

Spinocerebellar Ataxia type 2 (SCA2) is an autosomal dominant ataxia with no effective treatments. It is the second most prevalent form of dominant ataxia, accounting for approximately 15% of cases worldwide1, 2. Clinical features of SCA2 include gait ataxia, tremor, abnormal eye movements, peripheral neuropathy, and other multisystem features. SCA2 neuropathology is characterized by olivopontocerebellar atrophy with marked loss of Purkinje cells, and inferior olivary, pontocerebellar, and substantia nigra neuron loss3–6.
SCA2 belongs to a growing class of nucleotide repeat expansion disorders arising from unstable repeats in the genome7, 8. In this issue of Movement Disorders, Pan P. Li et al. add to an expanding body of literature indicating that repeat expansion disorders are often characterized by plural molecular pathogenic mechanisms9.
SCA2 is caused by expansion of CAG trinucleotide repeats in the first exon of the ATXN2 gene, encoding a polyglutamine (polyQ) repeat containing protein. In individuals with SCA2, repeat numbers are in the range of 37–39 repeats, as opposed to a normal range of 13–23 repeats10. Intermediate repeat lengths of 27–33 CAG repeats are associated with increased risk for Amyotrophic Lateral Sclerosis (ALS)11. The exact function of ATXN2 protein is not yet known, but it is implicated in RNA processing and translation, stress granule assembly, endoplasmic reticular (ER) calcium response, and cytoskeletal reorganization12. The expanded CAG repeat in ATXN2 encodes an abnormal stretch of polyglutamine (polyQ) in the N-terminal region. In multiple polyQ disorders, these canonically translated repeat harboring proteins show gain of function toxicities separate from their normal cellular functions13. These pathogenic protein species may exert their effects via multiple proximate mechanisms14. In the case of the polyQ disorder Huntington disease (HD), haploinsufficiency may also contribute to neurodegeneration7.
A second general molecular mechanism in nucleotide expansion disorders is the phenomenon of repeat-associated Non-AUG (RAN) translation, which gives rise to toxic repeat peptides due to aberrant translation from expanded repeat regions. RAN translation, however, from the expanded SCA2 transcript appears to be minimal and may not be a significant pathogenic process in SCA215.
Pan P. Li et al. evaluated a third potential pathogenic molecular mechanism involving repeat containing RNA transcripts. In some nucleotide expansion disorders, such as Type 1 Myotonic Dystrophy, expanded repeats exert neurotoxicity at the RNA level by disrupting mRNA processing. One of the mechanisms of RNA mediated toxicity is sequestration of key RNA binding proteins (RBPs), making them unavailable for their normal cellular functions16. RNA toxicity is often accompanied by formation of RNA foci. These authors find that expanded sense ATXN2 RNA transcripts cause neurotoxicity and disrupt ribosomal RNA processing. They present evidence that overexpression of an expanded ATXN2 transcript, not undergoing RAN translation, shows increased Caspase 3/7 activity in human neuroblastoma cells, and shows increased toxicity in nuclear condensation assays performed with primary mouse cortical neurons. The authors go on to show that the mutant transcripts bearing either 58 or 104 CAG triplets form more RNA foci when transfected into cells. RNA foci were also detected in cerebellar Purkinje cells of SCA2 transgenic mice and in one out of five postmortem human patient brains.
To examine the possibility of RBP sequestration, the authors performed in vitro pull down of the mutant ATXN2 transcript followed by Mass Spectrometry. They identified a number of RBPs with a predominant nuclear and nucleolar localization pattern, suggesting the nucleus as the possible site for aberrant interactions. Several of the sequestered RBPs are critical for maturation of the small rRNA subunit. The authors selected TBL3 (transducing β-like protein 3) for further analysis due to its known interaction with the expanded Huntingtin transcript, implicating a possible common disease mechanism in HD and SCA2. With a series of elegant biochemical studies, the authors demonstrate that TBL3 binds to the aberrant hairpin structure formed by continuous CAG repeats in the expanded repeat ATXN2 transcript.
The yeast homolog of TBL3 plays a role in 35S rRNA processing and 18S rRNA biogenesis17. Based on these findings, Pan P. Li et al tested the levels of 45S pre-rRNA (human equivalent of yeast 35S pre-rRNA) and the ratios of mature 18S and 28S rRNA after TBL3 knockdown in HEK293T cells. They observed an increase in 45S pre-rRNA levels and a decrease in the ratio of mature 18S and 28S rRNA, suggesting defective rRNA processing and maturation, respectively, in the absence of TBL3. Similar trends, albeit without statistical significance, were observed in postmortem HD and SCA2 brains. Defects in ribosome biogenesis are reported in HD, accompanied by nucleolar aggregates18, 19. This study opens up lines of investigation into the possible role of ribosomal abnormalities in another neurodegenerative disorder20. Taken together, the report by Pan Li. P et al., along with their earlier finding of a toxic antisense ATXN2 transcript, presents evidence for combinatorial protein-RNA driven pathology in SCA2 (Fig.1).
Figure 1. Multiple modes of SCA2 pathogenesis.
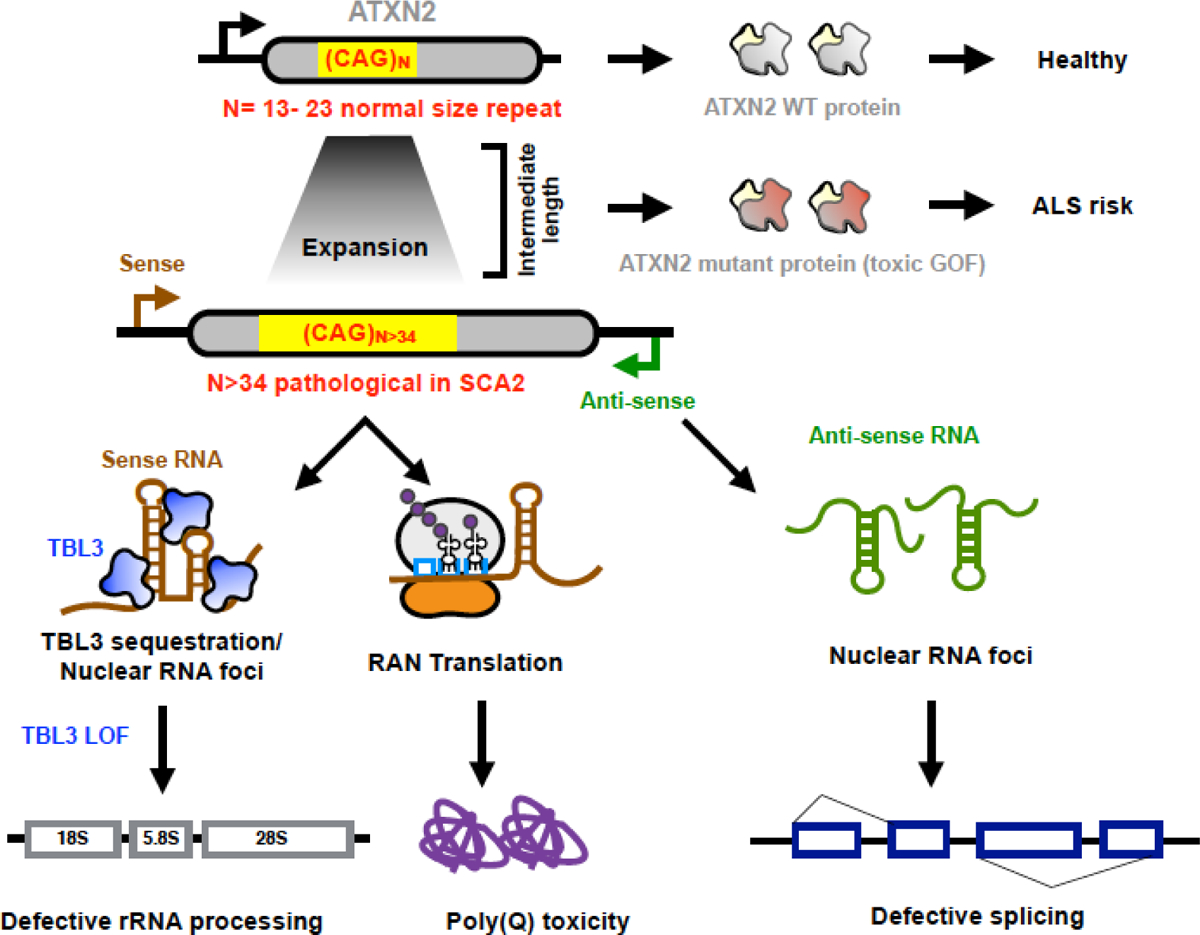
Normal CAG repeat number in the ATXN2 gene generates a WT protein that participates in a host of cellular functions. The intermediate repeat length is associated with ALS while the expanded CAG repeats in the ATXN2 gene causes toxicity by RNA binding protein sequestration, Repeat-associated Non-AUG translation or via the ATXN2- Antisense (AS) transcript. Each of these aberrant processes cause dysregulation of downstream molecular pathways and could collectively contribute to neurodegeneration.
Neurodegenerative disorders such as SCA2 typically exhibit age-related penetrance. The observation that ribosome biogenesis may be affected in SCA2 is intriguing and with implications for what might happen in aging brains, characterized by decreased global protein translation21, 22. Many age-associated neurodegenerative disorders are due to dysfunctions in core components of the translation machinery23. Ribosomal proteins, as well as rRNA levels, are affected during healthy aging24 and could conceivably change ribosomal assembly as well as mRNA translation dynamics. These age-associated changes could influence ribosomal and proteasomal subunit stoichiometry, altering global protein homeostasis25. These age-related changes might make neurons more susceptible to the toxic effects of expanded repeat transcripts.
Another characteristic aspect of neurodegenerative disorders, such as SCA2, is degeneration of specific neuronal subtypes. Single-cell transcriptomic analyses of the aging brain are beginning to unravel tissue and cell type specificities for components of the translation machinery, such as ribosomal proteins26, 27, suggesting distinct post-transcriptional outcomes based on neuronal cell type. This might explain the susceptibility of neuronal subtypes in specific disease contexts. In addition, high-energy neuronal subtypes, such as dopaminergic neurons, are particularly susceptible to mitochondrial dysfunction, an emerging factor in multiple neurodegenerative disorders.28, 29
Moreover, the most affected transcripts in the aging human cortex are involved in synaptic function30. Since activity-mediated dynamic regulation of mRNA translation at synapses is a key factor for neuronal function and survival31, sequestration of crucial molecules, such as TBL3 as reported here, could be particularly impactful for synaptic mRNA translation. Ultimately leading to neuronal death, the initial manifestations would be synaptic dysfunction and degeneration, which are widely believed to be common features of many neurodegenerative disorders.
Within the context of an aging neuronal environment, the accumulation of expanded repeat containing RNA and proteins could trigger cascades of events that affect both arms of the peptide life cycle - synthesis and degradation - precipitating neurodegeneration32.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr. Indranil Malik for assistance with the figure and Dr. Peter Todd for helpful suggestions. Dr. Geena Skariah receives support from the Claude D. Pepper Older Adults Independence Center (AG024824), Michigan Alzheimer Disease Center (AG053760) and NIH grants to Dr. Todd (R01NS086810, R01NS099280 and P50HD104463). Dr. Albin receives support from P50NS123067 and the Parkinson’s Foundation.
REFERENCES:
- 1.Pulst S-M, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nature Genetics 1996;14:269–276. [DOI] [PubMed] [Google Scholar]
- 2.Imbert G, Saudou F, Yvert G, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nature Genetics 1996;14:285–291. [DOI] [PubMed] [Google Scholar]
- 3.Filla A, De Michele G, Santoro L, et al. Spinocerebellar ataxia type 2 in southern Italy: a clinical and molecular study of 30 families. J Neurol 1999;246:467–471. [DOI] [PubMed] [Google Scholar]
- 4.Velázquez-Pérez L, Rodríguez-Labrada R, García-Rodríguez JC, Almaguer-Mederos LE, Cruz-Mariño T, Laffita-Mesa JM. A comprehensive review of spinocerebellar ataxia type 2 in Cuba. Cerebellum 2011;10:184–198. [DOI] [PubMed] [Google Scholar]
- 5.Estrada R, Galarraga J, Orozco G, Nodarse A, Auburger G. Spinocerebellar ataxia 2 (SCA2): morphometric analyses in 11 autopsies. Acta Neuropathologica 1999;97:306–310. [DOI] [PubMed] [Google Scholar]
- 6.Schöls L, Reimold M, Seidel K, et al. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain 2015;138:3316–3326. [DOI] [PubMed] [Google Scholar]
- 7.Lieberman AP, Shakkottai VG, Albin RL. Polyglutamine Repeats in Neurodegenerative Diseases. Annu Rev Pathol 2019;14:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Eyk CL, Richards RI. Dynamic mutations: where are they now? Adv Exp Med Biol 2012;769:55–77. [PubMed] [Google Scholar]
- 9.Li PP, Moulick R, Feng H, et al. RNA Toxicity and Perturbation of rRNA Processing in Spinocerebellar Ataxia Type 2. Mov Disord 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antenora A, Rinaldi C, Roca A, et al. The Multiple Faces of Spinocerebellar Ataxia type 2. Ann Clin Transl Neurol 2017;4:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elden AC, Kim H-J, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magaña JJ, Velázquez-Pérez L, Cisneros B. Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol Neurobiol 2013;47:90–104. [DOI] [PubMed] [Google Scholar]
- 13.Malik I, Kelley CP, Wang ET, Todd PK. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jafar-Nejad P, Ward CS, Richman R, Orr HT, Zoghbi HY. Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci U S A 2011;108:2142–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scoles DR, Ho MH, Dansithong W, et al. Repeat Associated Non-AUG Translation (RAN Translation) Dependent on Sequence Downstream of the ATXN2 CAG Repeat. PLoS One 2015;10:e0128769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo j 2000;19:4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dragon F, Gallagher JE, Compagnone-Post PA, et al. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 2002;417:967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsoi H, Chan HY. Roles of the nucleolus in the CAG RNA-mediated toxicity. Biochim Biophys Acta 2014;1842:779–784. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Hwang YJ, Ryu H, Kowall NW, Ryu H. Nucleolar dysfunction in Huntington’s disease. Biochim Biophys Acta 2014;1842:785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hetman M, Slomnicki LP. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J Neurochem 2019;148:325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ageing Tavernarakis N. and the regulation of protein synthesis: a balancing act? Trends Cell Biol 2008;18:228–235. [DOI] [PubMed] [Google Scholar]
- 22.Rattan SI. Synthesis, modifications, and turnover of proteins during aging. Exp Gerontol 1996;31:33–47. [DOI] [PubMed] [Google Scholar]
- 23.Kapur M, Ackerman SL. mRNA Translation Gone Awry: Translation Fidelity and Neurological Disease. Trends Genet 2018;34:218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 2007;3:e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelmer Sacramento E, Kirkpatrick JM, Mazzetto M, et al. Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation. Mol Syst Biol 2020;16:e9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davie K, Janssens J, Koldere D, et al. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 2018;174:982–998.e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ximerakis M, Lipnick SL, Innes BT, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 2019;22:1696–1708. [DOI] [PubMed] [Google Scholar]
- 28.Müller-Nedebock AC, van der Westhuizen FH, Kõks S, Bardien S. Nuclear Genes Associated with Mitochondrial DNA Processes as Contributors to Parkinson’s Disease Risk. Mov Disord 2021;36:815–831. [DOI] [PubMed] [Google Scholar]
- 29.Norat P, Soldozy S, Sokolowski JD, et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. npj Regenerative Medicine 2020;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dillman AA, Majounie E, Ding J, et al. Transcriptomic profiling of the human brain reveals that altered synaptic gene expression is associated with chronological aging. Sci Rep 2017;7:16890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapur M, Monaghan CE, Ackerman SL. Regulation of mRNA Translation in Neurons-A Matter of Life and Death. Neuron 2017;96:616–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skariah G, Todd PK. Translational control in aging and neurodegeneration. Wiley Interdiscip Rev RNA 2021;12:e1628. [DOI] [PMC free article] [PubMed] [Google Scholar]