: 50 μm. Data represent the mean ± SEM. *p<0.05, **p<0.01.
MAOA induces PNI through activation of class 3 semaphorin signaling in a cMET-dependent fashion
To delineate the mechanism underlying MAOA’s action on PNI, we showed a 4.4-fold increase of NGF protein secretion, which has been reported to be associated with PNI and promote tumor-associated neurogenesis in several cancer types [9, 23], in MAOA-overexpressing (OE) PC-3 cells relative to controls (Fig. 3a). Given the recent implication of class 3 semaphorin signaling in driving PC towards an invasive and metastatic phenotype [24, 25], we examined the expression levels of key members of this signaling cascade. Our qPCR-based gene expression profiling data identified significant upregulation of SEMA3C, PlexinA2 and NRP1, which coincidentally may form a ligand/receptor/co-receptor molecular complex, in MAOA-OE PC-3 cells compared to controls, which was confirmed at the protein level by Western blot (Figs. 3b and 3c). Consistently, we found reduced transcript levels of these genes in MAOA-knockdown LNCaP and 22Rv1 cells (Fig. 3d). Since SEMA3C was reported as a transcriptional target of androgen receptor (AR), the primary oncogenic driver of PC [26], we also compared the effects of MAOA versus AR on SEMA3C and found that forced expression of MAOA or AR induced SEMA3C mRNA levels to a similar extent in PC-3 cells (Supplemental Fig. 1). Examining a PC tissue microarray (TMA) as used to associate MAOA levels with PNI status (Fig. 1) by immunohistochemical (IHC) staining further revealed a positive co-expression correlation between tumor MAOA and SEMA3C in the clinical setting (Fig. 3e and Supplementary Table 1). To determine if SEMA3C-dependent signaling mediates MAOA’s effect on PNI, we showed that individual siRNA-based knockdown of SEMA3C, PlexinA2 and NRP1 suppressed MAOA OE-induced PNI of PC-3 cells when co-cultured with either PC-12 or 50B11 cells compared to controls (Fig. 3f).
Fig. 3.
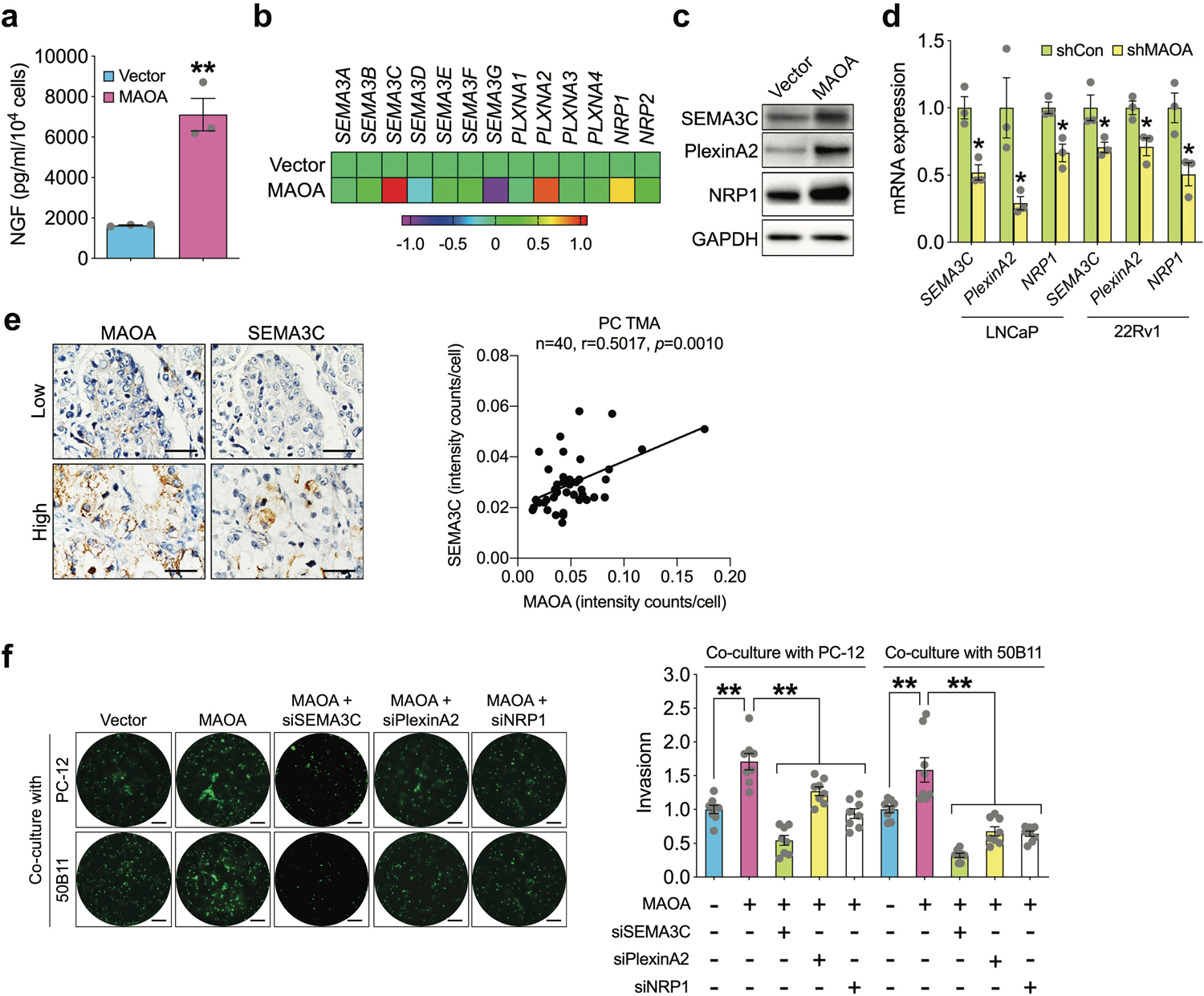
MAOA induces PNI of PC cells through activation of class 3 semaphorin signaling. a Quantification of NGF protein secretion in conditioned media of control and MAOA-OE PC-3 cells by ELISA (n=3). b Heatmap depicting the class 3 semaphorin family gene levels in control and MAOA-OE PC-3 cells by RT-qPCR (n=3 with averages used for plots). c Western blot analysis of SEMA3C, PlexinA2 and NRP1 protein expression in control and MAOA-OE PC-3 cells. d Quantification of SEMA3C, PlexinA2 and NRP1 mRNA levels in control and MAOA-knockdown LNCaP and 22Rv1 cells by RT-qPCR (n=3). e Co-expression correlation analysis of MAOA and SEMA3C in a PC TMA (n=40) by IHC. Average cell-based staining intensity counts for each protein were analyzed by inForm software to assess co-expression correlation. Images show individual protein staining in a representative tumor area of a low- or high-expression patient sample. Scale bars: 20 μm. f Quantification of PNI of control and MAOA-OE PC-3 cells transfected with or without SEMA3C, PlexinA2 or NRP1 siRNAs when co-cultured with PC-12 or 50B11 cells by fluorescence measurement (n=8). Cancer cell invasion in the control group was set as 1. Representative images of fluorescent cancer cells that invaded nerve cells from each group are shown. Scale bars: 50 μm. Data represent the mean ± SEM. *p<0.05, **p<0.01.
SEMA3C, through different combinations of plexins and neuropilins, stimulates multiple receptor tyrosine kinase (RTK) pathways such as EGFR, HER2/ErbB2, cMET, and SRC, some of which are involved in PNI during PC progression [27, 28]. We used a phospho-kinase antibody array coupled with a candidate approach to search for RTK(s) downstream of the MAOA/SEMA3C axis in MAOA-OE and control PC-3 cells. cMET stands out because it had the most significant activation of phosphorylation levels in MAOA-OE PC-3 cells compared to controls, with minimal changes observed for the other kinases (Fig. 4a and Supplementary Table 2). We showed that siRNA-mediated silencing of SEMA3C, PlexinA2 and NRP1 individually suppressed MAOA-activated phosphorylation of cMET in PC-3 cells (Fig. 4b), suggesting that MAOA signals through the SEMA3C/PlexinA2/NRP1 triad to regulate cMET. Since plexins and neuropilins have been shown to physically interact with RTKs, a mechanism for enhanced tyrosine phosphorylation and kinase activity of RTKs [27, 29, 30], we examined the direct interaction of PlexinA2 and NRP1 with cMET in a MAOA/SEMA3C-dependent context. An in situ proximity ligation assay visualized endogenous PlexinA2-cMET and NRP1-cMET protein complexes in the cytoplasm of PC-3 cells. Enforced MAOA expression strengthened PlexinA2-cMET and NRP1-cMET interactions in PC-3 cells compared to controls, which were attenuated by siRNA-mediated knockdown of SEMA3C (Fig. 4c). To determine if cMET mediates MAOA’s effect on PNI, we showed that siRNA-based silencing of cMET reduced MAOA OE-induced PNI of PC-3 cells when co-cultured with PC-12 or 50B11 cells compared to controls, which was similar to the effect exerted by SEMA3C siRNAs. Prior treatment with cMET siRNAs further abolished the suppressive effect of SEMA3C knockdown on MAOA-induced PNI of PC-3 cells, supporting the idea that cMET acts downstream of SEMA3C to mediate MAOA’s effect on PNI (Fig. 4d). Collectively, we concluded that MAOA induces PNI of PC cells through class 3 semaphorin signaling, particularly SEMA3C, PlexinA2 and NRP1, and subsequent activation of cMET.
Fig. 4.
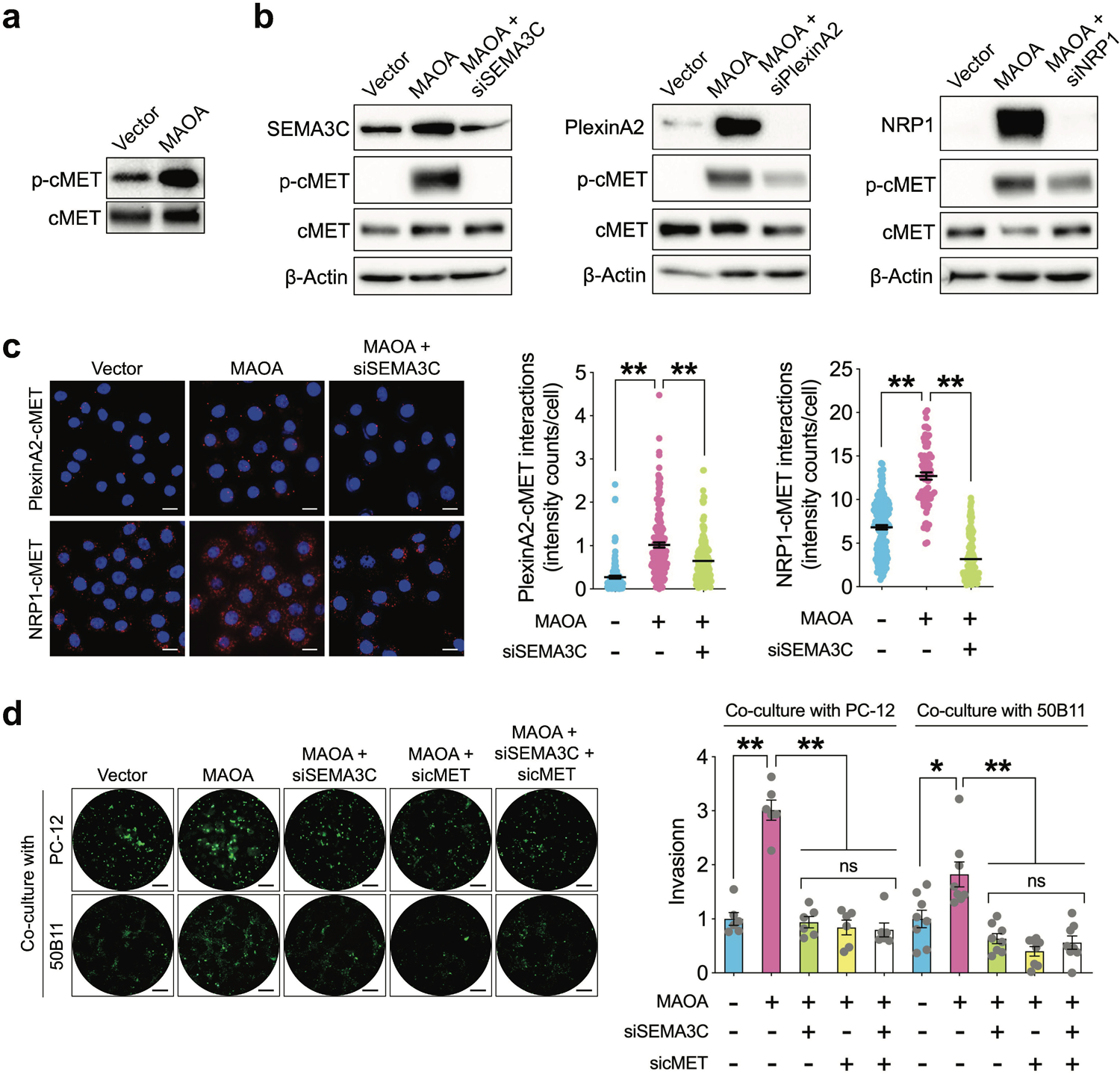
The SEMA3C/PlexnA2/NRP1 triad mediates MAOA’s effect on PNI of PC cells in a cMET-dependent manner. a Western blot analysis of phospho-cMET and total cMET in control and MAOA-OE PC-3 cells. b Western blot analysis of phospho-cMET and total cMET in control and MAOA-OE PC-3 cells with or without transfection of SEMA3C, PlexinA2 or NRP1 siRNAs. c Quantification of PlexinA2-cMET and NRP1-cMET interactions by per-cell cytoplasmic fluorescence signal intensity in control, MAOA-OE and MAOA-OE/siRNA-based SEMA3C-knockdown PC-3 cells (PlexinA2-cMET: n=128, 161 and 177 cells for control, MAOA-OE and MAOA-OE/SEMA3C-knockdown groups respectively; NRP1-cMET: n=171, 73 and 151 cells for control, MAOA-OE and MAOA-OE/SEMA3C-knockdown groups respectively). Representative fluorescence images in each group from proximity ligation assays are shown. Scale bars: 50 μm. d Quantification of PNI of control and MAOA-OE PC-3 cells transfected with or without SEMA3C and/or cMET siRNAs when co-cultured with PC-12 or 50B11 cells by fluorescence measurement (n=6–8). Cancer cell invasion in the control group was set as 1. Representative images of fluorescent cancer cells that invaded nerve cells from each group are shown. Scale bars: 50 μm. Data represent the mean ± SEM. *p<0.05, **p<0.01; ns, not significant.
MAOA activates SEMA3C expression through Twist1-dependent transcriptional upregulation
We next investigated the molecular basis for MAOA’s control of SEMA3C in PC cells. We demonstrated previously that MAOA actives Twist1, a basic helix-loop-helix transcription factor, via a reactive oxygen species-driven signaling pathway and utilizes Twist1 as a central mediator of MAOA function in PC cells [17, 18, 31], which allows us to speculate that Twist1 may mediate MAOA’s effect on SEMA3C gene expression. We showed that siRNA-mediated silencing of Twist1 downregulated MAOA activation of SEMA3C protein expression as well as cMET phosphorylation in PC-3 cells (Fig. 5a). Moreover, forced expression of Twist1 elevated SEMA3C protein expression, while siRNA-based knockdown of Twist1 repressed SEMA3C at the protein level, independent from the MAOA context (Fig. 5b). We also revealed that Twist1 knockdown suppressed MAOA-induced SEMA3C mRNA expression (Fig. 5c), implying that Twist1 may regulate SEMA3C at the transcriptional level. Using a 1.5-kb SEMA3C promoter-luciferase reporter construct, we further found that Twist1 siRNA treatment reversed MAOA activation of SEMA3C promoter activity (Fig. 5d). To determine whether Twist1 directly binds to the SEMA3C gene locus to mediate its transcriptional activation, we interrogated a Twist1 ChIP-seq dataset for Twist1 binding information in human neuroblastoma cells. We found Twist1 enrichment at the promoter region proximal to the transcription start site (−1~−280 with transcription start site of SEMA3C set as +1). Indeed, we were able to identify several consensus Twist1-binding E-box sites in this region (Fig. 5e). To confirm direct occupancy of Twist1 with SEMA3C promoter, we conducted chromatin immunoprecipitation assays coupled with quantitative PCR using primers that amplified the putative Twist1-binding region in control and MAOA-OE PC-3 cells. We demonstrated 1.1-fold higher association of endogenous Twist1 protein with the SEMA3C promoter region encompassing putative Twist1-binding sites in MAOA-OE cells compared to controls. Limited signals were detected from negative controls in which either a nonspecific IgG antibody was used in the immunoprecipitation step or the SEMA3C exon 2 was probed in order to confirm the targeting specificity of the primer set used in PCR (Fig. 5f). Finally, we showed that siRNA-mediated ablation of Twist1 suppressed MAOA-induced PNI of PC-3 cells in co-cultures with PC-12 or 50B11 cells, suggesting that Twist1 regulation of SEMA3C confers MAOA’s effect on PNI (Fig. 5g). Taken together, these observations establish that MAOA activates SEMA3C in a Twist1-dependent manner.
Fig. 5.
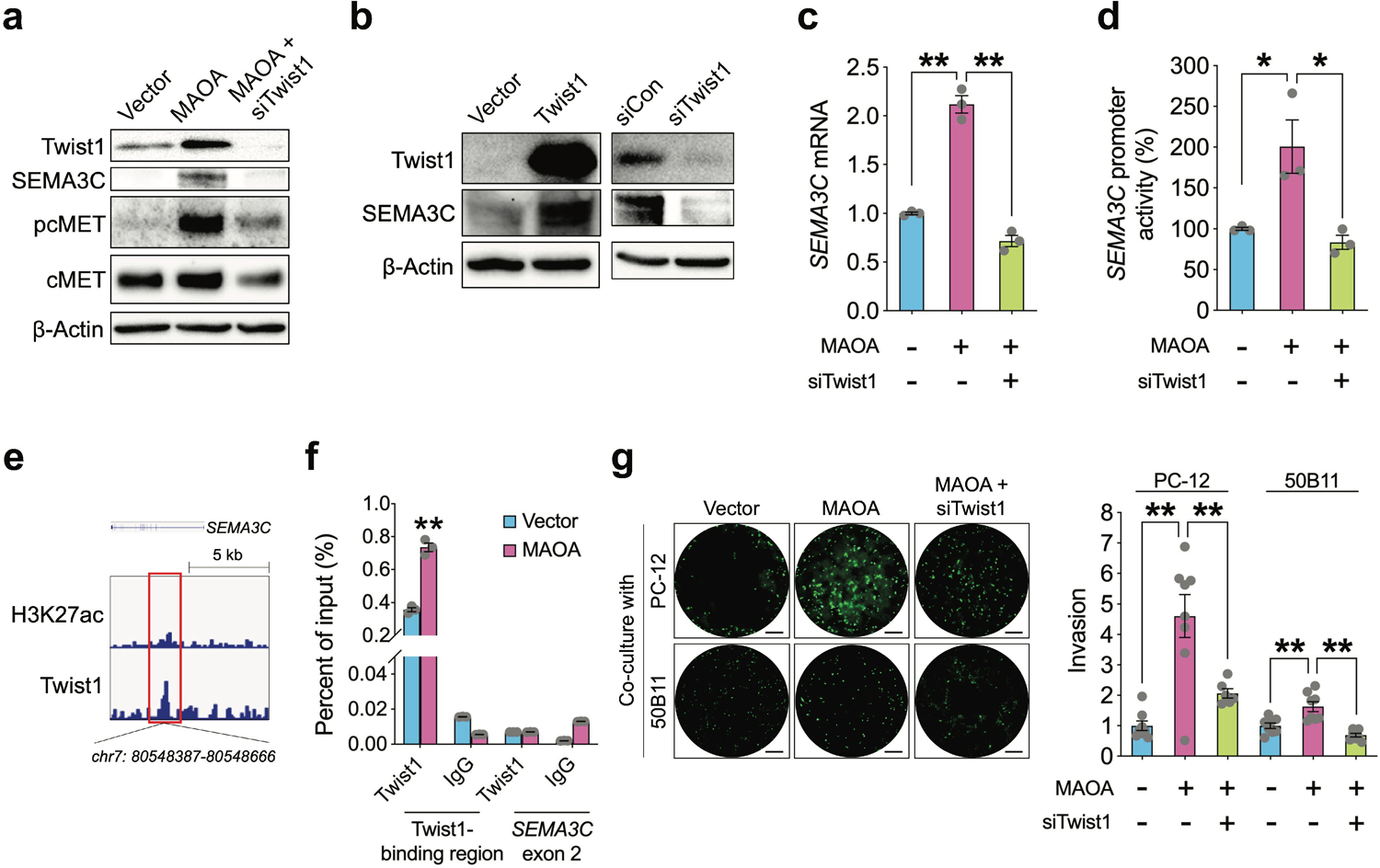
MAOA activates SEMA3C directly through Twist1 in PC cells. a Western blot analysis of Twist1, SEMA3C, phospho-cMET and total cMET in control, MAOA-OE and MAOA-OE/siRNA-mediated Twist1-knockdown PC-3 cells. b Western blot analysis of Twist1 and SEMA3C in control and Twist1-manipulated PC-3 cells. c RT-qPCR analysis of SEMA3C mRNA levels in control, MAOA-OE and MAOA-OE/Twist1-knockdown PC-3 cells (n=3). d Determination of SEMA3C 1.5-kb promoter activity in control, MAOA-OE and MAOA-OE/Twist1-knockdown PC-3 cells (n=3). e Genomic browser representation of H3K27ac and Twist1 binding in human neuroblastoma BE(2)C cells from GSE80151 in the proximal region of SEMA3C promoter that harbors a Twist1-binding sequence. f ChIP-qPCR analysis of control and MAOA-OE PC-3 cells immunoprecipitated by anti-Twist1 antibody or IgG followed by qPCR using 2 primers sets targeting the Twist1-binding sequence in SEMA3C promoter and SEMA3C exon 2. Data represent the percentage of input (n=3). g Quantification of PNI of control and MAOA-OE PC-3 cells transfected with or without Twist1 siRNA when co-cultured with PC-12 or 50B11 cells by fluorescence measurement (n=6–8). Cancer cell invasion in the control group was set as 1. Representative images of fluorescent cancer cells that invaded nerve cells from each group are shown. Scale bars: 50 μm. Data represent the mean ± SEM. *p<0.05, **p<0.01.
MAOA enhances tumor innervation during prostate tumor growth and progression
To further elucidate MAOA’s potential supportive role in cancer-nerve crosstalk in vivo, we established an orthotopic prostate tumor xenograft model allowing us to observe interaction of cancer cells with the neural microenvironment in vivo. We implanted luciferase-labeled control and MAOA-OE PC-3 cells into the prostates of immunodeficient mice and monitored growth, progression and metastasis of prostate tumors by bioluminescence imaging (BLI). Enforced expression of MAOA in PC-3 cells accelerated prostate tumor growth, particularly the incidence of metastasis to distant organs, including lung and liver, as confirmed by necropsy and ex vivo BLI, compared to controls (Figs. 6a–6d). Subsequently, we analyzed primary tumor samples by multiple quantum dot (QD) labeling analysis, a method capable of simultaneously detecting multiple markers in a single sample following sequential staining procedures. We found significant increases of Ki-67-positive proliferating tumor cells and tumor-infiltrating nerve fiber density characterized by neurofilament-L (NF-L) and neurofilament-H (NF-H) staining, which mark newly formed and mature nerve fibers respectively [32], in MAOA-OE primary tumor samples compared with controls (Fig. 6e). Conversely, shRNA-mediated stable silencing of MAOA in ARCaPM cells, a highly invasive and metastatic line with abundant levels of MAOA [18], suppressed prostate tumor growth, accompanied by lower Ki-67 and NF-L/NF-H expression in tumor samples compared to controls (Figs. 6f–6h). In addition, IHC analysis demonstrated declined staining of cleaved caspase 3, an apoptotic cell marker, and increased expression levels of NGF, SEMA3C and phosphorylated cMET in MAOA-OE PC-3 tumor samples compared to controls, with consistent observations further found in control and MAOA-knockdown ARCaPM tumor samples (Figure 6i). These data provide in vivo evidence that MAOA enhances prostate tumor-nerve interaction, particularly tumor-related neurogenesis, alongside tumor growth and progression.
Fig. 6.

MAOA enhances tumor innervation during prostate tumor growth and progression in mice. a Growth curves of luciferase-tagged control and MAOA-OE PC-3 cells implanted into the prostates of mice by weekly BLI monitoring (n=7 mice per group). b BLI images of a representative mouse from each group in (a). c Quantification of distant metastatic foci per mouse from each group in (a) (n=7). d BLI images of a representative mouse developing distant metastasis in MAOA-OE PC-3 cell group. Both whole-body in vivo (i) and organ-specific ex vivo (ii, prostate tumor; iii, lung metastasis) images are shown. BLI scales: i, 8 × 104−1 × 106; ii and iii, 1 × 106−2 × 107. e Quantification of Ki-67, NF-L and NF-H per-cell staining intensity in control and MAOA-OE orthotopic xenograft tumors by multiplexed QD labeling (n=3). A representative multiplexed QD labeling image of Ki-67 (pink), NF-L (green) and NF-H (red) in a PC-3 control xenograft prostate tumor is shown. Scale bars: 5 μm. f Growth curves of luciferase-tagged control (shCon) and shRNA-mediates stable MAOA-knockdown (shMAOA) ARCaPM cells implanted into the prostates of mice by weekly BLI monitoring (n=6 mice per group). g BLI images of a representative mouse from each group in (f). h Quantification of Ki-67, NF-L and NF-H per-cell staining intensity in control and MAOA-knockdown ARCaPM orthotopic xenograft tumors by multiplexed QD labeling (n=3). i Representative IHC images of cleaved caspase 3 (c-caspase 3), NGF, SEMA3C and phospho-cMET in orthotopic xenograft tumors from (a) and (f). Scale bars: 20 μm. Data represent the mean ± SEM. *p<0.05, **p<0.01.
Pharmacological inhibition of MAOA reduces PNI in vitro and tumor-associated neurogenesis in vivo in PC
Considering the current clinical use of MAOA inhibitors, we sought to determine whether pharmacological interference with MAOA effectively inhibits PNI and cancer-nerve interaction in PC. We showed that treatment with clorgyline, a MAOA small-molecule inhibitor, suppressed PNI of LNCaP and 22Rv1 cells when co-cultured with PC-12 cells (Fig. 7a). Using an orthotopic xenograft model, we found that clorgyline treatment significantly slowed both 22Rv1 and ARCaPM prostate tumor growth in mice (Figs. 7b–7e). Moreover, multiple QD labeling analysis demonstrated a decreased percentage of Ki-67 positivity reflecting tumor cell proliferation and NF-L/NF-H staining indicative of nerve fiber density in clorgyline-treated prostate tumors compared to controls in both 22Rv1 and ARCaPM tumors (Fig. 7f). Furthermore, clorgyline treatment led to increased levels of cleaved caspase 3 and lower expression of NGF, SEMA3C and phosphorylated cMET compared with controls, as revealed by IHC analysis of both 22Rv1 and ARCaPM xenograft tumors (Fig. 7g).
Fig. 7.
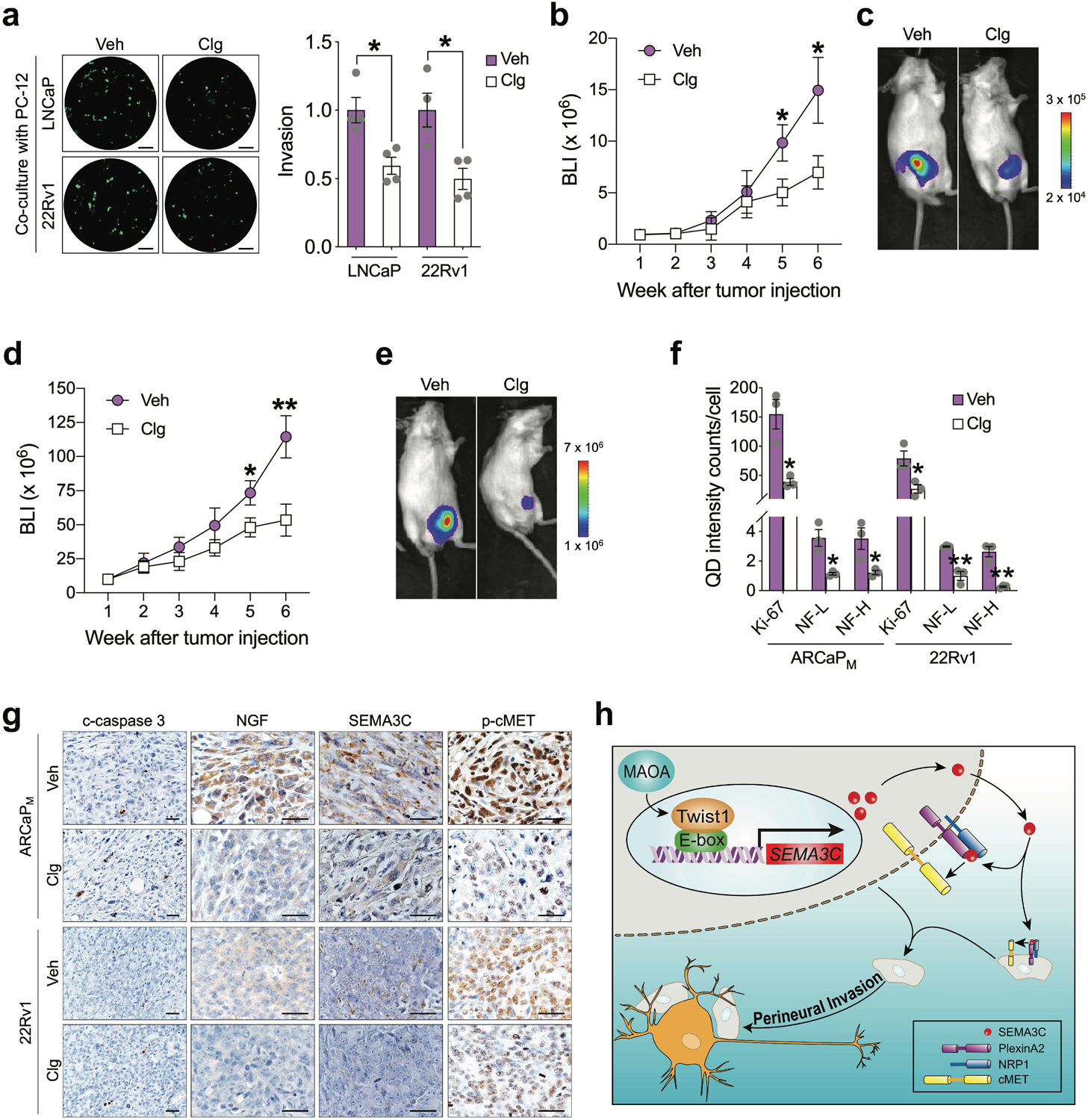
Pharmacological inhibition of MAOA reduces PNI and tumor infiltration by nerves in PC. a Quantification of PNI of LNCaP and 22Rv1 cells treated with vehicle (PBS, Veh) or clorgyline (Clg, 1 μM, 4 days) when co-cultured with PC-12 cells by fluorescence measurement (n=4). Cancer cell invasion in the control group was set as 1. Representative images of fluorescent cancer cells that invaded nerve cells from each group are shown. Scale bars: 50 μm. b BLI monitoring growth of luciferase-tagged ARCaPM cells implanted into the prostates of mice followed by saline (Veh) or clorgyline (10 mg/kg, i.p., daily) treatment one week after tumor cell injection (n=7 mice per group). c BLI images of a representative mouse from each group in (b). d BLI monitoring growth of luciferase-tagged 22Rv1 cells implanted into the prostates of mice followed by saline (Veh) or clorgyline (10 mg/kg, i.p., daily) treatment one week after tumor cell injection (n=7–8 mice per group). e BLI images of a representative mouse from each group in (d). f Quantification of Ki-67, NF-L and NF-H per-cell staining intensity in control and clorgyline-treated ARCaPM and 22Rv1 orthotopic xenograft tumors by multiplexed QD labeling (n=3). g Representative IHC images of c-caspase 3, NGF, SEMA3C and phospho-cMET in orthotopic xenograft tumors from (b) and (d). Scale bars: 20 μm. h Schematic depicting the mechanism by which MAOA promotes PNI of PC cells where MAOA upregulates SEMA3C in a Twist1-dependent transcriptional manner, which in turn via autocrine or paracrine interaction with coactivated PlexinA2 and NRP1 to stimulate cMET to promote PNI. Data represent the mean ± SEM. *p<0.05, **p<0.01.
In summary, our preclinical studies suggest that the increased intrinsic expression of MAOA associated with PNI promotes PNI of PC cells through a mechanism initiated by Twist1-dependent transcriptional upregulation of SEMA3C, which subsequently interacts with PlexinA2 and NRP1 to activate cMET in either an autocrine or a paracrine manner (Fig. 7h). We further provide evidence for using MAOA inhibitors for potential therapeutic targeting of neoplastic invasion of nerves and cancer-related neurogenesis in PC.
Discussion
PNI is a tropism of cancer cells toward surrounding nerves, after which cancer cells demonstrate the ability to invade and track along nerves. PNI is correlated with disease recurrence, metastasis and poor survival in multiple malignancies, including prostate, pancreatic, head and neck, salivary and colon cancers. In contrast to cancer metastasis through lymphatic or vascular conduits, PNI allows cancer cells to spread along nerve tracts beyond the predicted anatomic borders of a primary tumor, often resulting in incomplete surgical resection associated with elevated recurrence rates. Previously underrecognized, PNI has been brought into focus by these notorious clinical facts, encouraging mechanistic study of its pathogenesis in recent years [3]. PNI is an active rather than a passive process occurring within all three layers of the nerve sheath (epineurium, perineurium and endoneurium), suggesting that cancer cells actively communicate with nerves [3, 9]. Using in vitro and in vivo models coupled with clinical samples, we showed that the neural enzyme MAOA, which is highly induced throughout the PC disease trajectory as we previously reported, additionally contributes to PC PNI through activation of class 3 semaphorin signaling and cMET. These findings expand our understanding of the regulatory mechanisms for PNI and suggest a novel targeting pathway to interrupt this process. We previously established MAOA’s role promoting PC invasion and metastasis by activating Twist1 and associated paracrine Shh signaling in an intracardiac xenograft model, which rapidly develops distant metastasis through the bloodstream, suggesting MAOA’s capability to induce blood-borne metastasis in PC [17, 18]. Complementarily, our current study demonstrated that MAOA is also able to increase the invasive and metastatic potential of PC cells along nerves as an alternative route for cancer cell spreading using Twist1-directed SEMA3C-cMET signaling as the major driving mechanism. Collectively, these studies reinforce MAOA’s supportive action in PC invasion and metastasis, which is likely to be mediated by different molecular signaling depending on which metastatic route cancer cells utilize in the microenvironmental context.
Like many other solid organs, the prostate is innervated by sensory and autonomic nerve fibers, creating a favorable neural niche for nerves to communicate with cancer cells [22]. Indeed, a recent landmark study has demonstrated that autonomic nerve sprouting in prostate tumors is essential and necessary for supporting PC development and progression across nearly all phases in mouse models [33]. Intriguingly, PC also develops a neuronal phenotype by starting to express neuronal genes and exhibit neurogenic properties during disease progression, especially advanced, castration-resistant and metastatic stages [34], enhancing the likelihood of reciprocal crosstalk between cancer cells and nerves. Of many emerging neuronal genes relevant to PC, MAOA is a neural enzyme best known for degrading monoamine neurotransmitters in the brain, including serotonin, norepinephrine, epinephrine and dopamine [15]. A recent study has established that PC-nerve crosstalk invokes a mechanism in which neurotransmitters released from the autonomic nervous system, such as norepinephrine, elicit signaling cascades in cancer cells [33]. Thus, it would be interesting to pursue whether and how MAOA modulates other aspects of cancer-nerve interactions directly through its innate enzymatic nature, in addition to the mechanism we demonstrated here.
Our findings revealed that class 3 semaphorin signaling, particularly SEMA3C, PlexinA2 and NRP1 that may form a ternary complex, functionally mediates MAOA’s action on PC PNI. The class 3 semaphorin signaling molecule family genes were initially identified as evolutionarily conserved axon guidance cues that instruct the assembly of the neural circuity [13, 35]. Recent studies have shifted research interests in these molecules to the area of cancer research, demonstrating their previously unknown secretion by cancer cells in multiple tumor types, including PC, which supports the theory that PNI may result from cancer-initiated stimulation of axonogenesis or neurogenesis [36]. Despite the more frequent association of semaphorins and plexins with axonal guidance as repulsion signals, semaphorin-plexin interactions can be switched to an attraction mode depending on the context, for instance in the presence of neuropilins [13, 37]. Since we show that SEMA3C, PlexinA2 and NRP1 are all expressed in PC cells, it is plausible that the autocrine or paracrine signaling involving these proteins in PC cells influences PNI. However, we could not rule out the possibility that nerves also express these ligand/receptors for dynamic interaction with cancer cells and added perineural invasive potential, which warrants further investigation.
Seeking to uncover how MAOA induces SEMA3C, we demonstrated that Twist1 directly interacts with the SEMA3C promoter for transcriptional activation. Twist1 is a master mediator of MAOA function in PC, controlling the expression of different downstream genes at the transcriptional level, including repressing E-cadherin to promote epithelial-mesenchymal transition and activating Shh and IL-6 to facilitate tumor-stromal cell interactions [17, 18, 38]. Intriguingly, Shh has been found to induce semaphorin signaling response in axon guidance [39], which coincides with our recent findings on Twist1 activation of Shh in PC cells [18], further supporting the notion that Twist1 induces SEMA3C as we elucidated above. In a parallel comparison, we revealed that SEMA3C is inductive to both MAOA and AR to a similar extent in PC cells. Coincidentally, Twist1 was shown to be upregulated by androgen via two AR-regulated transcription factors, ETV1 and NKX3–1, and disruption of AR expression or activity abolishes Twist1 expression in PC cells [40, 41]. Although Tam et al. reported that AR transcriptionally activates SEMA3C through an androgen response element within the SEMA3C locus further in a GATA2-dependent manner [26], we argue for another possible scenario where AR indirectly activates SEMA3C via Twist1. On the other hand, a recent study indicated the capability of the MAOA inhibitor clorgyline to decrease AR expression in PC cells [42], which led us to reason that AR may also aid in MAOA induction of Twist1. These mechanistic possibilities interwinding MAOA, AR and Twist1 for SEMA3C gene regulation merit additional exploration.
cMET is emerging as a key regulator of a metastasis-promoting phenotype in multiple malignancies, including PC. cMET has actions in the neuronal system and in PC, but its possible role in regulating crosstalk between nerves and PC cells is not clear. cMET, a RTK, binds HGF, and following dimerization and autophosphorylation it drives cancer cell proliferation, survival, motility and invasion. Specifically, cMET overexpression is rare in primary tumors but observed in a high portion of metastatic PC lesions, which has led to several clinical trials testing the validity of cMET as a candidate for targeted therapeutic intervention in metastatic PC [43, 44]. Aside from well-characterized roles of cMET in cancer spreading, cMET is also implicated in neuronal contexts. For example, cMET through its ligand HGF promotes peripheral nerve regeneration by activating Schwann cell-mediated nerve repair, suggesting HGF gene transfer or cMET agonists as a potential option for treating peripheral neuropathy [45]. cMET also synergizes with CNTF to enhance motor neuron survival [46]. These favorable neural features may explain why cMET dictates a PNI-promoting role in PC. We demonstrated the capability of cMET to physically interact with PlexinA2 and NRP1, a mechanism conferring cMET activation, in PC cells primed for PNI, which is mirrored by recent evidence pointing out direct binding of cMET to PlexinB1 in the castration- and enzalutamide-resistant states in PC [27]. These results indicate a context-dependent switch of cMET affinity with different plexins, which is likely under the control of versatile upstream signal inputs related to disease phenotype.
In conclusion, we identified the active role of MAOA-Twist1-SEMA3C/PlexinA2/NRP1-cMET signaling in PC perineural invasion and infiltration. These findings contribute to a better understanding of the molecular events underlying PNI and also provide insights into developing future PNI-specific therapeutic strategies to improve clinical outcomes.
Materials and Methods
Clinical specimens
The PC clinical tissue samples for MAOA evaluation were assembled from TMAs PR483c, PR808a and PR809 containing a total of 113 cases of primary prostate adenocarcinoma with single or duplicate cores and 1.5 mm diameter size, which were obtained from US Biomax, and archived single tissue section slides including 3 cases of PNI-positive primary prostate adenocarcinoma obtained from the Biobank of West China Hospital.
Cell lines and cell culture
Human PC PC-3, LNCaP, 22Rv1, rat adrenal pheochromocytoma PC-12 and human embryonic kidney 293T cell lines were obtained from American Type Culture Collection. The human PC LAPC4 cell line was kindly provided by Michael Freeman (Cedars-Sinai Medical Center). The human PC ARCaPM cell line was kindly provided by Leland W.K. Chung (Cedars-Sinai Medical Center). The immortalized rat dorsal root ganglion sensory neuronal 50B11 cell line was kindly provided by Ahmet Hoke (Johns Hopkins University). All cells were regularly tested for Mycoplasma by the MycoProbe Mycoplasma Detection kit (R&D Systems) and used with the number of cell passages below 10. The PC-3 cell line was authenticated by STR profiling (Genetica) in August 2019. The ARCaPM cell line was cultured in T medium supplemented with 10% FBS (Atlanta Biologicals) and 1% penicillin/streptomycin (P/S, Corning). The PC-12 and 293T cell lines were cultured in DMEM medium supplemented with 10% FBS and 1% P/S. The 50B11 cell line was cultured as described previously [21]. The other human PC cell lines were cultured in RPMI-1640 medium (Corning) supplemented with 10% FBS and 1% P/S.
Perineural invasion assay
A 3D cancer-nerve cell co-culture model was established following a published protocol [47] with minor modifications. Briefly, the PC-12 or 50B11 cells were seeded in a 2 μl drop of growth factor-reduced Matrigel (BD Biosciences, 2 × 104 cells/drop), which were treated by recombinant NGF protein (100 ng/ml, 4 days) or forskolin (75 μM, 24 hrs) respectively to allow cell differentiation into a neuronal phenotype with neurite outgrowth. Subsequently, PC cells were labeled with 25 μM fluorescent CellTracker Green CMFDA (GeneCopoeia) for 1 hr at 37°C and seeded (1.2 × 105 cells/drop) to surround the Matrigel for co-culture with differentiated neuronal or neuronal-like cells for 4 days. A culture of PC cells surrounding a 2 μl drop of blank Matrigel under the same experimental conditions was performed in parallel for monitoring autonomous cancer cell invasion. At the experimental endpoints, the media were removed, and the Matrigel was washed by PBS 3 times and subjected to fluorescence image acquisition by a Leica DM IL microscope equipped with a MC190 HD camera. Nerve-invading PC cells were defined as fluorescent cancer cells in association with Matrigel-embedded neurites. The PNI extent of PC cells was calculated by the fluorescence signal generated from the neurite-bearing Matrigel in cancer-nerve cell co-culture after subtraction of the fluorescence signal generated from the blank Matrigel in parallel cancer cell culture alone by ImageJ software.
Animal studies
All animal studies received prior approval from the Cedars-Sinai Medical Center IACUC and complied with IACUC recommendations. Male 4- to 6-week-old SCID mice were purchased from Taconic, housed in the animal facility at Cedars-Sinai Medical Center, and fed a normal chow diet. For determining MAOA’s effect on tumor-nerve interactions alongside tumor growth and progression, 1 × 106 luciferase-tagged PC-3 or ARCaPM cells subjected to different manipulations of MAOA levels were mixed 1:1 with Matrigel (BD Biosciences) and injected into the dorsolateral lobe of the prostate in mice. Tumor growth and progression was monitored by BLI with a Xenogen IVIS Spectrum Imaging System (PerkinElmer) weekly after injection. Distant metastases were confirmed by necropsy and ex vivo BLI imaging at sacrifice. For determining the effect of clorgyline on disrupting tumor-nerve interactions in vivo, 1 × 106 luciferase-tagged ARCaPM or 22Rv1 cells were injected intraprostatically into mice. Mice were randomly assigned to two groups for treatment with vehicle or clorgyline based on BLI signals in prostates 1 week after tumor injection. Daily intraperitoneal injection of clorgyline (10 mg/kg) was given for 5 weeks, with saline injection for the control group. BLI signal data were acquired after background subtraction and analyzed by Living Image software (PerkinElmer).
Statistics
Data were presented as the mean ± SEM as indicated in figure legends. Comparisons were analyzed by unpaired 2-tailed Student’s t test. Correlations were determined by Pearson correlation using GraphPad Prism 7. A p value less than 0.05 was considered statistically significant. Sample size as indicated in the corresponding text and/or figure legends were chosen based on the power to detect significant differences (p<0.05) between either control versus experimental groups, control versus MAOA-OE/-KD tumor inoculated mice, or vehicle-treated versus clorgyline-treated mice. The investigators were not blinded to the group allocation and outcome assessment during the experiments.
Supplementary Material
Acknowledgements
We thank Gina Chu (Cedars-Sinai Medical Center) for technical help, Leland W.K. Chung (Cedars-Sinai Medical Center) for comprehensive support of this study, and Gary Mawyer for editorial assistance. This work was supported by DOD Prostate Cancer Research Program grants W81XWH-15-1-0493 and W81XWH-19-1-0279, NIH/NCI grant R37CA233658, and the WSU startup fund to B.J. Wu.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: a cancer journal for clinicians 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Deng X, He G, Liu J, Luo F, Peng X, Tang S et al. Recent advances in bone-targeted therapies of metastatic prostate cancer. Cancer treatment reviews 2014; 40: 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: a review of the literature. Cancer 2009; 115: 3379–3391. [DOI] [PubMed] [Google Scholar]
- 4.Maru N, Ohori M, Kattan MW, Scardino PT, Wheeler TM. Prognostic significance of the diameter of perineural invasion in radical prostatectomy specimens. Human pathology 2001; 32: 828–833. [DOI] [PubMed] [Google Scholar]
- 5.Villers A, McNeal JE, Redwine EA, Freiha FS, Stamey TA. The role of perineural space invasion in the local spread of prostatic adenocarcinoma. The Journal of urology 1989; 142: 763–768. [DOI] [PubMed] [Google Scholar]
- 6.Kraus RD, Barsky A, Ji L, Garcia Santos PM, Cheng N, Groshen S et al. The Perineural Invasion Paradox: Is Perineural Invasion an Independent Prognostic Indicator of Biochemical Recurrence Risk in Patients With pT2N0R0 Prostate Cancer? A Multi-Institutional Study. Adv Radiat Oncol 2019; 4: 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLancey JO, Wood DP Jr., He C, Montgomery JS, Weizer AZ, Miller DC et al. Evidence of perineural invasion on prostate biopsy specimen and survival after radical prostatectomy. Urology 2013; 81: 354–357. [DOI] [PubMed] [Google Scholar]
- 8.Ciftci S, Yilmaz H, Ciftci E, Simsek E, Ustuner M, Yavuz U et al. Perineural invasion in prostate biopsy specimens is associated with increased bone metastasis in prostate cancer. The Prostate 2015; 75: 1783–1789. [DOI] [PubMed] [Google Scholar]
- 9.Bakst RL, Wong RJ. Mechanisms of Perineural Invasion. J Neurol Surg B Skull Base 2016; 77: 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jobling P, Pundavela J, Oliveira SM, Roselli S, Walker MM, Hondermarck H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer research 2015; 75: 1777–1781. [DOI] [PubMed] [Google Scholar]
- 11.Ding Y, He D, Florentin D, Frolov A, Hilsenbeck S, Ittmann M et al. Semaphorin 4F as a critical regulator of neuroepithelial interactions and a biomarker of aggressive prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2013; 19: 6101–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binmadi NO, Yang YH, Zhou H, Proia P, Lin YL, De Paula AM et al. Plexin-B1 and semaphorin 4D cooperate to promote perineural invasion in a RhoA/ROK-dependent manner. The American journal of pathology 2012; 180: 1232–1242. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nat Rev Mol Cell Biol 2005; 6: 789–800. [DOI] [PubMed] [Google Scholar]
- 14.Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Advanced drug delivery reviews (Research Support, N.I.H., Extramural Review) 2008; 60: 1527–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annual review of neuroscience 1999; 22: 197–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.True L, Coleman I, Hawley S, Huang CY, Gifford D, Coleman R et al. A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 2006; 103: 10991–10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu JB, Shao C, Li X, Li Q, Hu P, Shi C et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J Clin Invest 2014; 124: 2891–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu JB, Yin L, Shi C, Li Q, Duan P, Huang JM et al. MAOA-Dependent Activation of Shh-IL6-RANKL Signaling Network Promotes Prostate Cancer Metastasis by Engaging Tumor-Stromal Cell Interactions. Cancer cell 2017; 31: 368–382. [DOI] [PubMed] [Google Scholar]
- 19.Flamand V, Zhao H, Peehl DM. Targeting monoamine oxidase A in advanced prostate cancer. Journal of cancer research and clinical oncology (Research Support, N.I.H., Extramural) 2010; 136: 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westerink RH, Ewing AG. The PC12 cell as model for neurosecretion. Acta Physiol (Oxf) 2008; 192: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen W, Mi R, Haughey N, Oz M, Hoke A. Immortalization and characterization of a nociceptive dorsal root ganglion sensory neuronal line. J Peripher Nerv Syst 2007; 12: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saloman JL, Albers KM, Rhim AD, Davis BM. Can Stopping Nerves, Stop Cancer? Trends Neurosci 2016; 39: 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pundavela J, Demont Y, Jobling P, Lincz LF, Roselli S, Thorne RF et al. ProNGF correlates with Gleason score and is a potential driver of nerve infiltration in prostate cancer. The American journal of pathology 2014; 184: 3156–3162. [DOI] [PubMed] [Google Scholar]
- 24.Herman JG, Meadows GG. Increased class 3 semaphorin expression modulates the invasive and adhesive properties of prostate cancer cells. Int J Oncol 2007; 30: 1231–1238. [PubMed] [Google Scholar]
- 25.Tam KJ, Hui DHF, Lee WW, Dong M, Tombe T, Jiao IZF et al. Semaphorin 3 C drives epithelial-to-mesenchymal transition, invasiveness, and stem-like characteristics in prostate cells. Sci Rep 2017; 7: 11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tam KJ, Dalal K, Hsing M, Cheng CW, Khosravi S, Yenki P et al. Androgen receptor transcriptionally regulates semaphorin 3C in a GATA2-dependent manner. Oncotarget 2017; 8: 9617–9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peacock JW, Takeuchi A, Hayashi N, Liu L, Tam KJ, Al Nakouzi N et al. SEMA3C drives cancer growth by transactivating multiple receptor tyrosine kinases via Plexin B1. EMBO Mol Med 2018; 10: 219–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He S, Chen CH, Chernichenko N, He S, Bakst RL, Barajas F et al. GFRalpha1 released by nerves enhances cancer cell perineural invasion through GDNF-RET signaling. Proceedings of the National Academy of Sciences of the United States of America 2014; 111: E2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swiercz JM, Kuner R, Offermanns S. Plexin-B1/RhoGEF-mediated RhoA activation involves the receptor tyrosine kinase ErbB-2. J Cell Biol 2004; 165: 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D et al. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nature cell biology 2002; 4: 720–724. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–939. [DOI] [PubMed] [Google Scholar]
- 32.Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VM. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci 1987; 7: 3489–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ et al. Autonomic nerve development contributes to prostate cancer progression. Science 2013; 341: 1236361. [DOI] [PubMed] [Google Scholar]
- 34.Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun 2016; 7: 10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chilton JK. Molecular mechanisms of axon guidance. Developmental biology 2006; 292: 13–24. [DOI] [PubMed] [Google Scholar]
- 36.Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2008; 14: 7593–7603. [DOI] [PubMed] [Google Scholar]
- 37.Chauvet S, Cohen S, Yoshida Y, Fekrane L, Livet J, Gayet O et al. Gating of Sema3E/PlexinD1 signaling by neuropilin-1 switches axonal repulsion to attraction during brain development. Neuron 2007; 56: 807–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Pu T, Yin L, Li Q, Liao CP, Wu BJ. MAOA-mediated reprogramming of stromal fibroblasts promotes prostate tumorigenesis and cancer stemness. Oncogene 2020. [DOI] [PubMed] [Google Scholar]
- 39.Parra LM, Zou Y. Sonic hedgehog induces response of commissural axons to Semaphorin repulsion during midline crossing. Nat Neurosci 2010; 13: 29–35. [DOI] [PubMed] [Google Scholar]
- 40.Khatiwada P, Kannan A, Malla M, Dreier M, Shemshedini L. Androgen up-regulation of Twist1 gene expression is mediated by ETV1. PeerJ 2020; 8: e8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eide T, Ramberg H, Glackin C, Tindall D, Tasken KA. TWIST1, A novel androgen-regulated gene, is a target for NKX3–1 in prostate cancer cells. Cancer Cell Int 2013; 13: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaur S, Gross ME, Liao CP, Qian B, Shih JC. Effect of Monoamine oxidase A (MAOA) inhibitors on androgen-sensitive and castration-resistant prostate cancer cells. The Prostate 2019; 79: 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nature reviews Cancer 2012; 12: 89–103. [DOI] [PubMed] [Google Scholar]
- 44.Knudsen BS, Gmyrek GA, Inra J, Scherr DS, Vaughan ED, Nanus DM et al. High expression of the Met receptor in prostate cancer metastasis to bone. Urology 2002; 60: 1113–1117. [DOI] [PubMed] [Google Scholar]
- 45.Ko KR, Lee J, Lee D, Nho B, Kim S. Hepatocyte Growth Factor (HGF) Promotes Peripheral Nerve Regeneration by Activating Repair Schwann Cells. Sci Rep 2018; 8: 8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong V, Glass DJ, Arriaga R, Yancopoulos GD, Lindsay RM, Conn G. Hepatocyte growth factor promotes motor neuron survival and synergizes with ciliary neurotrophic factor. The Journal of biological chemistry 1997; 272: 5187–5191. [DOI] [PubMed] [Google Scholar]
- 47.Huyett P, Gilbert M, Liu L, Ferris RL, Kim S. A Model for Perineural Invasion in Head and Neck Squamous Cell Carcinoma. J Vis Exp 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.